

Abstract
Advanced glycated end-products (AGEs) are ligands of the receptor for AGEs and increase in diabetic disease. MAPK organizer 1 (Morg1) via its binding partner prolyl-hydroxylase domain (PHD)-3 presumably plays a role in the regulation of hypoxia-inducible factor (HIF)-1α and HIF-2α transcriptional activation. The purpose of this study was to analyze the influence of AGEs on Morg1 expression and its correlation to PHD3 activity and HIF-transcriptional activity in various renal cell types. The addition of glycated BSA (AGE-BSA) significantly up-regulated Morg1 mRNA levels in murine mesangial cells and down-regulated it in murine proximal tubular cells and differentiated podocytes. These effects were reversible when the cells were preincubated with a receptor for α-AGE antibody. AGE-BSA treatment induced a relocalization of the Morg1 cellular distribution compared with nonglycated control-BSA. Analysis of PHD3 activity demonstrated an elevated PHD3 enzymatic activity in murine mesangial cells but an inhibition in murine proximal tubular cells and podocytes after the addition of AGE-BSA. HIF-transcriptional activity was also affected by AGE-BSA treatment. Reporter gene assays and EMSAs showed that AGEs regulate HIF- transcriptional activity under nonhypoxic conditions in a cell type-specific manner. In proximal tubular cells, AGE-BSA stimulation elevated mainly HIF-1α transcriptional activity and to a lesser extent HIF-2α. We also detected an increased expression of the HIF-1α and the HIF-2α proteins in kidneys from Morg1 heterozygous (HZ) placebo mice compared with the Morg1 wild-type (WT) placebo-treated mice, and the HIF-1α protein expression in the Morg1 HZ streptozotocin-treated mice was significantly higher than the WT streptozotocin-treated mice. Analysis of isolated mesangial cells from Morg1 HZ (±) and WT mice showed an inhibited PHD3 activity and an increased HIF-transcriptional activity in cells with only one Morg1 allele. These findings are important for a better understanding of the molecular mechanisms of diabetic nephropathy.
The formation of advanced glycated end-products (AGEs) occurs as a result of the Maillard reaction, a nonenzymatic modification of proteins, lipids, and nucleic acids by reducing sugars (1–4). The accumulation of AGEs is a physiological process associated with aging or is pathologically enhanced in diabetes mellitus types 1 and 2 due to continual high levels of glucose (5–7). The glycated proteins no longer function via their own signaling pathways but instead are recognized by the receptor for advanced glycation end-products (RAGE) and thereby contribute to the development and pathogenesis of diabetic complications (8–10), cardiovascular diseases (11, 12), and atherosclerosis (13). RAGE is a pattern recognition receptor present in diverse cell types (14), and it has been shown to bind as well to non-AGE-related ligands, such as S100/calgranulins (15) and HGMB1 (16, 17). These findings suggest a role of RAGE in inflammatory processes (17, 18). It has also been demonstrated that the RAGE/AGE ligand interaction is associated with the activation of the transcription factor nuclear factor-κB (NF-κB), therefore linking RAGE to the activation of the transcription machinery in cells expressing RAGE (19–21).
The discovery of the MAPK organizer 1 (Morg1) as a binding partner of a prolyl-hydroxylase domain-containing protein-3 (PHD3) raised the question of whether Morg1 is involved in the regulation of the hypoxia-inducible factor (HIF)-1α and/or HIF-2α, which are regulated by prolyl-hydroxylase domains (22, 23). Recently we demonstrated that in renal cells angiotensin II (ANG II) activation is associated with a down-regulation of Morg1 expression in murine proximal tubular cells (MTC) and differentiated podocytes and is correlated with an inhibition of PHD3 prolyl-hydroxylase activity (24). Moreover, reporter assays revealed an elevation of the HIF-transcriptional activation under normoxic conditions via ANG II (24). In addition, a role of Morg1 has been reported in studies of a renal ischemia-reperfusion model in mice because Morg1 heterozygote (±) mice exhibited less kidney injury compared with wild-type (WT) mice (25). Furthermore, in a mouse model of focal cerebral ischemia-reperfusion, the stroke areas were smaller in Morg1+/− mice compared with the WT (26). In this study, we have focused on how AGEs affect expression of Morg1 in kidney cells via binding to PHD3, impacting HIF-transcriptional activation. The aim is to better understand the molecular mechanisms underlying diabetic nephropathy.
Materials and Methods
Cells and cell culture
Mouse mesangial cells (27) and mouse proximal tubular cells (28) were established from mesangial, respectively, proximal tubular cells originating from kidneys of SJL/J (H-2s) mice, which were then for long-term culturing transformed with a nonreplicating, noncapsid-forming Simian virus-40 virus. Murine mesangial cells (MMCs), and MTCs cells were routinely cultured in DMEM medium (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (FCS) (PAN Biotech GmbH) and 100 U/mL penicillin/streptomycin (Sigma-Aldrich Chemie GmbH). Subconfluent MMCs and MTCs were synchronized in DMEM supplemented with 0.1% heat-inactivated FCS and 100 U/mL penicillin/streptomycin overnight followed by stimulation with 5 mg/mL control-BSA (Co-BSA) or glycated BSA (AGE-BSA) for the corresponding time period as shown in the figure legend.
Glomerular podocytes are terminally differentiated cells and do not proliferate in cell culture; therefore, for cell culture studies of the differentiated podocytes, we used the conditionally immortalized podocytes (29), which were provided by Peter Mundel (Florida University, Gainesville, Florida). The undifferentiated and differentiated state of the cells is controlled by the temperature sensitive Simian virus-40 T large antigen and the presence of γ-interferon (29). Briefly, for cell expansion the conditionally immortalized podocytes were cultured in RPMI 1640 medium (Invitrogen GmbH) supplemented with 10% heat-inactivated FCS and 10 U/mL mouse γ-interferon for 3 weeks at 33°C (permissive conditions) and 5% CO2. A temperature switch to 37°C (nonpermissive conditions) and removal of γ-interferon initiate the differentiation of the mouse podocytes, characterized by the expression of synaptopodin. For further experiments the podocytes were grown in nonpermissive conditions 37°C, 5% CO2 for 2 weeks to differentiate in RPMI 1640 medium (Invitrogen) supplemented with 10% heat-inactivated FCS, and 100 U/mL penicillin/streptomycin.
Podocyte differentiation was routinely tested and confirmed by immunological detection of synaptopodin expression using an antisynaptopodin (C-19) antibody (Sigma-Aldrich) (data not shown). The differentiated podocytes were further used for the analysis. Subconfluent differentiated podocytes were synchronized in RPMI 1640 medium supplemented with 0.1% heat-inactivated FCS and 100 U/mL penicillin/streptomycin overnight followed by stimulation with 5 mg/mL Co-BSA or AGE-BSA for the corresponding time period as shown in the figure legend. The isolation and culturing of the primary mesangial cells from Morg1+/− heterozygous (HZ) and WT mice was performed as previously described (24, 27). In brief, the mesangial cells were isolated from kidneys of 4-month-old WT (C57BL/6J background) or Morg1+/− HZ mice. The kidneys were washed several times in PBS, and the cortices were separated followed by sieving in 100- and 70-μm sieves, resulting in glomeruli purification. Isolated glomeruli were treated with collagenase (0.2 mg/mL) for 30 minutes at 37°C. Glomeruli were resuspended in RPMI 1640 medium containing 15% heat-inactivated serum to inhibit the collagenase activity, and the suspension was centrifuged for 5 minutes at 1000 rpm. The supernatant was discarded and the cells and the glomeruli were plated on 6-cm plates in RPMI 1640 supplemented with 10% heat-inactivated FCS. The glomeruli outgrowth spindle-like mesangial cells and the epithelial cells were further characterized and cloned. Isolated mesangial cells were grown in RPMI 1640 medium containing 10% heat-inactivated serum, and passages between 14 and 20 were used in the experiments performed with the primary mesangial cells.
Animals and experimental design
Animals were maintained in a pathogen-free facility, receiving a standard diet and water. All animal experiments were approved by the Ethics Committee of the State of Thuringen and the University of Jena. Diabetes mellitus was induced in WT (C57BL/6J background) and HZ Morg1+/− mice by five consecutively daily ip injections of streptozotocin (STZ; 50 mg/kg body weight; Sigma-Aldrich Chemicals) dissolved in 10 mmol/L sodium citrate buffer (pH 5.5). Control animals were ip injected with only sodium citrate buffer (30). The development of diabetes was analyzed by measurement of the blood glucose concentrations from tail-vein probes using a glucometer FreeStyle Lite (Abbgott Diabetes Care). After a period of 30 days, the animals were killed, and the kidneys were collected for further analysis as needed. Routinely, one kidney per mouse was fixed in 10% phosphate-buffered formalin and paraffin embedded for further histological and immunohistochemical studies. The remaining kidney was homogenized using the SpeedMill P12 homogenizer (Analytik Jena Bio Solutions), and total RNA or proteins were isolated as described below.
RNA isolation, reverse transcription, and real-time PCR
Total RNA was isolated 1 hour, 6 hours, 12 hours, 24 hours, and 48 hours after Co-BSA or AGE-BSA treatment using a RNA Easy kit (QIAGEN GmbH), and 1 μg total RNA underwent reverse transcription for 1 hour at 37°C using an murine leukemia virus-reverse transcription system (Invitrogen). The following primers designated from mouse mRNA sequences were used for real-time PCR analysis of mRNA: Morg1, forward primer, 5′-CCTATCACCTGCACCTGCTT-3′, reverse primer, 5′-CACTTTCCCGTCTTCAGAGC-3′, annealing temperature 58°C; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), forward primer, 5′-TGTCAGCAATGCATCCTGCA-3′, reverse primer 5′-ATGTCATCATACTTGGCAGGTT-3′, annealing temperature 58°C; and RAGE, forward primer, 5′-AGCCTGAAGGTGGAATAGTCG-3′, reverse primer, 5′-GCCGGTTTCTGTGACCCTGA-3′, annealing temperature 55°C. Expression of the gene of interest and GAPDH were performed using monoplex analysis (31, 32) in an Eppendorf Mastercycler realplex instrument (Eppendorf Instruments GmbH). The change in cycle threshold value method was used for quantitative analysis of mRNA expression (31).
Western blot analysis
Cells were lysed in Complete M lysis buffer (Roche Diagnostic) supplemented with cocktail inhibitors (Roche) and 100 mM Na3VO4 (Sigma). An equal amount of protein lysate was separated on a 10% SDS-PAGE, followed by semidry Western blot transfer for analysis of the protein expression. The primary antibodies used for the analysis are as follows: anti-Morg1 rabbit antibody (24) (1:1000 dilution), anti-PHD3 (Santa Cruz Biotechnology) (1:500 dilution), anti-HIF-2α (Santa Cruz Biotechnology) (1:500 dilution), anti-Nϵ-CML (Cell Biolabs, Inc) (1:2000 dilution), antivinculin (Santa Cruz Biotechnology) (1:2000 dilution), anti-β-actin (Sigma) (1:5000 dilution). The appropriate secondary, horseradish peroxidase-conjugated antibodies were from KPL and were diluted 1:10 000. The proteins were detected by enhanced chemiluminescence reagents (PerkinElmer) and LAS 3000 system (Fujifilm Life Science). The protein expression was normalized against the expression of vinculin respectively β-actin and presented as fold relative to the control conditions. Quantification of intensities was performed using the Multigauge software (Fujifilm Life Science).
Immunohistochemical analysis
For immunofluorescent staining, 1 × 105 cells were seeded in 4-well chamber permanox slides (NUNC; Thermo Fischer Scientific Inc). The cells were stimulated as indicated and fixed in 4% paraformaldehyde and 0.1% Triton X-100 in PBS for 20 minutes at room temperature, followed by blocking in 5% BSA in PBS for 1 hour. Routinely the primary antibodies were incubated at 4°C overnight in blocking buffer. The secondary antibody Alexa 488- or Alexa Cy3-conjugated were both from Invitrogen and were used in 1:500 dilutions. Nuclei were counterstained with 4′,6-diamidino 2-phenylindole (DAPI), and the slides were embedded with Kaisersgelatine (Merck). Slides were analyzed using Axioplan, a fluorescent microscope, and AxioVision Rel 4.6 software, both from Zeiss. Quantifications were done by measuring the relative intensity of at least 100 cells per treatment and expressed in percent relative to the controls. The experiments were repeated at least three times.
For immunohistochemistry performed on kidney sections from WT and Morg1+/− mice, 4-μm sections were deparaffinized, and the antigen retrieval was performed in citrate buffer (pH 6.0; Dako Target Retrieval Solution; DAKO North America Inc) using the microwave-based technique, and endogenous peroxidase was inactivated by incubation for 10 minutes at room temperature with 3% hydrogen peroxide. The sections were further subjected to immunohistochemical staining using a rabbit IgG VECTASTAIN Elite ABC kit (Vector Laboratories Inc) according to the manufacturer's instructions. The primary antibodies rabbit anti-HIF-1α (1:200 dilution) or rabbit anti-HIF-2-α (1:200 dilution) were both purchased from Abcam and added to the sections overnight at 4°C. To increase the staining specificity, the sections additionally blocked the kidney sections for 30 minutes at room temperature before the addition of the secondary antibody. As a peroxidase substrate 3–3′, diaminobenzidin in chromogen solution (DAKO North America Inc) was used, and the sections were mounted with aqueous mounting medium VectaMountAQ (Vector Laboratories Inc). The sections contained no nuclear counterstaining because it is well documented that the activated HIFs are localized into the nucleus of the cells.
In parallel, the control staining was performed where instead of the primary antibody, the sections were incubated with an isotype-specific rabbit IgG (Santa Cruz Biotechnology Inc). Images of the staining were recorded by a computer-assisted microscope with a digital camera, and AxioVision 4.6 Rel software was used (Zeiss). At least four mice per group were analyzed for HIF-1/2α immunohistological detection in kidney sections. Ten images of each individual kidney sample were scanned in the monochrome mode of the camera (magnification, ×400). All images were taken under constant conditions. The glomerular staining of the kidney sections was measured by highlighting the glomerular area, and the mean densitometric gray values were measured using AxioVision 4.6 software (Zeiss). The average of gray labels obtained for each individual kidney sample was used as an equivalent for the corresponding staining intensity.
HIF-1α EMSA
The EMSA assay was performed according to the protocol previously described (33). HIF-1α consensus oligonucleotide, 5′-TCTGTACGTGACCACACTCACCTC-3′ (sc-2625) and HIF-1α mutant oligonucleotide, 5′-TCTGTAAAAGACCACACTCACCTC-3′ (sc 2626) were purchased from Santa Cruz Biotechnology, Inc. Unlabeled cold competitor probe was added 15 minutes prior to the labeled probe. Supershift assays were performed using an anti-HIF-1α antibody (HIF-1α [C-19] X) or HIF-2α antibody (EPAS-1 [C-16] X) both purchased from Santa Cruz Biotechnology. The antibodies were added 15 minutes prior to the labeled oligonucleotides. The reactions were performed for 30 minutes in 20 μL final volume, and the DNA-protein complexes were resolved through 6% nondenaturing PAGE, dried, and exposed on a film for visualization or detected using a Fujufilm FLA 500 image analyzer.
HIF-transcriptional activity assay
HIF-1 promoter activity was analyzed using a reporter gene vector, pHIF-Luc, (Signosis), which contains a cis-element (DNA binding sequence), a minimal promoter, and a firefly luciferase gene. Activated HIF-1α or HIF-2α transcription factors bind to the cis-element and transactivate the expression of the luciferase gene correlating with the measured luciferase enzyme activity. Therefore, in this assay the luciferase activity represents the activation of both transcription factors HIF-1α and HIF-2α. Briefly, the cells were seeded in 1 × 105 cells/ well in 12-well plates and transfected the next day using 1 μg of the HIF-Luc reporter construct and 0.1 μg of β-galactosidase control vector (CLONTECH) with Lipofectamine 2000 reagent (Invitrogen) for MMCs and MTCs or Lipofectamine Plus reagent (Invitrogen) for differentiated podocytes in 0.1% FCS containing DMEM, or RPMI 1640 medium, respectively. The transfections were carried out for 24 hours, followed by stimulation with 5 mg/mL Co-BSA or 5 mg/mL AGE-BSA for 24 hours. Afterward the cells were washed with PBS and lysed in 100 μL of the 1× CCLR (Promega). The luciferase activity was assayed using the Luciferease reagent (Promega) and the β-galactosidase activity was measured via β-galactosidase assay kit (CLONTECH on a luminometer mode using an Infinite M200 microplate reader and i-control software both from Tecan). The luciferase activity was normalized for β-galactosidase and expressed as a percentage of the luciferase activity detected in cells treated with Co-BSA for each treatment and cell type.
Prolyl-hydroxylase activity assay
Analysis of PHD3 activity was performed using an equal amount of protein lysates lysed in Complete Lysis M (Roche) supplemented with a cocktail of proteinases inhibitors (Roche) and 100 mM Na3VO4. The HIF-PHD3 activity was measured by the end point method, based on hydroxylation-coupled decarboxylation of [1-14C]-2-oxoglutarate (PerkinElmer), and HIF-1α peptide DLDLEMLAPYIPMDDDFQL (Innovagen), corresponding to residues 556–574 from the human HIF-1α, was used as a substrate. The formed 14CO2 captured by alkaline solution was measured with a liquid scintillation counter (23).
The assays were performed exactly as previously described (24). Briefly, the PHD3 protein was immunoprecipitated from 300 μg protein lysates using an anti-PHD3 antibody (Abcam), and the immunocomplexes were collected with protein G/Plus A beads (Calbiochem). The beads were then washed with a buffer containing 80 mM K2HPO4, 5.0 mM Na2HPO4, and 0.10 μM CaCl2 (pH 7.4). The PHD3 activity was analyzed in 400 μL reaction washing buffer supplemented with 1.0 mM dithiothreitol, 2.0 mM ascorbate, 5.0 μM FeSO4, 0.16 mM 2-oxoglutarate (1.6 × 105 dpm/mL), 2.0 mg/mL BSA, 0.60 mg/mL catalase, 50 μM HIF-1α peptide, and 20 μL of immunoprecipitated PHD3 enzyme. All components except [1-14C]-2-oxoglutarate were mixed and kept on ice. Finally, [1-14C]-2-oxoglutarate (PerkinElmer) was added, and the reaction tubes were immediately sealed with rubber caps with filter paper (8 × 20 mm, soaked in freshly prepared 1 M NaOH) on a hook. The reactions were performed at 37°C for 30 minutes, followed by injection of 400 μL of 1.0 M NaH2PO4 (pH 4.0), and the probes were shaken at 300 rpm for 2 hours at room temperature. Filter papers with captured 14CO2 were placed into 10 mL Hionic-Fluor scintillation liquid (PerkinElmer) and the radioactivity was measured for 20 minutes by a scintillation counter LS 6500 Scintillation counter (Beckman Coulter). Similar reactions were performed in the absence of the HIF-1α peptide and the reactions were measured as above. The differences between the two reactions were subtracted and the activity of the PHD3 was calculated.
Statistical analysis
All data are reported as means ± SD. Statistical analysis was performed using the statistical package SPSS for Windows version 11.0 (SPSS, Inc). Results were analyzed with the Kruskal-Wallis test followed by the Mann-Whitney U test. Differences were considered significant when P < .05.
Results
RAGE expression in renal cells
The expression of the RAGE has already been demonstrated in the kidney, but its expression in different renal cell types in vivo is somewhat controversial (34–38). Initially, we investigated the presence of RAGE mRNA expression via semiquantitative RT-PCR using cDNAs from three different renal cell lines: MMC, MTCs, and differentiated murine podocytes treated for 24 hours with Co-BSA or AGE-BSA. As shown in Figure 1A, we detected a 245-bp PCR fragment as predicted for all three cell lines. We also compared the RAGE mRNA expression levels between the three renal cell lines treated with Co-BSA. As presented in Figure 1B, different expressions of RAGE mRNA were detected with real-time PCR. We found similar RAGE mRNA expression levels in differentiated podocytes and proximal tubular cells, whereas the RAGE mRNA in mesangial cells was significantly lower compared with podocytes or MTCs (Figure 1B). Nϵ-carboxymethyl-lysine (CML) is one major AGE-modified protein found in the serum proteins and tissues of diabetic patients (39, 40). We also detected CML-modified BSA in our AGE-BSA preparation by Western blot (data not shown). Therefore, we assessed the presence of CML-modified BSA by Western blot in protein lysates obtained from podocytes, MMCs, and MTCs treated with Co-BSA or AGE-BSA. We observed a CML-BSA-specific band only in the AGE-BSA-, but not in the Co-BSA-treated cells (Figure 1C). Thus, we demonstrated the uptake of CML-BSA in all three renal cell lines.
Figure 1.
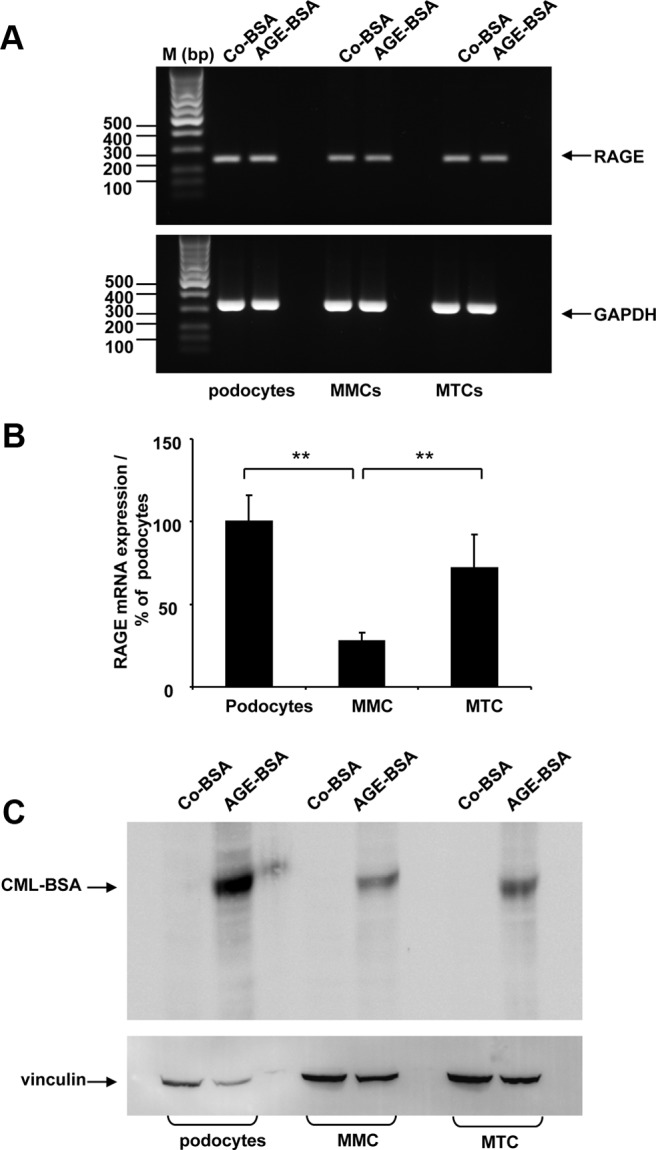
A–C, Expression of receptor of AGEs in renal cells. A, Semiquantitative RT-PCR analysis of RAGE expression in differentiated podocytes, MMCs, and MTCs. Cells were treated with Co-BSA and AGE-BSA for 24 hours. Upper panel, Detection of RAGE expression; lower panel, analysis of GAPDH expression performed in parallel reactions from the same cDNAs. RAGE expression was detected in all cell lines. The molecular weight of the bands from the DNA marker is shown. The experiment was repeated three times with similar results. One representative image from the agarose gel electrophoresis is shown. B, Comparison of RAGE mRNA expression in Co-BSA-treated differentiated podocytes, MMCs, and MTCs by real-time PCR analysis. The expression of RAGE in differentiated podocytes was arbitrarily considered as 100%. Differentiated podocytes and MTCs show similar levels of RAGE mRNA, whereas MMCs revealed a significant lower level of RAGE mRNA. **, P < .005 vs podocytes (n = 9). C, Detection of CML-modified BSA (CML-BSA) in protein lysates obtained from differentiated podocytes, MMCs, and MTCs treated with either Co-BSA or AGE-BSA analyzed by Western blot. Immunological reaction specific for CML-BSA protein was present only in AGE-BSA-treated cells (upper panel). Protein loading was controlled by vinculin protein expression (lower panel). A representative Western blot from three experiments is shown.
AGEs influence Morg1 expression in renal cells
The expression of RAGE in differentiated podocytes, MMCs, and MTCs prompted us to further elucidate the influence of AGEs on Morg1 expression in renal cells. Therefore, we challenged differentiated murine podocytes, MMCs, and MTCs with 5 mg/mL AGE-BSA or the same amount of Co-BSA and investigated the expression of Morg1 mRNA with real-time PCR in a time course-dependent manner. The cells were treated for 1 hour, 6 hours, 12 hours, 24 hours, and 48 hours with Co-BSA or AGE-BSA. We observed that the Morg1 mRNA level was significantly suppressed in differentiated podocytes from 6 hours up to 48 hours of treatment with AGE-BSA compared with Co-BSA (Figure 2A). In proximal tubular cells (Figure 2B) the addition of AGE-BSA inhibited significantly the Morg1 mRNA starting from 12 hours to 48 hours, whereas it was significantly increased in MMCs (Figure 2C) after 24 hours and 48 hours after AGE-BSA application to the cells.
Figure 2.

A–C, Influence of AGE-BSA treatment on Morg1 mRNA expression in renal cells. Morg1 mRNA expression was analyzed by real-time PCR in differentiated podocytes, MTCs, and MMCs in a time-dependent manner from cells treated for 1 hour, 6 hours, 12 hours, 24 hours, and 48 hours with Co-BSA or AGE-BSA. Morg1 mRNA expression is presented in percent relative to Co-BSA treated cells for each time point. A, Morg1 mRNA expression in differentiated podocytes. AGE-BSA treatment significantly inhibits Morg1 mRNA expression starting from 6 hours up to 48 hours. *, P < .05 vs Co-BSA (n = 6). B, Morg1 mRNA expression in MTCs. Morg1 expression was significantly down-regulated in MTCs after addition of AGE-BSA from 12 hours up to 48 hours. *, P < .05; **, P < .01 vs Co-BSA (n = 6). C, Morg1 mRNA expression in MMCs. Addition of AGE-BSA increased Morg1 expression in MMCs after 24 hours and 48 hours of treatment relative to Co-BSA. *, P < .05; **, P < .005 vs Co-BSA (n = 6).
Because the 24-hour treatment of differentiated podocytes, MTCs, and MMCs with AGE-BSA represented the influence of AGE-BSA on Morg1 expression, we considered the 24-hour treatment with Co-BSA or AGE-BSA as the most appropriate time period for our further analysis in all three cell lines. No apoptotic effects of AGE-BSA were detected in the cells after the 24-hour AGE-BSA incubation period (data not shown). Next, we tested whether AGEs affect Morg1 mRNA expression through the receptor for AGE-RAGE. We blocked the RAGE/AGE interaction by application of an α-RAGE antibody to the cells prior to AGE-BSA stimulation. We found that the RAGE blockade reversed Morg1 expression in differentiated podocytes, MMCs, and MTCs (Figure 3, A–C). We also tested the effect of RAGE antibody alone or in the presence of Co-BSA on Morg1 expression, but its impact on Morg1 expression was similar to Co-BSA treatment (Figure 3, A–C). Therefore, the observed AGE-BSA-induced changes in Morg1 mRNA expressions are likely RAGE dependent.
Figure 3.

A–C, AGE-BSA affects Morg1 mRNA expression in renal cells in a RAGE-dependent manner. Renal cells were stimulated with Co-BSA or AGE-BSA for 24 hours in the presence or absence of an α-RAGE antibody. In addition, the effect of the α-RAGE antibody alone on Morg1 expression was assessed. Where indicated, the α-RAGE antibody was added 1 hour prior to Co-BSA or AGE-BSA addition to the cells. Morg1 mRNA expression was analyzed by real-time PCR and is presented in percentage relative to Co-BSA-treated cells. A, Influence of RAGE blockade on Morg1 mRNA expression in differentiated podocytes. AGE-BSA treatment significantly inhibits Morg1 mRNA expression. *, P < .05 vs Co-BSA (n = 15). Blocking the RAGE receptor with an antibody (α-RAGE) partly abolished this suppression. B, Influence of RAGE blockade on Morg1 mRNA expression in MTCs. Morg1 expression was significantly down-regulated in MTCs after addition of AGE-BSA. #, P < .03 vs Co-BSA (n = 15). Addition of α-RAGE antibody prior to AGE-BSA treatment reversed the inhibitory effect of AGE-BSA on Morg1 expression. **, P < .005 vs AGE-BSA (n = 8). C, Influence of RAGE blockade on Morg1 mRNA expression in MMCs. Addition of AGE-BSA increased Morg1 expression in MMCs. *, P < .05 vs Co-BSA (n = 12). Blocking of the RAGE/AGE interaction with α-RAGE antibody antagonized the AGE-BSA-dependent increase of Morg1 expression. *, P < .05 vs AGE-BSA (n = 8).
AGE-BSA affects Morg1 cellular distribution in renal cells
It has already been shown that Morg1 is present in the nucleus as well as in the cytoplasm of cells (22, 24). Consequently, we examined whether AGE-BSA also affects the cellular distribution of the Morg1 protein in differentiated podocytes, MMCs, and MTCs using immunostaining. We found that in Co-BSA-treated podocytes, the Morg1 protein was mainly localized in the cell nucleus (Figure 4A), whereas AGE-BSA stimulation induced a nuclear export of the Morg1 protein into the cytoplasm (Figure 4B). Analysis of MTCs demonstrated that the Morg1 protein was present in the nucleus as well in the cytoplasm of the proximal tubular cells when Co-BSA was added to the cells (Figure 4C). On the other hand, AGE-BSA application was associated with a reduction of the overall fluorescence intensity of the cells (Figure 4D). Similar to differentiated podocytes was the Morg1 protein localization in MMCs. It was mostly detected in the nucleus under Co-BSA conditions (Figure 4E), whereas AGE-BSA treatment was associated predominantly with a cytoplasmic staining (Figure 4F). Measurements of the fluorescent intensity revealed that AGE-BSA treatment reduced Morg1 expression in differentiated podocytes and MTC cells but increased the Morg1 protein level in MMC cells compared with Co-BSA-incubated cells (Figure 4G).
Figure 4.

A–G, Cellular distribution of Morg1 protein in renal cells treated with Co-BSA and AGE-BSA. Morg1 cellular localization was analyzed in podocytes, MTCs, and MMCs. Cells were plated in chamber slides and were stimulated for 24 hours with Co-BSA or AGE-BSA. Morg1 expression was detected by immunofluorescence. Morg1 localization was analyzed using Zeiss, an Axioplan fluorescent microscope, and AxioVision software. Representative images of Co-BSA- and AGE-BSA-treated cells are shown. Bar, 20 μm (n = 6). A, Morg1 localization in podocytes stimulated with Co-BSA. B, Morg1 cellular distribution in AGE-BSA-treated podocytes (n = 6). C, Morg1 localization in MTCs stimulated with Co-BSA (n = 6). D, Morg1 cellular distribution in AGE-BSA-treated MTCs (n = 6). E, Morg1 localization in MMCs stimulated with Co-BSA (n = 6). F, Morg1 cellular distribution in AGE-BSA-treated MMCs (n = 6). G, Densitometry quantification of Morg1 immunological staining in differentiated podocytes, MTCs, and MMCs. The relative intensity of Morg1 protein is presented in percentage relative to Co-BSA. *, P < .005; **, P < .003 (n = 6).
AGE-BSA modulates PHD3 prolyl-hydroxylase activity
Morg1 binds to PHD3 (22). Therefore, we further tested the influence of AGE-BSA on PHD3 enzymatic activity in vitro, using a synthetic HIF-1α peptide as a PHD3 substrate. The PHD3 protein was immunoprecipitated from protein lysates of cells treated with Co-BSA or AGE-BSA. In parallel experiments, the binding of AGE-BSA to its receptor was prevented by preincubation with an anti-RAGE antibody. We measured a significant inhibition of PHD3 activity in differentiated podocytes (Figure 5A) and MTCs (Figure 5B) treated with AGE-BSA. On the other hand, pretreatment of differentiated podocytes and MTCs with the α-RAGE antibody abolished AGE-BSA-dependent changes in PHD3 activity (Figure 5, A and B). In contrast to differentiated podocytes and MTCs, in MMCs PHD3 activity was significantly elevated relative to Co-BSA-stimulated cells (Figure 5C). This increase was regulated via RAGE because the addition of the α-RAGE antibody suppressed AGE-BSA-dependent PHD3 activation (Figure 5C). We also tested the effect of the RAGE antibody when it was added alone to the cells, but it did not influence PHD3 activity in the cells compared with the Co-BSA (Figure 5, A–C). In addition, we investigated PHD3 protein expression in renal cells stimulated with Co-BSA or AGE-BSA by Western blot. We found an elevated PHD3 protein level in MMCs with AGE-BSA treatment, whereas in MTCs and differentiated podocytes, the protein level of PHD3 was significantly inhibited compared with the Co-BSA-stimulated cells (Figure 5D).
Figure 5.

A–D, PHD3 hydroxylase activity assay in renal cells treated with AGE-BSA. The cells were treated with 5 mg/mL Co-BSA or AGE-BSA for 24 hours. In an inhibitor study, the AGE receptor was blocked by addition of anti-RAGE antibody alone (α-RAGE) or together with AGE-BSA. An equal amount of protein lysate was subjected to the PHD3 hydroxylase activity assay using HIF-1α peptide as a substrate. A, PHD3 activity assay in differentiated podocytes. *, P < .05 vs Co-BSA (n = 8). B, PHD3 activity in MTCs. #, P < .01 vs Co-BSA (n = 8). C, PHD3 activity in MMCs. *, P < .05 vs Co-BSA; **, P < .001 vs AGE-BSA (n = 8). AGE-BSA treatment activates PHD3 prolyl-hydroxylase activity in MMCs and inhibits it in MTCs and podocytes relative to the cells treated with Co-BSA alone. D, Expression of PHD3 protein in differentiated podocytes, MMCs, and MTCs treated with Co-BSA or AGE-BSA analyzed by Western blot. A representative image of three independent experiments is shown. Upper panel, PHD3 Western blot; lower panel, β-actin protein expression as loading control. The graph presents a densitometry quantification of PHD3 protein expression in treated cells. The relative intensity of PHD3 protein is presented in percent relative to Co-BSA. *, P < .05 vs Co-BSA (n = 3).
Regulation of HIF-transcriptional activation via AGE-BSA in renal cells
We next asked whether AGE-BSA can affect HIF-1α or HIF-2α transcriptional activation in renal cells via altered PHD3 activity. We explored a pHIF-Luc reporter vector and assayed the firefly luciferase enzyme activity as a measurement for the transcriptional activation for HIF-1α/HIF-2α transcription factors. Transfection efficiency was monitored by β-galactosidase control plasmid cotransfection. Our data show that AGE-BSA significantly induced HIF-reporter activity in differentiated podocytes (Figure 6A) and proximal tubular cells (Figure 6B), whereas it inhibited the luciferase activity in MMCs (Figure 6C) relative to Co-BSA-incubated cells. We also investigated HIF activity using EMSAs from nuclear protein extracts (Figure 7, A–C). The EMSA experiment unveiled that the strongest HIF transcriptional activation was detected in MTCs stimulated with AGE-BSA, whereas Co-BSA stimulation showed no transcriptional activity of HIF-1α or HIF-2α (Figure 7C). We then performed a more detailed EMSA analysis using nuclear protein extracts of MTCs treated with AGE-BSA.
Figure 6.

A–C, AGE-BSA modulates HIF-transcriptional activity in renal cells as detected with reporter gene assay analysis. Luciferase activity was normalized to β-galactosidase activity and presented as percentage relative to Co-BSA-treated cells. A, HIF reporter activity in differentiated podocytes. **, P < .001 (n = 18). B, HIF reporter activity in MTC cells. **, P < .001 (n = 18). C, HIF reporter activity in MMCs. *, P < .005 (n = 12). HIF-transcriptional activity was significantly stimulated in differentiated podocytes and MTCs treated with AGE-BSA, whereas it was suppressed in MMCs.
Figure 7.

A–D, Influence of AGE-BSA on HIF-1/2α transcriptional activity analyzed by EMSA. Nuclear proteins were extracted from podocytes, MMCs, and MTCs treated with Co-BSA or AGE-BSA for 24 hours and were subjected to EMSA using radioactively labeled HIF-1α double-stranded consensus oligonucleotides. Representative images of the EMSAs are shown (n = 4 independent experiments with qualitatively similar results). A, EMSA analysis of podocyte nuclear extracts (NE) from Co-BSA- or AGE-BSA-treated cells. B, EMSA analysis of MMC nuclear extracts (NE) from Co-BSA- or AGE-BSA-treated cells. C, EMSA analysis of MTC nuclear extracts (NE) from Co-BSA- or AGE-BSA-treated cells. The lane labeled with − was only loaded with the oligonucleotides without protein extracts as a negative control. A specific band is already found in podocytes and MMCs under a basal (Co-BSA) condition. In these two cell lines, AGE-BSA failed to further induce binding of transcription factors in vitro. In contrast, in MTCs (C), AGE-BSA induced a strong binding of nuclear proteins to HIF-1α double-stranded consensus oligonucleotides. D, EMSA analysis of MTC nuclear protein extracts (NE) from AGE-BSA-treated cells. The specificity of the reaction was tested by incubation of the NEs with cold competitor oligonucleotides prior to the assay or by mutant (−) oligonucleotides. In addition, the NEs were subjected to a supershift assay in which the HIF-1α and HIF-2α antibodies were added alone or together to the corresponding reaction. The HIF-1α supershift is marked with a white arrowhead, and the additional bands detected in the EMSA when the HIF-1α antibody was added to the reactions are shown with black arrowheads. The same bands occurred as well when both antibodies were added together.
To determine whether HIF-1α or HIF-2α transcriptional activity is affected by the AGE-BSA stimulation, we performed a supershift analysis using either HIF-1α or HIF-2α antibodies or a combination of both (Figure 7D). Our data revealed that in MTCs a strong HIF-1α transcriptional activation was observed because the HIF-1α supershift assay demonstrated a band corresponding to the supershift of the HIF-1α transcription factor and the antibody complex (marked with a white arrowhead in Figure 7D) as well as additional lower bands (marked with black arrowheads in Figure 7D). The same HIF-1α-specific bands were present as well when both antibodies were added to the binding reactions. Only a moderate HIF-2α supershift was detected (marked with a white asterisk in Figure 7D), which was not detectable when both antibodies were added in the assay. This could be due to the fact that both HIF-transcription factors compete for the binding to the particular promoter sequences.
Localization of HIF-1α in renal cells after addition of AGE-BSA to the cells
Next, we investigated the nuclear localization of HIF-1α in all three cell lines after the addition of Co-BSA or AGE-BSA with immunostaining. Nuclei were counterstained with DAPI. An increased nuclear localization of HIF-1α in differentiated podocytes (Figure 8A) and MTCs (Figure 8B) was observed after the addition of AGE-BSA, which was more enhanced in the HIF-1α and DAPI merge images. On the other hand, HIF-1α nuclear staining in MMCs was detected in cells stimulated with Co-BSA as well as AGE-BSA (Figure 8C).
Figure 8.

A–C, Effect of AGE-BSA on HIF-1α nuclear localization in differentiated podocytes, MTCs, and MMCs. The localization of HIF-1α was investigated using immunological staining with a HIF-1α-specific antibody followed by secondary antibody, conjugated Alexa Cy3. The nuclei were visualized by DAPI staining. Representative images of Co-BSA- and AGE-BSA-treated cells stained for HIF-1α alone or a merge image of HIF-1α and DAPI staining are shown for each analyzed cell type. Bar, 20 μm (n = 4 independent experiments). A, Detection of HIF-1α and DAPI nuclear staining in differentiated podocytes. B, HIF-1α expression in MTCs. C, HIF-1α expression and DAPI nuclear staining in MMCs. HIF-1α nuclear translocation was induced in differentiated podocytes and MTCs treated with AGE-BSA, whereas under Co-BSA conditions, less nuclear HIF-1α was detected. A higher HIF-1α nuclear staining was detected in MMCs under control conditions (Co-BSA) than in cells treated with AGE-BSA.
Influence of AGE-BSA on PHD3 HIF-prolyl-hydroxylase activity and HIF-transcriptional activation in mesangial cells isolated from Morg1+/− and WT+/+ mice
Recently we generated a Morg1 knockout mouse (Morg1−/−) to study the physiological role of Morg1, but the mice were embryonic lethal due to massive defects in neuronal development (Wolf, G., unpublished data). On the other hand, Morg1 HZ mice (Morg1+/−) did not show phenotypic differences from the WT animals (25). Therefore, we analyzed the influence of AGE-BSA on PHD3 activity in primary mesangial cells isolated from Morg1 HZ and WT mice. For the experiments two different clones of primary mesangial cells isolated from Morg1 HZ and WT mice were used. Our data show that PHD3 activity in mesangial cells from WT mice treated with AGE-BSA was significantly increased compared with Co-BSA-stimulated cells, and a blocking of the RAGE receptor with a RAGE-specific antibody suppressed this effect (Figure 9A). On the other hand, in mesangial cells from Morg1 HZ mice, the PHD3 activity in Co-BSA-stimulated cells was lower compared with the WT mice, and the addition of AGE-BSA did not further elevate PHD3 activity (Figure 9A). Furthermore, we found that HIF-transcriptional activation was increased in mesangial cells from Morg1 HZ mice compared with mesangial cells isolated from WT animals (Figure 9B).
Figure 9.

A–D, Impact of AGE-BSA on PHD3 enzymatic activity and HIF-transcriptional activity in mesangial cells isolated from Morg1+/− HZ and WT (Morg1+/+) mice. A, PHD3 activity. AGE-BSA elevated the PHD3 enzymatic activity in mesangial cells isolated from WT compared with Co-BSA-stimulated cells. A significantly reduced PHD3 activity in Co-BSA as well as in AGE-BSA-treated mesangial cells obtained from Morg1+/− (Morg1 HZ) was measured relatively to WT mice. *, P < .05 vs Co-BSA WT; **, P < .005 vs Co-BSA WT (n = 9). Inhibitory studies demonstrated that PHD3 activity was regulated via the RAGE/AGE axis in WT cells. **, P < .005 vs AGE-BSA without α-RAGE (n = 9). In Morg1 HZ, AGE-BSA treatment failed to significantly induce PHD3 activity. B, Analysis of HIF-transcriptional activity by luciferase activity assay in WT and Morg1 HZ mesangial cells treated with Co-BSA and AGE-BSA (n = 18). HIF-luciferase reporter gene assays showed that AGE-BSA application to the WT mesangial cells significantly reduced HIF-DNA binding activity. In contrast, HIF-transcriptional activity was significantly increased in Morg1 HZ mesangial cells compared with WT cells. *, P < .05; **, P < .01 (n = 18). C, Protein lysates from Co-BSA, AGE-BSA, or AGE-BSA + α-RAGE antibody-treated mesangial cells obtained from Morg1 HZ or WT mice were subjected to Western blot analysis to monitor protein expression. Upper panel, Morg1 Western blot; lower panel, protein loading was controlled by detection of the β-actin protein expression. Representative Western blots are shown (n = 2 independent experiments). D, Protein expression of PHD3 and HIF-2α. The cells were treated as shown here and protein lysates were analyzed for protein expression. Upper panel, HIF-2α protein expression; middle panel, PHD3 protein Western blot; lower panel, detection of vinculin expression as a marker for protein loading. Clearly, less Morg1 and PHD3 protein expression was found in Morg1 HZ cells and no stimulation occurred with AGE-BSA. In contrast, AGE-BSA induced an increase in Morg1 and PHD 3 protein expression in WT cells that was reversed with the α-RAGE antibody. However, no major changes were seen in HIF-2α expression among the experimental conditions, probably due to the fact that cytoplasmatic lysates were used.
We also tested the expression of Morg1, PHD3, HIF-1α, and HIF-2α proteins in primary mesangial cells from Morg1 HZ and Morg1 WT mice stimulated with Co-BSA, AGE-BSA, or pretreated with the α-RAGE antibody before AGE-BSA addition using Western blots. As expected, we observed a reduced expression of Morg1 in the mesangial cells from Morg1 HZ mice compared with the cells isolated from WT mice (Figure 9C). Interestingly, we also detected a reduced PHD3 protein expression and increased HIF-2α protein levels in cells from Morg1 HZ mice relative to WT in which HIF-2α protein was not detectable (Figure 9D). The HIF-1α protein expression was not detected in either the mesangial cells isolated from the Morg1 HZ or from the cells of the WT mice (not shown).
Expression of HIF-1α and HIF-2α proteins in WT (Morg1+/+) and Morg1+/− HZ mice in STZ-induced diabetes
Morg1 WT and Morg1 HZ mice were treated with STZ to induce diabetes or were ip injected with sodium citrate buffer, respectively, WT placebo and Morg1 HZ placebo mice. Thirty days later the mice were killed and analyzed for the expression of HIF-1α and HIF-2α proteins in 4-μm kidney paraffin sections using immunohistology studies. We detected an increased expression of HIF-1α (Figure 10A) and HIF-2α (Figure 10B) in Morg1 HZ placebo mice compared with the WT placebo mice. Induction of diabetes increased significantly the nuclear amounts of HIF-1α in glomeruli of WT STZ mice (Figure 10B; **, P < .002 vs WT placebo), but not in Morg1 HZ mice because the basal HIF-1α levels were already significantly higher in Morg1 HZ placebo mice than in the WT placebo (Figure 10B). In addition, Morg1 STZ-treated mice showed increased HIF-1α protein levels above the WT STZ-treated mice. Furthermore, in tubuli from WT STZ and more obvious in tubuli of Morg1 STZ-treated mice was the nuclear accumulation of the HIF-1α (shown with arrows in the images). Induction of HIF is shown to be protective in ischemic kidney injury, and it reduced apoptosis in damaged renal tissues (25, 42, 43). We also detected less tubular damage in kidney sections from Morg1 HZ STZ mice than in the WT STZ, but this observation should be further analyzed. Analysis of HIF-2α expression in renal tissues of STZ-treated WT and Morg1 HZ mice unveiled that HIF-2α protein was also significantly higher in renal glomeruli of Morg1 HZ placebo mice vs the WT placebo mice (Figure 10C). These amounts were further elevated in Morg1 HZ STZ-treated mice, but there was not a significant difference in HIF-2α staining intensity between WT STZ- and Morg1 STZ-treated mice (Figure 10D).
Figure 10.

A–D, Immunohistological detection of HIF-1α and HIF-2α in Morg1+/− HZ and WT (Morg1+/+) mice after STZ-induced diabetes mellitus. At least four mice per group were analyzed for HIF-1/2α immunohistological detection in 4-μm paraffin kidney sections. Ten images of each individual kidney sample were scanned in the monochrome mode of the camera (magnification, ×400). All images were taken under constant conditions. The glomerular staining of the kidney sections was measured by highlighting the glomerular area, and the mean densitometric gray values were measured using AxioVision 4.6 software (Zeiss). The average of gray labels obtained for each individual kidney sample was used as an equivalent for the corresponding staining intensity. The staining intensity is normalized to the WT placebo mice and is presented in percentage. A, Staining intensity of HIF-1α in kidney sections of WT placebo- (n = 4), WT STZ- (n = 4), Morg1 HZ STZ- (n = 4), and Morg1 HZ placebo-treated mice (n = 4). Representative images are shown in monochrome mode. Scale bar, 20 μm. B, Staining intensity of HIF-1α in renal glomeruli, and semiquantitative analysis staining intensity is presented in percentage relatively to the WT placebo-treated nice. The area used for measurements is highlighted. Forty glomeruli were quantified per group. HIF-1α is induced in diabetes. WT STZ-treated mice showed a higher HIF-1α staining intensity vs WT placebo. **, P < .002. HIF-1α is significantly induced in Morg1 HZ placebo in comparison with the WT placebo mice as well as in Morg1 HZ STZ-induced diabetes relative to the WT STZ-induced mice. **, P < .002. C, Detection of HIF-2α in kidney sections. Representative images are shown in monochrome mode. The HIF-2α nuclear localization is shown with an arrow in the tubular region of the sections. Scale bar, 20 μm. D, Semiquantitative analysis of HIF-2α expression in kidney sections. Forty glomeruli were quantified per group. Morg1 down-regulation also induced the HIF-2α protein expression in vehicle-treated Morg1 HZ mice compared with the WT placebo mice. *, P < .05 as well as in Morg1 HZ STZ vs Morg1 placebo; *, P < .05.
Discussion
The Morg1 was recently isolated by a yeast two-hybrid system as a binding partner of PHD3 (22). Under in vitro conditions, the Morg1/PHD3 association correlated with an inhibition of HIF-transcriptional activity (22, 23). Morg1 is also characterized as a scaffold protein of the MAPK cascade as it binds as well to a number of proteins of the MAPK signaling pathway (44). We and others have demonstrated that it is activated by G protein coupled-receptor agonists such as ANG II (24) and lysophosphatidic acid (44), but its function is independent of tyrosine kinase receptors such as epithelial growth factor receptor and platelet-derived growth factor receptor (44, 45). On the other hand, Morg1 is an interesting molecule because it provides a link between the MAPK cascade and the oxygen-sensing proteins of the PHD3/HIF-1/2α pathway.
Recently we reported that Morg1 expression in renal cells was differentially regulated by ANG II, and a role for the ANG II type 1 receptor in mesangial cells and ANG II type 2 receptor in tubular cells was suggested. In addition, our current data confirm earlier findings (22) that elevated Morg1 expression is associated with an increased PHD3 activity and reduced HIF-1α and/or HIF-2α transcriptional activities (24). Therefore, Morg1 could be a new target molecule in the search for new mechanisms to modulate HIF-activity. ANG II is elevated in diabetic disease and is a risk factor for diabetic nephropathy (46–48). AGEs include a diverse group of proteins and lipids, which undergo nonenzymatic modifications due to the prolonged action of reducing sugars (1–3, 49). AGEs are another risk factor for the development of diabetic complications because AGEs accumulate with age (50) and in diabetes mellitus types 1 and 2 as a result of continuing high glucose levels (5, 36). Therefore, the AGE/RAGE interaction plays a major role in the development of diabetic complications (40, 51), and the regulation of this association could represent a helpful tool to modulate the severity of renal damage in diabetic disease. RAGE is a pattern recognition receptor, and therefore, in addition to binding AGEs, it also binds to non-AGE-related agonists such as the high mobility group box protein 1 (16), which links RAGE to inflammatory signaling cascades; via activation of NF-κB transcriptional machinery, it is involved in the regulation of gene expression (19, 52). It has been reported that RAGE expression is elevated in the glomeruli of diabetic mice compared with healthy animals (53). Its expression was found to be regulated by the NF-κB transcription factor (19), and a recent report demonstrated that the RAGE promoter has at least one HIF- consensus binding site, which is active in hypoxic conditions (54). We also reported that ANG II induced RAGE expression via the ANG II type 2 receptors in cultured differentiated murine podocytes (55).
The focus of the present study was to investigate the influence of AGE-BSA on Morg1 expression and determine potential functional consequences in different renal cell lines. We found that AGEs affect Morg1 mRNA expression in a cell type-specific manner. Treatment with AGE-BSA significantly reduced Morg1 mRNA expression in differentiated podocytes and MTCs, whereas MMCs showed an elevated Morg1 mRNA expression. These effects were regulated via RAGE-transduced signals because blocking of the RAGE receptor with a specific α-RAGE antibody abolished the observed changes in Morg1 mRNA expression. On the other hand, we tested for the presence of RAGE mRNA with semiquantitative and real-time PCR. Although RAGE expression has been previously reported in the renal tissues, the data are somewhat controversial, and it was assumed that RAGE is present in glomerular differentiated podocytes (41, 56). We found RAGE mRNA in all analyzed cells. On the other hand, the expressions of RAGE mRNA in the differentiated podocytes, MMCs, and MTCs differs. It was expressed more strongly in differentiated podocytes and about 4-fold less in MMCs. The RAGE ligand CML-BSA, the major BSA-modified product in our AGE-BSA preparation, was also detected via Western blot in protein lysates from cells treated with AGE-BSA.
Morg1 has been detected in the cytoplasm as well as the nucleus of cells (22, 24). We show that in Co-BSA-treated podocytes and MMCs, the protein was mostly detected in the nucleus of the cells and the perinuclear space. In the cytoplasm the immunogenic signal was extremely weak and the protein was detected in speckle-like structures in the cytoplasm. In MTCs under control conditions, Morg1 was found in the cytoplasm and the cell nucleus. With AGE-BSA stimulation, Morg1 protein expression was significantly reduced in differentiated podocytes and MTCs, and a translocation from the nucleus to the cell body was observed. In MTC cells there was a reduction of the overall Morg1 protein staining. In MMCs similar to the differentiated podocytes Morg1 protein was enhanced in the cytoplasm after addition of AGE-BSA to the cells. Morg1 protein does not have a nuclear localization signal or an export signal. It was also reported that PHD3 is found in the cell nucleus, and HIF- transcription factors are also present in the nucleus in an activated state. Nevertheless, the protein(s) that are involved in the Morg1 nuclear translocation are currently unknown.
Analysis of PHD3 enzyme activity showed a correlation between the level of Morg1 protein expression and PHD3 hydroxylase activity. Moreover, analysis of the PHD3 protein expression by Western blot revealed that the PHD3 protein level was also reduced in differentiated podocytes and MTCs and increased in MMCs treated with AGE-BSA. Therefore, the reduced PHD3 enzymatic activity was presumably due to the lower PHD3 protein levels in the cells exposed to AGE-BSA treatment. Inhibition of PHD3 activity was in turn associated with increased HIF-1α and/or HIF-2α transcriptional activation in podocytes and MTCs and a significant reduction of HIF-reporter activity in MMCs as demonstrated by HIF- reporter assay analysis. EMSA assays revealed that in MTCs no DNA binding activity was detected in those treated with Co-BSA.
This finding differs from the reporter assays. The supershift analysis showed more active HIF-1α than HIF-2α DNA binding activity. Similar results were observed with immunostaining analysis of HIF-1α nuclear localization in differentiated podocytes, MMCs, and MTCs. An activated transcription factor was detected in the nucleus of the MTCs and podocytes treated with AGE-BSA, and only a low level was seen in the cytoplasm of these cells. In Co-BSA-treated cells, a perinuclear localization was observed. An opposite effect was detected in the MMCs, which showed a nuclear staining for HIF-1α under control conditions; therefore, one can conclude that MMCs may require HIF-1α activation, even under basal conditions.
We studied the PHD3 enzymatic activity and HIF-promoter activity in primary mesangial cells from Morg1+/− and WT (Morg1+/+) mice. In addition to the predicted reduced Morg1 protein expression, suggesting haplotype insufficiency, an inhibited PHD3 protein expression correlating with lower hydroxylase enzymatic activity was found in mesangial cells isolated from Morg1+/− mice. Interestingly, we detected a protein band of HIF-2α but not of HIF-1α in the protein lysates of Morg1+/− mice and an increased basal HIF-promoter activity, which was not further activated or reduced by AGE-BSA treatment. Increased HIF-2α is associated with an elevated HIF-2α promoter activity in mesangial cells from Morg1+/− mice. Because it has been previously reported that PHD3 mainly hydroxylates HIF-2α (54), this finding is reasonable. We also detected an increased expression of the HIF-1α and HIF-2α proteins in kidneys from Morg1 HZ placebo mice compared with the Morg1 WT placebo-treated mice and the HIF-1α protein expression in Morg1 HZ STZ-treated mice was significantly higher than the WT STZ-treated mice. It has been shown that induction of HIF could be protective in ischemic kidney injury and hypoxic preconditioning reduced apoptosis in renal tissues (25, 42, 43). Nevertheless, the exact mechanism and the particular role of HIF are still not well understood. In this regard an elevated HIF-1/2α levels as a preconditioning could have a protective and antiapoptotic function in renal tissue, questions that could be addressed using a Morg1 HZ mice model because of the stabilization of the HIF-1/2α proteins.
We have reported here for the first time on a link between the AGE/RAGE axis and the Morg1/PHD3/HIF- complex, which seems to play an important role in the pathophysiology of diabetic nephropathy. The connection between elevated ANG II levels and the stimulated AGE/RAGE axis, both of which are characteristic of diabetic nephropathy, may converge at the level of the Morg1 scaffold protein and accelerate the Morg1/PHD3/HIF signal cascade.
Acknowledgments
We thank S. Franke, PhD, for the preparation of the AGE-BSA and CML determinations.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Wo 460/14-2, 17-1).
Disclosure Summary: We have nothing to disclose.
Funding Statement
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Wo 460/14-2, 17-1).
Footnotes
- AGE
- advanced glycated end-product
- AGE-BSA
- addition of glycated BSA to AGE
- ANG II
- angiotensin II
- CML
- Nϵ-carboxymethyl-lysine
- Co-BSA
- control, nonglycated BSA
- DAPI
- 4′,6-diamidino-2-phenylindole
- FCS
- fetal calf serum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HIF
- hypoxia-inducible factor
- HZ
- heterozygous
- MMC
- murine mesangial cell
- MTC
- murine proximal tubular cell
- Morg1
- MAPK organizer 1
- NF-κB
- nuclear factor-κB
- PHD3
- prolyl-hydroxylase domain-containing protein-3
- RAGE
- receptor for advanced glycation end-products
- STZ
- streptozotocin
- WT
- wild-type.
References
- 1. Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med 1988;318:1315–1321. [DOI] [PubMed] [Google Scholar]
- 2. Brownlee M, Cerami A, Vlassara H. Advanced products of nonenzymatic glycosylation and the pathogenesis of diabetic vascular disease. Diabetes Metab Rev. 1988;4:437–451. [DOI] [PubMed] [Google Scholar]
- 3. Fu MX, Wells-Knecht KJ, Blackledge JA, Lyons TJ, Thorpe SR, Baynes JW. Glycation, glycoxidation, and cross-linking of collagen by glucose. Kinetics, mechanisms, and inhibition of late stages of the Maillard reaction. Diabetes. 1994;43:676–683. [DOI] [PubMed] [Google Scholar]
- 4. Busch M, Franke S, Ruster C, Wolf G. Advanced glycation end-products and the kidney. Eur J Clin Invest. 2010;40:742–755. [DOI] [PubMed] [Google Scholar]
- 5. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. [DOI] [PubMed] [Google Scholar]
- 6. Kilhovd BK, Giardino I, Torjesen PA, et al. Increased serum levels of the specific AGE-compound methylglyoxal-derived hydroimidazolone in patients with type 2 diabetes. Metabolism. 2003;52:163–167. [DOI] [PubMed] [Google Scholar]
- 7. Schleicher E, Wieland OH. Protein glycation: measurement and clinical relevance. J Clin Chem Clin Biochem. 1989;27:577–587. [PubMed] [Google Scholar]
- 8. Cohen MP, Ziyadeh FN. Role of Amadori-modified nonenzymatically glycated serum proteins in the pathogenesis of diabetic nephropathy. J Am Soc Nephrol. 1996;7:183–190. [DOI] [PubMed] [Google Scholar]
- 9. Bierhaus A, Humpert PM, Morcos M, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl). 2005;83:876–886. [DOI] [PubMed] [Google Scholar]
- 10. Semba RD, Fink JC, Sun K, Bandinelli S, Guralnik JM, Ferrucci L. Carboxymethyl-lysine, an advanced glycation end product, and decline of renal function in older community-dwelling adults. Eur J Nutr. 2009;48:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakaguchi T, Yan SF, Yan SD, et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest. 2003;111:959–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Semba RD, Najjar SS, Sun K, Lakatta EG, Ferrucci L. Serum carboxymethyl-lysine, an advanced glycation end product, is associated with increased aortic pulse wave velocity in adults. Am J Hypertens. 2009;22:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piarulli F, Lapolla A, Ragazzi E, et al. Role of endogenous secretory RAGE (esRAGE) in defending against plaque formation induced by oxidative stress in type 2 diabetic patients. Atherosclerosis. 2013;226:252–257. [DOI] [PubMed] [Google Scholar]
- 14. Lipscomb EA, Sarmiere PD, Freeman RS. SM-20 is a novel mitochondrial protein that causes caspase-dependent cell death in nerve growth factor-dependent neurons. J Biol Chem. 2001;276:5085–5092. [DOI] [PubMed] [Google Scholar]
- 15. Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. [DOI] [PubMed] [Google Scholar]
- 16. van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008;11:91–99. [DOI] [PubMed] [Google Scholar]
- 17. Lin L, Park S, Lakatta EG. RAGE signaling in inflammation and arterial aging. Front Biosci. 2009;14:1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cipollone F, Iezzi A, et al. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation. 2003;108:1070–1077. [DOI] [PubMed] [Google Scholar]
- 19. Haslbeck KM, Schleicher E, Bierhaus A, et al. The AGE/RAGE/NF-κB pathway may contribute to the pathogenesis of polyneuropathy in impaired glucose tolerance (IGT). Exp Clin Endocrinol Diabetes. 2005;113:288–291. [DOI] [PubMed] [Google Scholar]
- 20. Haslbeck KM, Friess U, Schleicher ED, et al. The RAGE pathway in inflammatory myopathies and limb girdle muscular dystrophy. Acta Neuropathol. 2005;110:247–254. [DOI] [PubMed] [Google Scholar]
- 21. Franke S, Sommer M, Ruster C, Bondeva T, Marticke J, Hofmann G, Hein G, Wolf G. Advanced glycation end products induce cell cycle arrest and proinflammatory changes in osteoarthritic fibroblast-like synovial cells. Arthritis Res Ther. 2009;11:R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hopfer U, Hopfer H, Jablonski K, Stahl RA, Wolf G. The novel WD-repeat protein Morg1 acts as a molecular scaffold for hypoxia-inducible factor prolyl hydroxylase 3 (PHD3). J Biol Chem. 2006;281:8645–8655. [DOI] [PubMed] [Google Scholar]
- 23. Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–30780. [DOI] [PubMed] [Google Scholar]
- 24. Bondeva T, Heinzig J, Franke S, Wolf G. Angiotensin II differentially regulates Morg1 expression in kidney cells. Am J Nephrol. 2012;35:442–455. [DOI] [PubMed] [Google Scholar]
- 25. Hammerschmidt E, Loeffler I, Wolf G. Morg1 heterozygous mice are protected from acute renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2009;297:F1273–F1287. [DOI] [PubMed] [Google Scholar]
- 26. Stahr A, Frahm C, Kretz A, Bondeva T, Witte OW, Wolf G. Morg1(+/−) heterozygous mice are protected from experimentally induced focal cerebral ischemia. Brain Res. 2012;1482:22–31. [DOI] [PubMed] [Google Scholar]
- 27. Wolf G, Haberstroh U, Neilson EG. Angiotensin II stimulates the proliferation and biosynthesis of type I collagen in cultured murine mesangial cells. Am J Pathol. 1992;140:95–107. [PMC free article] [PubMed] [Google Scholar]
- 28. Haverty TP, Kelly CJ, Hines WH, et al. Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J Cell Biol. 1988;107:1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mundel P, Kriz W. Cell culture of podocytes. Exp Nephrol. 1996;4:263–266. [PubMed] [Google Scholar]
- 30. Tesch GH, Allen TJ. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology (Carlton). 2007;12:261–266. [DOI] [PubMed] [Google Scholar]
- 31. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-δ δ C(T)] method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 32. Bondeva T, Roger T, Wolf G. Differential regulation of Toll-like receptor 4 gene expression in renal cells by angiotensin II: dependency on AP1 and PU.1 transcriptional sites. Am J Nephrol. 2007;27:308–314. [DOI] [PubMed] [Google Scholar]
- 33. Bondeva T, Wolf G. Advanced glycation end products suppress neuropilin-1 expression in podocytes by a reduction in Sp1-dependent transcriptional activity. Am J Nephrol. 2009;30:336–345. [DOI] [PubMed] [Google Scholar]
- 34. Neeper M, Schmidt AM, Brett J, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 35. Brett J, Schmidt AM, Yan SD, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 36. Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann NY Acad Sci. 2008;1243:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lindsey JB, Cipollone F, Abdullah SM, McGuire DK. Receptor for advanced glycation end-products (RAGE) and soluble RAGE (sRAGE): cardiovascular implications. Diab Vasc Dis Res. 2009;6:7–14. [DOI] [PubMed] [Google Scholar]
- 38. Tekabe Y, Luma J, Li Q, Schmidt AM, Ramasamy R, Johnson LL. Imaging of receptors for advanced glycation end products in experimental myocardial ischemia and reperfusion injury. JACC Cardiovasc Imaging. 2012;5:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schleicher ED, Wagner E, Nerlich AG. Increased accumulation of the glycoxidation product Nϵ-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest. 1997;99:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Z, Jiang Y, Liu N, et al. Advanced glycation end-product Nϵ-carboxymethyl-lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis. 2012;221:387–396. [DOI] [PubMed] [Google Scholar]
- 41. Tekabe Y, Li Q, Rosario R, et al. Development of receptor for advanced glycation end products-directed imaging of atherosclerotic plaque in a murine model of spontaneous atherosclerosis. Circ Cardiovasc Imaging. 2008;1:212–219. [DOI] [PubMed] [Google Scholar]
- 42. Bernhardt WM, Campean V, Kany S, et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol. 2006;17:1970–1978. [DOI] [PubMed] [Google Scholar]
- 43. Weidemann A, Bernhardt WM, Klanke B, et al. HIF activation protects from acute kidney injury. J Am Soc Nephrol. 2008;19:486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vomastek T, Schaeffer HJ, Tarcsafalvi A, Smolkin ME, Bissonette EA, Weber MJ. Modular construction of a signaling scaffold: MORG1 interacts with components of the ERK cascade and links ERK signaling to specific agonists. Proc Natl Acad Sci USA. 2004;101:6981–6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. [DOI] [PubMed] [Google Scholar]
- 46. Wolf G. Angiotensin II as a renal growth factor. Contrib Nephrol. 2001;(135):92–110. [DOI] [PubMed] [Google Scholar]
- 47. Xu ZG, Yoo TH, Ryu DR, et al. Angiotensin II receptor blocker inhibits p27Kip1 expression in glucose-stimulated podocytes and in diabetic glomeruli. Kidney Int. 2005;67:944–952. [DOI] [PubMed] [Google Scholar]
- 48. Wolf G. Growth factors and the development of diabetic nephropathy. Curr Diab Rep. 2003;3:485–490. [DOI] [PubMed] [Google Scholar]
- 49. Kimura T, Takamatsu J, Ikeda K, Kondo A, Miyakawa T, Horiuchi S. Accumulation of advanced glycation end products of the Maillard reaction with age in human hippocampal neurons. Neurosci Lett. 1996;208:53–56. [DOI] [PubMed] [Google Scholar]
- 50. Wang M, Takagi G, Asai K, et al. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41:1308–1316. [DOI] [PubMed] [Google Scholar]
- 51. Bucciarelli LG, Kaneko M, Ananthakrishnan R, et al. Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury. Circulation. 2006;113:1226–1234. [DOI] [PubMed] [Google Scholar]
- 52. Schiekofer S, Andrassy M, Chen J, et al. Acute hyperglycemia causes intracellular formation of CML and activation of ras, p42/44 MAPK, and nuclear factor κB in PBMCs. Diabetes. 2003;52:621–633. [DOI] [PubMed] [Google Scholar]
- 53. Tanji N, Markowitz GS, Fu C, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11:1656–1666. [DOI] [PubMed] [Google Scholar]
- 54. Pichiule P, Chavez JC, Schmidt AM, Vannucci SJ. Hypoxia-inducible factor-1 mediates neuronal expression of the receptor for advanced glycation end products following hypoxia/ischemia. J Biol Chem. 2007;282:36330–36340. [DOI] [PubMed] [Google Scholar]
- 55. Ruster C, Bondeva T, Franke S, Tanaka N, Yamamoto H, Wolf G. Angiotensin II upregulates RAGE expression on podocytes: role of AT2 receptors. Am J Nephrol. 2009;29:538–550. [DOI] [PubMed] [Google Scholar]
- 56. Guo J, Ananthakrishnan R, Qu W, et al. RAGE mediates podocyte injury in adriamycin-induced glomerulosclerosis. J Am Soc Nephrol. 2008;19:961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]