

Abstract
CC Chemokine Receptor 2 (CCR2) and its endogenous ligand CCL2 are involved in a number of diseases, including atherosclerosis. Several CCR2 antagonists have been developed as potential therapeutic agents, however their in vivo clinical efficacy was limited. In this report, we aimed to determine whether 15a, an antagonist with a long residence time on the human CCR2, is effective in inhibiting the development of atherosclerosis in a mouse disease model. First, radioligand binding assays were performed to determine affinity and binding kinetics of 15a on murine CCR2. To assess the in vivo efficacy, western-type diet fed apoE−/− mice were treated daily with 15a or vehicle as control. Treatment with 15a reduced the amount of circulating CCR2+ monocytes and the size of the atherosclerotic plaques in both the carotid artery and the aortic root. We then showed that the long pharmacokinetic half-life of 15a combined with the high drug concentrations ensured prolonged CCR2 occupancy. These data render 15a a promising compound for drug development and confirms high receptor occupancy as a key parameter when targeting chemokine receptors.
Introduction
The chemokine system comprises more than 20 different chemokine receptors, which belong to the class A or rhodopsin-like family of G protein-coupled receptors (GPCRs). Almost 50 chemokine ligands play a critical role in the immune system, mediating the migration and differentiation of immune cells during homeostasis and inflammation1. Dysregulation of this system can lead to a variety of different pathologies, including inflammatory and autoimmune diseases2, 3. For instance, preclinical evidence suggests that CC Chemokine Receptor 2 (CCR2) and its high-affinity ligand CCL2 are involved in the pathogenesis of atherosclerosis4–6, neuropathic pain7 and multiple sclerosis8. Genetic knockout of CCR2 (CCR2−/−) in the Apolipoprotein E-deficient (apoE−/−) mouse model of atherosclerosis resulted in a significant decrease in lesion size compared to apoE−/− controls, which was caused by a reduction in monocyte/macrophage recruitment to the atherosclerotic lesion4–6. Similar results have been reported in studies with genetic knockout of CCL2 in both the low density lipoprotein receptor-deficient (LDLr −/−) and the human Apolipoprotein B transgenic mouse models9, 10. These findings suggest that the CCR2/CCL2 axis is critically involved in the mobilization and recruitment of monocytes to the early atherosclerotic plaque.
The involvement of CCR2 in pathologies such as atherosclerosis has resulted in many efforts to develop biologic and small molecule antagonists targeting this receptor. However, despite inhibitory effects on for example monocyte recruitment11, a very limited number of compounds has shown in vivo efficacy in inhibiting atherosclerosis. In this regard, the characterization of the drug-target residence time (RT)—a measure of the ligand-receptor complex lifetime—has been proposed as a good predictor of in vivo efficacy and safety12–15. Although the link from in vitro kinetics to in vivo outcomes has only been studied in few drug targets, these studies confirm the importance of characterizing the RT of drug candidates16–21. Yet, the impact of RT to prolong the duration of effect can only be assessed when considering the whole target biology and pharmacokinetic properties of the drug of interest22, 23. Inclusion of RT in early hit-to-lead optimization has also been recently used for the development of high-affinity and long-RT CCR2 antagonists24, 25. The combination of structure-affinity and structure-kinetics optimization resulted in the discovery of compound 15a (Fig. 1), an orthosteric CCR2 small molecule antagonist with high affinity of 2.4 nM and a RT of 714 min for human CCR2 (hCCR2)24. With such a prolonged CCR2 inhibition, 15a emerges as a potential candidate to evaluate the in vivo effects of long RT.
Figure 1.
Chemical structure of CCR2 antagonist 15a.
In this study, we aimed to evaluate the binding kinetics and pharmacokinetics of 15a for murine CCR2 (mCCR2) and determine whether this CCR2 antagonist is effective in an apoE−/− mouse model of atherosclerosis. Using a combination of in vitro radioligand binding assays and in vivo studies, we show that prolonged CCR2 antagonism with 15a, due to high target occupancy, is linked to a robust and significant inhibition of atherogenesis in mice. These results support the need of achieving more than 90% continuous inhibition when developing chemokine receptor antagonists26. This study also highlights the importance of in vitro characterization of drug candidates in all relevant species for in vivo pre-clinical studies, in order to improve the translational value of animal models, and thus to reduce the attrition in drug discovery programs.
Results
Characterization of [3H]INCB3344 and 15a in mouse CCR2 (mCCR2)
To determine the binding affinity and the kinetic profile of 15a at mCCR2, the radioligand [3H]INCB3344 was first characterized by performing association and dissociation binding assays in membranes of CHO cells transiently expressing mCCR2. At 25 °C, binding of [3H]INCB3344 to mCCR2 reached equilibrium around 20 min and was best fit with a one-phase exponential model that yielded an association (k on) rate of 0.030 nM−1 min−1. Dissociation of the radioligand from mCCR2 was also best fit with a one-phase model that yielded a dissociation rate (k off) of 0.227 min−1 (Fig. 2 and Table 1). These values resulted in a kinetic dissociation binding constant (K D) of 7.6 nM and a RT of 4.4 min for this receptor (Table 1).
Figure 2.
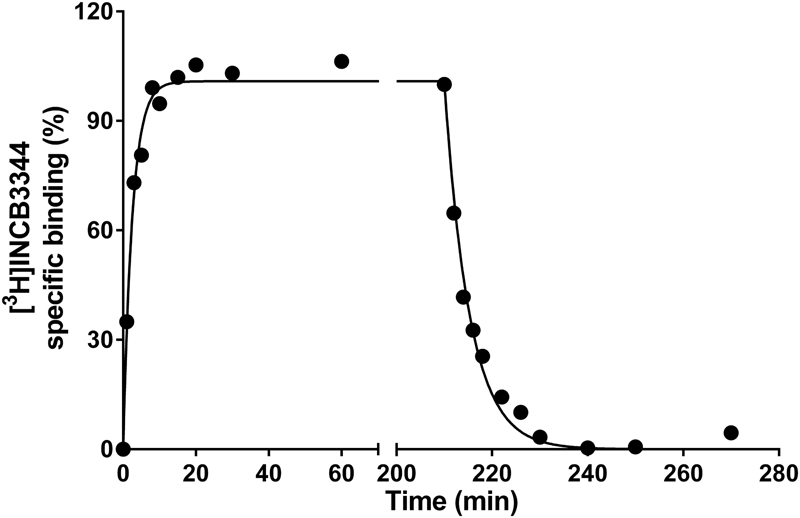
Kinetic characterization of [3H]INCB3344 in murine CCR2. Association and dissociation kinetics of 5 nM [3H]INCB3344 binding to membranes of CHO cell membranes transiently expressing murine CCR2 at 25 °C. Dissociation was initiated by the addition of 10 µM INCB3344. Association and dissociation data were fitted using a monophasic exponential association, or exponential decay model, respectively. Graphs shown are representative from one experiment performed in duplicate. Data for all association and dissociation experiments (n = 3) are presented in Table 1.
Table 1.
Kinetic characterization of [3H]INCB3344 and 15a in membranes of CHO cells transiently expressing murine CCR2 (mCCR2).
| [3H]INCB3344 (hCCR2b*) | 15a (hCCR2b†) | |
|---|---|---|
| KRI‡ | 1.0 (1.0) | 1.6 (1.7) |
| k on (nM−1 min−1) | 0.030 ± 0.003 (0.054) | 0.007 ± 0.001 (0.008) |
| k off (min−1) | 0.227 ± 0.031 (0.013) | 0.051 ± 0.008 (0.0014) |
| K D § (nM) | 7.6 ± 1.3 (0.23) | 7.4 ± 1.5 (0.175) |
| RT|| (min) | 4.4 ± 0.6 (76) | 19.7 ± 3.1 (714) |
Values are means ± S.E.M of three separate experiments performed in duplicate.
*Values of [3H]INCB3344 for hCCR2b were taken from Vilums et al. 25.
†Values of 15a for hCCR2b were taken from Vilums et al. 24.
‡Kinetic rate index (KRI) = Bt1/Bt2 (see Materials and Methods).
§ K D = k off/k on.
||RT = 1/k off.
After the characterization of [3H]INCB3344, we performed displacement experiments at 25 °C with 15a to determine its affinity for mCCR2. 15a was able to displace [3H]INCB3344 binding in a concentration-dependent way, with a Ki value of 10.6 nM (Fig. 3a). In addition, a [3H]INCB3344 competition association assay was performed in order to determine the kinetic profile of unlabeled 15a. Figure 3b shows a representative competition association curve of [3H]INCB3344 to mouse CCR2, in the absence or presence of 30 nM 15a (approximately 3-fold its Ki value). We used 30 nM 15a as this concentration provided a sufficient window for data analysis. Association curves of [3H]INCB3344 in the presence of 15a in mCCR2 resulted in the typical overshoot characteristic for long RT compounds (Fig. 3b) and a KRI > 1.0 (KRI = 1.6). By fitting the curves to the competition association model of Motulsky and Mahan27, it was possible to determine the k on and k off values of 15a: 0.007 ± 0.001 nM−1 min−1 and 0.051 ± 0.008 min−1, respectively. These results allowed the calculation of the kinetic K D (7.4 ± 1.5 nM), which was comparable to the obtained Ki value, and confirmed a slower dissociation—and a longer RT of 20 min—compared to [3H]INCB3344 (RT = 4.4 min, Table 1). For comparison, the kinetic parameters of 15a and [3H]INCB3344 on human CCR2 (hCCR2) are also shown in Table 1, as previously published by Vilums et al.24, 25.
Figure 3.

Characterization of 15a in murine CCR2. (a) Displacement of [3H]INCB3344 binding from CHO cell membranes transiently expressing murine CCR2 at 25 °C, upon addition of increasing concentrations of compound 15a. (b) Competition association of 5 nM [3H]INCB3344 binding to membranes of CHO cells transiently expressing murine CCR2 at 25 °C, in the absence or presence of 30 nM 15a. Data were fitted to the competition association model of Motulsky and Mahan27. Graphs shown are representative from one experiment performed in duplicate. Data for kinetic experiments (n = 3) are presented in Table 1.
15a reduces circulating CCR2+ monocyte levels
In this study, apoE−/− mice were treated with the orthosteric antagonist 15a or vehicle control for four weeks. During the study, body weight, body weight gain and plasma total cholesterol levels were not affected by administration of the CCR2 antagonist (Supplementary Fig. S1). Liver morphology was similar between the controls and the 15a treated mice, and 15a did not affect liver mRNA expression of the toxicity markers ALT, apoH and GC. mRNA levels of AST were even somewhat decreased in the 15a treated group (P = 0.02, Supplementary Fig. S2). Interestingly, treatment with 15a resulted in increased plasma CCL2 levels, while plasma IL-6 levels remained unaffected (Supplementary Fig. S3). After two weeks of treatment, we determined CCR2+ monocyte levels (defined as CD11b+Ly6Glow cells) in blood of control and 15a treated mice at two hours after injection by means of flow cytometry. At that time point, we observed a significant reduction in the percentage of circulating CCR2+ monocytes in the 15a treated mice (Fig. 4a, controls: 8.7 ± 1.2%, 15a: 1.8 ± 0.2%, P = 0.0004).
Figure 4.

Effect of 15a treatment on the amount of monocytes. (a) At 2 hours after injection and two weeks of treatment, 15a reduced the relative amount of circulating CCR2+ monocytes (defined as CD11b+Ly6Glow cells). (b) At 18 hours after the last injection (after 4 weeks of treatment), the number of circulating CD11b+Ly6GlowCCR2+ cells was significantly reduced by 15a treatment. (c) The ratio of patrolling monocytes and pro-inflammatory monocytes was significantly increased upon treatment with 15a (*P < 0.05, **P < 0.01, ***P < 0.001). Graphs shown are representative from one experiment with n = 5 per group in panel (a) and n = 6 per group in panels (b) and (c).
At the time of sacrifice, the total number of monocytes was significantly reduced in the mice treated with 15a, while total lymphocyte and neutrophil numbers remained unaffected (Supplementary Fig. S4). Flow cytometry analysis further confirmed that 15a did not affect the number of neutrophils, CD19+ B cells, CD4+ or CD8+ T cells in blood or spleen, nor did we detect any effects of the antagonist on splenic or circulating Tregs (data not shown). Of note, these analyses were performed at 18 hour after the last injection of the antagonist. At that time-point, we still observed that the number of circulating CCR2+ monocytes was significantly reduced in the 15a treated mice compared to controls (Fig. 4b, controls: 14.9 ± 3.2*103 cells/mL versus 15a: 4.5 ± 1.0*103 cells/mL, P = 0.01). The ratio of patrolling CD11b+Ly6ClowCX3CR1+ monocytes versus inflammatory CD11b+Ly6ChighCCR2+ monocytes in blood increased upon treatment with 15a (Fig. 4c), which was not only due to a reduction in the number of CCR2+ monocytes, but also caused by a significant 1.5-fold increase in the amount of CD11b+Ly6ClowCX3CR1+ monocytes. We did not observe any change in the percentage of circulating CCR5+ monocytes in both the treated and untreated mice, which suggests a selective interaction of 15a with CCR2 (Supplementary Fig. S5).
CCR2 antagonism inhibits atherosclerotic plaque development
Atherosclerotic plaque size in the carotid artery at the site of maximal stenosis was reduced from 64.4 ± 11.8*103 µm2 in control mice to 17.6 ± 4.1*103 µm2 in mice treated with 15a (Fig. 5a, −73%, P = 0.002). Furthermore, as depicted in Fig. 5b,c, plaque volume throughout the carotid arteries was reduced from 1.75 ± 0.33*107 µm3 in control mice to 0.46 ± 0.13*107 µm3 in 15a treated mice (P = 0.003). In addition, relative necrotic area was reduced from 35.3 ± 8.2% in controls to 7.6 ± 4.5% in the 15a treated mice (P = 0.01).
Figure 5.

Inhibition of lesion development in the carotid artery by 15a. (a) The CCR2 antagonist 15a significantly reduced atherosclerotic plaque size in the carotid artery at the site of maximal stenosis. (b) Similarly, plaque volume, given as plaque size at increasing distance from the collar and (c) as total volume in µm3 was significantly reduced by 15a. Micrographs show representative images of the individual groups (100X magnification). *P < 0.05, **P < 0.01, ***P < 0.001. Graphs shown are representative from one experiment with n = 10 controls versus n = 9 15a-treated mice.
In the aortic root, average plaque size was significantly reduced by treatment with 15a (Fig. 6a, controls: 215.9 ± 24.6*103 µm2, 15a: 157.0 ± 15.7*103 µm2, P = 0.005) and also aortic root plaque volume was lower in the 15a treated mice (Fig. 6b,c, controls: 11.8 ± 1.2*107 µm3, 15a: 7.4 ± 0.7*107 µm3, P = 0.005).
Figure 6.

Inhibition of lesion development in the aortic root by 15a. (a) Also in the aortic root, 15a inhibited atherosclerotic lesion development after 6 weeks of Western type diet. (b) Lesion area in the aortic roots presented for each group every 90 μm of the aortic root. (c) Plaque volume was also significantly reduced in the 15a-treated group. *P < 0.05, **P < 0.01. Graphs shown are representative from one experiment with n = 10 controls versus n = 9 15a-treated mice.
The macrophage content as measured by MOMA2 staining revealed that the macrophage positive area of the carotid artery plaques was significantly reduced in the 15a treated mice (Fig. 7a, left panel; controls: 22.3 ± 4.1*103 µm2, 15a: 6.2 ± 2.5*103 µm2, P = 0.005). Similarly, relative macrophage content (corrected for lesion size) was reduced in the 15a treated group (Fig. 7a, right panel; controls: 46 ± 4%, 15a: 25 ± 8%, P = 0.02). In the aortic root, macrophage positive area was also significantly lower in the 15a treated mice (Fig. 7b, controls: 92.0 ± 15.6*103 µm2, 15a: 37.2 ± 8.7*103 µm2, P = 0.008).
Figure 7.

Macrophage content in carotid artery and aortic root atherosclerotic lesions. (a) Macrophage content in the carotid artery lesions as measured by MOMA2 staining was reduced upon treatment with 15a, both in total macrophage positive area (left panel) and relatively as percentage of lesion size (right panel). Micrographs show representative images of the individual groups (100X magnification). (b) Also in the aortic root, 15a significantly reduced the macrophage+ area (left panel). Relative macrophage content did not significantly differ between the groups (right panel). Micrographs show representative images of the individual groups (40X magnification). *P < 0.05, **P < 0.01. Graphs shown are representative from one experiment with n = 10 controls versus n = 9 15a-treated mice.
Pharmacokinetics-based prediction of target occupancy
To investigate the role of pharmacokinetics in the generation of the effective drug response of 15a, we measured the plasma levels of 15a over a 24 hour period using a LC-MS method. Following a single intraperitoneal dose of 5 mg/kg, plasma levels of 15a varied from 12 to 1456 ng/mL 1 h after administration; 2 h after dosing, plasma levels were above 600 ng/mL in all individual mice (613 to 1048 ng/mL), which remained relatively constant at 6 h after administration (182 to 1024 ng/mL) and fell to 32 to 331 ng/mL at 24 h after administration (Fig. 8a). Pharmacokinetic analysis at this dose revealed a relatively slow pharmacokinetic profile (Fig. 8a, elimination half-life = 10.7 hours), with an elimination half-life that exceeds by far the determined RT in mCCR2. Using these plasma concentrations, we predicted the target occupancy attained by 15a using two different approaches: i) calculation of “equilibrium” target occupancy using equation (1) as shown in Materials and Methods, which accounts for both the pharmacokinetics and affinity of 15a, and ii) simulation of target occupancy using equations (3–5), which account for the pharmacokinetics and binding kinetics of this compound. These calculations demonstrated that the combination of slow pharmacokinetics (Fig. 8a) and high drug concentrations relative to affinity leads to a high target occupancy of >90% for 24 hours (Fig. 8b, dots). This was confirmed by the simulations of receptor occupancy (Fig. 8b, line), indicating that a simpler calculation method (equation (1)) is sufficient to predict receptor occupancy in this study. Comparing the long duration of target occupancy for equilibrium binding with the RT of 15a (of 24 minutes) makes clear that RT is not a contributing factor in prolonging target occupancy under these conditions. In this regard, equation (2) allowed us to determine the maximal value of k off that could contribute to a prolonged duration of target occupancy. As previously demonstrated, the level of target saturation can be used to compare the plasma elimination with the k off, by multiplying the pharmacokinetic elimination rate with a factor (1-BF), where BF represents the percentage of target occupancy divided by 10028. This means that if the target occupancy needs to be above 90% for effective treatment, the k off of the drug candidate needs to be ten times lower than the pharmacokinetic elimination rate.
Figure 8.

Total plasma concentrations, calculated and simulated target occupancy. (a) Observed plasma concentrations (dots), linear fit of the data (line) and 95% confidence interval of the linear regression (shaded area). (b) Equilibrium occupancy derived from the experimentally observed plasma concentrations (dots) and simulated occupancy on basis of the observed binding kinetics and plasma elimination rate (line). Graphs shown are representative from one experiment (n = 3 per time point).
Discussion and Conclusions
In this study, we investigated the efficacy of the novel CCR2 antagonist 15a—designed to display a long residence time (RT) on human CCR2 (hCCR2)—in inhibiting the development of atherosclerosis in apoE−/− mice. Characterization of the drug-target complex lifetime or RT in early phases of the drug discovery and development process might help to improve the prediction of in vivo efficacy and safety12–15. Evidence from retrospective studies has shown that several marketed drugs exhibit a long RT on their target13, 29, prompting the prospective optimization of both affinity and kinetic properties of compounds for a variety of targets17, 25, 30–32. In this regard, 15a was discovered by optimizing a reported lead in the development of MK-0812, a Merck CCR2 antagonist that has failed in clinical trials due to lack of efficacy33. Although the RT of MK-0812 in mCCR2 is not known, in hCCR2 it exhibited an almost 8-fold shorter RT than compound 15a (92 min versus 714 min, respectively)24. Thus, our selection of 15a was based on the need of advancing drug candidates with high affinity and favorable binding kinetics. In this study, we first determined the kinetic profile of 15a on mouse CCR2 (mCCR2), as the pharmacological properties of drug candidates, including the kinetic profile, can differ between species orthologues34. This renders it necessary to characterize the binding kinetics of lead compounds in all relevant species for preclinical testing12. Using [3H]INCB3344 we were able to determine the binding affinity and the kinetic profile of 15a in mCCR2. The determination of RT using the competition association model of Motulsky and Mahan27 confirmed the longer RT of 15a in comparison with the control [3H]INCB3344, a CCR2 receptor antagonist with similar affinity for mCCR2. This is in line with previous studies in hCCR2, where 15a showed a RT of 714 min compared with 76 min of [3H]INCB3344 (Table 1)24. However, a clear difference was found between species, as 15a dissociates much faster from mCCR2 than from hCCR2: 15a has a lifetime of less than 30 min on mCCR2, but more than 10 hours on hCCR2. This poor translation of kinetic parameters between species represents a clear limitation in understanding the effect of RT in in vivo efficacy, adding an extra level of complexity when selecting relevant animal models for preclinical studies.
Next we showed that 15a significantly reduced the number of circulating inflammatory CCR2+ monocytes and that 15a was very effective in inhibiting atherosclerotic plaque development in both the carotid artery and the aortic root of apoE−/− mice. In addition, relative macrophage content in the lesion was reduced in the 15a treated mice at both sites of lesion development. CCR2 was previously shown to be predominantly involved in lesion initiation4, 6, but seemed less effective in inhibiting lesion progression. For therapeutic application, halting lesion progression would be favorable to prevent an existing plaque to progress to an advanced and unstable lesion. In this study, 15a almost completely blocked lesion initiation in the carotid artery, as treatment was commenced immediately from time of collar placement and thus of lesion development at that site. In the aortic root, early atherosclerosis had already developed before treatment was initiated, and treatment was also effective in inhibiting plaque progression in this location. Aiello et al. showed that treatment with the CCR2 antagonist INCB3344 failed to decrease the size of early and advanced atherosclerotic lesions in apoE−/− mice11, which may be related to both the shorter RT of this drug in mCCR2, and the use of an ineffective dose. Although the plasma levels of INCB3344 were not investigated in this study, previous data suggest that a single oral dose of 50 mg/kg leads to approximately 75% CCR2 inhibition in mice35. In the study by Aiello et al., inflammatory monocyte levels were significantly reduced up to 6 hours after oral infusion, which was lost at 9 hours, while we still observed a >60% reduction in circulating CCR2+ monocyte numbers at 18 hours after treatment, suggestive of a longer therapeutic effect. Interestingly, we also observed an increase in the relative amount of Ly6ClowCX3CR1+ monocytes, which is indicative of a more patrolling response36. Similarly as in the study by Aiello et al., we did not observe any effects on bone marrow monocyte subpopulations upon treatment (data not shown), suggesting that the reduction observed on circulating CCR2+ monocytes is not due to changes in migration of these cells from the bone marrow. Further studies should shed more light on the exact mechanisms of depletion of this specific monocyte subset from the circulation, and how this affects the immune system, including effects on other chemokine receptors expressed by monocytes. In both studies however, plasma cholesterol levels were not affected, while an increase in plasma CCL2, one of the endogenous ligands for CCR2, was observed, which may be explained by the fact that clearance of CCL2 is mediated by CCR237. Noteworthy, treatment with 15a resulted in more than 70% reduction in carotid artery lesion size, which even exceeds the effects of the CCR2/CCR5/CXCR3 triple antagonist TAK-779 in inhibiting atherogenesis38, suggesting that inhibition of a single receptor, CCR2, might be sufficient for the therapeutic effect in atherosclerosis.
Finally we also investigated the pharmacokinetics of 15a in apoE−/− mice, which allowed us to determine drug exposure and predict the target occupancy achieved by 15a after a single dose. For a drug to exert its pharmacological effect, it must be bound to its target, making target occupancy a requisite for any in vivo effect. Previous analysis of clinical trials with CCR1 antagonists led to the conclusion that a receptor occupancy of >90% is required for an effective anti-inflammatory response26, 39. Calculation of CCR2 occupancy levels in our study revealed that, after a single dose, 15a was able to block more than 90% of the receptors for over 24 hours. Such high level of target engagement can be achieved by slow drug-target dissociation or by target-saturating drug concentrations. In our case, the calculated high receptor occupancy is the result of saturating concentrations and slow plasma elimination of 15a. For RT to play a key role in increasing the target occupancy, the k off of the drug candidate must at least surpass its pharmacokinetic elimination rate22, 23. However, the direct comparison of the pharmacokinetic elimination rate and the k off is only informative when the target occupancy is low, since it assumes that the declines of the target occupancy and the unbound drug concentration are parallel lines on a semi-logarithmic scale. For high levels of target occupancy (i.e. high target saturation), a steep decline of unbound drug concentrations is not directly reflected in a steep decline of target occupancy28, as we have predicted. This reasoning was used to determine the k off value required to prolong target occupancy even further: at least a ten-fold difference between the dissociation rate and the plasma elimination rate is required. Although the contribution of target saturation is not always recognized in literature19, we provide evidence that the use of saturating concentrations can result in the required occupancy levels for an anti-inflammatory effect. However, caution must be taken in order to avoid safety risks and toxicity with high doses. These results highlight the need of obtaining pharmacokinetic data and using experimental or mathematical approaches to determine receptor occupancy in early preclinical studies. In this regard, we have established that a simple “equilibrium” occupancy calculation can be sufficient to explain the in vivo efficacy in this study, providing a simple method that can be readily used in a range of in vitro and in vivo preclinical studies. This calculation method allows for further optimization towards a clinical candidate, for example by developing an oral dosing regimen to improve patient compliance.
Although the role of the CCR2:CCL2 axis in the development of atherosclerosis has been recognized in literature, previous studies with CCR2 small-molecule antagonists failed to successfully inhibit atherosclerosis11, 40. We are the first to prove that CCR2 antagonism with a selective small molecule antagonist can result in effective atherogenesis inhibition. The results of our in vivo study provide direct evidence that high receptor occupancy might be one of the key factors to improve the translation from in vitro findings into in vivo efficacy in inflammatory diseases. In addition, our findings in the in vitro study support the need of characterizing the affinity and kinetic profile of drug candidates in multiple species, as a way to improve preclinical and clinical translation of efficacy and safety findings12, 41. To conclude, the CCR2 antagonist 15a emerges as a potential candidate for further drug development to inhibit atherosclerotic lesion development and progression, and may also be of therapeutic value for other diseases involving CCR2.
Materials and Methods
Chemicals and reagents
CCR2 antagonists INCB3344, 15a ((1S,3R)-3-(((R)-5-bromo-2,3-dihydro-1H-inden-1-yl)amino)-1-isopropylcyclopentyl)(7-(trifluoromethyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone) and the internal standard 15b ((1S,3R)-3-(((S)-5-bromo-2,3-dihydro-1H-inden-1-yl)amino)-1-isopropylcyclopentyl)(7-(trifluoromethyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone) were synthesized in-house as described previously24, 42, 43. [3H]INCB3344 (specific activity 32 Ci mmol−1) was custom-labeled by Vitrax (Placentia, CA) as a racemic mixture of the two isomers N-(2-(((3S,4S)-1-((1r,4S)-4-(benzo[d][1,3]dioxol-5-yl)-4-hydroxycyclohexyl)-4-ethoxypyrrolidin-3-yl)amino)-2-oxoethyl)-3-(trifluoromethyl)benzamide and N-(2-(((3R,4R)-1-((1r,4R)-4-(benzo[d][1,3]dioxol-5-yl)-4-hydroxycyclohexyl)-4-ethoxypyrrolidin-3-yl)amino)-2-oxoethyl)-3-(trifluoromethyl)benzamide. Bovine serum albumin (BSA, fraction V) was purchased from Sigma (St. Louis, MO, USA). Bicinchoninic acid (BCA) and BCA protein assay reagent were obtained from Pierce Chemical Company (Rockford, IL, USA). Acetonitrile (LC-MS grade), trifluoroacetic acid (TFA) and ULC-MS water were from Biosolve (Valkenswaard, the Netherlands); and ethyl acetate from Baker (Deventer, The Netherlands). pcDNA3.1+ plasmid containing the murine CCR2 (mCCR2) with an hemagglutinin (HA) epitope tag at the N-terminus was cloned in-house. All other chemicals were obtained from standard commercial sources.
Cell culture
Chinese hamster ovary (CHO) cells were cultured at 37 °C and 5% CO2 in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12) supplemented with 10% (v/v) newborn calf serum, 50 IU/mL penicillin and 50µg/mL streptomycin. Cells were subcultured twice a week on 10-cm ø plates, at a ratio of 1:20 to 1:40 using trypsinization.
Transfections
Transient transfection of CHO cells with HA-tagged mouse CCR2 (mCCR2) was performed using a polyethyleneimine (PEI) method, as described previously44. Briefly, CHO cells were seeded at 50–60% confluence on 15-cm ø plates, transfected with 10 µg of plasmid DNA per plate, and incubated for 48 hrs at 37 °C and 5% CO2. Before transfection, the plasmid DNA was diluted in sterile 150 mM NaCl solution and mixed with PEI solution (1 mg/mL) to obtain a DNA:PEI mass ratio of 1:6. Finally, the culture medium of the cells was refreshed and 1 mL of DNA/PEI mixture was added to the cells, after incubation of the mixture for 20 min at room temperature.
Cell membrane preparation
Membranes were prepared as described before44. Briefly, cells were detached from 15-cm ø plates by scraping them with 5 ml phosphate-buffered saline (PBS), and the membranes were separated from the cytosolic fractions by several centrifugation and homogenization steps. The remaining membrane pellet was resuspended in ice-cold Tris buffer (50mM Tris-HCl, pH 7.4) containing 5 mM MgCl2. Membrane protein concentrations were measured using a bicinchoninic acid (BCA) protein determination with bovine serum albumin (BSA) as a standard45.
[3H]INCB3344 Binding assays
Radioligand binding assays were performed in a 100 µL reaction volume containing 50 mM Tris-HCl buffer (pH 7.4), 5 mM MgCl2, 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonic acid (CHAPS), 5 nM [3H]INCB3344 and 50 to 60 µg of membrane protein at 25 °C. Non-specific binding was determined with 10 μM of the cold ligand INCB3344. Displacement assays were carried out using 9 concentrations of competing ligand, ranging from 0.1 nM to 100 μM, for 120 min of incubation. The kinetic parameters of 15a were also determined at 25 °C using a competition association assay as previously described25, 27. For association and competition association experiments, CHO-mCCR2 membranes were added to the reaction volume at different time points, in the absence or presence of 30 nM of competing ligand. For dissociation experiments, CHO-mCCR2 membranes were first incubated with 5 nM [3H]INCB3344 for 60 min. Dissociation was initiated by addition of 10 µM of INCB3344 at different time points. For all experiments, incubation was terminated by dilution with ice-cold 50 mM Tris-HCl buffer supplemented with 5 mM MgCl2 and 0.05% CHAPS. Separation of bound from free radioligand was performed by rapid filtration over a 96-well GF/B filter plate using a Perkin Elmer Filtermate-harvester (Perkin Elmer, Groningen, the Netherlands) in the case of displacement assays; or over GF/B filters using a Brandel harvester 24 (Brandel, Gaithersburg, MD, USA) for kinetic assays. Filter-bound radioactivity was determined by scintillation spectrometry after addition of Microscint scintillation cocktail (Perkin-Elmer, Groningen, the Netherlands). The P-E 2450 Microbeta2 plate counter (Perkin Elmer, Groningen, The Netherlands) was used for displacement assays, and the TRI Carb 2900 TR counter (Perkin Elmer, Groningen, The Netherlands) for kinetic assays. In all cases, total radioligand binding did not exceed 10% of the amount of radioligand added to prevent ligand depletion.
Animals
This study was performed in compliance with Dutch government guidelines and the Directive 2010/63/EU of the European Parliament. All animal experiments were approved by the animal welfare committee of Leiden University (approval reference number 13213).
Male apoE−/− mice (10–12 weeks old), obtained from the local animal breeding facility (Gorlaeus Laboratories, Leiden, The Netherlands), were fed a Western type diet, containing 0.25% cholesterol and 15% cocoabutter (SDS, Sussex, UK).
To measure plasma concentrations of 15a over time, Western type diet fed mice were injected with 5 mg/kg 15a, after which blood samples were taken at fixed time points (1, 2, 6 and 24 hours after injection). Blood samples were processed as described below under “Measurement of 15a plasma levels”. The final dose of 5 mg/kg/day was based on a pilot study in which two different doses (1.5 and 5 mg/kg/day) were tested using leucocyte migration towards the peritoneal cavity as a read-out. Results from this pilot study showed that 1.5 mg/kg per day was not effective in inhibiting migration, while 5 mg/kg per day was (data not shown). In addition, the dose of 5 mg/kg was also based on a previous study with TAK-77938, in which a dose of 5 mg/kg was used.
To determine the effect of the CCR2 antagonist on atherosclerotic lesion development, mice were fed a Western type diet, containing 0.25% cholesterol and 15% cocoabutter (SDS, Sussex, UK) throughout the experiment starting two week before surgery. Carotid artery plaque formation was induced by perivascular collar placement as described previously46 and from the time of surgery, the mice received daily intraperitoneal injections containing 15a (5 mg/kg/day) or vehicle control (n = 9–10 per group). During the experiment, total body weight and weight gain were measured. Serum total cholesterol levels were determined using an enzymatic colorimetric assay according to manufacturer’s protocol (Roche Diagnostics). Precipath (standardized serum; Boehringer Mannheim, Germany) was used as an internal standard. Plasma cytokine levels were measured by ELISA according to manufacturer’s protocol (BD Biosciences). Blood was collected weekly via tail cut. Total cell count and cellular differentiation patterns in blood were analyzed using an automated XT-2000iV veterinary hematology analyzer (Sysmex Europe GMBH, Norderstedt, Germany). After erythrocyte lysis, blood leukocyte suspensions were stained for surface markers, after which their expression was determined by FACS analysis (FACS Canto, BD Biosciences). After 4 weeks of lesion development, the animals were anaesthetized and blood was collected. After in situ perfusion through the left cardiac chamber, the carotid arteries and hearts were harvested.
Measurement of 15a plasma levels
Blood plasma from mice was analyzed by LC-MS to determine 15a plasma levels. 20 μL of internal standard (compound 15b, a diastereomer of 15a24) and 20 μL of solvent C (20% (v/v) Acetonitrile and 0.1% (v/v) TFA in LC-MS water) were first mixed with 20 µL of plasma. A calibration curve from 2 to 200 ng/ml of 15a was prepared using solvent C. Calibration samples were prepared as normal samples using blanc plasma and calibration solution instead of solvent C. Aliquots of 40 µL of 1% TFA were added to the samples and mixed well. After addition of 900 µL of ethyl acetate, mixing was performed for 5 minutes at a titramax plate shaker (Heidolph, Swabach, Germany). Samples were then centrifuged at 15,000 × g during 10 minutes and 800 µL of the supernatant was evaporated in a vacuum centrifuge (Labconco, Kansas City, Missouri). The sample was reconstituted by mixing with 10 µL of acetonitrile, followed by addition of 40 µL solvent C. After a second centrifugation at 15,000 × g for 10 minutes, 35 µL of supernatant were transferred to LC vials and inserted into the LC-MS system consisting of a Nexera X2 UHPLC system with two UHPLC pumps (Shimadzu, ’s Hertogenbosch, The Netherlands) connected to a TSQ Quantum Ultra (Thermo Fisher Scientific, Breda, The Netherlands). Separation of 15a was accomplished in a Gemini 3 µm C18 reversed phase HPLC column (50 × 4.6 mm) (Phenomenex, Amstelveen, The Netherlands) using an injection volume of 10 µL and a flow rate of 0.6 ml/min at 40 °C. After elution of 15a with a 9:11 (v/v) ratio of acetonitrile in water with 0.1% TFA, the column was flushed with a gradient up to 90% acetonitrile with 0.1% TFA. The column was re-equilibrated with a 9:11 (v/v) ratio of acetonitrile in water with 0.1% TFA before the next injection. Samples were analyzed by mass spectrometry using Electro Spray Ionization at a voltage of 3000 V. 15a and the internal standard 15b showed the same fragmentation pattern and were quantified by monitoring the transition of 549.17 to 202.08 (m/z). The collision energy used was 26 V at a skimmer offset of 16 V, the scan peak width was 0.5 m/z, while the scan time was 0.3 seconds. Sheath gas and auxiliary gas pressure (Nitrogen) were set to 60 and 5 respectively (arbitrary units), and the collision gas Argon was set at 1.0 atm.
Histology and morphometry
Carotid artery cryosections of 5 µm thick were prepared, which were stained with hematoxylin-eosin to determine plaque size and necrotic core size. Morphometric analysis was performed on sections throughout the atherosclerotic lesion (100 µm apart) and at the site of maximal stenosis using Leica Qwin image analysis software. The relative necrotic area was defined as the a-cellular, debris-rich plaque area as percentage of total plaque area, and also quantified using Leica QWin software.
The hearts were dissected just below the atria and sectioned perpendicular to the axis of the aorta, starting within the heart and working in the direction of the aortic arch. Once the aortic root was identified by the appearance of aortic valve leaflets, 10 µm sections were collected. Mean lesion area (in µm2) of the aortic root was calculated from six Oil-Red-O stained sections in distal direction starting at the point where all three aortic valve leaflets first appeared.
Macrophage content of the lesions in carotid arteries and aortic roots was stained using a rat monoclonal MOMA2 antibody (1:1000, Serotec, Kidlington, Oxford, UK). Macrophage positive areas were analyzed using computer-assisted colour-gated analysis, and related to the total intimal surface area (Leica QWin). Liver cryosections of 10 µm thick were prepared and subsequently stained with hematoxylin-eosin to analyze liver morphology, which was assessed using a Leica DMRE microscope (Leica Ltd., Cambridge, England).
RNA isolation and gene expression analysis
Total RNA was extracted from homogenized liver tissue with the guanidium thiocyanate-phenol-bromochloropropane extraction method38. RNA concentration, purity and integrity were examined by nanodrop (Nanodrop® Technologies). RNA was reverse transcribed by M-MuLV reverse transcriptase (RevertAid, MBI Fermentas, Landsmeer, The Netherlands) and used for quantitative analysis of the mouse genes ALT, AST, apoH and GC with an ABI PRISM 7700 Taqman apparatus (Applied Biosystems). For qPCR primer pairs refer to Supplementary Table S1.
Data analysis
All radioligand binding assays were analyzed using Prism 6.0 (GraphPad software, San Diego, CA, USA). All data shown represent means ± SEM of at least three independent experiments performed in duplicate. Ki values were calculated from IC50 values obtained by non-linear regression analysis of the displacement assays and by using the Cheng-Prusoff equation47. Dissociation rate constants k off were determined by fitting the dissociation data using an exponential decay model. Association rate constants k on were obtained by converting the apparent association rate constants k obs according to the equation k on = (k obs − k off)/[L], in which L is the radioligand concentration used for the experiments, k obs is the constant rate determined using exponential association analysis and k off represents the mean k off value obtained independently in the dissociation experiments. The kinetic profile of compound 15a was determined by calculation of its kinetic rate index (KRI) value48. The KRI values were calculated according to the equation KRI = Bt1/Bt2, in which Bt represents the specific radioligand binding measured at t1 = 15 min and t2 = 210 min. In addition, the association and dissociation rates of 15a were obtained by nonlinear regression analysis of the competition association data using the equations described by Motulsky and Mahan27. The association and dissociation rates obtained with this model were used to calculate the kinetic K D, according to the equation K D = k off/k on. In addition, the RT was calculated by obtaining the reciprocal of the k off. Data obtained from in vivo experiments are expressed as mean ± SEM. An unpaired two-tailed Student’s t-test was used to compare individual groups. Non-Gaussian distributed data were analyzed using a two-tailed Mann-Whitney U test. A level of P < 0.05 was considered significant.
Pharmacokinetics-based prediction of target occupancy
The equilibrium target occupancy was calculated by assuming that binding was fast and the drug-target residence time did not prolong the target occupancy. The target occupancy was calculated for each measurement of the drug concentration, according to equation (1), in which [C] is the concentration of the drug in the blood and KD is the drug target affinity.
| 1 |
In addition, a simple mathematical model was used to simulate target occupancy for the observed in vivo pharmacokinetics and in vitro binding kinetics according to the equations in the next section (equations (3–5)).
To determine the maximal value of k off that would prolong the receptor occupancy even further, we used equation (2)28, in which BF represents the percentage of target occupancy divided by 100 and t 1/2el represents the plasma elimination half-life (0.693/k el). In this case we set BF to 0.90, as 90% represents the minimum value of target occupancy to achieve efficacy. To obtain the elimination rate constant of compound 15a, a linear regression analysis of the natural logarithm of the concentrations versus time was performed, and the slope coefficient was used as the elimination rate constant k el.
| 2 |
Differential equations for the simulations of target occupancy
The concentrations of unbound drug in the blood [C] and target-bound drug [LR] were modeled according to equations (3–5). In these equations, k el is the first-order elimination rate constant, k on is the second-order association rate constant, k off is the first-order dissociation rate constant, [R free] is the concentration of unbound receptor and [R tot] is the concentration of bound plus unbound receptor, which is assumed to be constant.
| 3 |
| 4 |
The concentration of the target-bound drug is calculated according to equation (5):
| 5 |
Parameter values: for the simulations in Fig. 8b, k el was set to 0.0647 hr−1, based on the slope of the linear regression of the pharmacokinetic data (Fig. 8a). On basis of in-house measurements, k off was set to 3.06 hr−1 (corresponding to 0.051 min−1, as shown in Table 1) and the K D was set to 10 nM, which corresponds to the Ki value of 15a determined in [3H]INCB3344 displacement assays. The Ki value was used instead of the K D (from Table 1) as this represents a more conservative choice with regard to target saturation. The receptor concentration was set to an arbitrarily low number of 1 pM.
Electronic supplementary material
Acknowledgements
We want to thank T. Mulder-Krieger for technical assistance regarding the cloning and purification of the plasmid used. I.B. is supported by a Dr. Dekker Senior Postdoc grant from the Netherlands Heart Foundation (2012T083). We acknowledge the support from the Netherlands CardioVascular Research Initiative: “the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organization for Health Research and Development and the Royal Netherlands Academy of Sciences” for the GENIUS project “Generating the best evidence-based pharmaceutical targets for atherosclerosis” (CVON2011-19).
Author Contributions
I.B., N.V.O.Z., H. de V., P.J. van S., D. van der V., M.J.K., D. van der B., and W.E.A. de W. performed the research; I.B., J.K., N.V.O.Z., W.E.A. de W., E.C.M. de L., D.S., A.P.I. and L.H.H. designed the research study; I.B., N.V.O.Z. and W.E.A. de W. analyzed the data; I.B., N.V.O.Z., W.E.A. de W., E.C.M. de L., J.K., A.P.I. and L.H.H. wrote the paper.
Competing Interests
The authors declare no competing financial interests.
Footnotes
Ilze Bot and Natalia V. Ortiz Zacarías contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-00104-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 2.Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annual review of pharmacology and toxicology. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- 3.White GE, Iqbal AJ, Greaves DR. CC chemokine receptors and chronic inflammation–therapeutic opportunities and pharmacological challenges. Pharmacological reviews. 2013;65:47–89. doi: 10.1124/pr.111.005074. [DOI] [PubMed] [Google Scholar]
- 4.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 5.Dawson TC, Kuziel WA, Osahar TA, Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. doi: 10.1016/S0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 6.Guo J, et al. Transplantation of monocyte CC-chemokine receptor 2-deficient bone marrow into ApoE3-Leiden mice inhibits atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:447–453. doi: 10.1161/01.ATV.0000058431.78833.F5. [DOI] [PubMed] [Google Scholar]
- 7.Abbadie C, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proceedings of the National Academy of Sciences. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. The Journal of experimental medicine. 2000;192:899–906. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gosling J, et al. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. The Journal of clinical investigation. 1999;103:773–778. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu L, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Molecular cell. 1998;2:275–281. doi: 10.1016/S1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 11.Aiello RJ, et al. CCR2 receptor blockade alters blood monocyte subpopulations but does not affect atherosclerotic lesions in apoE(−/−) mice. Atherosclerosis. 2010;208:370–375. doi: 10.1016/j.atherosclerosis.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Guo, D., Heitman, L. H. & IJzerman, A. P. The Role of Target Binding Kinetics in Drug Discovery. Chem. Med. Chem. (2015). [DOI] [PubMed]
- 13.Copeland RA. The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert opinion on drug discovery. 2010;5:305–310. doi: 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- 14.Zhang R, Monsma F. Binding kinetics and mechanism of action: toward the discovery and development of better and best in class drugs. Expert opinion on drug discovery. 2010;5:1023–1029. doi: 10.1517/17460441.2010.520700. [DOI] [PubMed] [Google Scholar]
- 15.Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- 16.Ramsey SJ, Attkins NJ, Fish R, van der Graaf PH. Quantitative pharmacological analysis of antagonist binding kinetics at CRF1 receptors in vitro and in vivo. British journal of pharmacology. 2011;164:992–1007. doi: 10.1111/j.1476-5381.2011.01390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleck BA, Hoare SR, Pick RR, Bradbury MJ, Grigoriadis DE. Binding kinetics redefine the antagonist pharmacology of the corticotropin-releasing factor type 1 receptor. Journal of Pharmacology and Experimental Therapeutics. 2012;341:518–531. doi: 10.1124/jpet.111.188714. [DOI] [PubMed] [Google Scholar]
- 18.Lu H, et al. Slow-onset inhibition of the FabI enoyl reductase from Francisella tularensis: residence time and in vivo activity. ACS chemical biology. 2009;4:221–231. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindström E, et al. Neurokinin 1 receptor antagonists: correlation between in vitro receptor interaction and in vivo efficacy. Journal of Pharmacology and Experimental Therapeutics. 2007;322:1286–1293. doi: 10.1124/jpet.107.124958. [DOI] [PubMed] [Google Scholar]
- 20.Seow V, et al. Receptor residence time trumps drug-likeness and oral bioavailability in determining efficacy of complement C5a antagonists. Sci Rep. 2016;6:24575. doi: 10.1038/srep24575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calbet M, et al. Pharmacological characterization of CRTh2 antagonist LAS191859: Long receptor residence time translates into long-lasting in vivo efficacy. Pharmacol Res. 2016;111:208–216. doi: 10.1016/j.phrs.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 22.Dahl G, Akerud T. Pharmacokinetics and the drug-target residence time concept. Drug Discov Today. 2013;18:697–707. doi: 10.1016/j.drudis.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 23.Vauquelin G, Van Liefde I. Slow antagonist dissociation and long-lasting in vivo receptor protection. Trends Pharmacol Sci. 2006;27:356–359. doi: 10.1016/j.tips.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 24.Vilums M, et al. When structure-affinity relationships meet structure-kinetics relationships: 3-((Inden-1-yl)amino)-1-isopropyl-cyclopentane-1-carboxamides as CCR2 antagonists. European journal of medicinal chemistry. 2015;93:121–134. doi: 10.1016/j.ejmech.2015.01.063. [DOI] [PubMed] [Google Scholar]
- 25.Vilums M, et al. Structure-kinetic relationships–an overlooked parameter in hit-to-lead optimization: a case of cyclopentylamines as chemokine receptor 2 antagonists. Journal of medicinal chemistry. 2013;56:7706–7714. doi: 10.1021/jm4011737. [DOI] [PubMed] [Google Scholar]
- 26.Schall TJ, Proudfoot AE. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nature reviews. Immunology. 2011;11:355–363. doi: 10.1038/nri2972. [DOI] [PubMed] [Google Scholar]
- 27.Motulsky HJ, Mahan LC. The kinetics of competitive radioligand binding predicted by the law of mass action. Molecular pharmacology. 1984;25:1–9. [PubMed] [Google Scholar]
- 28.de Witte WE, Danhof M, van der Graaf PH, de Lange EC. In vivo Target Residence Time and Kinetic Selectivity: The Association Rate Constant as Determinant. Trends Pharmacol Sci. 2016;37:831–842. doi: 10.1016/j.tips.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Guo D, Hillger JM, IJzerman AP, Heitman LH. Drug‐Target Residence Time—A Case for G Protein‐Coupled Receptors. Medicinal research reviews. 2014;34:856–892. doi: 10.1002/med.21307. [DOI] [PubMed] [Google Scholar]
- 30.Yu Z, et al. Structure-Affinity Relationships (SARs) and Structure-Kinetics Relationships (SKRs) of Kv11.1 Blockers. Journal of medicinal chemistry. 2015;58:5916–5929. doi: 10.1021/acs.jmedchem.5b00518. [DOI] [PubMed] [Google Scholar]
- 31.Louvel J, et al. Agonists for the adenosine A1 receptor with tunable residence time. A case for nonribose 4-amino-6-aryl-5-cyano-2-thiopyrimidines. Journal of medicinal chemistry. 2014;57:3213–3222. doi: 10.1021/jm401643m. [DOI] [PubMed] [Google Scholar]
- 32.Andrés M, et al. Structure–activity relationships (SAR) and structure–kinetic relationships (SKR) of pyrrolopiperidinone acetic acids as CRTh2 antagonists. Bioorganic & medicinal chemistry letters. 2014;24:5111–5117. doi: 10.1016/j.bmcl.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 33.Struthers M, Pasternak A. CCR2 antagonists. Current topics in medicinal chemistry. 2010;10:1278–1298. doi: 10.2174/156802610791561255. [DOI] [PubMed] [Google Scholar]
- 34.Kohout TA, et al. Trapping of a nonpeptide ligand by the extracellular domains of the gonadotropin-releasing hormone receptor results in insurmountable antagonism. Molecular pharmacology. 2007;72:238–247. doi: 10.1124/mol.107.035535. [DOI] [PubMed] [Google Scholar]
- 35.Brodmerkel CM, et al. Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344. Journal of immunology. 2005;175:5370–5378. doi: 10.4049/jimmunol.175.8.5370. [DOI] [PubMed] [Google Scholar]
- 36.Auffray C, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 37.Tylaska LA, et al. Ccr2 regulates the level of MCP-1/CCL2 in vitro and at inflammatory sites and controls T cell activation in response to alloantigen. Cytokine. 2002;18:184–190. doi: 10.1006/cyto.2002.1031. [DOI] [PubMed] [Google Scholar]
- 38.van Wanrooij EJ, et al. HIV entry inhibitor TAK-779 attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:2642–2647. doi: 10.1161/01.ATV.0000192018.90021.c0. [DOI] [PubMed] [Google Scholar]
- 39.Dairaghi DJ, et al. Pharmacokinetic and pharmacodynamic evaluation of the novel CCR1 antagonist CCX354 in healthy human subjects: implications for selection of clinical dose. Clin Pharmacol Ther. 2011;89:726–734. doi: 10.1038/clpt.2011.33. [DOI] [PubMed] [Google Scholar]
- 40.Olzinski AR, et al. Pharmacological inhibition of C-C chemokine receptor 2 decreases macrophage infiltration in the aortic root of the human C-C chemokine receptor 2/apolipoprotein E−/− mouse: magnetic resonance imaging assessment. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:253–259. doi: 10.1161/ATVBAHA.109.198812. [DOI] [PubMed] [Google Scholar]
- 41.Cusack KP, et al. Design strategies to address kinetics of drug binding and residence time. Bioorganic & medicinal chemistry letters. 2015;25:2019–2027. doi: 10.1016/j.bmcl.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 42.Xue, C. B. et al. 3-aminopyrrolidine derivatives as modulators of chemokine receptors. (2004).
- 43.Xue CB, et al. Discovery of INCB3344, a potent, selective and orally bioavailable antagonist of human and murine CCR2. Bioorganic & medicinal chemistry letters. 2010;20:7473–7478. doi: 10.1016/j.bmcl.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 44.Zweemer AJ, et al. Multiple binding sites for small-molecule antagonists at the CC chemokine receptor 2. Molecular pharmacology. 2013;84:551–561. doi: 10.1124/mol.113.086850. [DOI] [PubMed] [Google Scholar]
- 45.Smith PK, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 46.von der Thusen JH, van Berkel TJ, Biessen EA. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation. 2001;103:1164–1170. doi: 10.1161/01.CIR.103.8.1164. [DOI] [PubMed] [Google Scholar]
- 47.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 48.Guo D, et al. Dual-point competition association assay: a fast and high-throughput kinetic screening method for assessing ligand-receptor binding kinetics. J Biomol Screen. 2013;18:309–320. doi: 10.1177/1087057112464776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.