

Abstract
The microglia, once thought only to be supporting cells of the central nervous system (CNS), are now recognized to play essential roles in many pathologies. Many studies within the last decades indicated that the neuro-immune interaction underlies the generation and maintenance of neuropathic pain. Through a large number of receptors and signaling pathways, the microglial cells communicate with neurons, astrocytes and other cells, including those of the immune system. A disturbance or loss of CNS homeostasis causes rapid responses of the microglia, which undergo a multistage activation process. The activated microglia change their cell shapes and gene expression profiles, which induce proliferation, migration, and the production of pro- or antinociceptive factors. The cells release a large number of mediators that can act in a manner detrimental or beneficial to the surrounding cells and can indirectly alter the nociceptive signals. This review discusses the most important microglial intracellular signaling cascades (MAPKs, NF-κB, JAK/STAT, PI3K/Akt) that are essential for neuropathic pain development and maintenance. Our objective was to identify new molecular targets that may result in the development of powerful tools to control the signaling associated with neuropathic pain.
Keywords: Microglia, NF-κB, MAPK, ERK1/2, p38, STAT, PI3K/Akt, neuropathic pain
NEUROPATHIC PAIN
According to the International Association for the Study of Pain (IASP) taxonomy, pain is “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage”. Pain plays protective and adaptive roles in the nervous system by alerting an individual to a possible injury [1]. Under healthy conditions, pain occurs in response to the activation of nociceptors, a specific high-threshold subset of the peripheral sensory neurons that conducts action potentials from the injured tissue to the central nervous system (CNS). The functional constituents of the pain pathway work together to protect the organism. However, long-lasting pain may lead to substantial suffering. Damage or diseases of the nervous system may lead to intense pain signals that persist for weeks or even years. In this pathological situation, pain loses its protective function and becomes a disease per se. The problem of chronic pain has a social dimension, by virtue of its contributions to a lower quality of life and the increased cost of social and medical care. The statistical surveys conducted by the IASP and its European department (EFIC) show that one in five Europeans suffers from a chronic pain condition, and this state makes it impossible for them to maintain an independent lifestyle. Chronic pain may be associated with tissue inflammation, which reduces the nociceptive threshold (inflammatory pain), but may also result from trauma, cancer, or ischemic or metabolic disorders of the peripheral or central nervous system (neuropathic pain) [2-5]. The main symptom of neuropathic pain is a long-lasting abnormal sensitivity that can spontaneously be manifested to normally innocuous stimuli; this disorder is known as allodynia. In addition, an exaggerated response to a noxious stimulus is observed during neuropathy: this is called hyperalgesia or hyperpathia.
An important advance in the study of neuropathic pain was the concept that mechanism-based rather than etiology-based approaches should be utilized to classify the neuropathic pain states. However, the mechanisms underlying these pathological conditions are complicated. One of the consequences of nerve damage is ineffective, unregulated nerve regeneration resulting in a tangled mass of nerve fibers, which is called a neuroma. Changes in the electrophysiological properties of the primary afferents are observed during neuropathy. These changes include spontaneous activity, a lowered excitability threshold, and an ongoing increase in the sensitivity to chemical, thermal or mechanical stimuli at the site of the neuroma, as well as in the cell soma of the dorsal root ganglion (DRG) neurons and the surrounding, uninjured afferents [6-8]. Recent studies have demonstrated considerable progress in understanding the molecular mechanisms that underlie nociceptor sensitization. Many researches focused on understanding the changes in the nociceptors’ activity under pathological conditions. These studies are concentrated on the roles of G protein-coupled receptors (GPCRs), ligand-gated ion channels, and tyrosine kinases receptors [9-12]. Many nociceptive mediators, e.g., bradykinin, serotonin, prostaglandins, opioids, glutamate, and chemokines, act via GPCRs, which in turn depend on the type of G protein activation and lead to the production of second messengers (cAMP, cGMP, diacylglycerol and phospholipase C). Another group of inflammatory factors acts by directly gating the ion channels, which depolarize the sensory neurons when they are open and lead to neuronal firing. The third class includes the cytokine receptors, including those for interleukins (ILs), interferons (IFNs), and tumor necrosis factor α (TNFα), and various tyrosine kinase receptors for neurotrophic factors, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and glial cell line-derived neurotrophic factor (GDNF). The activation of these receptors leads to the mobilization of intracellular cascades of events that affect the gene transcription and may modulate the function of the sensory neurons. Currently, many studies are attempting to define the roles of these intracellular pathways, which may be involved in the development and maintenance of neuropathic pain.
A prolonged afferent influx of nociceptive information may result in the spatial and temporal summation of the impulses, which can lead to excessive depolarization and the expansion of the neuronal receptive fields. The changes induced by peripheral nerve injury include both the up-regulation and down-regulation of the expression of various genes in the DRG. These changes may contribute to the hypersensitivity of the sensory neurons [13-16]. The development of neuropathic pain is also accompanied by an inflammatory component. Inflammatory mediators such as pro-inflammatory cytokines (IL-1β, TNFα), prostaglandins, nitric oxide, and nerve growth factor are released at the site of the nerve injury. These factors are involved in the generation of spontaneous activity and may potentiate pain hypersensitivity. The activity of the primary afferents is believed to drive the plasticity of the nociceptive neurons at the CNS level. This process is known as central sensitization [17-19]. The mechanisms underlying these phenomena include enhanced excitatory and decreased inhibitory synaptic transmission.
The ROLE OF GLIAL CELLS
For many years, most investigators focused on the roles of neurons in neuropathic pain without considering the contributions of other cells. Synaptic plasticity, which is known to be one of the neuronal mechanisms participating in neuropathic pain development, contribute to potentiated sensory responses after injury [20]. It is believed that long-term changes in synaptic plasticity within nociceptive pathway (from peripheral nociceptors, as well as spinal and supraspinal levels) leads to sensitization during neuropathic pain [20, 21]. The altered synaptic connectivity can be strongly modulated by both the classical neuron-derived neurotransmitters and by immune mediators released from the CNS-resident microglia and astrocytes as well as from infiltrating cells, e.g., macrophages (Fig. 1). A recent study indicated that the neuro-immune interaction in the central nervous system is an essential phenomenon that underlies the pathology of neuropathy [14-16,22-38]. Increasing evidence indicates that the numerous immunocompetent cells in the CNS, especially glia, are not silent components of the nervous system but instead play important roles in the modulation of neurotransmission. It is well documented that glial cells actively communicate with the neurons through direct junctions [39-41], synapses [42-45] and by the release of neuromodulatory chemicals, all of which may influence neuronal firing and intracellular signaling [46-48]. The role of glia was first noticed when increased levels of glial fibrillary acidic protein (GFAP; an astrocyte marker) and integrin αM (OX-42; a microglia marker) were observed in the spinal cords of rats under
Fig. (1).
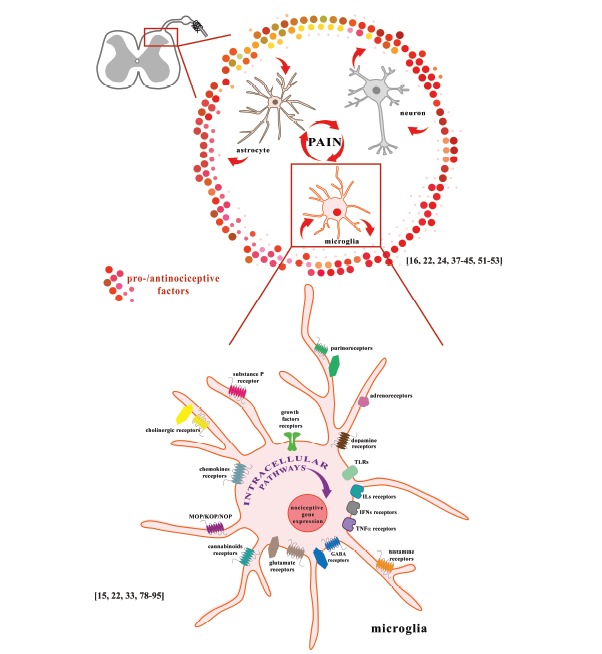
A. The interactions of neurons, astrocytes and microglia at the spinal cord level participate in the development and maintenance of neuropathic pain. B. Activation of microglial receptors leads to activation of numerous intracellular cascades. The consequence of the activation of these cells is the production of pro- and anti-nociceptive factors that are important for pain development. Microglial cells express a wide spectrum of neurotransmitter receptors: glutamate receptors (NMDA, AMPA, mGluRI, mGluRII, and mGluRIII), GABA receptors (GABAA and GABAB), cholinergic receptors (α7nAChR, M3), adrenoreceptors (α1A, α2A, β1, β2), dopamine receptors (D1-5), and purinoceptors (A1, A2A, A2B, A3, P2X1, P2X4, P2X7, P2Y2, P2Y6, P2Y12, P2Y13); receptors for hormones and modulators: histamine (H1, H2, H3, H4), opioids (MOP, KOP, NOP), cannabinoids (CB1, CB2), substance P (NK-1), neurotrophins (TrkB, TrkC), chemokines (CCR1-7, CXCR1-5, CX3CR1), interleukins (IL-1R1/IL-1R2, IL-2R, IL-4R, IL-5R, IL-6R, IL-8R, IL-9R, IL-10R, IL-12R, IL-13R, IL-15R, IL-18R), interferons (IFNγR, IFNAR type I), and TNFα (TNFR1, TNFR2); and pathogen-associated microbial patterns (TLR1-9).
neuropathic conditions [49,50]. A gene expression analysis revealed that neuropathic pain is not only correlated with the activation of neurons but also with the responses from immune cells, including microglial activation [16,22,24,37,38,51-53]. It was documented that activation of spinal microglia contribute to the induction of long-term potentiation (LTP). In this phenomena an activation of microglial ATP receptors (P2X4 or P2X7) and p38 MAPK phosphorylation are involved [54,55].
A morphological changes such as somatic hypertrophy, proliferation, thickened branches, are observed after spinal cord injury (SCI). Those features are correlated with behavioral, electrophysiological and molecular changes within the altered somatosensory system [56]. The patterns of activation of glial cells during neuropathy is quite different. The peak activation of the spinal microglia occurs in the first few days following injury [52,57] and persists from 3 days to 2 weeks before subsequently decreasing, although their activity remains elevated [2,58-61]. In contrast, the spinal activation of the astrocytes is maintained for longer periods [60,61]. Gwak et al. [56] observed activation of microglial and astrocytic cells at the early (2 hour to a few days) and late (over 3 months) stage of neuropathic pain within along the entire spinal axis after spinal cord injury. In a previous study, it was demonstrated that the number of activated microglia is significantly higher than the number of activated astrocytes 7 days after sciatic nerve injury [52].
Although dysfunction of microglia and astrocyte has been linked with neuropathic pain, very little is known about the role of other glial cells, oligodendrocytes, in modulation of nociceptive transmission. In 2014, Gritsch et al. reported that genetic ablation of oligodendrocytes was not associated with spinal microglia of astrocyte activation but leads to pathology of axons in the spinal dorsal horn and spinothalamic tract [62]. Those results suggest that oligodendrocyte functions during neuropathic pain is independent of immune contributions. However, the report published in 2016 revealed that oligodendrocyte are an important participant in neuro-immune interaction during neuropathic pain [63]. As observed by the authors oligodendrocytes are the main source of IL-33, pro-nociceptive interleukin which is the latest member of the IL-1 family of cytokines. IL-33 exerts its biological effects by binding to its receptor ST2, which in CNS is mainly expressed on astrocytes and neurons [64]. IL-33/ST2 triggers activation of intracellular signaling such as PI3K, MAPKs, and NF-κB, resulting in the production of TNFα and IL-1β, which potentiate nociceptive transmission [63].
Based on these results, investigators suggested that the microglia are more crucial at the early stages of neuropathic pain, while astrocytes play a role in persistent pain [57,65,66]. Astrocytes represent the most abundant cell population in the nervous system and are critical in maintaining the homeostasis of their surrounding environment by regulating the concentrations of the neurotransmitters, ions and proteins in the synaptic cleft. Activated astrocytes are an important source of inflammatory factors, such as nitric oxide [67], prostaglandins [68,69], excitatory amino acids [70], cytokines [71,72] and ATP [73]. The primary participant in pain development is the microglial cells. A microarray study has shown that most of the regulated genes following a nerve injury are expressed in the spine [16,74]. The microglia represent 5-20% of all glial cells under normal physiological conditions, but this situation is dramatically changed after nerve injury. Pathological conditions, such as neuropathic pain states, evoke rapid and profound changes in the morphology, proliferative potential, gene expression and function of the microglial cells. The morphological changes are manifested as an altered cell shape (the microglia revert to an amoeboid appearance) as well as the appearance and rearrangement of surface molecules [16,29,52,75-78]. The microglia respond to various pain-related neuromodulators, but are also capable of directly altering the pain pathways because they express specific receptors that play roles in pain sensitization. Microglial cells express a wide spectrum of receptors for neurotransmitters, including metabotropic receptors, which belong to GPCR class and ionotropic receptors. The most important neurotransmitter receptors are the glutamate receptors (NMDA, AMPA, mGluRI, mGluRII, and mGlu-RIII), GABA receptors (GABAA and GABAB), cholinergic receptors (α7nAChR, M3), adrenoreceptors (α1A, α2A, β1, β2), dopamine receptors (D1-5), and purinoceptors (A1, A2A, A2B, A3, P2X1, P2X4, P2X7, P2Y2, P2Y6, P2Y12, P2Y13) [33,79-89].
Another crucial group includes the microglial receptors for hormones and modulators, including the receptors for histamine (H1, H2, H3, H4), opioids (MOP, KOP, NOP), cannabinoids (CB1, CB2), substance P (NK-1), neurotrophins (TrkB, TrkC), chemokines (CCR1-7, CXCR1-5, CX3CR1), interleukins (IL-1R1/IL-1R2, IL-2R, IL-4R, IL-5R, IL-6R, IL-8R, IL-9R, IL-10R, IL-12R, IL-13R, IL-15R, IL-18R), interferons (IFNγR, IFNAR type I), TNFα (TNFR1, TNFR2), and pathogen-associated microbial patterns (Toll-like receptor 1-9 (TLR1-9)) [15, 22, 78, 81, 89-92]. Some of these are mainly (e.g., TLR2, TLR4, P2X4) or exclusively (e.g., CX3CR1) expressed by microglia [93,94]. The CX3CL1 that is produced by neurons influences the microglia through the activation of CX3CR1. This process seems to play an essential role in the microglial-neuronal cross-talk in neuropathic pain [95].
Activation of the microglial receptors leads to cellular signal transduction through many intracellular pathways, e.g., MAPKs, NF-κB, STATs, PI3K. Recent studies have revealed that targeting those cascades is an effective way to diminish neuropathic pain states. Moreover, inhibition of some microglial pathways up-regulates the effectiveness of analgesics, which are already in clinical use [15,16,22-24].
Additional elements of the activation process include the production of inflammatory mediators by the microglia [16,24,29,77,90,96-101]. During development, the microglial cells are derived from primitive myeloid progenitors in the yolk sac, and the microglia represent the resident tissue macrophages in the CNS [102]. After CNS injury, the activated microglia and macrophages cannot be distinguished by their morphology or antigenic markers and are generally designated macrophages/microglia. Many lines of evidence have revealed the phenotype-specific roles of macrophages. Depending on the local tissue milieu, macrophages undergo specific differentiation into classical (M1) or alternative (M2) polarization states [103-105]. However, the limited knowledge about microglial cell polarization is the result of a small number of recent studies concerning stroke, depression, neuroinflammation or Parkinson’s disease [77,90,106,107]. By analogy to that of the peripheral macrophages, microglial cell polarization is categorized into the classical activation state (M1), which exhibits harmful properties, and an alternative activation state (M2), which demonstrates protective and reparative functions. Depending on the environmental influences, the microglia shift their functions to maintain tissue homeostasis (Fig. 2). In vitro studies have revealed that stimulation with harmful factors (e.g., lipopolysaccharide (LPS)) moves the cells toward the M1 phenotype and induces the expression of proinflammatory mediators with pro-nociceptive properties. These mediators include IL-1𝛽, IL-6, IL-12, IL-15, IL-18, IFN-γ, TNFα, chemokine (C-C motif) ligands 2, 3, 4, 5, 7 (CCL2, CCL3, CCL4, CCL5, CCL7), cyclooxygenase 2 (COX-2) and inducible nitric oxide synthase (iNOS) [16, 24, 78, 90, 101, 108-112]. However, stimulation with anti-inflam-matory compounds, such as interleukins 4 and 13 (IL-4 and IL-13), promotes the M2 state, deactivates the pro-inflammatory cell phenotype, restores homeostasis and induces the increased expression of mediators with analgesic features (such as IL-1α, IL-1ra, IL-3, IL-10, IL-18BP, tissue inhibitors of metalloproteinases 1 (TIMP1), Arg1, NGF, TGFβ) [16, 24, 90, 109, 111, 113, 114]. The diverse forms of microglial cell activation and the associated inflammatory responses have been suggested to be new targets for the development of an effective therapy against the pathological state of neuropathic pain. However, the polarization process for microglial cells during neuropathic pain is still unclear. In recent published paper Xu et al. observed that activation of both M1 and M2 microglia-related genes occur 1 day after chronic constriction injury (CCI). However, only M1 markers remained elevated till day 7 and 14 after injury [115]. In our studies we demonstrated that the plant-derived compound, parthenolide, influences some intracellular pathways to shift the balance between the neuroprotective and neurotoxic microglial phenotypes at day 7 after CCI. This shift into M2 phase leads to the attenuation of neuropathic pain and improved morphine analgesia [23,24].
Fig. (2).
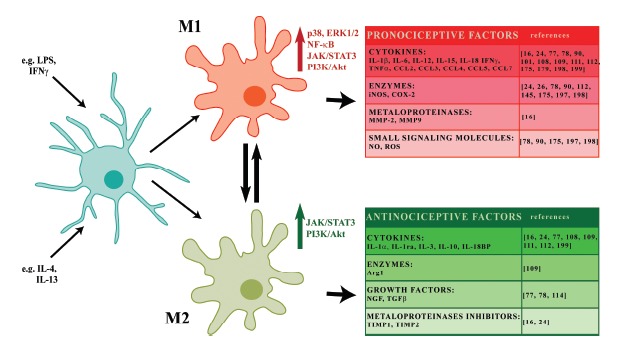
Polarization of microglia. Microglial cell polarization is categorized into the classical activation state (M1), which exhibits harmful properties, and an alternative activation state (M2), which demonstrates protective and reparative functions. Depending on the environmental influences, the microglia shift their functions to maintain tissue homeostasis. Stimulation with harmful factors (e.g. LPS or IFNγ) moves the cells toward the M1 phenotype and induces the expression of proinflammatory mediators with pro-nociceptive properties (IL-1β, IL-6, IL-12, IL-15, IL-18, CCL2, CCL3, CCL4, CCL5, CCL7, IFNγ, TNFα, iNOS, COX-2, MMP-2, MMP-9, NO, ROS). However, stimulation with anti-inflammatory compounds, such as IL-4 and IL-13, promotes the M2 state, deactivates the pro-inflammatory cell phenotype, restores homeostasis and induces the increased expression of mediators with analgesic features (such as IL-1α, IL-1ra, IL-3, IL-10, IL-18BP, TIMP1, Arg1, NGF, TGFβ. Microglial cells polarization is differentially activated by intracellular signaling cascades (MAPKs, NF-κB, JAK/STAT, PI3K/Akt) that are essential for neuropathic pain development and maintenance.
THE ROLE OF ESSENTIAL MICROGLIAL CELL SIGNALING IN THE MODULATION OF NEUROPATHIC PAIN PROCESSING
The nociceptive factors released by microglia during nerve injury may promote many of the behavioral states of neuropathic pain. Many studies over last years have revealed the importance of the intracellular neuronal pathways that are associated with neuropathic pain states. However, there is still a paucity of information regarding the participation of the microglial pathways in this pathology. The wide spectrum of receptors expressed by microglia allows those cells rapidly and effectively respond to microenvironmental changes. Activation of the microglia leads to the mobilization of numerous intracellular cascades. These cascades may participate in the alteration of the functions of these cells as well as those of neighboring cells. It is necessary to define how activation of microglial intracellular cascades work to program gene expression and how modulation of those pathways changes nociception and the action of analgesics. This will help us to understand the mechanisms underlying neuropathic pain and to generate new therapies.
MITOGEN-ACTIVATED PROTEIN KINASE (MAPK)
One of the essential participants in the intracellular signaling pathways that modulate the nociceptive response is the mitogen-activated protein kinase (MAPK) family. The MAPKs are evolutionarily conserved molecules that consists of three primary members: extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK). The MAPKs are crucial players in cell signaling and transmit a broad range of extracellular signals to mediate various intracellular responses. Recent progress in pain research has indicated that the MAPK pathway intermediates contribute to the development and maintenance of neuropathy. Early studies on the role of MAPK in pain had focused on the neuronal mechanisms of action [116-119]. However, several studies demonstrated profound MAPK activation in microglia [78,120-124] and astrocyte [61,122,123,125]. This process promotes intracellular events that contribute to the peripheral and central sensitization that is manifested at the behavioral and cellular levels [61,120,121,123-127] (Fig. 3).
Fig. (3).
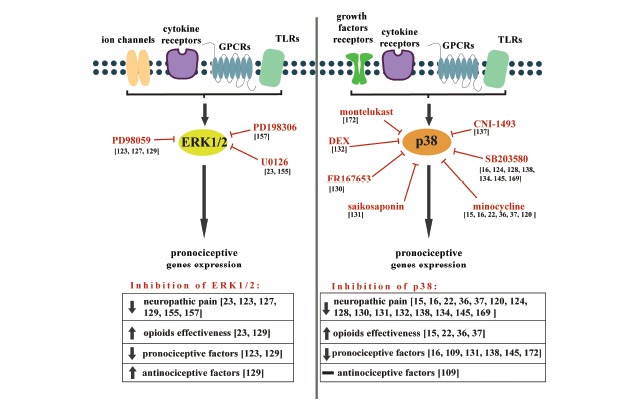
The effects of ERK1/2 and p38 inhibition on neuropathic pain development and opioid effectiveness, as well as the expression of pro- and anti-nociceptive factors.
Several studies have indicated that nerve injury is due to p38 kinase activation within the spinal cord [24,124,127-132]. Immunofluorescence studies have shown that the levels of p-p38 (phosphorylated p38) are particularly increased in the spinal microglia after nerve injury but are not increased in the neurons and astrocytes [124,127,128,130,132]. Importantly, the data obtained from animal studies, including those from our group, revealed that intrathecal injection of p38 inhibitors (SB203580 [124,128,133-135], FR167653 [136], CNI-1493 [137], skepinone [135] or minocycline [15,22,25,120,133,138]) prevent and/or reverse the neuropathic pain symptoms. These results confirm the role of the microglial p38 kinase in neuropathic pain sensitization. Interestingly, investigators demonstrated that the p38 inhibitors diminish the symptoms of neuropathic pain more effective when they are administered before the nerve injury [139,140].
An interesting report was published in 2015 by the Sorge’s group. The authors revealed that injections of minocycline or SB203580 had no significant effects on mechanical allodynia in females, contrary to male during neuropathy [141]. Those data suggest that there is sexual dimorphism in the microglia action in mechanical pain hypersensitivity. In 2015 another studies confirmed this interesting, however surprising, thesis. Taves et al. found that the sex difference is not based on microglial activation per se but rather on p38 signaling [135]. Authors shown that, intrathecal injection of an inhibitors of this kinase (skepinone and SB203580) reduced symptoms of neuropathy in male mice, while one years earlier Berta et al. revealed that both inhibitors diminished formalin-induced phase II in males, although microglia activation is not observed in this acute inflammatory pain model [142]. This new topic in the field of neuropathic pain research remain many questions and need to be analyzed in future studies.
The MAPKs are crucial players in cell signaling and transmit a broad range of extracellular signals to mediate various intracellular responses, which contribute to the development and maintenance of neuropathy. Inhibition of ERK1/2 kinase activation reduces symptoms of neuropathic pain, increases opioid effectiveness, diminishes pronociceptive and increases antinociceptive factors expression. Inhibition of p38 kinase activation reduce symptoms of neuropathic pain, increase opioid effectiveness, diminishes pronociceptive factors expression, however do not change antinociceptive factors expression.
The upstream mechanisms that induce p38 kinase activation have been explored [143-145]. It was shown that intrathecal administration of IL-1β resulted in an up-regulation of the spinal p-p38 levels [145]. Svensson’s group (2003) revealed that microglial p38 kinase activation is initiated by the spinal NMDA receptors and participates in hyperalgesia and prostaglandin release [146]. As described above, the microglia are the source of inflammatory factors, and, as reported by some studies, p38 kinase is involved in the synthesis of many of these factors via transcriptional regulation [16,121,127,138,145,147]. The activation of the microglial p38 following injury initiates a cascade of iNOS expression that causes NO-mediated degeneration of the neurons in the spinal cord [127]. It has been shown that in neuropathic pain conditions minocycline diminished the level of spinal pro-nociceptive factors such as IL-6, IL-18, and iNOS [16,26]. This effect was correlated with an inhibition of microglial cell activation and a reduction in the symptoms of neuropathy. However, cell culture studies showed that minocycline selectively inhibits the microglial polarization to a proinflammatory state [109]. Importantly, p38 kinase participates in the intracellular crosstalk and in the activation of various pathways. In particular, it has been implicated in the signaling by nuclear factor κB (NF-κB) in cultured microglia [109,148,149].
Other members of the MAPK family, ERK1/2, are also strongly activated within the glia during neuropathic pain. The ERK1/2 enzymes are phosphorylated by the upstream mitogen-activated protein kinase kinase (MEK1/2) in response to membrane depolarization, calcium influx, and NMDA receptor stimulation, as well as exposure to cytokines and LPS [78,116,150-152]. It was observed that a noxious electrical stimulus induced ERK phosphorylation, primarily within the lamina I and II of the ipsilateral dorsal horn of the spinal cord, which contributed to pain hypersensitivity [116]. Some studies have indicated that the action of the ERK signaling pathway involves the phosphorylation of the A-type potassium channel Kv4.2 [153] and tonic up-regulation of the voltage-dependent calcium channels in the rat sensory neurons [154]. However, ERK expression is not restricted to neural tissue. In 2005, Zhuang et al. showed that ERK is sequentially activated in both neuronal and glial cells after nerve injury, and this activation contributes to mechanical allodynia [123]. The authors observed immediate induction of ERK phosphorylation in neurons, while activation within microglia had a peak between day 1 and 3 after injury and in astrocytes predominantly at day 21. Spinal cord injury causes strong phosphorylation of ERK1/2 in the lesion area as soon as 6 hours after damage, and this is restricted to the microglial cells [127]. The activation of ERK signaling is also observed in the spinal cords of neuropathic rats 1 day after CCI [155]. In our experiments, we have shown that the ERK1/2 phosphorylation is increased on day 7 after CCI [24,129], and this is correlated with enhanced glial cell activation [24]. These results suggest that ERK is essential for the intracellular signaling that leads to the production of various nociceptive factors, including cytokines such as TNFα and IL-1β and the enzymes COX-2, iNOS, and nNOS, that participate in the intensification of the neuropathic pain sensations [24,127, 129,156]. Previous studies have demonstrated that the administration of MEK-ERK pathway inhibitors such as PD198306 [157], U0126 [23,155], and PD98059 [123,127,129] reduce the nociceptive behavior in animal models of neuropathic pain, such as CCI, SNL and SCI. These results suggest an important contribution of this signaling cascade to the perception of pain. Intrathecal, but not intraplantar, injection of PD198306 blocked static allodynia in the streptozocin, as well as CCI model 2 weeks after induction of neuropathy [157]. Using U0126, which was injected preemptively and then one daily for 3 days after injury, Han and colleagues (2011) revealed that ERK1/2 activation is essential to the initiation of CCI-induced pain hypersensitivity and the inhibition of this kinase significantly diminished the symptoms of neuropathy on the fourth day after the injury. In our studies, we demonstrated that intrathecal injection of U0126 [23], as well as another inhibitor PD98059 [129], adminis-trated preemptively and then once daily for 7 days not only relieves pain but also increase the effects of morphine. This effect is correlated with the up-regulation of the level of the opioid receptor mRNA at the spinal cord level. The pattern of ERK activation observed by Zhuang et al. revealed that this kinase is necessary for development and maintenance of neuropathic pain, while PD98059 injected intrathecally on day 2, 10 or 21 reduces SNL-induced mechanical allodynia, which suggest that both type of glial cells are affected.
The third member of MAPK family, c-Jun N-terminal kinase (JNK) also participates in the neuropathic pain pathology [61,129,158]. However, it is primarily expressed in astrocytes [61,125,158-160]. In 2006, Zhuang and collaborators demonstrated that a spinal injection of a JNK inhibitor, D-JNKI-1JNK, prevented mechanical allodynia in SNL-exposed animals, and this effect was correlated with astrocyte suppression [61]. The researchers further investigated how JNK regulates neuropathic pain. Using astrocytic cell cultures they revealed that TNFα activates pro-nociceptive JNK/MCP-1 (monocyte chemoattractant protein-1) cascade and it was dose dependently inhibited by the JNK inhibitors (SP600125 and D-JNKI-1). Spinal administration of TNFα produced JNK-dependent pain hypersensitivity and MCP-1 up-regulation, which was primarily induced in spinal cord astrocytes after injury. Activation of JNK within astrocyte during neuropathy was lately confirmed by Lu et al., authors revealed that up-regulation of spinal astrocyte in the late phase after injury was triggered by JNK/MCP-1 cascade activation within those cells [159]. However, recently published data revealed that after CCI of the median nerve profound activation of p-JNK is observed within microglia in cuneate nucleus as early as 1 day after injury with peak at day 7 [160].
NUCLEAR FACTOR KAPPA-LIGHT-CHAIN-ENHANCER OF ACTIVATED B CELLS (NF-κB)
The NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) is a pleiotropic transcription factor that is activated in the responses to a wide variety of neuroinflammation-associated stimuli. The NF-κB members exert effects in almost all cell types, where they play important roles in inflammation, immune responses, cell cycle and survival [161,162]. A growing number of reports reveals a crucial role for the NF-κB pathway in nociception [23,24,27,129,163-165]. The NF-κB family includes five members (p50, p52, p65 (Rel-A), c-Rel and Rel-B). However, within the nervous system, NF-κB is generally composed of a p50/p65 heterodimer [161,166,167]. All of these factors share a characteristic DNA-binding domain, the RHD (Rel homology domain), in their N-termini [168]. The activation of NF-κB is mainly mediated via two distinct kinase-dependent pathways, the classical (canonical) and the alternative (non-canonical). The best studied activation pathway of NF-κB within the nervous system is the canonical pathway, which is initiated through a variety of cell surface receptors, including cytokine receptors and TLRs (Fig. 4). Under normal conditions, the NF-κB dimers form complexes with members of a family of inhibitors (inhibitor of κB, IκB), which mask the nuclear localization signal of NF-κB and retain it in an inactive state within the cytoplasm. The phosphorylation of IκB by the IκB kinase triggers inhibitor degradation, releasing NF-κB and promoting its nuclear translocation and the modulation of gene expression. Although they have been the subjects of an increasing number of studies, the specific mechanisms by which the NF-κB pathway exerts its effects on nociception are not clear [27,163-165,169]. The expression of the genes that are regulated by NF-κB is known to increase following nerve injury and may contribute to the exacerbation of pain [24,163,165, 169,170]. Several studies have shown increased NF-κB activity within the DRG and spinal cords in various animal models of neuropathy [24,27,129,163,165, 171,172]. This pattern of activation indicates that NF-κB participates in the early stages of pain development as well as during prolonged spinal nociceptive signaling. In 2007, Meunier et al. showed that the selective inhibition of glial NF-κB results in diminished neuropathic symptoms and reduced expression of IL-6 and iNOS [163]. A few years later, Pan et al. (2010) revealed that NF-κB inhibition down-regulates the microglial activation and, in parallel, the CX3CR1 expression, and these effects were correlated with the elimination of the symptoms of neuropathy [170]. In vitro studies using primary cultures of microglial cells have shown that the inhibition of this pathway leads to reduced levels of the pro-nociceptive factors [78,173-175]. In the experiments performed by our group, a commercially available NF-κB inhibitor, parthenolide, significantly diminished allodynia and hyperalgesia and also potentiated the morphine analgesia [23,24]. Surprisingly, the parthenolide-induced analgesia was correlated with up-regulated activation of the spinal cord microglial cells in the CCI-exposed rats [24]. Additionally, we analyzed the changes in the M1 and M2 polarization factors of the spinal microglia. We observed that the parthenolide action was mediated by reduced levels of the M1 proteins (IL-1β, IL-18, and iNOS) and enhancement of the M2 factors (IL-10, TIMP1). Using primary microglial cell cultures, we have shown that both M1 and M2 factors are microglia-derived factors which might modulate nociception. In addition, parthenolide down-regulated the phosphorylated forms of the NF-κB, p38 MAPK, and ERK1/2 proteins, but, in contrast, up-regulated the signal transducer and activator of transcription 3 (STAT3). In summary, a growing body of evidence suggests that NF-κB inhibitors can directly or indirectly attenuate the symptoms of neuropathy and promote neuroprotective M2 polarization of the microglia/macrophages.
Fig. (4).
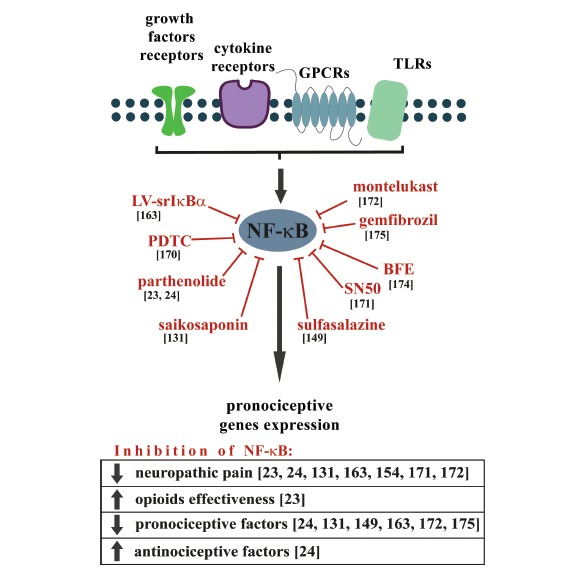
The effects of NF-κB inhibition on neuropathic pain development and opioid effectiveness, as well as the expression of pro- and anti-nociceptive factors. The NF-κB is a pleiotropic transcription factor that is activated in the responses to a wide variety of neuroinflammation-associated stimuli. The expression of the genes that are regulated by NF-κB is known to be increased following nerve injury and may contribute to the exacerbation of pain. Inhibition of NF-κB activation reduces symptoms of neuropathic pain, increases opioid effectiveness, diminishes pronociceptive and increases antinociceptive factors expression.
JANUS TYOSINE KINASE (JAK)/SIGNAL TRA-NSDUCER AND ACTIVATOR OF TRANSCRIPTION (STAT)
The Janus tyrosine kinase (JAK) and signal transducer and activator of transcription (STAT) pathway is one of the most important cascades for the cellular transduction of signals in response to many pain modulators including both the pro- (IL-6, IFN-γ) and antinociceptive (IL-10) factors. Binding of various cytokines to their cell surface receptors induces the dimerization and activation of the JAK proteins, which subsequently phosphorylate the STATs in the cytoplasm. Activated STATs form stable homo- and/or heterodimers and then are translocated into the nucleus where they function as transcription factors. The STAT proteins are implicated in the control of cell growth, differentiation and survival. Several lines of evidence have shown that one of the members of the STAT family, STAT3, is particularly important in nociceptive transmission and microglial action [24,28,112,176-180] (Fig. 5). Up-regulation of the JAK/STAT3 signaling after nerve in-jury has been observed in animal models of neuropathic pain [24,28,176,177,180], and the activation of this cascade during neuropathy occurs in various cell types as a function of the post-injury period [180]. However, some authors have suggested that this activation is mainly associated with the microglia [28,176, 178]. Inhibition of the microglial JAK/STAT3 pathway attenuated both mechanical allodynia and thermal hyperalgesia during the SNL-induced neuropathy in rats [28]. The activation of the microglial JAK/STAT3 cascade also impacts the function of neighboring cells, including promoting the proliferation of the astrocytes and changing the morphology of the neurons [178]. However, recent studies have revealed that STAT3
Fig. (5).
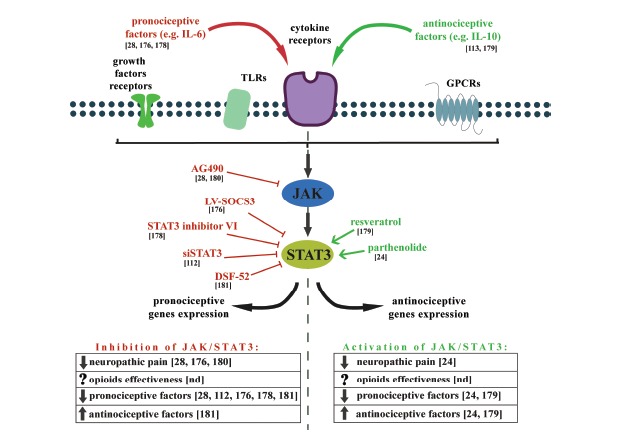
The effects of JAK/STAT3 modulation on neuropathic pain development and opioid effectiveness, as well as the expression of pro- and anti-nociceptive factors.
The JAK/STAT3 pathway is one of the most important cascades for the cellular transduction of signals in response to many pain modulators. Inhibition of JAK/STAT3 pathway activation reduces symptoms of neuropathic pain, diminishes pronociceptive factors and increases antinociceptive expression. However, it was shown that also activation of JAK/STAT3 pathway reduces symptoms of neuropathic pain, diminishes pronociceptive and increases antinociceptive factors.
plays an important role in the polarization of the microglia/macrophages that leads to the potentially neuroprotective “alternative activation” of those cells [24,113,179]. In our experiments, we showed that the microglial polarization process induced by parthenolide is correlated with a profound activation of STAT3 and increases in the antinociceptive factors (IL-10 and TIMP1) [24]. These conflicting reports could be partly explained by the dual nature of the activation of this pathway. As described above, the JAK/STAT3 cascade activation is induced by both pro- (IL-6) and anti-nociceptive (IL-10) factors. These two forms of activation lead to the transcription of different pools of genes that might change the polarization state of the microglia [112,178,179,181-183]. BV2 microglial cells transfected with constructs that encode a constitutively active STAT3 protein expressed high levels of both pro-nociceptive (CCL5, IL-6, TNF-α) and anti-nociceptive (IL-10) factors [112]. Moreover, the authors revealed that silencing the STAT3 expression prevented the LPS-induced expression of harmful genes (iNOS, IL-6, CCL5). Another recently published study revealed that the IL-6-induced microglial STAT3 activation leads to profound expression of many pro-nociceptive factors, including iNOS, IL-18, TNFα, and CCL2. Furthermore, inhibition of this pathway efficiently prevents the expression of the M1-state markers [178]. However, in resveratrol-treated microglial cells, the levels of the pro-nociceptive factors (IL-1β, TNFα, IL-6) were reduced whereas the expression of IL-10 was increased [179]. The authors demonstrated that the observed anti-inflammatory effects of resveratrol were mediated by activation of the JAK/STAT3 cascade.
As presented above, the JAK/STAT3 pathway seems to be sufficient to activate the pro-nociceptive microglial-derived factors. However, it is also involved in a complex physiological response that involves the termination of inflammation and promotion of the M2 state of microglial polarization.
PHOSPHOINOSITIDE 3-KINASE (PI3K)/Akt
Recent studies have focused substantial attention on another intracellular pathway that involves phosphoinositide 3-kinase (PI3K) and the Akt kinase (PI3K/Akt). The activation of this signaling cascade is initiated by many cytokines and growth factors as well as by insulin and LPS (Fig. 6). Ligand binding leads to a cascade of intracellular events beginning with activation of PI3K followed by the conversion of the lipid phosphatidylinositol (3,4)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which in turn leads to Akt trafficking and activation. Akt has a number of downstream effectors including the mammalian target of rapamycin (mTOR) [184] that play roles as regulators of nociceptor sensitivity during neuropathic pain [185]. It is well documented that the PI3K/Akt cascade participates in many cell functions including metabolism, cell growth, and proliferation. The dysregulation of this pathway is therefore implicated in a number of conditions including cancer, diabetes, cardiovascular disease and neurological diseases [186]. In the central nervous system, PI3K regulates synaptic plasticity and LTP [187-189]. It is also well established that the PI3K/Akt pathway modulates nociceptive information and mediates the central sensitization induced by a noxious stimuli [190-194]. The inhibition of PI3K significantly attenuates inflammatory and neuropathic pain [175,195-201]. Recent studies have revealed an important role of the PI3K/Akt cascade in the functions of microglial cells. In particular, in 2015 Jin and colleague demonstrated that using microglial cell cultures that stimulation of those cells by well known pro-nociceptive chemokine MCP-1 leads to profound PI3K/Akt activation [195]. In vivo studies revealed that intrathecal injection of PI3K inhibitor, LY29400, significantly decrease the level of spinal microglia in animal model of bone cancer pain (BCP). Moreover, LY294002 reduced BCP-induced mechanical allodynia [195]. It was shown that plantar incision (the animal model of post-operative pain) induced a time-dependent activation of PI3K/Akt pathway in spinal cord and DRG. At the spinal cord level this activation was colocalized with the neuronal and microglial markers, but not with astrocytes [202].
Fig. (6).
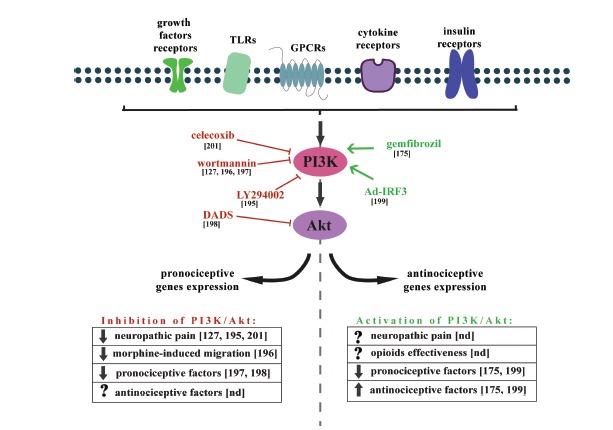
The effects of PI3K/Akt modulation on neuropathic pain development and morphine-induced microglia migration, as well as the expression of pro- and anti-nociceptive factors. The activation of PI3K/Akt pathway signaling cascade is initiated by many cytokines and growth factors as well as by insulin and LPS. This cascade modulates nociceptive information and mediates the central sensitization induced by a noxious stimuli. Inhibition of PI3K/Akt pathway activation reduces symptoms of neuropathic pain, reduces morphine-induced migration and diminishes pronociceptive factors expression. However, it was shown that also activation of PI3K/Akt pathway diminishes pronociceptive and increases antinociceptive factors.
In 2009 Horvath and DeLeo demonstrated that the PI3K/Akt pathway underlies the morphine-induced microglial migration via an interaction between the MOP and P2X4 receptors [196]. These data provide evidence that microglial activation plays a role in various morphine-induced side effects such as tolerance and hyperalgesia. Several lines of evidence have indicated that inhibition of the LPS-activated microglial PI3K/Akt pathway leads to a diminished level of pro-inflammatory factors [197,198]. In contrast, another study conducted by Jana and colleagues (2007) demonstrated an important role of PI3K pathway activation in mediating the anti-inflammatory effect of gemfibrozil, an activator of PPAR-α, in microglia [175]. In 2011 Tarassishin et al. showed that the PI3K/Akt pathway is involved in the promotion of the beneficial M2 microglial polarization state [199]. The activation of this signaling cascade suppresses the M1 state and enhances the M2 state after interferon regulatory factor 3 (IRF3) stimulation. The authors suggested that inhibition of the pro-inflammatory genes (M1 markers) correlates, at least in part, to the induction of the anti-inflammatory factors (M2 markers) such as IL-1ra and IL-10 and that this process is mediated by the PI3K/Akt pathway. The discrepancies in the effects of the activation of the intracellular pathways in microglial cells may depend on the different input signals.
CONCLUSION
The microglia monitor the environment of the CNS by interpreting and processing stimuli through a wide range of surface receptors that lead to the activation of numerous intracellular cascades. However, dysregulation and overactivation of the microglial cells may result in pathological conditions of the CNS as in the case of neuropathic pain. In the present review, we tried to shed light on the unresolved questions regarding the mechanisms by which microglial cells either prevent or facilitate the progression of neuropathic pain. The signaling cascades (MAPKs, NF-κB, JAK/ STAT, PI3K/Akt) discussed above seem to be promising molecular targets for new strategies for neuropathic pain therapy. A safe extrapolation from animal studies to the clinic is still challenging. Therefore, it is important to test compounds that can be safely used in human therapy in basic research experiments. It is important to realize that these intracellular pathways underlie many physiological functions, and their unselective and complete inhibition might lead to serious side effects.
To date, in several clinical trials researchers have targeted a members of signaling pathways discussed in our review. MAPK inhibitors emerge as an attractive antinociceptive drugs, because they are capable of blocking pro-nociceptive cytokine signaling. As we presented in our review, a vast number of MAPK inhibitors has been characterized in in vitro studies as well as in animal models, and now several substances have been advanced into clinical trials [203-205]. In 2011 Anand et al. in double-blind, placebo-controlled trial evaluate the effect of dilmapimod (SB-681323), a selective p38 MAPK inhibitor, on neuropathic pain symptoms [203]. The authors observed significant reduction in the primary endpoint of average daily pain score after two weeks of treatment, moreover dilmapimod was well-tolerated [203]. Another p38 kinase inhibitor, SCIO-469, also shown antinociceptive effects in patient with postsurgical dental impaction pain, however it had several adverse events, like dizziness, headache, nausea, vomiting, dry socket, and hematoma [205]. The drug which alert p38 activation and is already available in clinic is minocycline, however it is used as an antibacterial drug in acne therapy. One of ongoing clinical trial is based on inhibitory action of minocycline on microglial cells [ClinicalTrials.gov NCT02359006]. The researchers from Yale University will examine the effects of this drug in opioid-maintained patients.
NF-κB inhibitor, parthenolide, which was examined in our animal studies, is already used in the clinic. Parthenolide is used as an ingredient of feverfew-based drugs, which are utilized to treat migraines, arthritis, and digestive ailments. It is also used to reduce fevers and menstrual pain, and it has anticancer properties [206-208]. Prthenolide-based herbal remedies are well-tolerated by patients. In the newest Cochrane review (April 2015), the authors declare that there are only a few sides effects associated with feverfew-induced migraine therapy and that these treatments are not encumbered by any major safety concerns [208]. These facts further substantiate our suggestion that parthenolide might be an effective drug in multimodal neuropathic pain therapy.
Clinical trials with another discussed pathway, namely JAK/STAT3, concern mainly inhibition of this cascade in cancer therapies [207-210, ClinicalTrials. gov NCT02417753], however in ongoing studies there are plans to assess pain sensations [ClinicalTrials.gov NCT02531633]. Presently, there is no information about human-based studies using inhibitors of JAK/ STAT3 in the context of neuropathic pain.
In the light of newest evidenced which revealed sex-dependent p38 kinase activation during neuropathy, it should be considered how neuro-immune cross-talk might be modulated for the treatment of neuropathic pain in male versus female. The thesis postulated in our work that pain therapy should be based on drugs that selectively inhibit the pain-related intracellular pathway activities without affecting their basal activation, has strong support in the ongoing preclinical and clinical studies. Such selective agents would be of considerable clinical benefit. Changing the microglial activation by modifying the intracellular pathways could permit the expression of anti-inflammatory factors capable of altering the progression of neuropathic pain. This approach will provide the basis for the development of compounds that have improved therapeutic efficacy in the pathology of neuropathic pain. Future research will need to focus on the details of the mechanisms of activation of microglial intracellular pathways, especially those that play roles in the switching between the polarization states of the microglia.
ACKNOWLEDGEMENTS
This work was supported by the National Science Centre, Poland, via grants Preludium 2012/07/ N/NZ3/ 00379, Harmonia 5 2013/10/M/NZ4/00261 and by statutory funds from the Department of Pain Pharmacology, Institute of Pharmacology PAS. The manuscript was corrected by American Journal Experts.
List of abbreviations
- BCP
bone cancer pain
- BDNF
brain-derived neurotrophic factor
- CCI
chronic constriction injury
- CCL
chemokine (C-C motif) ligand
- CNS
central nervous system
- COX-2
cyclooxygenase 2
- DRG
dorsal root ganglion
- GDNF
glial cell line-derived neurotrophic factor
- GFAP
glial fibrillary acidic protein
- GPCRs
G protein-coupled receptors
- IFNs
interferons
- ILs
interleukins
- IRF3
interferon regulatory factor 3
- iNOS
inducible nitric oxide synthase
- JAK/STAT
Janus tyrosine kinase/signal transducer and activator of transcription
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- LTP
long-term potentiation
- MAPK
mitogen-activated protein kinase
- MCP-1
monocyte chemoattractant protein-1
- NF-κB
nuclear factor κ-light-chain-enhancer of activated B cells
- NGF
nerve growth factor
- PI3K
phosphoinositide 3-kinase
- SCI
spinal cord injury
- TGFβ
transforming growth factor β
- TIMPs
tissue inhibitors of metalloproteinases
- TLRs
Toll-like receptors
- TNFα
tumor necrosis factor α
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Merskey H., Bogduk N. Classification of Chronic Pain. Seattle: IASP Press; 1994. [Google Scholar]
- 2.Coyle D.E. Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia. 1998;23:75–83. [PubMed] [Google Scholar]
- 3.Dworkin R.H., O’Connor A.B., Audette J., Baron R., Gourlay G.K., Haanpää M.L., Kent J.L., Krane E.J., Lebel A.A., Levy R.M., Mackey S.C., Mayer J., Miaskowski C., Raja S.N., Rice A.S., Schmader K.E., Stacey B., Stanos S., Treede R.D., Turk D.C., Walco G.A., Wells C.D. Recommendations for the pharma-cological management of neuropathic pain: an overview and literature update. Mayo Clin. Proc. 2010;85:S3–S14. doi: 10.4065/mcp.2009.0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalso E., Allan L., Dellemijn P.L., Faura C.C., Ilias W.K., Jensen T.S., Perrot S., Plaghki L.H., Zenz M. Recommendations for using opioids in chronic non-cancer pain. Eur. J. Pain. 2003;7:381–386. doi: 10.1016/S1090-3801(02)00143-X. [DOI] [PubMed] [Google Scholar]
- 5.Kalso E., Edwards J.E., Moore R.A., McQuay H.J. Opioids in chronic non-cancer pain: Systematic review of efficacy and safety. Pain. 2004;112:372–380. doi: 10.1016/j.pain.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 6.Amir R., Kocsis J.D., Devor M. Multiple interacting sites of ectopic spike electrogenesis in primary sensory neurons. J. Neurosci. 2005;25:2576–2585. doi: 10.1523/JNEUROSCI.4118-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devor M. Sodium channels and mechanisms of neuropathic pain. J. Pain. 2006;7:S3–S12. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Wu G., Ringkamp M., Hartke T.V., Murinson B.B., Campbell J.N., Griffin J.W., Meyer R.a. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J. Neurosci. 2001;21:21. doi: 10.1523/JNEUROSCI.21-08-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colombo E., Francisconi S., Faravelli L., Izzo E., Pevarello P. Ion channel blockers for the treatment of neuropathic pain. Future Med. Chem. 2010;2:803–842. doi: 10.4155/fmc.10.19. [DOI] [PubMed] [Google Scholar]
- 10.Khan N., Smith M. Neurotrophins and neuropathic pain: role in pathobiology. Molecules. 2015;20:10657–10688. doi: 10.3390/molecules200610657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obara I., Goulding S.P., Hu J.H., Klugmann M., Worley P.F., Szumlinski K.K. Nerve injury-induced changes in Homer/glutamate receptor signaling contribute to the development and maintenance of neuropathic pain. Pain. 2013;154:1932–1945. doi: 10.1016/j.pain.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woolf C.J., Ma Q. Nociceptors-Noxious Stimulus Detectors. Neuron. 2007;55:353–364. doi: 10.1016/j.neuron.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 13.Ji R.R., Strichartz G. Cell signaling and the genesis of neuropathic pain. 2004. [DOI] [PubMed]
- 14.Mika J., Rojewska E., Makuch W., Przewlocka B. Minocycline reduces the injury-induced expression of prodynorphin and pronociceptin in the dorsal root ganglion in a rat model of neuropathic pain. Neuroscience. 2010;165:1420–1428. doi: 10.1016/j.neuroscience.2009.11.064. [DOI] [PubMed] [Google Scholar]
- 15.Mika J., Popiolek-Barczyk K., Rojewska E., Makuch W., Starowicz K., Przewlocka B. Delta-opioid receptor analgesia is independent of microglial activation in a rat model of neuropathic pain. PLoS One. 2014;9:e104420. doi: 10.1371/journal.pone.0104420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rojewska E., Popiolek-Barczyk K., Jurga A.M., Makuch W., Przewlocka B., Mika J. Involvement of pro- and antinociceptive factors in minocycline analgesia in rat neuropathic pain model. J. Neuroimmunol. 2014;277:57–66. doi: 10.1016/j.jneuroim.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 17.Ji R.R., Kohno T., Moore K.A., Woolf C.J. Central sensitization and LTP: Do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Mantyh P.W., Clohisy D.R., Koltzenburg M., Hunt S.P. Molecular mechanisms of cancer pain. Nat. Rev. Cancer. 2002;2:201–209. doi: 10.1038/nrc747. [DOI] [PubMed] [Google Scholar]
- 19.Woolf C.J., Salter M.W. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 20.Zhuo M., Wu G., Wu L.J. Neuronal and microglial mechanisms of neuropathic pain. Mol. Brain. 2011;4:31. doi: 10.1186/1756-6606-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuo M. Neuronal mechanism for neuropathic pain. Mol. Pain. 2007;3:14. doi: 10.1186/1744-8069-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Popiolek-Barczyk K., Rojewska E., Jurga A.M., Makuch W., Zador F., Borsodi A., Piotrowska A., Przewlocka B., Mika J. Minocycline enhances the effectiveness of nociceptin/ orphanin FQ during neuropathic pain. . Biomed. Res. Int. 2014. [DOI] [PMC free article] [PubMed]
- 23.Popiolek-Barczyk K., Makuch W., Rojewska E., Pilat D., Mika J. Inhibition of intracellular signaling pathways NF-κB and MEK1/2 attenuates neuropathic pain development and enhances morphine analgesia. Pharmacol. Rep. 2014;66:845–851. doi: 10.1016/j.pharep.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 24.Popiolek-Barczyk K., Kolosowska N., Piotrowska A., Makuch W., Rojewska E., Jurga A.M., Pilat D., Mika J. Parthenolide relieves pain and promotes M2 microglia/ macrophage polarization in rat model of neuropathy. . Neural Plast., 2015. [DOI] [PMC free article] [PubMed]
- 25.Osikowicz M., Skup M., Mika J., Makuch W., Czarkowska-Bauch J., Przewlcka B. Glial inhibitors influence the mRNA and protein levels of mGlu2/3, 5 and 7 receptors and potentiate the analgesic effects of their ligands in a mouse model of neuropathic pain. Pain. 2009;147:175–186. doi: 10.1016/j.pain.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 26.Makuch W., Mika J., Rojewska E., Zychowska M., Przewlocka B. Effects of selective and non-selective inhibitors of nitric oxide synthase on morphine- and endomorphin-1-induced analgesia in acute and neuropathic pain in rats. Neuropharmacology. 2013;75:445–457. doi: 10.1016/j.neuropharm.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 27.Miyoshi K., Obata K., Kondo T., Okamura H., Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J. Neurosci. 2008;28:12775–12787. doi: 10.1523/JNEUROSCI.3512-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dominguez E., Rivat C., Pommier B., Mauborgne A., Pohl M. JAK/STAT3 pathway is activated in spinal cord microglia after peripheral nerve injury and contributes to neuropathic pain development in rat. J. Neurochem. 2008;107:50–60. doi: 10.1111/j.1471-4159.2008.05566.x. [DOI] [PubMed] [Google Scholar]
- 29.Mika J., Zychowska M., Popiolek-Barczyk K., Rojewska E., Przewlocka B. Importance of glial activation in neuropathic pain. Eur. J. Pharmacol. 2013;716:106–119. doi: 10.1016/j.ejphar.2013.01.072. [DOI] [PubMed] [Google Scholar]
- 30.Zychowska M., Rojewska E., Przewlocka B., Mika J. Mechanisms and pharmacology of diabetic neuropathy - experimental and clinical studies. Pharmacol. Rep. 2013;65:1601–1610. doi: 10.1016/s1734-1140(13)71521-4. [DOI] [PubMed] [Google Scholar]
- 31.Austin P.J., Moalem-Taylor G. The neuro-immune balance in neuropathic pain: Involvement of inflammatory immune cells, immune-like glial cells and cytokines. J. Neuroimmunol. 2010;229:26–50. doi: 10.1016/j.jneuroim.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Austin P.J., Berglund A.M., Siu S., Fiore N.T., Gerke-Duncan M.B., Ollerenshaw S.L., Leigh S.J., Kunjan P.A., Kang J.W., Keay K.A. Evidence for a distinct neuro-immune signature in rats that develop behavioural disability after nerve injury. J. Neuroinflammation. 2015;12:96. doi: 10.1186/s12974-015-0318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buchanan M.M., Hutchinson M., Watkins L.R., Yin H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010;114:13–27. doi: 10.1111/j.1471-4159.2010.06736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grace P.M., Hutchinson M.R., Maier S.F., Watkins L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014;14:217–231. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Machelska H. Dual peripheral actions of immune cells in neuropathic pain. Arch. Immunol. Ther. Exp. (Warsz.) 2011;59:11–24. doi: 10.1007/s00005-010-0106-x. [DOI] [PubMed] [Google Scholar]
- 36.Mika J., Osikowicz M., Makuch W., Przewlocka B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur. J. Pharmacol. 2007;560:142–149. doi: 10.1016/j.ejphar.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Mika J., Wawrzczak-Bargiela A., Osikowicz M., Makuch W., Przewlocka B. Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain Behav. Immun. 2009;23:75–84. doi: 10.1016/j.bbi.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Rojewska E., Korostynski M., Przewlocki R., Przewlocka B., Mika J. Expression profiling of genes modulated by minocycline in a rat model of neuropathic pain. Mol. Pain. 2014;10:47. doi: 10.1186/1744-8069-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 40.Roh D.H., Yoon S.Y., Seo H.S., Kang S.Y., Han H.J., Beitz A.J., Lee J.H. Intrathecal injection of carbenoxolone, a gap junction decoupler, attenuates the induction of below-level neuropathic pain after spinal cord injury in rats. Exp. Neurol. 2010;224:123–132. doi: 10.1016/j.expneurol.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Zündorf G., Kahlert S., Reiser G. Gap-junction blocker carbenoxolone differentially enhances NMDA-induced cell death in hippocampal neurons and astrocytes in co-culture. J. Neurochem. 2007;102:508–521. doi: 10.1111/j.1471-4159.2007.04509.x. [DOI] [PubMed] [Google Scholar]
- 42.Haber M., Murai K.K. Reshaping neuron-glial communi-cation at hippocampal synapses. Neuron Glia Biol. 2006;2:59–66. doi: 10.1017/S1740925X06000032. [DOI] [PubMed] [Google Scholar]
- 43.Haber M., Zhou L., Murai K.K. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J. Neurosci. 2006;26:8881–8891. doi: 10.1523/JNEUROSCI.1302-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jourdain P., Bergersen L.H., Bhaukaurally K., Bezzi P., Santello M., Domercq M., Matute C., Tonello F., Gundersen V., Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 2007;10:331–339. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- 45.Oliet S.H., Panatier A., Piet R., Mothet J.P., Poulain D.A., Theodosis D.T. Neuron-glia interactions in the rat supraoptic nucleus. Prog. Brain Res. 2008;170:109–117. doi: 10.1016/S0079-6123(08)00410-X. [DOI] [PubMed] [Google Scholar]
- 46.Hatori K., Nagai A., Heisel R., Ryu J.K., Kim S.U. Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 2002;69:418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- 47.Ren K., Dubner R. Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr. Opin. Anaesthesiol. 2008;21:570–579. doi: 10.1097/ACO.0b013e32830edbdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rocha S.M., Pires J., Esteves M., Graça B., Bernardino L. Histamine: a new immunomodulatory player in the neuron-glia crosstalk. Front. Cell. Neurosci. 2014;8:120. doi: 10.3389/fncel.2014.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eriksson N.P., Persson J.K., Aldskogius H., Svensson M. A quantitative analysis of the glial cell reaction in primary sensory termination areas following sciatic nerve injury and treatment with nerve growth factor in the adult rat. Exp. Brain Res. 1997;114:393–404. doi: 10.1007/pl00005649. [DOI] [PubMed] [Google Scholar]
- 50.Garrison C.J., Dougherty P.M., Kajander K.C., Carlton S.M. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565:1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- 51.Dreger M., Mika J., Bieller A., Jahnel R., Gillen C., Schaefer M.K., Weihe E., Hucho F. Analysis of the dorsal spinal cord synaptic architecture by combined proteome analysis and in situ hybridization. J. Proteome Res. 2005;4:238–249. doi: 10.1021/pr049870w. [DOI] [PubMed] [Google Scholar]
- 52.Mika J., Osikowicz M., Rojewska E., Korostynski M., Wawrzczak-Bargiela A., Przewlocki R., Przewlocka B. Differential activation of spinal microglial and astroglial cells in a mouse model of peripheral neuropathic pain. Eur. J. Pharmacol. 2009;623:65–72. doi: 10.1016/j.ejphar.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez Parkitna J., Korostynski M., Kaminska-Chowaniec D., Obara I., Mika J., Przewlocka B., Przewlocki R. Comparison of gene expression profiles in neuropathic and inflammatory pain. J. Physiol. Pharmacol. 2006;57:401–414. [PubMed] [Google Scholar]
- 54.Gong Q.J., Li Y.Y., Xin W.J., Zhang Y., Ren W.J., Wei X.H., Li Y.Y., Zhang T., Liu X.G. ATP induces long-term potentiation of C-fiber-evoked field potentials in spinal dorsal horn: The roles of P2X4 receptors and p38 MAPK in microglia. Glia. 2009;57:583–591. doi: 10.1002/glia.20786. [DOI] [PubMed] [Google Scholar]
- 55.Chu Y.X., Zhang Y., Zhang Y.Q., Zhao Z.Q. Involvement of microglial P2X7 receptors and downstream signaling pathways in long-term potentiation of spinal nociceptive responses. Brain Behav. Immun. 2010;24:1176–1189. doi: 10.1016/j.bbi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 56.Gwak Y.S., Kang J., Unabia G.C., Hulsebosch C.E. Spatial and temporal activation of spinal glial cells: Role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp. Neurol. 2012;234:362–372. doi: 10.1016/j.expneurol.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanga F., Raghavendra V., DeLeo J.A. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem. Int. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 58.Clark A.K., Yip P.K., Grist J., Gentry C., Staniland A.a, Marchand F., Dehvari M., Wotherspoon G., Winter J., Ullah J., Bevan S., Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc. Natl. Acad. Sci. USA. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark A.K., Gentry C., Bradbury E.J., McMahon S.B., Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur. J. Pain. 2007;11:223–230. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 60.Zhang J., De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J. Neurochem. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- 61.Zhuang Z.Y., Wen Y.R., Zhang D.R., Borsello T., Bonny C., Strichartz G.R., Decosterd I., Ji R.R. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J. Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gritsch S., Lu J., Thilemann S., Wörtge S., Möbius W., Bruttger J., Karram K., Ruhwedel T., Blanfeld M., Vardeh D., Waisman A., Nave K.A., Kuner R. Oligodendrocyte ablation triggers central pain indepen-dently of innate or adaptive immune responses in mice. Nat. Commun. 2014;5:5472. doi: 10.1038/ncomms6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zarpelon A.C., Rodrigues F.C., Lopes A.H., Souza G.R., Carvalho T.T., Pinto L.G., Xu D., Ferreira S.H., Alves-Filho J.C., McInnes I.B., Ryffel B., Quesniaux V.F., Reverchon F., Mortaud S., Menuet A., Liew F.Y., Cunha F.Q., Cunha T.M., Verri W.A. Spinal cord oligodendrocyte-derived alarmin IL-33 mediates neuro-pathic pain. FASEB J. 2016;30:54–65. doi: 10.1096/fj.14-267146. [DOI] [PubMed] [Google Scholar]
- 64.Liu S., Mi W.L., Li Q., Zhang M.T., Han P., Hu S., Mao-Ying Q.L. Spinal IL-33/ST2 signaling contributes to neuropathic pain via neuronal CaMKII-CREB and astroglial JAK2-STAT3 cascades in mice. Anesthesiology. 2015 doi: 10.1097/ALN.0000000000000850. [DOI] [PubMed] [Google Scholar]
- 65.Colburn R.W., Rickman A.J., DeLeo J.A. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp. Neurol. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- 66.Romero-Sandoval E.A., Horvath R.J., DeLeo J.A. Neuroimmune interactions and pain: focus on glial-modulating targets. Curr. Opin. Investig. Drugs. 2008;9:726–734. [PMC free article] [PubMed] [Google Scholar]
- 67.Liu B., Neufeld A.H. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia. 2000;30:178–186. doi: 10.1002/(sici)1098-1136(200004)30:2<178::aid-glia7>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 68.Dirig D.M., Yaksh T.L. Spinal synthesis and release of prostanoids after peripheral injury and inflammation. Adv. Exp. Med. Biol. 1999:401–408. doi: 10.1007/978-1-4615-4793-8_58. [DOI] [PubMed] [Google Scholar]
- 69.Ghilardi J.R. Constitutive spinal cyclooxygenase-2 participates in the initiation of tissue injury-induced hyperalgesia. J. Neurosci. 2004;24:2727–2732. doi: 10.1523/JNEUROSCI.5054-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duan S., Anderson C.M., Keung E.C., Chen Y., Chen Y., Swanson R.A. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 2003;23:1320–1328. doi: 10.1523/JNEUROSCI.23-04-01320.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao Y.J., Zhang L., Samad O.A., Suter M.R., Yasuhiko K., Xu Z.Z., Park J.Y., Lind A.L., Ma Q., Ji R.R. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J. Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kawasaki Y., Xu Z.Z., Wang X., Park J.Y., Zhuang Z.Y., Tan P.H., Gao Y.J., Roy K., Corfas G., Lo E.H., Ji R.R. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat. Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Queiroz G., Gebicke-Haerter P., Schobert A., Starke K., von Kügelgen I. Release of ATP from cultured rat astrocytes elicited by glutamate receptor activation. Neuroscience. 1997;78:1203–1208. doi: 10.1016/s0306-4522(96)00637-9. [DOI] [PubMed] [Google Scholar]
- 74.Griffin R.S., Costigan M., Brenner G.J., Him Eddie Ma C., Scholz J., Moss A., Allchorne A.J., Stahl G.L., Woolf C.J. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersen-sitivity. J. Neurosci. 2007;27:8699–8708. doi: 10.1523/JNEUROSCI.2018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Graeber M.B. Changing face of microglia. Science. 2010;330:783–788. doi: 10.1126/science.1190929. [DOI] [PubMed] [Google Scholar]
- 76.McMahon S.B., Cafferty W.B., Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp. Neurol. 2005;192:444–462. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 77.Slusarczyk J., Trojan E., Glombik K., Budziszewska B., Kubera M., Lason W., Popiolek-Barczyk K., Mika J., Wedzony K., Basta-Kaim A. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front. Cell. Neurosci. 2015;9:1–14. doi: 10.3389/fncel.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Slusarczyk J., Trojan E., Glombik K., Piotrowska A., Budziszewska B., Kubera M., Popiolek-Barczyk K., Lason W., Mika J., Basta-Kaim A. Anti-inflammatory properties of tianeptine on lipopolysaccharide-induced changes in microglial cells involve Toll-like receptor-related pathways. J. Neurochem. 2016;136(5):958–970. doi: 10.1111/jnc.13452. [DOI] [PubMed] [Google Scholar]
- 79.Bédard A., Tremblay P., Chernomoretz A., Vallières L. Identification of genes preferentially expressed by microglia and upregulated during cuprizone-induced inflammation. Glia. 2007;55:777–789. doi: 10.1002/glia.20477. [DOI] [PubMed] [Google Scholar]
- 80.Lehnardt S., Massillon L., Follett P., Jensen F.E., Ratan R., Rosenberg P.A., Volpe J.J., Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kettenmann H., Kirchhoff F., Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77:10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 82.Jurga A.M., Piotrowska A., Starnowska J., Rojewska E., Makuch W., Mika J. Treatment with a carbon monoxide-releasing molecule (CORM-2) inhibits neuropathic pain and enhances opioid effectiveness in rats. Pharmacol. Rep. 2016;68:206–213. doi: 10.1016/j.pharep.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 83.Verkhratsky A., Kettenmann H. Calcium signalling in glial cells. Trends Neurosci. 1996;19:346–352. doi: 10.1016/0166-2236(96)10048-5. [DOI] [PubMed] [Google Scholar]
- 84.Inoue K. The functions of ATP receptors in the hippo-campus. Pharmacol. Res. 1998;38:323–331. doi: 10.1006/phrs.1998.0382. [DOI] [PubMed] [Google Scholar]
- 85.Hide I., Tanaka M., Inoue A., Nakajima K., Kohsaka S., Inoue K., Nakata Y. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J. Neurochem. 2000;75:965–972. doi: 10.1046/j.1471-4159.2000.0750965.x. [DOI] [PubMed] [Google Scholar]
- 86.Shigemoto-Mogami Y., Koizumi S., Tsuda M., Ohsawa K., Kohsaka S., Inoue K. Mechanisms underlying extracellular ATP-evoked interleukin-6 release in mouse microglial cell line, MG-5. J. Neurochem. 2001;78:1339–1349. doi: 10.1046/j.1471-4159.2001.00514.x. [DOI] [PubMed] [Google Scholar]
- 87.Tsuda M., Shigemoto-Mogami Y., Koizumi S., Mizokoshi A., Kohsaka S., Salter M.W., Inoue K. P2X(4) receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 88.Pannell M., Meier M.A., Szulzewsky F., Matyash V., Endres M., Kronenberg G., Prinz V., Waiczies S., Wolf S.A., Kettenmann H. The subpopulation of microglia expressing functional muscarinic acetylcholine receptors expands in stroke and Alzheimer’s disease. Brain Struct. Funct. 2014;19:19. doi: 10.1007/s00429-014-0962-y. [DOI] [PubMed] [Google Scholar]
- 89.Lee Y.B., Nagai A., Kim S.U. Cytokines, chemokines, and cytokine receptors in human microglia. J. Neurosci. Res. 2002;69:94–103. doi: 10.1002/jnr.10253. [DOI] [PubMed] [Google Scholar]
- 90.Malek N., Popiolek-Barczyk K., Mika J., Przewlocka B., Starowicz K. Anandamide, acting via CB2 receptors, alleviates LPS-induced neuroinflammation in rat primary microglial cultures. . Neural Plast. 2015. [DOI] [PMC free article] [PubMed]
- 91.Dong H., Zhang W., Zeng X., Hu G., Zhang H., He S., Zhang S. Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol. Neurobiol. 2014;49:1487–1500. doi: 10.1007/s12035-014-8697-6. [DOI] [PubMed] [Google Scholar]
- 92.Elkabes S., Peng L., Black I.B. Lipopolysaccharide differentially regulates microglial Trk receptor and neurotrophin expression. J. Neurosci. Res. 1998;54:117–122. doi: 10.1002/(SICI)1097-4547(19981001)54:1<117::AID-JNR12>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 93.Harrison J.K., Jiang Y., Chen S., Xia Y., Maciejewski D., McNamara R.K., Streit W.J., Salafranca M.N., Adhikari S., Thompson D.a, Botti P., Bacon K.B., Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wolf Y., Yona S., Kim K.W., Jung S. Microglia, seen from the CX3CR1 angle. Front. Cell. Neurosci. 2013;7:26. doi: 10.3389/fncel.2013.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clark A.K., Malcangio M. Fractalkine/CX3CR1 signaling during neuropathic pain. Front. Cell. Neurosci. 2014;8:121. doi: 10.3389/fncel.2014.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boddeke E.W. Involvement of chemokines in pain. Eur. J. Pharmacol. 2001;429:115–119. doi: 10.1016/s0014-2999(01)01311-5. [DOI] [PubMed] [Google Scholar]
- 97.Johnston I., Milligan E., Wieseler-Frank J. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J. Neurosci. 2004;24:7353–7365. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Oprée A., Kress M. Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J. Neurosci. 2000;20:6289–6293. doi: 10.1523/JNEUROSCI.20-16-06289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pilat D., Rojewska E., Jurga A.M., Piotrowska A., Makuch W., Przewlocka B., Mika J. IL-1 receptor antagonist improves morphine and buprenorphine efficacy in a rat neuropathic pain model. Eur. J. Pharmacol. 2015;764:240–248. doi: 10.1016/j.ejphar.2015.05.058. [DOI] [PubMed] [Google Scholar]
- 100.Yezierski R.P., DeLeo J.A. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 101.Kwiatkowski K., Piotrowska A., Rojewska E., Makuch W., Jurga A., Slusarczyk J., Trojan E., Basta-Kaim A., Mika J. Beneficial properties of maraviroc on neuropathic pain development and opioid effectiveness in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2016;64:68–78. doi: 10.1016/j.pnpbp.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 102.Ginhoux F., Greter M., Leboeuf M., Nandi S., See P., Gokhan S., Mehler M.F., Conway S.J., Ng L.G., Stanley E.R., Samokhvalov I.M., Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 104.Martinez F.O. Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:1–13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Steinman R., Idoyaga J. Features of the dendritic cell lineage. Immunol. Rev. 2010;234:5–17. doi: 10.1111/j.0105-2896.2009.00888.x. [DOI] [PubMed] [Google Scholar]
- 106.Pan J., Jin J., Ge H., Yin K., Chen X., Han L., Chen Y., Qian L., Li X., Xu Y. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARγ-dependent manner. J. Neuroinflammation. 2015;12:51. doi: 10.1186/s12974-015-0270-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pisanu A., Lecca D., Mulas G., Wardas J., Simbula G., Spiga S., Carta A.R. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis. 2014;71:280–291. doi: 10.1016/j.nbd.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 108.Burke N.N., Kerr D.M., Moriarty O., Finn D.P., Roche M. Minocycline modulates neuropathic pain behaviour and cortical M1-M2 microglial gene expression in a rat model of depression. Brain Behav. Immun. 2014:147–156. doi: 10.1016/j.bbi.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 109.Kobayashi K., Imagama S., Ohgomori T., Hirano K., Uchimura K., Sakamoto K., Hirakawa A., Takeuchi H., Suzumura A., Ishiguro N., Kadomatsu K. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013;4:e525. doi: 10.1038/cddis.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Labuzek K., Liber S., Gabryel B., Okopień B. Metformin has adenosine-monophosphate activated protein kinase (AMPK)-independent effects on LPS-stimulated rat primary microglial cultures. Pharmacol. Rep. 2010;62:827–848. doi: 10.1016/s1734-1140(10)70343-1. [DOI] [PubMed] [Google Scholar]
- 111.Orihuela R., McPherson C.A., Harry G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016;173(4):649–665. doi: 10.1111/bph.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Przanowski P., Dabrowski M., Ellert-Miklaszewska A., Kloss M., Mieczkowski J., Kaza B., Ronowicz A., Hu F., Piotrowski A., Kettenmann H., Komorowski J., Kaminska B. The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia. J. Mol. Med. 2014;92:239–254. doi: 10.1007/s00109-013-1090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koscsó B., Csóka B., Kókai E., Németh Z.H., Pacher P., Virág L., Leibovich S.J., Haskó G. Adenosine augments IL-10-induced STAT3 signaling in M2c macrophages. J. Leukoc. Biol. 2013;94:1309–1315. doi: 10.1189/jlb.0113043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou X., Spittau B., Krieglstein K. TGFβ signalling plays an important role in IL4-induced alternative activation of microglia. J. Neuroinflammation. 2012;9:210. doi: 10.1186/1742-2094-9-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu F., Huang J., He Z., Chen J., Tang X., Song Z., Guo Q., Huang C. Microglial polarization dynamics in dorsal spinal cord in the early stages following chronic sciatic nerve damage. Neurosci. Lett. 2016;617:6–13. doi: 10.1016/j.neulet.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 116.Ji R.R., Baba H., Brenner G.J., Woolf C.J. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat. Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- 117.Ji R.R., Samad T.A., Jin S.X., Schmoll R., Woolf C.J. P38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 118.Karim F., Wang C.C., Gereau R.W. Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J. Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pezet S., Malcangio M., Lever I.J., Perkinton M.S., Thompson S.W., Williams R.J., McMahon S.B. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol. Cell. Neurosci. 2002;21:684–695. doi: 10.1006/mcne.2002.1205. [DOI] [PubMed] [Google Scholar]
- 120.Hains B.C., Waxman S.G. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J. Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ji R.R., Suter M.R. P38 MAPK, microglial signaling, and neuropathic pain. Mol. Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Piotrowska A., Kwiatkowski K., Rojewska E., Makuch W., Mika J. Maraviroc reduces neuropathic pain through polarization of microglia and astroglia - evidence from in vivo and in vitro studies. Neuropharmacology. 2016:207–219. doi: 10.1016/j.neuropharm.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 123.Zhuang Z.Y., Gerner P., Woolf C.J., Ji R.R. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 124.Tsuda M., Mizokoshi A., Shigemoto-Mogami Y., Koizumi S., Inoue K. Activation of p38 Mitogen-Activated Protein Kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- 125.Ma W., Quirion R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–184. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- 126.Ma W., Quirion R. The ERK/MAPK pathway, as a target for the treatment of neuropathic pain. Expert Opin. Ther. Targets. 2005;9:699–713. doi: 10.1517/14728222.9.4.699. [DOI] [PubMed] [Google Scholar]
- 127.Xu Z., Wang B.R., Wang X., Kuang F., Duan X.L., Jiao X.Y., Ju G. ERK1/2 and p38 mitogen-activated protein kinase mediate iNOS-induced spinal neuron degeneration after acute traumatic spinal cord injury. Life Sci. 2006;79:1895–1905. doi: 10.1016/j.lfs.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 128.Jin S.X., Zhuang Z.Y., Woolf C.J., Ji R.R. P38 Mitogen-Activated Protein Kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rojewska E., Popiolek-Barczyk K., Kolosowska N., Piotrowska A., Zychowska M., Makuch W., Przewlocka B., Mika J. PD98059 influences immune factors and enhances opioid analgesia in model of neuropathy. PLoS One. 2015;10:e0138583. doi: 10.1371/journal.pone.0138583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wen Y., Suter M., Kawasaki Y., Huang J., Pertin M., Kohno T., Berde C., Decosterd I., Ji R. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–321. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- 131.Zhou X., Cheng H., Xu D., Yin Q., Cheng L., Wang L., Song S., Zhang M. Attenuation of neuropathic pain by saikosaponin a in a rat model of chronic constriction injury. Neurochem. Res. 2014:2136–2142. doi: 10.1007/s11064-014-1407-y. [DOI] [PubMed] [Google Scholar]
- 132.Zhou T.T., Wu J.R., Chen Z.Y., Liu Z.X., Miao B. Effects of dexmedetomidine on P2X4Rs, p38-MAPK and BDNF in spinal microglia in rats with spared nerve injury. Brain Res. 2014;1568:21–30. doi: 10.1016/j.brainres.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 133.Rojewska E., Popiolek-Barczyk K., Jurga A.M., Makuch W., Przewlocka B., Mika J. Involvement of pro- and antinociceptive factors in minocycline analgesia in rat neuropathic pain model. J. Neuroimmunol. 2014;277:57–66. doi: 10.1016/j.jneuroim.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 134.Obata K., Yamanaka H., Dai Y., Mizushima T., Fukuoka T., Tokunaga A., Noguchi K. Differential activation of MAPK in injured and uninjured DRG neurons following chronic constriction injury of the sciatic nerve in rats. Eur. J. Neurosci. 2004;20:2881–2895. doi: 10.1111/j.1460-9568.2004.03754.x. [DOI] [PubMed] [Google Scholar]
- 135.Taves S., Berta T., Liu D.L., Gan S., Chen G., Kim Y.H., Van de Ven T., Laufer S., Ji R.R. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: Sex-dependent microglial signaling in the spinal cord. Brain Behav. Immun. 2016;55:70–81. doi: 10.1016/j.bbi.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wen Y., Suter M., Kawasaki Y., Huang J., Pertin M., Kohno T., Berde C., Decosterd I., Ji R. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–321. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- 137.Milligan E.D., Twining C., Chacur M., Biedenkapp J., O’Connor K., Poole S., Tracey K., Martin D., Maier S.F., Watkins L.R. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J. Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mei X.P., Sakuma Y., Xie C., Wu D., Ho I., Kotani J., Xu L.X. Depressing interleukin-1β contributed to the synergistic effects of tramadol and minocycline on spinal nerve ligation-induced neuropathic pain. Neurosignals. 2013;710032:1–13. doi: 10.1159/000355071. [DOI] [PubMed] [Google Scholar]
- 139.Raghavendra V., Tanga F., DeLeo J.A. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J. Pharmacol. Exp. Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- 140.Ledeboer A., Sloane E.M., Milligan E.D., Frank M.G., Mahony J.H., Maier S.F., Watkins L.R. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 141.Sorge R.E., Mapplebeck J.C., Rosen S., Beggs S., Taves S., Alexander J.K., Martin L.J., Austin J.S., Sotocinal S.G., Chen D., Yang M., Shi X.Q., Huang H., Pillon N.J., Bilan P.J., Tu Y., Klip A., Ji R-R., Zhang J., Salter M.W., Mogil J.S. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 2015;18:1–5. doi: 10.1038/nn.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Berta T., Park C.K., Xu Z.Z., Xie R.G., Liu T., Lu N., Liu Y.C., Ji R.R. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-α secretion. J. Clin. Invest. 2014;124:1173–1186. doi: 10.1172/JCI72230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kaminska B., Gozdz A., Zawadzka M., Ellert-Miklaszewska A., Lipko M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat. Rec. 2009;292:1902–1913. doi: 10.1002/ar.21047. [DOI] [PubMed] [Google Scholar]
- 144.Lin X., Wang M., Zhang J., Xu R. P38 MAPK: a potential target of chronic pain. Curr. Med. Chem. 2014;21:4405–4418. doi: 10.2174/0929867321666140915143040. [DOI] [PubMed] [Google Scholar]
- 145.Sung C.S., Wen Z.H., Chang W.K., Chan K.H., Ho S.T., Tsai S.K., Chang Y.C., Wong C.S. Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1beta-induced thermal hyperalgesia and inducible nitric oxide synthase expression in the spinal cord. J. Neurochem. 2005;94:742–752. doi: 10.1111/j.1471-4159.2005.03226.x. [DOI] [PubMed] [Google Scholar]
- 146.Svensson C.I., Hua X.Y., Protter A.A., Powell H.C., Yaksh T.L. Spinal p38 MAP kinase is necessary for NMDA-induced spinal PGE(2) release and thermal hyperalgesia. Neuroreport. 2003;14:1153–1157. doi: 10.1097/00001756-200306110-00010. [DOI] [PubMed] [Google Scholar]
- 147.Kumar S., Boehm J., Lee J.C. P38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 148.Jana M., Dasgupta S., Saha R.N., Liu X., Pahan K. Induction of tumor necrosis factor-α (TNF-α) by interleukin-12 p40 monomer and homodimer in microglia and macrophages. J. Neurochem. 2003;86:519–528. doi: 10.1046/j.1471-4159.2003.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wilms H., Rosenstiel P., Sievers J., Deuschl G., Zecca L., Lucius R. Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson’s disease. FASEB J. 2003;17:500–502. doi: 10.1096/fj.02-0314fje. [DOI] [PubMed] [Google Scholar]
- 150.Bading H., Greenberg M.E. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- 151.Fiore R.S., Murphy T.H., Sanghera J.S., Pelech S.L., Baraban J.M. Activation of p42 mitogen-activated protein kinase by glutamate receptor stimulation in rat primary cortical cultures. J. Neurochem. 1993;61:1626–1633. doi: 10.1111/j.1471-4159.1993.tb09796.x. [DOI] [PubMed] [Google Scholar]
- 152.Rosen L.B., Ginty D.D., Weber M.J., Greenberg M.E. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 153.Morozov A., Muzzio I.a., Bourtchouladze R., Van-Strien N., Lapidus K., Yin D., Winder D.G., Adams J.P., Sweatt J.D., Kandel E.R. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron. 2003;39:309–325. doi: 10.1016/s0896-6273(03)00404-5. [DOI] [PubMed] [Google Scholar]
- 154.Fitzgerald E.M. Regulation of voltage-dependent calcium channels in rat sensory neurones involves a Ras — mitogen-activated protein kinase pathway. J. Physiol. 2000;527(3):433–444. doi: 10.1111/j.1469-7793.2000.00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Han M., Huang R.Y., Du Y.M., Zhao Z.Q., Zhang Y.Q. Early intervention of ERK activation in the spinal cord can block initiation of peripheral nerve injury-induced neuropathic pain in rats. Sheng Li Xue Bao. 2011;63:106–114. [PubMed] [Google Scholar]
- 156.Dai Z., Lin T., Liou J., Cheng K., Chen J., Chu L., Chen I., Wu B. Xanthine derivative KMUP-1 reduces inflammation and hyperalgesia in a bilateral chronic constriction injury model by suppressing MAPK and NFκB activation. Mol. Pharm. 2014;11:1621–1631. doi: 10.1021/mp5000086. [DOI] [PubMed] [Google Scholar]
- 157.Ciruela A., Dixon A.K., Bramwell S., Gonzalez M.I., Pinnock R.D., Lee K. Identification of MEK1 as a novel target for the treatment of neuropathic pain. Br. J. Pharmacol. 2003;138:751–756. doi: 10.1038/sj.bjp.0705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tang J., Zhu C., Li Z., Liu X., Sun S., Zhang T., Luo Z., Zhang H., Li W. Inhibition of the spinal astrocytic JNK/MCP-1 pathway activation correlates with the analgesic effects of tanshinone IIA sulfonate in neuropathic pain. J. Neuroinflammation. 2015;12:57. doi: 10.1186/s12974-015-0279-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lu Y., Jiang B.C., Cao D.L., Zhang Z.J., Zhang X., Ji R.R., Gao Y.J. TRAF6 upregulation in spinal astrocytes maintains neuropathic pain by integrating TNF-alpha and IL-1beta signaling. Pain. 2014;155:2618–2629. doi: 10.1016/j.pain.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huang C.T., Tsai Y.J. Docosahexaenoic acid confers analgesic effects after median nerve injury via inhibition of c-Jun N-terminal kinase activation in microglia. J. Nutr. Biochem. 2016;29:97–106. doi: 10.1016/j.jnutbio.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 161.Kaltschmidt B., Kaltschmidt C. NF-kappaB in the nervous system. Cold Spring Harb. Perspect. Biol. 2009;1(3):a001271. doi: 10.1101/cshperspect.a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shih R.H., Wang C.Y., Yang C.M. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front. Mol. Neurosci. 2015;8:1–8. doi: 10.3389/fnmol.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Meunier A., Latrémolière A., Dominguez E., Mauborgne A., Philippe S., Hamon M., Mallet J., Benoliel J., Pohl M. Lentiviral-mediated targeted NF-κB blockade in dorsal spinal cord glia attenuates sciatic nerve injury - induced neuropathic pain in the rat. Mol. Ther. 2007;15:687–697. doi: 10.1038/sj.mt.6300107. [DOI] [PubMed] [Google Scholar]
- 164.Tchivileva I.E., Nackley A.G., Qian L., Wentworth S., Conrad M., Diatchenko L.B. Characterization of NF-kB-mediated inhibition of catechol-O-methyltransferase. Mol. Pain. 2009;5:13. doi: 10.1186/1744-8069-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ma W., Bisby M.A. Increased activation of nuclear factor kappa B in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Brain Res. 1998;797:243–254. doi: 10.1016/s0006-8993(98)00380-1. [DOI] [PubMed] [Google Scholar]
- 166.Bakalkin G., Yakovleva T., Terenius L. NF-kappa B-like factors in the murine brain. Developmentally-regulated and tissue-specific expression. Brain Res. Mol. Brain Res. 1993;20:137–146. doi: 10.1016/0169-328x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- 167.Kaltschmidt C., Kaltschmidt B., Baeuerle P.A. Brain synapses contain inducible forms of the transcription factor NF-kappa B. Mech. Dev. 1993;43:135–147. doi: 10.1016/0925-4773(93)90031-r. [DOI] [PubMed] [Google Scholar]
- 168.Baeuerle P.A., Henkel T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 169.Lee K.M., Kang B.S., Lee H.L., Son S.J., Hwang S.H., Kim D.S., Park J.S., Cho H.J. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflam-matory pain hypersensitivity. Eur. J. Neurosci. 2004;19:3375–3381. doi: 10.1111/j.0953-816X.2004.03441.x. [DOI] [PubMed] [Google Scholar]
- 170.Pan Y., Guo Q., Wang E., Ye Z., He Z., Zou W., Cheng Z., Wang Y. Intrathecal infusion of pyrrolidine dithiocarbamate for the prevention and reversal of neuropathic pain in rats using a sciatic chronic constriction injury model. Reg. Anesth. Pain Med. 2010;35:231–237. doi: 10.1097/AAP.0b013e3181df245b. [DOI] [PubMed] [Google Scholar]
- 171.Lee M.K., Han S.R., Park M.K., Kim M.J., Bae Y.C., Kim S.K., Park J.S., Ahn D.K. Behavioral evidence for the differential regulation of p-p38 MAPK and p-NF-κB in rats with trigeminal neuropathic pain. Mol. Pain. 2011;7:57. doi: 10.1186/1744-8069-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhou C., Shi X., Huang H., Zhu Y., Wu Y. Montelukast attenuates neuropathic pain through inhibiting p38 mitogen-activated protein kinase and nuclear factor-kappa B in a rat model of chronic constriction injury. Anesth. Analg. 2014;118:1090–1096. doi: 10.1213/ANE.0000000000000174. [DOI] [PubMed] [Google Scholar]
- 173.Zhou H., Chen S., Wang W., Wang Z., Wu X., Zhang Z. Nanog inhibits lipopolysaccharide-induced expression of pro-inflammatory cytokines by blocking NF-kappaB transcriptional activity in rat primary microglial cells. Mol. Med. Rep. 2012;5:842–846. doi: 10.3892/mmr.2011.719. [DOI] [PubMed] [Google Scholar]
- 174.Olajide O.A., Aderogba M.A., Okorji U.P., Fiebich B.L. Bridelia ferruginea produces antineuroinflammatory activity through inhibition of nuclear factor-kappa B and p38 MAPK signalling. . Evid. Based. Complement. Alternat. Med. 2012. [DOI] [PMC free article] [PubMed]
- 175.Jana M., Jana A., Liu X., Ghosh S., Pahan K. Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of IkappaB in anti-inflammatory effect of gemfibrozil in microglia. J. Immunol. 2007;179:4142–4152. doi: 10.4049/jimmunol.179.6.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dominguez E., Mauborgne A., Mallet J., Desclaux M., Pohl M. SOCS3-mediated blockade of JAK/STAT3 signaling pathway reveals its major contribution to spinal cord neuroinflammation and mechanical allodynia after peripheral nerve injury. J. Neurosci. 2010;30:5754–5766. doi: 10.1523/JNEUROSCI.5007-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang N., Liang H., Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014;5:1–9. doi: 10.3389/fimmu.2014.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Molet J., Mauborgne A., Diallo M., Armand V., Geny D., Villanueva L., Boucher Y., Pohl M. Microglial Janus kinase/signal transduction and activator of transcription 3 pathway activity directly impacts astrocyte and spinal neuron characteristics. J. Neurochem. 2015;•••:133–147. doi: 10.1111/jnc.13375. [DOI] [PubMed] [Google Scholar]
- 179.Cianciulli A., Dragone T., Calvello R., Porro C., Trotta T., Lofrumento D.D., Panaro M.A. IL-10 plays a pivotal role in anti-inflammatory effects of resveratrol in activated microglia cells. Int. Immunopharmacol. 2015;24:369–376. doi: 10.1016/j.intimp.2014.12.035. [DOI] [PubMed] [Google Scholar]
- 180.Yamauchi K., Osuka K., Takayasu M., Usuda N., Nakazawa A., Nakahara N., Yoshida M., Aoshima C., Hara M., Yoshida J. Activation of JAK/STAT signalling in neurons following spinal cord injury in mice. J. Neurochem. 2006;96:1060–1070. doi: 10.1111/j.1471-4159.2005.03559.x. [DOI] [PubMed] [Google Scholar]
- 181.Zeng K.W., Wang S., Dong X., Jiang Y., Tu P.F. Sesquiterpene dimer (DSF-52) from Artemisia argyi inhibits microglia-mediated neuroinflammation via supp-ression of NF-κB, JNK/p38 MAPKs and Jak2/Stat3 signaling pathways. Phytomedicine. 2014;21:298–306. doi: 10.1016/j.phymed.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 182.Murray P.J. STAT3-mediated anti-inflammatory signa-lling. Biochem. Soc. Trans. 2006;34:1028–1031. doi: 10.1042/BST0341028. [DOI] [PubMed] [Google Scholar]
- 183.Scheller J., Chalaris A., Schmidt-Arras D., Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. . Biochim. Biophys. Acta - Mol. Cell Res. . 2011; 1813 :878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 184.Hay N., Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 185.Obara I., Tochiki K.K., Géranton S.M., Carr F.B., Lumb B.M., Liu Q., Hunt S.P. Systemic inhibition of the mammalian target of rapamycin (mTOR) pathway reduces neuropathic pain in mice. Pain. 2011;152:2582–2595. doi: 10.1016/j.pain.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 186.Hemmings B.A., Restuccia D.F. PI3K-PKB / Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012;4:1–4. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kelly Á., Lynch M. Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology. 2000;39:643–651. doi: 10.1016/s0028-3908(99)00169-0. [DOI] [PubMed] [Google Scholar]
- 188.Lin X., Bohle A.S., Dohrmann P., Leuschner I., Schulz A., Kremer B., Fandrich F. Overexpression of phosphati-dylinositol 3-kinase in human lung cancer. Langenbecks Arch. Surg. 2001;386:293–301. doi: 10.1007/s004230100203. [DOI] [PubMed] [Google Scholar]
- 189.Man H.Y., Wang Q., Lu W.Y., Ju W., Ahmadian G., Liu L., D’Souza S., Wong T.P., Taghibiglou C., Lu J., Becker L.E., Pei L., Liu F., Wymann M.P., MacDonald J.F., Wang Y.T. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 190.Sun R., Yan J., Willis W.D. Activation of protein kinase B/Akt in the periphery contributes to pain behavior induced by capsaicin in rats. Neuroscience. 2007;144:286–294. doi: 10.1016/j.neuroscience.2006.08.084. [DOI] [PubMed] [Google Scholar]
- 191.Xu J.T., Tu H.Y., Xin W.J., Liu X.G., Zhang G.H., Zhai C.H. Activation of phosphatidylinositol 3-kinase and protein kinase B/Akt in dorsal root ganglia and spinal cord contributes to the neuropathic pain induced by spinal nerve ligation in rats. Exp. Neurol. 2007;206:269–279. doi: 10.1016/j.expneurol.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 192.Carvalho T.T., Flauzino T., Otaguiri E.S., Batistela A.P., Zarpelon A.C., Cunha T.M., Ferreira S.H., Cunha F.Q., Verri W.A. Granulocyte-Colony Stimulating Factor (G-CSF) induces mechanical hyperalgesia via spinal activation of MAP kinases and PI3K in mice. Pharmacol. Biochem. Behav. 2011;98:188–195. doi: 10.1016/j.pbb.2010.12.027. [DOI] [PubMed] [Google Scholar]
- 193.Pezet S., Marchand F., D’Mello R., Grist J., Clark A.K., Malcangio M., Dickenson A.H., Williams R.J., McMahon S.B. Phosphatidylinositol 3-kinase is a key mediator of central sensitization in painful inflammatory conditions. J. Neurosci. 2008;28:4261–4270. doi: 10.1523/JNEUROSCI.5392-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yu L.N., Zhou X.L., Yu J., Huang H., Jiang L.S., Zhang F.J., Cao J.L., Yan M. PI3K contributed to modulation of spinal nociceptive information related to ephrinBs/EphBs. PLoS One. 2012;7:e40930. doi: 10.1371/journal.pone.0040930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jin D., Yang J.P., Hu J.H., Wang L.N., Zuo J.L. MCP-1 stimulates spinal microglia via PI3K/Akt pathway in bone cancer pain. Brain Res. 2015;1599:158–167. doi: 10.1016/j.brainres.2014.12.043. [DOI] [PubMed] [Google Scholar]
- 196.Horvath R.J., DeLeo J.A. Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J. Neurosci. 2009;29:998–1005. doi: 10.1523/JNEUROSCI.4595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Saponaro C., Cianciulli A., Calvello R., Dragone T., Iacobazzi F., Panaro M.A. The PI3K/Akt pathway is required for LPS activation of microglial cells. Immunopharmacol. Immunotoxicol. 2012;34:858–865. doi: 10.3109/08923973.2012.665461. [DOI] [PubMed] [Google Scholar]
- 198.Park H.Y., Kim N.D., Kim G.Y., Hwang H.J., Kim B.W., Kim W.J., Choi Y.H. Inhibitory effects of diallyl disulfide on the production of inflammatory mediators and cytokines in lipopolysaccharide-activated BV2 microglia. Toxicol. Appl. Pharmacol. 2012;262:177–184. doi: 10.1016/j.taap.2012.04.034. [DOI] [PubMed] [Google Scholar]
- 199.Tarassishin L., Suh H.S., Lee S.C. Interferon regulatory factor 3 plays an anti-inflammatory role in microglia by activating the PI3K/Akt pathway. J. Neuroinflammation. 2011;8:187. doi: 10.1186/1742-2094-8-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jiang Z., Wu S., Wu X., Zhong J., Lv A., Jiao J., Chen Z. Blocking mammalian target of rapamycin alleviates bone cancer pain and morphine tolerance via µ-opioid receptor. Int. J. Cancer. 2016;138(8):2013–2020. doi: 10.1002/ijc.29927. [DOI] [PubMed] [Google Scholar]
- 201.Jiang S., Zhang Z., Kang L., Wang Q., Zhang L., Chen H. Celecoxib reverts oxaliplatin-induced neuropathic pain through inhibiting PI3K/Akt2 pathway in the mouse dorsal root ganglion. Exp. Neurol. 2016;275(Pt 1):11–16. doi: 10.1016/j.expneurol.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 202.Xu B., Guan X.H., Yu J.X., Lv J., Zhang H.X., Fu Q.C., Xiang H.B., Bu H.L., Shi D., Shu B., Qin L.S., Manyande A., Tian Y.K. Activation of spinal phosphatidylinositol 3-kinase/protein kinase B mediates pain behavior induced by plantar incision in mice. Exp. Neurol. 2014;255:71–82. doi: 10.1016/j.expneurol.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 203.Anand P., Shenoy R., Palmer J.E., Baines A.J., Lai R.Y., Robertson J., Bird N., Ostenfeld T., Chizh B.A. Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur. J. Pain. 2011;15:1040–1048. doi: 10.1016/j.ejpain.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 204.Singh D., Smyth L., Borrill Z., Sweeney L., Tal-Singer R. A randomized, placebo-controlled study of the effects of the p38 MAPK inhibitor SB-681323 on blood biomarkers of inflammation in COPD patients. J. Clin. Pharmacol. 2010;50:94–100. doi: 10.1177/0091270009347873. [DOI] [PubMed] [Google Scholar]
- 205.Tong S.E., Daniels S.E., Black P., Chang S., Protter a., Desjardins P.J. Novel p38 Mitogen-Activated Protein Kinase Inhibitor Shows Analgesic Efficacy in Acute Postsurgical Dental Pain. J. Clin. Pharmacol. 2012;52:717–728. doi: 10.1177/0091270011405496. [DOI] [PubMed] [Google Scholar]
- 206.Pareek A., Suthar M., Rathore G., Bansal V. Feverfew (Tanacetum parthenium L.): A systematic review. Pharmacogn. Rev. 2011;5:103. doi: 10.4103/0973-7847.79105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Studzińska-Sroka E., Znajdek-Awizeń P., Gawron-Gzella A. Studies on the antimigraine action of Feverfew (Tanacetum parthenium (L.) Sch. Bip.). Wiad. Lek. 2013;6:195–199. [PubMed] [Google Scholar]
- 208.Wider B., Pittler M.H. E.E. Feverfew for preventing migraine. Cochrane Database Syst. Rev. 2015:CD002286. doi: 10.1002/14651858.CD002286.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sen M., Thomas S.M., Kim S., Yeh J.I., Ferris R.L., Johnson J.T., Duvvuri U., Lee J., Sahu N., Joyce S., Freilino M.L., Shi H., Li C., Ly D., Rapireddy S., Jonathan P., Li P., Wang L., Chiosea S., Seethala R.R., Gooding W.E. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2:694–705. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bendell J.C., Hong D.S., Burris H.A., Naing A., Jones S.F., Falchook G., Bricmont P., Elekes A., Rock E.P., Kurzrock R. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother. Pharmacol. 2014;74:125–130. doi: 10.1007/s00280-014-2480-2. [DOI] [PubMed] [Google Scholar]