

Abstract
Most tumors undergo metabolic reprogramming towards glycolysis, the so-called Warburg effect, to support growth and survival. Overexpression of IF1, the physiological inhibitor of the F0F1ATPase, has been related to this phenomenon and appears to be a relevant marker in cancer. Environmental contributions to cancer development are now widely accepted but little is known about the underlying intracellular mechanisms. Among the environmental pollutants humans are commonly exposed to, benzo[a]pyrene (B[a]P), the prototype molecule of polycyclic aromatic hydrocarbons (PAHs), is a well-known human carcinogen. Besides apoptotic signals, B[a]P can also induce survival signals in liver cells, both likely involved in cancer promotion. Our previous works showed that B[a]P elicited a Warburg-like effect, thus favoring cell survival. The present study aimed at further elucidating the molecular mechanisms involved in the B[a]P-induced metabolic reprogramming, by testing the possible involvement of IF1. We presently demonstrate, both in vitro and in vivo, that PAHs, especially B[a]P, strongly increase IF1 expression. Such an increase, which might rely on β2-adrenergic receptor activation, notably participates to the B[a]P-induced glycolytic shift and cell survival in liver cells. By identifying IF1 as a target of PAHs, this study provides new insights about how environmental factors may contribute to related carcinogenesis.
Introduction
The mitochondrial H+-ATP synthase, also called F0F1ATPase or complex V, is a master regulator of energy production and cell fate1–3. Indeed, besides its well-recognized physiological role in oxidative phosphorylation (OXPHOS) as the major cell producer of ATP, this enzyme has been implicated in the morphogenesis of mitochondrial cristae4, in the formation of the mitochondrial permeability transition pore (mPTP) during cell death5, and the metabolic reprogramming of tumor cells6. Regarding this latter point, a decreased OXPHOS capacity and subsequent drop in ATP synthesis due to complex V inhibition, appears to be responsible for a metabolic shift towards aerobic glycolysis, which is better known as the Warburg effect1. In this context, OXPHOS inhibition is often linked to an apoptotic-resistant phenotype, and complex V regulation thus appears to be essential for tumor progression1, 6.
Among the known regulators of the H+-ATP synthase, the physiological inhibitor ATP Inhibitory Factor 1 (IF1) has been implicated in the short-term regulation of energy metabolism by directly interacting with the βF1 subunit of this pump, whereby inhibiting its ATP hydrolysis activity7. IF1 protein, in its native form, is present as a tetramer in mitochondrial matrix at a physiological matrix pH of ∼8.0. When matrix acidifies, a release of the active dimeric form of IF1 occurs, triggering the IF1 binding to the β-F1 subunit, thus preventing complex V reverse activity8. Indeed, complex V activity is able to switch from ATP synthase activity to ATP hydrolase activity under certain circumstances in order to sustain mitochondrial membrane potential (Δψm), and hence mitochondria integrity; however, this could inexorably sentence cell to death due to ATP hydrolysis if no complex V inhibition occurs9, 10. Interestingly, an increased IF1 level has been involved in the establishment of a high Δψm and the conservation of ATP11, 12. Besides, Sánchez-Aragó and coworkers have evidenced high IF1 levels in diverse human cancers, thus highlighting its relevance as a predictive marker for clinical outcome13. In line with this, a recent paper described an increased expression of IF1 in human hepatocellular carcinoma (HCC), such a high increase being predictive of poor survival14. Regarding IF1 roles in tumorigenesis, its overexpression has been involved in the acquisition by cells of several cancer phenotype hallmarks, including metabolic reprogramming11, 14, 15, increased proliferation and invasion14, and cell evasion from death16, 17. Angiogenesis would also be targeted by IF114. Altogether these observations emphasize the key role IF1 might play in tumor development. However, the precise mechanisms underlying the IF1 increase during tumorigenesis remain poorly described.
One clue might come from exposure to environmental carcinogens such as polycyclic aromatic hydrocarbons (PAHs). These widespread contaminants are notably found in cigarette smoke, exhaust fumes, grilled meat, among others, and have been related to tumor development, notably in lung and liver18, 19. We recently demonstrated that a low concentration of benzo[a]pyrene (B[a]P), the prototype molecule of PAHs which is classified as human carcinogen of group 1 by IARC20, not only hyperpolarized mitochondria21, but also induced both a mitochondrial matrix acidification, a glycolytic shift, and an EMT/migration phenotype of liver cells22. Furthermore, a reverse activity of complex V upon B[a]P exposure, was also previously suggested23. As all these cell responses have been linked to IF1, the present study therefore aimed at testing the impact of B[a]P on IF1 level, and at evaluating its role in the survival process elicited by this contaminant in the F258 rat liver epithelial cell line. Here we show that PAHs can increase in vivo the IF1 content in rat liver. We further demonstrate that the IF1 up-regulation observed in vitro upon B[a]P exposure would rely on the activation of the β2 adrenergic receptor pathway, and that it would be determinant in both the glycolytic shift and cell survival elicited by this compound.
Methods
Chemicals
Benzo[a]pyrene (B[a]P), α-naphthoflavone (α-NF), propranolol, ICI-118,551 and 1-methyl – N - [2 – methyl – 4 -[2 - (2 - methylphenyl) diazenyl] phenyl - 1H – pyrazole – 5 -carboxamide (CH223191), were all purchased from Sigma Chemical Co. (St. Louis, MO). Hoechst 33342 and MitoTracker Red CMXROS were purchased from Life Technologies (Saint-Aubin, France). All these products were used as a stock solution in DMSO; final concentration of this vehicle in culture medium was <0.00005% (v/v), and control cultures received the same concentration of vehicle as treated cultures.
Monoclonal mouse anti-ATP synthase subunit beta (A-21351) antibody was purchased from Life Technologies (Saint-Aubin, France). Polyclonal rabbit anti-ATPIF1 antibody (#8528; Cell Signalling) and monoclonal rabbit anti-COX IV antibody (#4850; Cell Signalling) were purchased from Ozyme (Montigny-le-Bretonneux, France). Monoclonal mouse anti-HSC70 antibody (sc-7298) was purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Polyclonal rabbit anti-AhR antibody (BML-SA550) was purchased from Enzo Life Sciences (Lyon, France). Secondary antibodies conjugated with horseradish peroxidase were purchased from DAKO (Les Ulis, France).
The sixteen PAHs (naphthalene, fluorene, acenaphthene, acenaphthylene, anthracene, phenanthrene, fluoranthene, pyrene, benzo[a]anthracene, chrysene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, benzo[g,h,i]perylene, indeno[1,2,3-cd]pyrene and dibenzo[a,h]anthracene) used for the animal experiment model were purchased from Sigma Aldrich (Bornem, Belgium).
Gene expression microarray and Gene Set Enrichment Analyses (GSEA)
We used GSEA24 to identify pathways and gene sets associated with variation in ATPIF1 mRNA expression levels across 91 hepatocellular carcinomas25 (GSE20238). Genes were sorted by their concordance (Pearson correlation) with ATPIF1 mRNA expression levels across tumors, and GSEA was used to evaluate gene sets enriched for either negatively or positively correlated genes. Published GSE20238 was downloaded from the InSilico DB Genomic Datasets Hub26 (https://insilicodb.com/), and analyzed by using the GSEA v2.07 software (http://www.broad.mit.edu/gsea), as previously described24. To account for gene-gene correlations in the enrichment analysis, GSEA p values were computed with respect to a null distribution obtained from 1,000 randomizations of the patient-phenotype labels. Enriched gene sets were selected on the basis of statistical significance (false discovery rate FDR q value < 0.25, and normalized p value < 0.05). Heatmap visualization was performed using GENE-E (http://www.broadinstitute.org/).
Animal Experimentation
Animal housing
Fifteen Long Evans rats (female of 180–200 g, Elevage Janvier, St Berthevin, France) were housed in plastic cages under controlled environment (12 h light/dark cycle, light on at 7 am, temperature of 22 ± 2 °C and relative humidity of 40 ± 5%). Food and water were available ad libitum. The water, food and oil were tested according to NF ISO 15302 to confirm that all these matrices were PAH-free down to a detection limit of 10 ng/L of water and 1 ng/g of fat. Rats were acclimatized to the animal facility for 2 weeks prior to experiment onset.
Animal treatment
The mix of PAHs was composed of the 16 compounds pointed out by the US-Environmental Protection Agency (US-EPA) for their toxicity, and prepared in vegetable oil weekly (ISIO4, Lesieur, Neuilly-sur-Seine, France). Five rats were randomly allocated to each of the experimental groups receiving 0.04 and 0.8 mg/kg body weight of each compound included in the mix, by oral administration, 3 times per week over a 90-day period. The exposure levels were far below the LD50 for all the molecules tested and determined on the basis of a previous study27. Control rats received only the vehicle. At the end of the 90 days-experiment, the rats were euthanized 3 hours after the last gavage by using carbon dioxide. A cardiac puncture was performed after the 3 minutes of unconsciousness and just before the heart stopped beating. All procedures were conducted in accordance with European Communities Council Directive of 22 September 2010 (2010/63/EU) and approved by the Ministry of Agriculture, Grand-Duchy of Luxembourg.
Tissue collections
Livers were dissected and weighed. The base of the left lateral liver lobe was divided into 5 equivalent pieces of 100 mg each which were placed in cryogenic tubes, frozen in liquid nitrogen and stored at −80 °C until analysis.
Liver sample preparation
These 100 mg of tissue were lysed on ice, using a potter, in 600 µl of RIPA buffer supplemented with 1 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol, 1 mM orthovanadate, and a cocktail of protein inhibitors (Roche, Meylan, France), and were sonicated for 10 seconds. Lysates were then centrifuged at 14,000 g for 15 min at 4 °C. The resulting supernatants were collected and frozen at −80 °C or used immediately. Samples were finally diluted 1/50 before protein quantification for western blotting.
Cell culture
The F258 rat liver epithelial cell line was cultured in Williams’ E medium supplemented with 10% fetal calf serum, 2 mM L-glutamine, at 37 °C under a 5% CO2 atmosphere and treated 24 h following seeding as previously described21, 28. F258 cells were treated with B[a]P for 24, 48 and 72 h. Cells were seeded at a density of 2.5× 103 cells/cm2 one day before treatment, and the medium changed before exposures. When using inhibitors, these molecules were added 1 h before B[a]P treatment, for the indicated time points.
RNA isolation and reverse transcription – real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNAs were isolated from F258 cells using the TRIzol method (InVitrogen) and were then subjected to RT-qPCR analysis, as previously described29. Gene-specific primers for ATPIF1 and 18S were purchased from Sigma, and the sequences used for each gene were as follows: ATPIF1: forward, ACGCCGAAGATAATGGCAGG – reverse, ATCCATGCTCTCCGACGAGT; 18S: forward, CCGGTACAGTGAAACTGCGA – reverse, GATAAATGCACGCGTTCCCC. The amplification curves of the PCR products were analyzed with the ABI Prism SDS software using the comparative cycle threshold method. To assess the successful amplification of each target gene, a standard curve was performed for each primer set. The expression levels of target genes were normalized relative to the expression of an 18S RNA endogenous reference and were plotted as fold change compared to control with vehicle (DMSO).
Mitochondrial purification
Briefly, after treatments cells were washed twice with cold PBS. Then, cells of each 150 mm petri dishes were directly scratched in 1.5 ml of cold hypotonic buffer (20 mM HEPES pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 250 mM sucrose) supplemented with 100 µM PMSF and complete protease inhibitor cocktails (Roche, Meylan, France). Lysates were transferred into 1.5 ml tubes before starting lysis with a 26G needle syringe. Samples were kept under stirring at 4 °C for 1 hour to improve lysis efficiency. A first 10 min, 750 g centrifugation was performed at 4 °C. Supernatants were collected and a second 25 min, 10,000 g centrifugation was performed at 4 °C. Mitochondria pellets were then lysed in RIPA buffer supplemented with 1 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol, 1 mM orthovanadate, and a cocktail of protein inhibitors (Roche). Mitochondrial fractions were finally centrifuged at 13,000 g for 15 min at 4 °C. The resulting supernatants were collected and frozen at −80 °C or used immediately.
Western Blot immunoassays
See Supplementary Information.
Intracellular Calcium Measurements
F258 cells were cultured on glass coverslips and incubated with 2.5 μM Fura-2-AM for 20 min at 37 °C in Hepes-buffered medium (10 mM Hepes, 134.8 mM NaCl, 4.7 mM KCl, 1 mM MgCl2, 1.2 mM KH2PO4, 1 mM CaCl2, 10 mM glucose, pH 7.4, at 37 °C) supplemented with 0.006% pluronic acid. Fura-2-loaded cells were placed in a continuously perfused recording chamber mounted on the stage of an epifluorescence microscope (Nikon Diaphot). Calcium measurements were then performed as previously described30.
Extracellular lactate measurement
Cell supernatants were collected and directly frozen. Quantification of L-lactate levels was based on two enzymatic reactions. Lactate dehydrogenase (LDH; Roche, Meylan, France) catalyzed the NAD+-mediated oxidation of lactate into pyruvate. Glutamate-pyruvate transaminase (GPT; Roche, Meylan, France) was then used to shift first reaction equilibrium by transforming the entire pyruvate into alanine and α-ketoglutarate. The amount of formed NADH was related to the quantity of lactate processed by these reactions. Briefly, 20 µL of each sample were added to 200 µL of reaction buffer (620 mM sodium carbonate, 78.7 mM L-glutamate, 0.92 mM NAD, 2 µg GPT and 2 µg LDH). The standard range was performed using lithium lactate (Sigma Aldrich). The 96 multiwell plates were then incubated at 37 °C for 30 minutes before quantifying extracellular lactate production by monitoring the increase in absorbance of NADH at 355 nm on a spectrophotometer (SPECTROstar nano, BMG LABTECH, France).
Transfection and RNA interference (siRNA)
ON-TARGETplus Rat IF1 siRNA SMARTpool (si IF1), and ON-TARGETplus Non-Targeting Pool siRNA negative control (si CTL) were purchased from GE Dharmacon. Basic Small interfering RNA (siRNA) resuspension was performed according to manufacturer’s recommendations. Transfections of siRNA were performed in 60 mm dishes on 60% confluent F258 cells, in the presence of TransFectin Lipid Reagent (BioRad). Per dish, siRNA (100 nM) and 12.5 µl TransFectin lipid reagent were applied in a final volume of 2.5 ml Opti-MEM. Six hours later, the medium was renewed with the normal medium. Cells were then passaged in order to be treated during exponential phase, as described above.
Glucose oxidation assay
Glucose oxidation levels were monitored as previously described22.
Cell toxicity estimation
Cell toxicity was measured by analysis of chromatin condensation and fragmentation in the cell nucleus as previously described21.
Statistical analysis
All data were obtained from a minimum of three independent experiments. They were quoted as mean ± SD. Analysis of variance followed by Newman–Keuls test was used to test the effects of B[a]P. Differences were considered significant at the level of P < 0.05. All statistical analyses were performed using GraphPad Prism 5.01 Software (GraphPad Software, San Diego, USA).
Results
High IF1 expression is correlated with liver cancer progression and gene expression related to glycolytic metabolism in human hepatocarcinoma
We first explored by GSEA the potential contribution of ATPIF1 expression in human liver cancer progression (91 human HCV related-hepatocarcinoma, GSE20238, as previously described25). As shown in Fig. 1A, data mining revealed a positive correlation between high IF1 mRNA expression and the progression of non-tumor (L1) to well differentiated liver tumor (G1) (with a normalized enrichment score of 1.668, and a p value of 0.025). Consistent with the reported role of ATPIF1 in HCC metabolism in vitro 14, further GSEA analysis of GSE20238 pointed to a positive correlation between high IF1 expression and high glycolytic metabolism in human HCC (p value of 0.02 and normalized enrichment score = 1.61; Fig. 1B and Supplementary Fig. S1). The false-discovery rate (FDR) q-value of 0.175 is considered significant24. Altogether, these studies indicated that a high expression of IF1 was associated with both liver cancer progression and high glycolysis in human.
Figure 1.
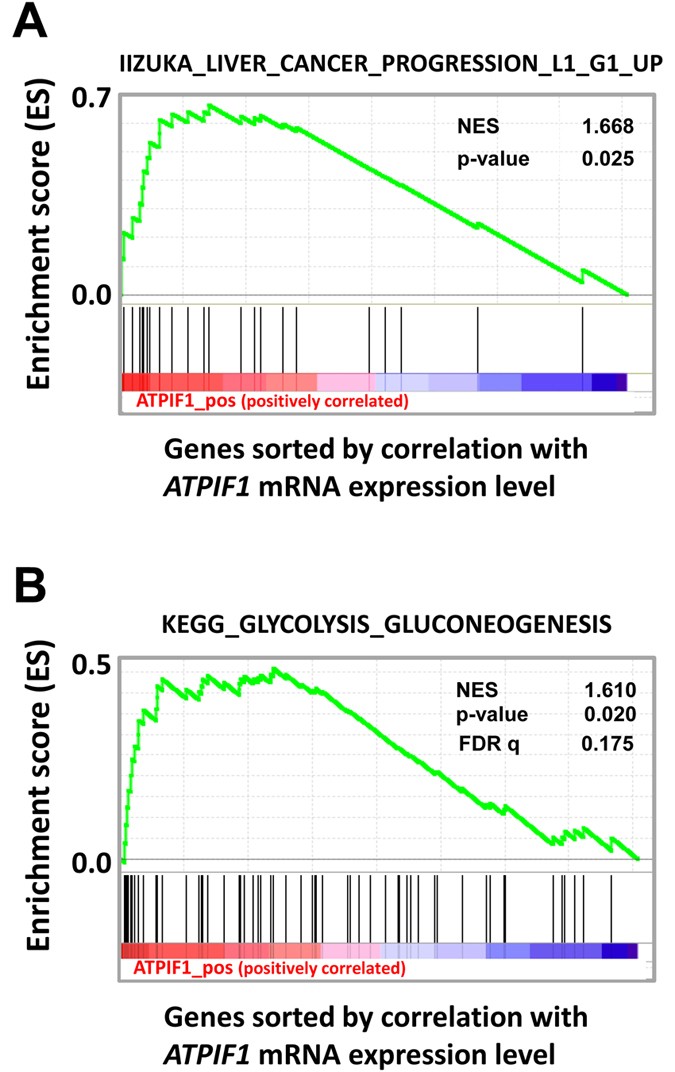
GSEA profiles showing a significant enrichment of gene sets associated with liver cancer progression (A), and glycolysis (KEGG, (B)) in human HCC with high levels of ATPIF1 mRNA (GSE20238). See heat-map in Supplementary Fig. S1.
In vivo effect of the 16 US-EPA PAH mixture and in vitro effect of B[a]P on hepatic IF1 expression
Based upon the relevance of IF1 in liver tumor progression, we decided to test the in vivo effect of a mixture of the 16 PAHs listed as “priority” compounds by the US-EPA, because of their occurrence in the environment and their potential toxicity. Thus, the hepatic IF1 protein level was evaluated in liver tissue samples collected from rats exposed through diet for 90 days at two doses (0.04 mg/kg and 0.8 mg/kg), and compared to untreated animals. Interestingly, an increase in IF1 level was detected with a dose-dependent effect (Fig. 2A). As B[a]P is the prototype of these PAHs and as it is a well-known carcinogen for human20, the effect of this molecule was tested on IF1 mRNA expression in rat liver epithelial F258 cells. Indeed, B[a]P was recently found to induce a metabolic reprogramming related to survival signal notably in these cells22. After a treatment of 24 and 48 h with two concentrations of B[a]P, a significant induction of IF1 mRNA expression was observed as soon as 24 h; an even stronger effect was detected at 48 h with B[a]P 1 µM (Fig. 2B). Regarding protein level, IF1 was detected mainly in the mitochondrial fraction compared to the cytosolic fraction in F258 cells (Supplementary Fig. S2A). Cell exposure to both concentrations of B[a]P for 48 h resulted in a dose-dependent increase in the mitochondrial IF1 protein level, as confirmed by the densitometric analysis of the ratio between IF1 and its target β-F1 subunit (Fig. 2C). Interestingly, a band corresponding to IF1 dimers was also detected, with a strong intensity at 1 µM, suggesting an increased proportion of the active form of IF1 upon B[a]P exposure. A dose-dependent effect of B[a]P (48 h) on IF1 protein level was also detected in total lysates from Hepa1c1c7 cells (Supplementary Fig. S3A), a mouse liver hepatoma cell line in which B[a]P was also found to increase glycolysis22. It is also noteworthy that another carcinogenic PAH, the 7,12-Dimethylbenz(a)anthracene (DMBA), also elicited an increase in IF1 protein level in F258 cells (Supplementary Fig. S3B). Altogether, these results clearly pointed to IF1 as a new target of PAHs, in particular of B[a]P, in liver.
Figure 2.
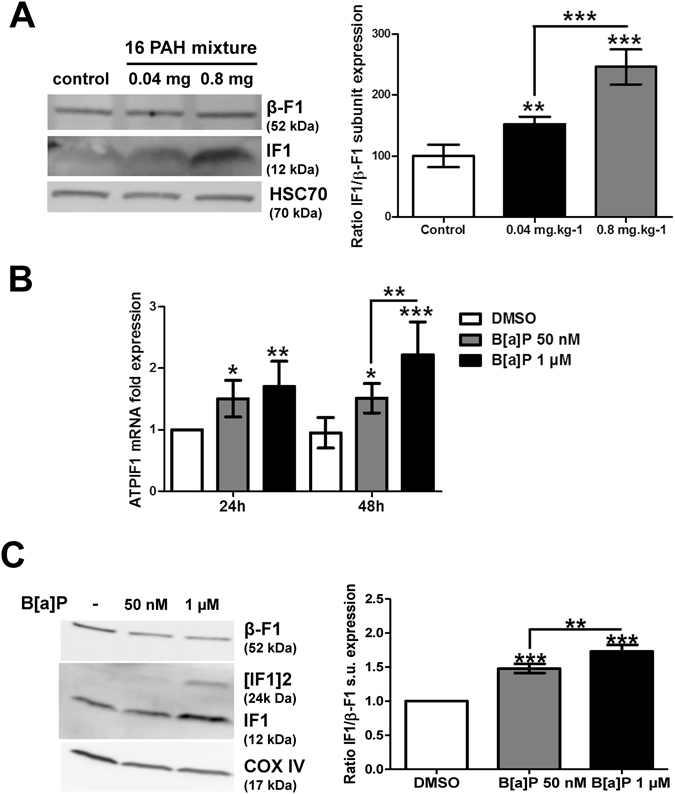
Effects of PAHs on IF1 expression both in vivo and in vitro. (A) The levels of IF1 protein and of the β-F1 subunit of the F0F1ATPase were analyzed by western blotting in liver tissues from rats exposed to 0.04 or 0.8 mg.kg−1 of a 16 PAH mixture. HSC70 was used as loading control. (B) IF1 mRNA expression was monitored in F258 cells treated with B[a]P (50 nM or 1 µM) for 24 or 48 hours. (C) The IF1 protein content of mitochondrial fractions of B[a]P-exposed F258 cells was evaluated by western blotting. Histogram gives the results of densitometric analysis of IF1 protein level relative to β-F1 subunit protein level. COXIV was used as loading control. Number of independent experiments = 3 for all conditions. *p < 0.05, **p < 0.01, ***p < 0.001, DMSO vs B[a]P-treated cells, unless otherwise quoted.
B[a]P metabolism would not be responsible for up-regulation of IF1 protein level
Having demonstrated that IF1 was a new molecular target for B[a]P, we next sought to identify the origin of IF1 up-regulation. We previously demonstrated that mitochondrial hyperpolarization as well as glycolytic shift induced by B[a]P in F258 cells were dependent upon B[a]P metabolism22, 31. We thus tested the effect of α-naphthoflavone (α-NF; 10 µM), an inhibitor of B[a]P metabolism on the mitochondrial IF1 protein level. α-NF did not prevent IF1 up-regulation but rather enhanced it (Fig. 3). As α-NF is known to inhibit both the cytochromes P450 involved in B[a]P metabolism as well as AhR (a cytosolic receptor known to be activated by B[a]P), CH223191 (10 µM; to inhibit AhR) was tested; as illustrated in Supplementary Fig. S2A, it did not prevent the B[a]P-increased mitochondrial IF1 protein level. In contrast, it prevented the B[a]P-triggered induction of IF1 mRNA expression (Supplementary Fig. S2B). Moreover, TCDD which is a strong activator of AhR, was ineffective towards IF1 protein level when used alone (Supplementary Fig. S2C). In total, these results showed that B[a]P enhanced the mitochondrial IF1 protein level by a pathway that would involve neither B[a]P metabolism, nor AhR activation.
Figure 3.
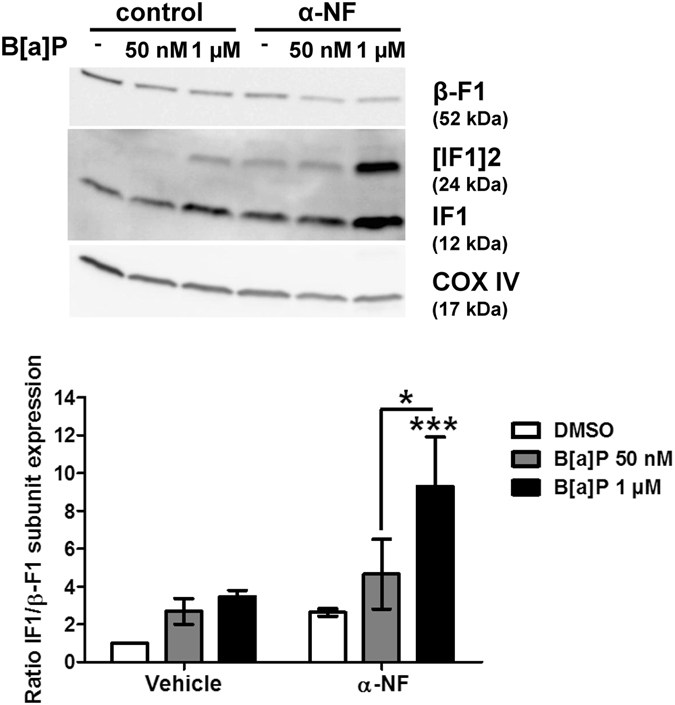
B[a]P metabolism was not involved in IF1 up-regulation in response to B[a]P. F258 cells were pre-treated for 1 hour with α-NF (10 µM) prior to co-exposure to B[a]P for 48 hours. The involvement of B[a]P metabolism in IF1 up-regulation was assessed by analyzing IF1 protein level on mitochondrial fractions by western blotting. Histogram gives the results of densitometric analysis of IF1 protein level relative to β-F1 subunit protein level. COXIV was used as loading control. Results were representative of 3 independent experiments. *p < 0.05, ***p < 0.001, DMSO vs B[a]P-treated cells, unless otherwise quoted.
The β2-adrenergic pathway would be involved in IF1 up-regulation upon B[a]P exposure
The next set of experiments was performed in order to test the possible involvement of the β2-adrenergic pathway in the regulation of the protein level of IF1 by B[a]P. As an increase in intracellular calcium concentration has been previously observed upon B[a]P-induced β2ADR activation32, the effect of B[a]P (50 nM) on calcium concentration was first assayed under our experimental conditions, by using Fura-2-AM as calcium sensitive fluoroprobe, and microspectrofluorimetry. As shown in Fig. 4A, an increase in calcium was triggered by B[a]P in F258 cells following 10 min of exposure. This then led us to test the possible involvement of β2ADR in the B[a]P effects on IF1 protein level; propranolol, a non-selective β blocker, was thus used to inhibit this pathway. We first showed that a pre-treatment (1 h) with propranolol (10 µM) followed by co-treatment with B[a]P (48 h), prevented not only the release of lactate, especially at the lowest B[a]P concentration (Fig. 4B), but also the increase in IF1 protein level (Fig. 4C). The use of a selective β2-blocker, ICI-118,551, gave similar results on IF1 level (n = 2; data not shown). Finally, the effect of B[a]P was assayed in HEK293 cells overexpressing β2ADR. Indeed, these cells are known to express a very low basal level of β2ADR33; they were then transfected either with control plasmid (HEKwt) or with a β2ADR plasmid (HEKβ2), as previously described32. As shown in Fig. 4D, B[a]P was responsible for a dose-dependent release of lactate from HEKβ2 cells, with no effect in HEKwt. When looking at the IF1 protein level in these two cell lines, the effect of B[a]P on IF1 was more marked in HEKβ2 compared to HEKwt cells (Fig. 4E). Altogether, these results strongly suggested a role for β2ADR in the regulation of IF1 protein level by B[a]P.
Figure 4.
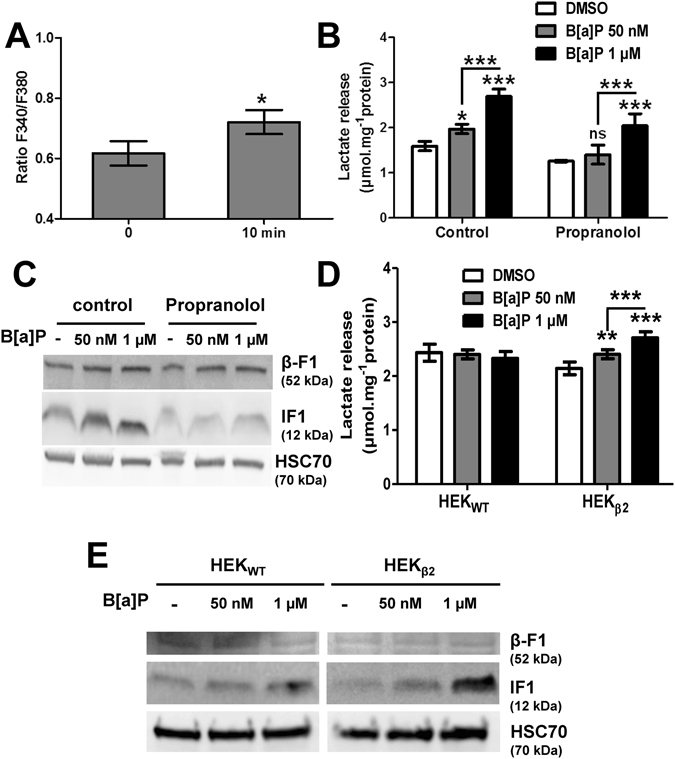
B[a]P-induced IF1 induction could occur through β2-adrenergic pathway stimulation. (A) Effects of B[a]P (50 nM) on calcium was assessed by microspectrofluorimetry after staining F258 cells with the Fura-2-AM probe. The ratio F340/F380 reflects intracellular Ca2+ concentration. *p < 0.05, DMSO vs B[a]P-treated cells. (B,C) F258 cells were pre-treated or not with the β-receptor inhibitor propranolol (10 µM) for 1 hour prior to co-exposure to B[a]P for 48 hours. The B[a]P-induced glycolytic shift (B) was investigated by monitoring extracellular lactate, and the IF1 protein level (C) was analyzed on total lysates by western blotting. A role for β2-adrenergic receptor in the B[a]P (50 nM or 1 µM, 48 h)-induced extracellular lactate (D) or total IF1 protein level (E), was evaluated in both HEKWT cells (not expressing β2-adrenergic receptor), and HEKβ2 (in which the receptor is overexpressed). HSC70 was used as loading control. Number of independent experiments = 3 for all conditions. *p < 0.05, **p < 0.01, ***p < 0.001, DMSO vs B[a]P-treated cells, unless otherwise quoted.
IF1 exhibits a pivotal role in B[a]P propensity to induce cell survival
An increase in IF1 protein level has been previously related to Warburg effect and cell survival15. In order to test whether the B[a]P-increased mitochondrial IF1 protein level was involved in both the glycolytic shift and cell survival signaling we recently reported21, 22, we decided to test the role of IF1 towards both glycolytic shift and cell death by using a siRNA approach. First, western blotting experiments were carried out in order to validate the siRNA targeting IF1 (siIF1) used. As shown in Fig. 5A, transfecting cells with siIF1 markedly reduced the expression level of this protein, compared to conditions with control siRNA (siCTL). Glucose oxidation, previously shown to be enhanced by B[a]P22, was then evaluated in siIF1-or siCTL-transfected cells. Data from Fig. 5B indicated that the B[a]P (50 nM, 48 h)-increased glucose oxidation did not rely on IF1. In contrast, silencing IF1 fully prevented the increase in lactate release induced by B[a]P (48 h), whatever the concentration used (Fig. 5C). Finally, analysis of cell death by cell nucleus staining with Hoechst 33342 showed a significant increase in B[a]P-induced cell death when cells were transfected with siIF1 (Fig. 5D). Such an increase did not seem to rely upon an over-activation of the p53 pathway (Supplementary Fig. S4). Altogether, these results pointed to a crucial role for IF1 in B[a]P-induced glycolytic shift and cell survival.
Figure 5.
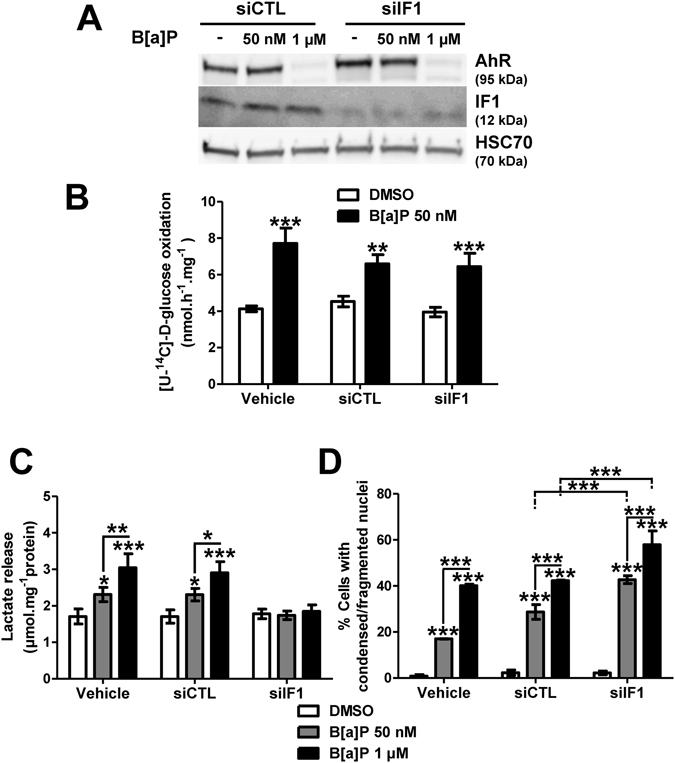
IF1 upregulation following B[a]P exposure acts to promote F258 cell survival. (A) Western blotting analysis was performed to control siIF1 inhibitory efficacy on IF1 protein level expression compared to the Non Targeting siRNA (siCTL). AhR expression was analyzed to ensure the specificity of Si targeting. HSC70 was used as loading control. (B) Impact of IF1 silencing on glucose oxidation was quantified by measuring transfer of 14C radioactivity from glucose to CO2. Cells were treated with B[a]P (50 nM) for 48 h. (C) Role for IF1 in the B[a]P-induced glycolytic shift was evaluated by measuring extracellular lactate upon IF1 silencing by siRNA. (D) B[a]P toxicity was evaluated in IF1 silenced cells by counting cells with fragmented or condensed chromatin following Hoechst 33342 staining. Number of independent experiments = 3 for all conditions. *p < 0.05, **p < 0.01, ***p < 0.001, DMSO vs B[a]P-treated cells, unless otherwise quoted.
Discussion
High overexpression of IF1 has been reported in numerous human cancers, such as gastric cancer34, colon, lung, breast and ovarian carcinomas13, as well as hepatocarcinoma14, which makes it as an important cancer biomarker. Regarding its role in carcinogenesis, such a high expression has been clearly evidenced as notably inducing metabolic reprogramming towards glycolysis and cell survival11, 15, 35. However, as stressed by Sánchez-Arago et al.13, the question that still remains to be fully solved is how IF1 is upregulated in cancer. This is in this context that our study was performed in order to evaluate the possible impact of known environmental carcinogens, namely PAHs. The present data clearly evidenced that rat exposure (90 days) to a low dose of the 16 US-EPA PAH mixture, which is known to have deleterious effects, was capable of increasing in vivo the IF1 protein level in liver, as shown by the marked increase in the monomeric 12 kDa form. Furthermore, we found that the PAH prototype B[a]P, which is a well-recognized human carcinogen20, up-regulated IF1 level in vitro in both F258 and Hepa1c1c7 cells. Note that 7,12-Dimethylbenz(a)anthracene, another known carcinogenic PAH, also upregulated IF1 in F258 cells. In this context, our study is the first one to identify environmental carcinogens as exogenous regulators of IF1 level.
Whereas some intracellular regulators of IF1 mRNA expression have been reported, such as the transcription factor NFκB notably in hepatocarcinoma14, or the nuclear receptor pregnane X receptor (PXR) in the liver from rats treated by the synthetic steroid pregnenolone 16α-carbonitrile36, little information is available regarding the involvement of other endogenous transcriptional regulators. HIF-1α would control IF1 protein level in Clone 9 cells, a non-transformed rat hepatic epithelial cell line, upon hypoxia; however, whereas a decrease in IF1 mRNA expression was paralleled by a decrease in HIF-1α mRNA expression in rat liver during sepsis, direct evidence for the transcriptional regulation of ATPIF1 gene by this transcription factor was not sought37. Our present study would point to AhR as possibly involved in the transcriptional regulation of ATPIF1 gene. Indeed, by using the specific AhR inhibitor CH223191, induction of IF1 mRNA expression by B[a]P was found to be fully inhibited. However, this possible transcriptional regulation would not play any role in the increase of mitochondrial IF1 protein level induced by B[a]P exposure, since inhibiting AhR was ineffective on this increase. In line with this result, AhR activation by TCDD alone had no effect under control conditions (Supplementary Fig. S2C). Our previous work indicated that B[a]P metabolism by CYP1B1 could play a role in metabolism reprogramming21, 22. However, as for CH223191, α-naphthoflavone did not prevent IF1 increase. In this context, B[a]P metabolism would not be involved, even though this will need to be firmly validated.
Direct activation of β2ADR by B[a]P has been previously described, thus leading to a rapid increase in intracellular calcium concentration32; moreover, this increase in intracellular calcium was found to be AhR-independent30. Besides, β2ADR is known to control mitochondria biogenesis and function38, 39. Regarding this latter aspect, it is worth emphasizing that β2ADR was reported to afford cardioprotection by interfering with mitochondrial dysfunction upon oxidative stress induced by doxorubicin38. In this context, regulation by β2ADR of the B[a]P-elicited increase in IF1 protein level was tested by using chemical inhibitors in F258 cells or by using β2ADR-expressing HEK293 cells. From our results, it appeared that early activation of the β2ADR pathway by B[a]P might be involved in the control of mitochondrial IF1 protein level (Fig. 6). Regarding that point, it is noteworthy that a recent study has revealed a role for the β-adrenergic receptor/cAMP/PKA pathway in controlling the phosphorylation state of IF1, thereby preventing its binding to the F0F1-ATPase40.
Figure 6.

A proposed model for the B[a]P-mediated metabolic reprogramming and its role in cell fate determination in F258 rat hepatic epithelial cells. B[a]P metabolism, via the constitutively expressed CYP1B1, leads to the activation of NHE1 transporter and p53 pathway, both triggering apoptotic cell death. Note that AhR activation by B[a]P, along with the reactive oxygen species (ROS) production related to CYP metabolism, are involved in NHE1 activation via membrane remodeling49. Beside promoting apoptosis, activation of NHE1 and p53 also appeared to be involved in the B[a]P-elicited metabolic reprogramming22, thus inducing cell survival. B[a]P would also induce a β2-adrenergic receptor-dependent activation of IF1, the physiological inhibitor of the F1F0ATPase, which appears to be involved in the related glycolytic shift and cell survival. Activation of AhR by B[a]P might be involved in the upregulation of IF1 mRNA level, but without any incidence on the mitochondrial IF1 protein level. Along with its classical and well described transcription factor activity, a mitochondrial fraction of AhR might also participate to the overall metabolic reprogramming.
IF1 activation has been previously related to a drop in mitochondrial matrix pH, thus allowing the appearance of a dimeric form, which then binds to the βF1 subunit of the F1F0ATPase, thereby inhibiting its hydrolase activity8. As B[a]P was reported to trigger a mitochondrial matrix acidification, from approximately 8 to 6.5, in F258 cells22, this could explain the appearance of a 24 kDa band presently detected after electrophoresis, thus revealing the presence of active IF1 dimers. Despite the IF1 up-regulation and dimer appearance, only a trend towards decrease for complex V activity was found (decrease by about 12%; data not shown). However, it would be worth measuring specifically its hydrolase activity upon B[a]P exposure; indeed, a decrease in this activity of complex V is known to afford some cell protection towards cell death by limiting ATP consumption41. Previous studies have revealed that IF1 overexpression leads to Warburg effect, a well-known hallmark of cancer11, 15. Besides, our recent work has shown that B[a]P triggers a Warburg effect involved in cell survival in F258 cells22. Here we further show that silencing IF1 blocks the B[a]P-induced enhanced extracellular lactate release, and would thus been involved in the B[a]P-induced glycolytic shift. However, the boost in glucose oxidation elicited by B[a]P22 does not seem to rely on IF1. As we have previously shown that (i)-NHE1 controls both lactate release and glucose oxidation induced by B[a]P, and that (ii)-AhR might also be involved22, it then appears that the overall metabolic reprogramming induced by B[a]P might be more complex than first believed (Fig. 6), and would thus deserve further investigation. Besides being involved in the B[a]P-induced glycolytic reprogramming, IF1 might also exert its pro-survival effects through regulating the mitochondrial permeability transition pore PTP. Indeed, Antoniel et al.42 recently pointed out some striking similarities between inhibition of the permeability transition pore (PTP) opening and IF1 regulation, both known for example to rely on mitochondrial matrix pH. Indeed, matrix acidification has been shown to prevent the PTP opening43. In support to such a possible effect of IF1 on PTP, it is worth stressing that the B[a]P-induced apoptosis was found to occur without any cytochrome c release in F258 cells23. Nevertheless, IF1 effects are still controversial and seem to be highly dependent on the biological context. In particular, Fujikawa et al.44, and more recently Barbato et al.45 rather demonstrate a role for IF1 overexpression in mitochondrial membrane depolarization related to an enhancement of OXPHOS rate. Finally, overexpression of IF1 is also known to greatly impact on mitochondria network dynamics16, 17, 44, 46. Hyperfused mitochondria, whose formation depends on maintenance of Δψm, can transiently buffer the effects of respiratory chain dysfunction; such mitochondria would not be processed by mitophagy, thereby promoting a selective rescue mechanism47, 48. Regarding that point, mitotracker Red staining (Supplementary Fig. S5A) and TEM (Supplementary Fig. S5B), evidenced elongated mitochondria in B[a]P-treated F258 cells. In this context, one might also suggest a role for IF1 in this enhanced mitochondrial fusion process, modeled by a marked mitochondrial elongation, as previously reported16, 17.
In conclusion, although B[a]P is known to be involved in the different phases of tumor development, contribution of B[a]P-induced mitochondrial dysfunction to cell transformation, and in the following onset of oncogenesis still needs to be clarified. Here we show that IF1, the physiological inhibitor of the F0F1ATPase is a new target for PAHs, even at low concentrations, and that PAH-regulated IF1 is linked to glycolytic shift and survival. Interestingly, our datamining analysis from transcriptomics data issued from human hepatocarcinoma clearly reveals positive correlations between high IF1 expression on one hand, and glycolysis and tumor progression on the other hand. In this context, IF1 upregulation might then be a new means for PAHs to drive carcinogenesis. We therefore suggest that IF1 might be a suitable marker for PAH-induced carcinogenesis.
Electronic supplementary material
Acknowledgements
We wish to thank the MRic facilities (SFR Biosit) for microscopy experiments, especially Marie-Thérèse Lavault and Stéphanie Dutertre for their technical assistance. We also wish to thank Dr. Henri Schroeder for valuable scientific discussion, Drs Pierre Rustin and Paule Bénit for complex V activity measurements, and Elise Saunier for glucose oxidation experiments. KH was a recipient of a fellowship from French Ministry for Education and Research. We are very grateful to the Faculté des Sciences Pharmaceutiques et Biologiques de Rennes (University of Rennes 1) for further financial support to KH. We also wish to thank the Ligue Contre le Cancer (committees 22,35,49,85; DLG, OS), the French National Academy of Medicine (DLG), ANR (STEATOX project; “ANR-13-CESA-0009”; DLG, OS, BM), the ITMO-cancer Plan Cancer 2009–2013 (SB), “Region Provence Alpes Côte d’Azur”, “Agence régionale santé Provence Alpes Côte d’Azur”, “Direction régionale de l’Environnement, de l’aménagement et du logement Provence Alpes Côte d’Azur” (BM, plan régional santé environnement PRSE PACA n°6.3.3.3 and 6.3.3.4), for financial support to our work.
Author Contributions
D.L.G., K.H., L.H., S.B., B.M. and O.S. conceived the study and designed the experiments; K.H., M.F., I.G., N.P., E.L.F., N.G., B.A., A.B. and M.C. performed the experiments; D.L.G., L.H., S.B., K.H. and B.M. analyzed the data; D.L.G., K.H., N.G., B.A. and B.M. wrote the manuscript in close collaboration with all the other authors. All authors reviewed the manuscript. All authors finally approved this version to be published.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-00269-7
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sánchez-Aragó M, Formentini L, Cuezva JM. Mitochondria-mediated energy adaption in cancer: the H(+)-ATP synthase-geared switch of metabolism in human tumors. Antioxid. Redox. Signal. 2013;19:285–298. doi: 10.1089/ars.2012.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long Q, Yang K, Yang Q. Regulation of mitochondrial ATP synthase in cardiac pathophysiology. Am. J. Cardiovasc. Dis. 2015;5:19–32. [PMC free article] [PubMed] [Google Scholar]
- 3.García-Bermúdez J, Cuezva JM. The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochim. Biophys. Acta. 2016;1857(8):1167–1182. doi: 10.1016/j.bbabio.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Paumard P, et al. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO. J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernardi P, Di Lisa F, Fogolari F, Lippe G. From ATP to PTP and Back: A Dual Function for the Mitochondrial ATP Synthase. Circ. Res. 2015;116:1850–62. doi: 10.1161/CIRCRESAHA.115.306557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martínez-Reyes I, Cuezva JM. The H(+)-ATP synthase: a gate to ROS-mediated cell death or cell survival. Biochim. Biophys. Acta. 2014;1837:1099–1112. doi: 10.1016/j.bbabio.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Zanotti F, Raho G, Gaballo A, Papa S. Inhibitory and anchoring domains in the ATPase inhibitor protein IF1 of bovine heart mitochondrial ATP synthase. J. Bioenerg. Biomembr. 2004;36:447–457. doi: 10.1023/B:JOBB.0000047327.68173.9b. [DOI] [PubMed] [Google Scholar]
- 8.Cabezon E, Butler PJ, Runswick MJ, Walker JE. Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH. J. Biol. Chem. 2000;275:25460–25464. doi: 10.1074/jbc.M003859200. [DOI] [PubMed] [Google Scholar]
- 9.Hassinen IE, Vuorinen KH, Ylitalo K, Ala-Rämi A. Role of cellular energetics in ischemia-reperfusion and ischemic preconditioning of myocardium. Mol. Cell. Biochem. 1998;184:393–400. doi: 10.1023/A:1006818708565. [DOI] [PubMed] [Google Scholar]
- 10.Campanella M, Parker N, Tan CH, Hall AM, Duchen MR. IF(1): setting the pace of the F(1)F(o)-ATP synthase. Trends. Biochem. Sci. 2009;34:343–350. doi: 10.1016/j.tibs.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Formentini L, Sánchez-Aragó M, Sánchez-Cenizo L, Cuezva JM. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol. Cell. 2012;45:731–742. doi: 10.1016/j.molcel.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Green DW, Grover GJ. The IF(1) inhibitor protein of the mitochondrial F(1)F(0)-ATPase. Biochim. Biophys. Acta. 2000;1458:343–355. doi: 10.1016/S0005-2728(00)00085-2. [DOI] [PubMed] [Google Scholar]
- 13.Sánchez-Aragó M, et al. Expression, regulation and clinical relevance of the ATPase inhibitory factor 1 in human cancers. Oncogenesis. 2013;2:e46. doi: 10.1038/oncsis.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song R, et al. Reciprocal activation between ATPase inhibitory factor 1 and NF-κB drives hepatocellular carcinoma angiogenesis and metastasis. Hepatology. 2014;60:1659–1673. doi: 10.1002/hep.27312. [DOI] [PubMed] [Google Scholar]
- 15.Sánchez-Cenizo L, et al. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J. Biol. Chem. 2010;285:25308–25313. doi: 10.1074/jbc.M110.146480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faccenda, D., Tan, C. H., Duchen, M. R. & Campanella, M. 2013a Mitochondrial IF1 preserves cristae structure to limit apoptotic cell death signaling. Cell. Cycle. 12, 2530–2532 (2013). [DOI] [PMC free article] [PubMed]
- 17.Faccenda D, Tan CH, Seraphim A, Duchen MR, Campanella M. IF1 limits the apoptotic-signalling cascade by preventing mitochondrial remodelling. Cell. Death. Differ. 2013;20:686–697. doi: 10.1038/cdd.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Motorykin O, Matzke MM, Waters KM, Massey Simonich SL. Association of carcinogenic polycyclic aromatic hydrocarbon emissions and smoking with lung cancer mortality rates on a global scale. Environ. Sci. Technol. 2013;47:3410–3416. doi: 10.1021/es305295d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wester PW, et al. Carcinogenic activity of benzo[a]pyrene in a 2 year oral study in Wistar rats. Food. Chem. Toxicol. 2012;50:927–935. doi: 10.1016/j.fct.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 20.IARC Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC Monogr. Eval. Carcinog. Risks. Hum. 2010;92:111–144. [PMC free article] [PubMed] [Google Scholar]
- 21.Hardonnière K, et al. Benzo[a]pyrene-induced nitric oxide production acts as a survival signal targeting mitochondrial membrane potential. Toxicol. In. Vitro. 2015;29:1597–1608. doi: 10.1016/j.tiv.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Hardonnière K, et al. The environmental carcinogen benzo[a]pyrene induces a Warburg-like metabolic reprogramming dependent on NHE1 and associated with cell survival. Sci. Rep. 2016;6:30776. doi: 10.1038/srep30776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huc L, et al. Multiple apoptotic pathways induced by p53-dependent acidification in benzo[a]pyrene-exposed hepatic F258 cells. J. Cell. Physiol. 2006;208:527–537. doi: 10.1002/jcp.20686. [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mínguez B, et al. Gene-expression signature of vascular invasion in hepatocellular carcinoma. J. Hepatol. 2011;55:1325–1331. doi: 10.1016/j.jhep.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coletta A, et al. InSilico DB genomic datasets hub: an efficient starting point for analyzing genome-wide studies in GenePattern, Integrative Genomics Viewer, and R/Bioconductor. Genome. Biol. 2012;13:R104. doi: 10.1186/gb-2012-13-11-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grova N, Salquebre G, Appenzeller BM. Gas chromatography-tandem mass spectrometry analysis of 52 monohydroxylated metabolites of polycyclic aromatic hydrocarbons in hairs of rats after controlled exposure. Anal. Bioanal. Chem. 2013;405:8897–8911. doi: 10.1007/s00216-013-7317-z. [DOI] [PubMed] [Google Scholar]
- 28.Huc L, et al. Identification of Na+/H+ exchange as a new target for toxic polycyclic aromatic hydrocarbons. FASEB. J. 2004;18:344–346. doi: 10.1096/fj.03-0316fje. [DOI] [PubMed] [Google Scholar]
- 29.Podechard N, et al. Interleukin-8 induction by the environmental contaminant benzo(a)pyrene is aryl hydrocarbon receptor-dependent and leads to lung inflammation. Toxicol. Lett. 2008;177:130–137. doi: 10.1016/j.toxlet.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Mayati A, Le Ferrec E, Lagadic-Gossmann D, Fardel O. Aryl hydrocarbon receptor-independent up-regulation of intracellular calcium concentration by environmental polycyclic aromatic hydrocarbons in human endothelial HMEC-1 cells. Environ. Toxicol. 2012;27:556–562. doi: 10.1002/tox.20675. [DOI] [PubMed] [Google Scholar]
- 31.Huc L, et al. Apoptotic mitochondrial dysfunction induced by benzo(a)pyrene in liver epithelial cells: role of p53 and pHi changes. Ann. N. Y. Acad. Sci. 2003;1010:167–170. doi: 10.1196/annals.1299.028. [DOI] [PubMed] [Google Scholar]
- 32.Mayati A, et al. Induction of intracellular calcium concentration by environmental benzo(a)pyrene involves a β2-adrenergic receptor/adenylyl cyclase/Epac-1/inositol 1,4,5-trisphosphate pathway in endothelial cells. J. Biol. Chem. 2012;287:4041–4052. doi: 10.1074/jbc.M111.319970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Zastrow M, Kobilka BK. Ligand-regulated internalization and recycling of human beta 2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J. Biol. Chem. 1992;267:3530–3538. [PubMed] [Google Scholar]
- 34.Yin T, Lu L, Xiong Z, Wei S, Cui D. ATPase inhibitory factor 1 is a prognostic marker and contributes to proliferation and invasion of human gastric cancer cells. Biomed. Pharmacother. 2015;70:90–96. doi: 10.1016/j.biopha.2014.12.036. [DOI] [PubMed] [Google Scholar]
- 35.Yadav N, Chandra D. Mitochondrial and postmitochondrial survival signaling in cancer. Mitochondrion. 2014;16:18–25. doi: 10.1016/j.mito.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiménez BD, Quattrochi LC, Yockey CB, Guzelian PS. Identification by differential display of the IF1 inhibitor peptide of ATP synthase/ATPase as a gene inducible in rat liver by pregnenolone 16alpha-carbonitrile. Life. Sci. 2000;67:1825–1832. doi: 10.1016/S0024-3205(00)00769-4. [DOI] [PubMed] [Google Scholar]
- 37.Huang LJ, Chuang IC, Dong HP, Yang RC. Hypoxia-inducible factor 1α regulates the expression of the mitochondrial ATPase inhibitor protein (IF1) in rat liver. Shock. 2011;36:90–96. doi: 10.1097/SHK.0b013e318219ff2a. [DOI] [PubMed] [Google Scholar]
- 38.Fajardo G, et al. β2-adrenergic receptors mediate cardioprotection through crosstalk with mitochondrial cell death pathways. J. Mol. Cell. Cardiol. 2011;51:781–789. doi: 10.1016/j.yjmcc.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wills LP, et al. The β2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J Pharmacol. Exp. Ther. 2012;342:106–118. doi: 10.1124/jpet.112.191528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.García-Bermúdez J, et al. PKA Phosphorylates the ATPase Inhibitory Factor 1 and Inactivates Its Capacity to Bind and Inhibit the Mitochondrial H+-ATP Synthase. Cell. Rep. 2015;12:1–13. doi: 10.1016/j.celrep.2015.08.052. [DOI] [PubMed] [Google Scholar]
- 41.Banerjee A, et al. Induction of an ATPase inhibitor protein by propylthiouracil and protection against paracetamol (acetaminophen) hepatotoxicity in the rat. Br. J. Pharmacol. 1998;124:1041–1047. doi: 10.1038/sj.bjp.0701917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Antoniel M, et al. The oligomycin-sensitivity conferring protein of mitochondrial ATP synthase: emerging new roles in mitochondrial pathophysiology. Int. J. Mol. Sci. 2014;15:7513–7536. doi: 10.3390/ijms15057513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mio Y, Uezono S, Kitahata H. Anesthetic cardioprotection in relation to mitochondria: basic science. Curr. Pharm Des. 2014;20:5673–5680. doi: 10.2174/1381612820666140204110101. [DOI] [PubMed] [Google Scholar]
- 44.Fujikawa M, Imamura H, Nakamura J, Yoshida M. Assessing actual contribution of IF1, inhibitor of mitochondrial FoF1, to ATP homeostasis, cell growth, mitochondrial morphology, and cell viability. J. Biol. Chem. 2012;287:18781–18787. doi: 10.1074/jbc.M112.345793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barbato S, Sgarbi G, Gorini G, Baracca A, Solaini G. The inhibitor protein (IF1) of the F1F0-ATPase modulates human osteosarcoma cell bioenergetics. J. Biol. Chem. 2015;290:6338–48. doi: 10.1074/jbc.M114.631788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campanella M, Seraphim A, Abeti R, Casswell E, Echave P, Duchen MR. IF1, the endogenous regulator of the F(1)F(o)-ATPsynthase, defines mitochondrial volume fraction in HeLa cells by regulating autophagy. Biochim. Biophys. Acta. 2009;1787:393–401. doi: 10.1016/j.bbabio.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 47.Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox. Signal. 2011;14:1939–1951. doi: 10.1089/ars.2010.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell. Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hardonnière, K., Huc, L., Sergent, O., Holme, J. A. & Lagadic-Gossmann D. Environmental carcinogenesis and pH homeostasis: Not only a matter of dysregulated metabolism. Semin. Cancer Biol. Jan 11. pii: S1044-579X(17)30001-9. doi:10.1016/j.semcancer.2017.01.001. [Epub ahead of print] Review (2017). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.