

Supplemental Digital Content is Available in the Text.
Key Words: tenofovir alafenamide, HIV, proteinuria, osteopenia
Abstract:
In a double-blind, phase 3 trial, 663 HIV-infected, virologically suppressed adults were randomized to switch to tenofovir alafenamide (TAF; n = 333) vs. remain on tenofovir disoproxil fumarate (TDF; n = 330), each coformulated with emtricitabine (FTC), while continuing their third agent (boosted protease inhibitor or unboosted third agent). At week 96, 88.6% on FTC/TAF and 89.1% on FTC/TDF had HIV-1 RNA <50 copies per milliliter [adjusted difference −0.5% (95% confidence interval: −5.3 to 4.4%)]. Proteinuria, albuminuria, proximal renal tubular function, and bone mineral density improved after switching to TAF- from TDF-containing regimens. These longer-term data support FTC/TAF as a safe, well-tolerated, and durable nucleotide reverse transcriptase inhibitor backbone.
INTRODUCTION
Although tenofovir disoproxil fumarate (TDF), a prodrug of tenofovir (TFV), is a potent and generally well-tolerated nucleotide analog, it has been associated with an increased risk of nephrotoxicity and greater reductions in bone mineral density (BMD) compared with other nucleotide reverse transcriptase inhibitors.1–6 Tenofovir alafenamide (TAF) is a novel TFV prodrug that is associated with 91% lower plasma TFV levels compared with TDF,7 which leads to less adverse impact on bone and kidneys.8,9 Recent HIV treatment guidelines have either replaced TDF with TAF or include both as part of recommended initial regimens.10,11
In this large, double-blind, multicenter trial, we compared the switch to fixed-dose emtricitabine (FTC) with TAF versus continued use of fixed-dose FTC with TDF in virologically suppressed HIV-infected patients (ClinicalTrials.gov number NCT02121795). Results from week 48 were previously reported and demonstrated that switching to FTC/TAF was noninferior to continued use of FTC/TDF while remaining on the same third agent in maintaining viral suppression and led to improvements in markers of bone and renal safety.12 We present safety and efficacy data through 96 weeks, with a focus on outcomes depending on third agent.
METHODS
Study Design and Participants
The design and inclusion criteria of this randomized, double-blind, multicenter, active-controlled phase 3 trial have been previously described.12 Briefly, we enrolled HIV-infected adults (aged ≥18 years) who were virologically suppressed (HIV-1 RNA <50 copies/mL) for ≥6 months on regimens containing fixed-dose FTC with TDF and had estimated glomerular filtration rate (eGFR) >50 mL/min (calculated by the Cockcroft–Gault equation). Eligible participants were randomized (1:1) to either switch to coformulated FTC/TAF or to continue FTC/TDF without changing their third agents. Participants on boosted protease inhibitors (PIs) (atazanavir, darunavir, and lopinavir all boosted with ritonavir) who were randomly assigned to switch treatment received coformulated 200 mg FTC with 10 mg TAF; those on other third agents received coformulated 200 mg FTC with 25 mg TAF. Participants also received placebo tablets matching the alternative treatment. The study remained blinded until the last participant receiving study drug completed week 96.
Postbaseline study visits occurred at weeks 2, 4, 8, 12, 16, 24, 36, and 48, after which participants continued treatment with visits every 12 weeks until week 96. Laboratory tests included hematological analysis, serum chemistry tests, fasting lipid parameters, CD4+ cell counts, measures of renal function, including eGFR, urine protein-to-creatinine ratio, urine albumin-to-creatinine ratio, tubular proteinuria (retinol-binding protein-to-creatinine ratio, β2-microglobulin-to-creatinine ratio, fractional excretion of uric acid, and fractional excretion of phosphate) (Covance Laboratories, Indianapolis, IN), and measurement of HIV RNA concentration (Roche TaqMan 2.0; Roche Diagnostics, Rotkreuz, Switzerland). Participants with confirmed virologic failure (2 consecutive HIV RNA >50 copies/mL) and an HIV RNA >400 copies per milliliter at week 8 or later had the second, confirmatory sample sent for resistance analysis by GeneSeq Integrase, PhenoSense GT, and PhenoSense Integrase (Monogram Biosciences, South San Francisco, CA). Dual-energy x-ray absorptiometry of the hip and lumbar spine was conducted at baseline and weeks 24, 48, 72, and 96 [analyzed centrally by BioClinica (Newton, PA)].
The study was conducted in accordance with the Declaration of Helsinki and approved by central or site-specific review boards or ethics committees. Each participant provided written informed consent.
Statistical Analyses
The primary endpoint was the proportion of participants who had plasma HIV-1 RNA <50 copies per milliliter at week 48 as defined by the US Food and Drug Administration (FDA) snapshot algorithm.13,14 Secondary endpoints included the proportion of participants with virologic success (maintenance of HIV-1 RNA <50 copies/mL) at week 96. The percentage differences and the associated 95% confidence intervals (CIs) were constructed with Mantel–Haenszel proportion stratified by previous treatment regimens. Noninferiority between treatment groups was assessed using a conventional 95% CI approach and a margin of 10% (1-sided 0.025 level). Safety outcomes included renal, bone, and metabolic endpoints. Changes from baseline were summarized by visit using descriptive statistics, and median change from baseline was analyzed by a 2-sided Wilcoxon signed-rank test. Subgroup analyses were performed for select endpoints by third agent (boosted PI or unboosted third agent). Adverse events (AEs) were coded with the Medical Dictionary for Regulatory Activities (version 19).
RESULTS
We randomized 668 participants; 663 received at least 1 dose of study drug (333 switched to FTC/TAF and 330 remained on FTC/TDF). Baseline characteristics were previously reported, with a median age of 49 years.12 The distributions of third agents were similar between the 2 treatment groups (boosted PI: FTC/TAF 47%, FTC/TDF 45%; unboosted third agent: FTC/TAF 53%, FTC/TDF 55%). Median (interquartile range) time of FTC/TDF use before dosing was 5.1 years (3.0–7.2 years) overall.
Switching to an FTC/TAF-containing regimen continued to be noninferior to continuing the FTC-/TDF-containing regimen at week 96 for the secondary efficacy outcome of proportion of participants with HIV-1 RNA <50 copies per milliliter [295/333 (88.6%) vs. 294/330 (89.1%), adjusted difference −0.5%, 95% CI: −5.3 to 4.4%] (Supplemental Digital Content, Table 1, http://links.lww.com/QAI/A980). Of the 38 (of 333) participants remaining in the FTC/TAF group who did not have HIV-1 RNA <50 copies per milliliter at week 96, 33 discontinued study drug because of reasons other than lack of efficacy (16 after week 48); 5 with HIV-1 RNA ≥50 copies per milliliter at week 96 (all were < 200 copies/mL) were later suppressed while continuing study drug. Of the 36 (of 330) participants remaining in the FTC/TDF group who did not have HIV-1 RNA <50 copies per milliliter at week 96, 33 discontinued study drug because of reasons other than lack of efficacy (15 after week 48); 1 who was suppressed before missing the week 96 visit, and 2 with HIV-1 RNA ≥50 copies per milliliter at week 96 (both were <200 copies/mL) were later suppressed while continuing study drug. Noninferiority was also demonstrated regardless of third agent [boosted PI: 85.8% vs. 88.1%; difference −2.3% (95% CI: −9.8 to 5.3%) or unboosted third agent: 91.0% vs. 89.9%; difference 1.1% (95% CI: −5.0 to 7.2%)]. The median (interquartile range) increase from baseline in CD4 cell count at week 96 was 44 (−54 to 144) cells per microliter for the FTC/TAF group and 33 (−44 to 134) cells per microliter for the FTC/TDF group. Other than the previously reported participant receiving FTC/TAF plus darunavir boosted by ritonavir who had emergent resistance M184V mutation, no other participants have developed HIV resistance since week 48.
Both regimens were well tolerated through a median exposure of 96 weeks. AEs leading to study drug discontinuation occurred in 8 (2%) participants in the FTC/TAF group vs. 4 (1%) in FTC/TDF group (1 in each group after week 48). For the FTC/TAF group, these AEs included affective disorder/acquired lipodystrophy (after week 48), atrial fibrillation, diarrhea, dysphagia, insomnia/altered mood, lymphoma, overdose, and peripheral edema. For the FTC/TDF group, these AEs included headache, increased blood creatinine, rectal tenesmus, and renal tubular disorder (after week 48). The type and frequency of treatment-emergent AEs were similar between groups. The incidence of serious AEs was similar in both groups, FTC/TAF 8% vs. FTC/TDF 9%. Two participants in the FTC/TAF group died [1 due to lymphoma and elevated lipase and 1 due to respiratory failure (after week 48)]; 1 participant in the FTC/TDF group died because of drowning (as reported by the investigator) (after week 48). None of the deaths were deemed related to study drugs.
We noted increases from baseline in eGFR for participants who switched to FTC/TAF, mostly occurring in the first 24 weeks, as compared with minimal changes among those who remained on an FTC/TDF regimen (10.0 vs. 4.0 mL/min). We observed significant differences between groups favoring FTC/TAF in changes of total proteinuria, albuminuria, and tubular proteinuria (urine retinol-binding protein/creatinine ratio and urine β2-microglobulin/creatinine ratio) (Fig. 1). Of note, in the FTC/TDF group, albuminuria and tubular proteinuria continued to worsen in the second year. Improvement of these markers of renal safety in the FTC/TAF group was similar regardless of third agent (Table 1). No participants in the FTC/TAF group and 2 in the FTC/TDF group discontinued study drug because of renal AEs. One participant with underlying hypertension on FTC/TDF plus ritonavir-boosted darunavir had an increase in serum creatinine that led to discontinuation of study drug. The second participant on FTC/TDF plus ritonavir-boosted atazanavir had laboratory findings consistent with proximal tubulopathy (proteinuria, normoglycemic glycosuria, hypophosphatemia, and an increase in serum creatinine). No cases of proximal tubulopathy or Fanconi syndrome were reported in the FTC/TAF group.
FIGURE 1.

Changes in BMD and renal markers, through week 96. RBP, retinol-binding protein.
TABLE 1.
Changes in Quantitative Measures of Proteinuria and Lipid Parameters at Week 96*
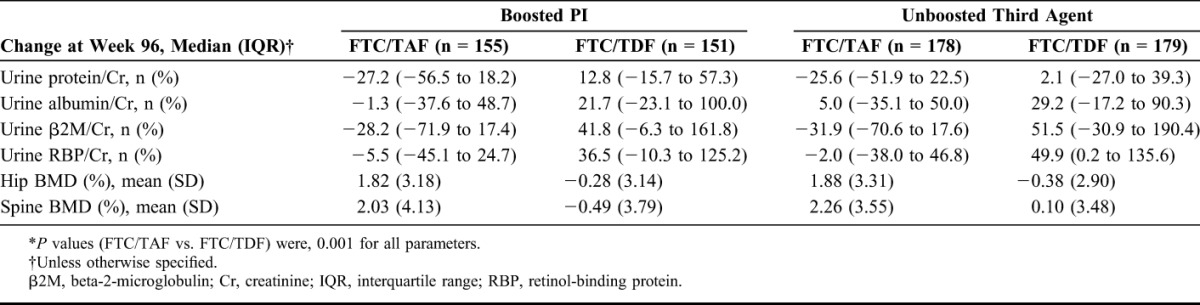
From baseline to week 96, BMD increased in the FTC/TAF group but not in the FTC/TDF group [median change: hip 1.78% vs. −0.17% (P < 0.001) and spine 1.85% vs. −0.33% (P < 0.001)] (Fig. 1). Of note, in the FTC/TAF group, BMD continued to increase in the second year. Results were similar regardless of third agent (Table 1). More participants in the FTC/TAF group had increase in BMD of at least 3% [spine 40% vs. 18% (P < 0.001) and hip 29% vs. 11% (P < 0.001)]. There were 4 fractures (1 on FTC/TAF and 3 on FTC/TDF), all related to mechanical trauma and considered by the investigator to be unrelated to study drug.
The incidence of laboratory abnormalities through week 96 was similar for both treatment groups. Fasting lipid values increased from baseline in the FTC/TAF group while remaining stable in the FTC/TDF group at week 96 [total cholesterol, FTC/TAF 14 vs. FTC/TDF 1 mg/dL, P < 0.001; low-density lipoprotein 14 vs. 4 mg/dL, P < 0.001; high-density lipoprotein (HDL) 1 vs. −1 mg/dL, P = 0.023]; however, median changes were minimal from a clinical standpoint and the median changes in total cholesterol to HDL ratio were similar (0.1 vs. 0.1; P = 0.26). The rate of initiating lipid-modifying medications was similar between the groups at week 96 (FTC/TAF 7.2%, FTC/TDF 6.4%, P = 0.76).
DISCUSSION
Our findings demonstrated that switching to FTC/TAF was noninferior to continuing FTC/TDF in maintaining virological suppression at 96 weeks in individuals with HIV receiving a large variety of third agents. Although overall safety was similar, renal parameters improved in patients who switched to FTC/TAF, with an increase in eGFR and a reduction in proteinuria, especially in the excretion of β2-microglobulin and retinol-binding protein, which are considered specific markers of proximal tubulopathy.15 BMD also improved in patients who switched, and changes were significantly greater in the FTC/TAF group at 96 weeks, with a greater likelihood of improvement in clinical bone density status (osteoporosis, osteopenia, or normal). The efficacy and safety results, including those of bone and renal, did not differ by third agent; particularly, improvement was also seen in patients receiving a ritonavir-boosted PI.
The results for the markers of renal safety were consistent with recent data from studies comparing TAF vs. TDF as part of 2 single-tablet regimens also containing elvitegravir, cobicistat, and FTC (E/C/F/TAF vs. E/C/F/TDF). Importantly, these data corroborate the clinical data, as there were no renal discontinuations or proximal renal tubulopathy in patients receiving FTC/TAF in this study (n = 333) nor in treatment-naive patients receiving E/C/F/TAF (n = 866) through week 96.16 By contrast, 2 patients receiving FTC/TDF in this study and 6 receiving E/C/F/TDF in the treatment-naive study16 had renal discontinuations; 1 in each group had proximal renal tubulopathy. The BMD results are also reassuring in their consistency with those from E/C/F/TAF studies. One interesting aspect is the fact that there is continuous improvement over the 96-week period, with no plateau effect, and the BMD increase by week 96 (around + 2% for both hip and spine) is clinically important in this population of patients with a median previous exposure to TDF of over 5 years.
Lipids increased in the FTC/TAF group while remaining stable in the FTC/TDF group. Because TDF is associated with lower lipids, it is likely that lower TFV exposures through switching TDF to TAF is leading to increase in lipids.17–20 However, no differences in total cholesterol to HDL ratio or initiation of lipid-modifying medications were noted between groups, suggesting that the increases in lipids on switching to FTC/TDF from FTC/TDF are probably of minimal clinical relevance.
FTC/TAF is now part of recommended initial regimens in treatment guidelines; E/C/F/TAF is also one of the recommended initial regimens.10,11 In addition, FTC/TAF is available as part of rilpivirine/FTC/TAF and is being developed as part of darunavir/COBI/FTC/TAF and bictegravir/FTC/TAF. In addition, FTC/TAF uniquely provides clinicians with the flexibility to combine with other agents that are not part of currently available single-tablet regimens. Lastly, FTC/TAF is also being developed for preexposure prophylaxis of HIV infection.
The longer-term data in this study confirm the potential of FTC/TAF to be an important nucleotide reverse transcriptase inhibitor backbone in the treatment of patients with HIV, with the flexibility to be combined with a variety of third agents and safety advantages over FTC/TDF. In patients exposed to FTC/TDF for many years, who represent a large majority of patients with HIV currently in care, and in those who are aging with potential for renal and or bone comorbidities from various causes, proactive switching from FTC/TDF to FTC/TAF can improve renal and bone parameters, while maintaining antiviral efficacy.
ACKNOWLEDGMENTS
The authors thank the patients of the Study GS-US-311-1089 and their partners and families. They also thank Sandra Friborg, Cecilia Tran-Muchowski, Lijie Zhong, Wei Dong, Ling Guo, and the complete GS-US-311-1089 study team, as well as Anna Kido for providing editorial assistance.
The authors also thank the principal investigators and their study staff for Study GS-US-311-1089: t J. Angel (Ottawa Hospital), N.B. (Southwest Infectious Disease Associates), P. Benson (Be Well Medical Center), C. Brinson (Central Texas Clinical Research), J. Brunetta (Maple Leaf Research), J. Chas (Hôpital Tenon), A.C. (Brighton and Sussex University Hospitals NHS Trust), N. Clumeck (CHU St-Pierre), B. Conway (Vancouver ID, Research & Care Centre Society), D. Coulston (Premier Clinical Research), G. Crofoot (The Crofoot Research Center), E.D. (Los Angeles Biomedical Research Institute at Harbor-UCLA), E. DeJesus (Orlando Immunology Center), C. Dietz (Kansas City CARE Clinic), W. Drummond (National Jewish Health), H. Edelstein (Alameda Health System—Highland Hospital), J. Flamm (Kaiser Permanente Medical Group), J.G. (Southwest CARE Center), J. Gathe (Therapeutic Concepts), D. Goldstein (Whitman-Walker Health), R. Grossberg (Montefiore Medical Center), C. Hare (Kaiser Permanente Medical Center, Research Unit), W.H. (Hennepin County Medical Center, Positive Care Center), R. Hsu (Dr. Ricky Hsu), M. Johnson (Royal Free London), C. Kinder (AHF Kinder Medical Group), D. Klein (Kaiser Permanente, Infectious Diseases Department), A. LaMarca (Therafirst Medical Center), A. Lazzarin (IRCCS Ospedale San Raffaele), C. Lucasti (South Jersey Infectious Disease), F. Maggiolo (AO Papa Giovanni XXIII), C. McDonald (Tarrant County, Infectious Disease Associates), J. McGowan (North Shore University Hospital, Division of Infectious Diseases), A. Meybeck (Centre Hospitalier de Tourcoing), A. Mills (Southern California Men's Medical Group), M. Mogyoros (Kaiser Permanente Colorado), R. Morales Ramirez (Clinical Research Puerto Rico, Inc.), G. Moyle (Chelsea and Westminster Hospital), H. Olivet (Community Research Initiative of New England), C.O. (Royal London Hospital), O. Osiyemi (Triple O Research Institute PA), M. Para (The Ohio State University, Medical Center), A. Petroll (Medical College of Wisconsin), G. Pierone (AIDS Research and Treatment Center of the Treasure Coast), C. Polk (ID Consultants), F. A. Post (Kings College Hospital), D. Prelutsky (Southampton Healthcare, Inc.), F.R. (CHU—Hôtel Dieu), M. Ramgopal (Midway Immunology and Research Center), B. Rashbaum (Capital Medical Associates), J. Reynes (Hôpital Gui de Chauliac), G. Richmond (Gary J. Richmond, MD, PA), A. Roberts (George Washington University), P. Ruane (Peter J. Ruane, MD, Inc.), M. Saag (University of Alabama at Birmingham), J.S.-B. (University of Puerto Rico, Proyecto ACTU, School of Medicine, UPR), L. Santiago (Hope Clinical Research), P. E. Sax (Brigham and Womens Hospital), A.S. (Pacific Oaks Medical Group), G. Schembri (Central Manchester University Hospitals), S. Segal-Maurer (New York Presbyterian/Queens), P. Shalit (Peter Shalit, MD), D. Shamblaw (La Playa Medical Group), L.S. (Hotel Dieu University Hospital), J. Slim (Saint Michael's Medical Center), L. Sloan (North TX Infectious Diseases Consultants PA), M. Sokol-Anderson (Saint Louis University), D.S. (Jacobi Medical Center), J. Stephens (Mercer University School of Medicine), M. Thompson (Aids Research Consortium of Atlanta Inc), T. Vanig (Spectrum Medical Group), G. Voskuhl (AIDS Arms, Inc.), B. Wade (Infectious Diseases Associates of NW FL), S. Walmsley (University Health Network, Princess Margaret Hospital), D. Ward (Dupont Circle Physicians Group), M. Wohlfeiler (AIDS Healthcare Foundation—South Beach), Y. Yazdanpanah (CHU Bichat), B. Young (Apex Research Institute), and C. Zurawski (Atlanta Infectious Disease Group PC).
Footnotes
This study was sponsored by Gilead Sciences, Inc. (Gilead). Presented at the HIV Glasgow Drug Therapy Congress; October 23–26, 2016; Glasgow, United Kingdom.
C.O. has received honoraria for lectures and for advisory boards and educational grants, travel scholarships and research grants from Gilead, AbbVie, Merck Sharp and Dome, Bristol-Myers Squibb, ViiV, Janssen, Boehringer Ingelheim, Abbott, and GlaxoSmithKline. L.S. reports personal fees from ViiV, Bristol-Myers Squibb, Gilead, and the AIDS International Education Project. V-AF reports grants from Agence Nationale de Recherche sur le Sida (ANRS) and conference fees from Gilead. E.D. is a consultant/advisor for Bristol-Myers Squibb, Gilead, Janssen, Teva, and ViiV and received research support from Gilead, Merck, and ViiV. K.H. has received research support from Gilead Sciences and ViiV Healthcare. J.S.-B. reports grant support from NIAID/NIDA, Merck, and Gilead, personal fees from Merck, Bristol-Myers Squibb, ViiV, and Gilead, and other support from Gilead. N.B. has received consultancy fees, speaking honoraria, and research support from Abbott, GlaxoSmithKline, and Tiobtec. M.Y., M.E.A., SF, A.C., and M.S.R. are employees of Gilead and hold stock interest in the company. All other authors declare no conflicts of interests.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Hall AM, Hendry BM, Nitsch D, et al. Tenofovir-associated kidney toxicity in HIV-infected patients: a review of the evidence. Am J Kidney Dis. 2011;57:773–780. [DOI] [PubMed] [Google Scholar]
- 2.Gupta SK. Tenofovir-associated Fanconi syndrome: review of the FDA adverse event reporting system. AIDS Patient Care STDS. 2008;22:99–103. [DOI] [PubMed] [Google Scholar]
- 3.Post FA, Moyle GJ, Stellbrink HJ, et al. Randomized comparison of renal effects, efficacy, and safety with once-daily abacavir/lamivudine versus tenofovir/emtricitabine, administered with efavirenz, in antiretroviral naive, HIV-1-infected adults: 48-week results from the ASSERT study. J Acquir Immune Defic Syndr. 2010;55:49–57. [DOI] [PubMed] [Google Scholar]
- 4.Stellbrink HJ, Orkin C, Arribas JR, et al. Comparison of changes in bone density and turnover with abacavir–lamivudine versus tenofovir–emtricitabine in HIV-infected adults: 48-week results from the ASSERT study. Clin Infect Dis. 2010;51:963–972. [DOI] [PubMed] [Google Scholar]
- 5.Schafer JJ, Manlangit K, Squires KE. Bone health and human immunodeficiency virus infection. Pharmacotherapy. 2013;33:665–682. [DOI] [PubMed] [Google Scholar]
- 6.McComsey GA, Kitch D, Daar ES, et al. Bone mineral density and fractures in antiretroviral-naive persons randomized to receive abacavir-lamivudine or tenofovir disoproxil fumarate-emtricitabine along with efavirenz or atazanavir-ritonavir: Aids Clinical Trials Group A5224s, a substudy of ACTG A5202. J Infect Dis. 2011;203:1791–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet. 2015;385:2606–2615. [DOI] [PubMed] [Google Scholar]
- 8.Ray AS, Cihlar T, Robinson KL, et al. Mechanism of active renal tubular efflux of tenofovir. Antimicrob Agents Chemother. 2006;50:3297–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bam RA, Yant SR, Cihlar T. Tenofovir alafenamide is not a substrate for renal organic anion transporters (OATs) and does not exhibit OAT-dependent cytotoxicity. Antivir Ther. 2014;19:687–692. [DOI] [PubMed] [Google Scholar]
- 10.U. S. Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER). Human Immunodeficiency Virus-1 Infection: Developing Antiretroviral Drugs for Treatment. Guidance for Industry. Latest revision November 2015. Available at https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm355128.pdf. Accessed September 1, 2016.
- 11.Günthard HF, Saag MS, Benson CA, et al. Antiretroviral drugs for treatment and Prevention of HIV infection in adults: 2016 recommendations of the international antiviral society–USA panel. JAMA. 2016;316:191–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallant JE, Daar ES, Raffi F, et al. Efficacy and safety of tenofovir alafenamide versus tenofovir disoproxil fumarate given as fixed-dose combinations containing emtricitabine as backbones for treatment of HIV-1 infection in virologically suppressed adults: a randomised, double-blind, active-controlled phase 3 trial. Lancet HIV. 2016;3:e158–e165. [DOI] [PubMed] [Google Scholar]
- 13.Smith F, Hammerstorm T, Soon G, et al. A meta-analysis to assess the FDA DAVP's TLOVR algorithm in HIV submissions. Drug Inf J. 2011;45:291–300. [Google Scholar]
- 14.U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for Industry Human Immunodeficiency Virus-1 Infection: Developing Antiretroviral Drugs for Treatment. DRAFT GUIDANCE. 2013;1–43. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM355128.pdf. Accessed September 1, 2016. [Google Scholar]
- 15.European AIDS Clinical Society. European Guidelines for Treatment of HIV-infected Adults in Europe 8.0. 2015. Available at: http://www.eacsociety.org/fi les/2015_eacsguidelines_8.0-english_rev-20151221.pdf. Accessed September 6, 2016. [Google Scholar]
- 16.Wohl D, Oka S, Clumeck N, et al. Brief report: a randomized, double-blind comparison of tenofovir alafenamide versus tenofovir disoproxil fumarate, each coformulated with elvitegravir, cobicistat, and emtricitabine for initial HIV-1 treatment: week 96 results. J Acquir Immune Defic Syndr. 2016;72:58–64. [DOI] [PubMed] [Google Scholar]
- 17.Tungsiripat M, Kitch D, Glesby MJ, et al. A pilot study to determine the impact on dyslipidemia of adding tenofovir to stable background antiretroviral therapy: ACTG 5206. AIDS. 2010;24:1781–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos JR, Saumoy M, Curran A, et al. The lipid-lowering effect of tenofovir/emtricitabine: a randomized, crossover, double-blind, placebo-controlled trial. Clin Infect Dis. 2015;61:403–408. [DOI] [PubMed] [Google Scholar]
- 19.Behrens G, Maserati R, Rieger A, et al. Switching to tenofovir/emtricitabine from abacavir/lamivudine in HIV-infected adults with raised cholesterol: effect on lipid profiles. Antivir Ther. 2012;17:1011–1020. [DOI] [PubMed] [Google Scholar]
- 20.Mulligan K, Glidden DV, Anderson PL, et al. Decreases in cholesterol in HIV-seronegative men using emtricitabine/tenofovir pre-exposure prophylaxis: lipid results of iPrEx. 15th International Workshop on Co-morbidities and Adverse Drug Reactions in HIV; October 15–17, 2013; Brussels, Belgium. Available at: http://www.intmedpress.com/serveFile.cfm?sUID=bc75b62c-c206-437a-b725-a75baa67f5b2. Accessed January 4, 2017.