

Abstract
Renal ischemia-reperfusion injury (IRI) is a major risk factor for delayed graft function in renal transplantation. Compelling evidence exists that the stress-responsive enzyme, heme oxygenase-1 (HO-1) mediates protection against IRI. However, the role of myeloid HO-1 during IRI remains poorly characterized. Mice with myeloid-restricted deletion of HO-1 (HO-1M-KO), littermate (LT), and wild-type (WT) mice were subjected to renal IRI or sham procedures and sacrificed after 24 hours or 7 days. In comparison to LT, HO-1M-KO exhibited significant renal histological damage, pro-inflammatory responses and oxidative stress 24 hours after reperfusion. HO-1M-KO mice also displayed impaired tubular repair and increased renal fibrosis 7 days after IRI. In WT mice, HO-1 induction with hemin specifically upregulated HO-1 within the CD11b+ F4/80lo subset of the renal myeloid cells. Prior administration of hemin to renal IRI was associated with significant increase of the renal HO-1+ CD11b+ F4/80lo myeloid cells in comparison to control mice. In contrast, this hemin-mediated protection was abolished in HO-1M-KO mice. In conclusion, myeloid HO-1 appears as a critical protective pathway against renal IRI and could be an interesting therapeutic target in renal transplantation.
Introduction
Ischemia-reperfusion injury (IRI) is inherent to renal transplantation and leads to delayed graft function (DGF) of transplanted kidneys from deceased donors in up to 20 to 50% of cases1, 2. Later, DGF contributes to the reduced longevity of the kidney allografts, notably because of a higher risk of acute and chronic rejection3, 4. Basically, IRI is a two-phase phenomenon, including ischemia in the donor and reperfusion injury in the recipient. It combines major ischemia-induced cell stress, significant burst of free radicals, and intense inflammatory immune responses that lead to extensive cell injury, necrosis, and late interstitial fibrosis of the kidney allograft1, 5. Today, the increasing demand for renal allografts implies more frequent use of organs issued from extended criteria donors and this change leads to an increased risk of DGF6, 7. So far, there is no specific treatment of IRI and a better understanding of underlying mechanisms might lead to a better prevention of DGF and successful transplantations even with transplant from extended criteria donors.
Several natural cellular mechanisms can confer resistance against IRI, including the ubiquitous heme oxygenase (HO) cytoprotective pathway8. Upon cellular stress, the expression of HO isoform 1 (HO-1, encoded by Hmox1) is significantly induced. The main action of HO-1 is the enzymatic degradation of free heme, a source of reactive species massively released during IRI. HO-1 metabolizes free heme into carbon monoxide, biliverdin that can both provide resistance against IRI8. Interestingly, HO-1-deficient mice exhibit severe acute kidney injury (AKI) and death upon renal IRI9, 10. In humans, reduced capacity of the donor to express HO-1 was associated with poorer kidney allograft survival11–13. At the opposite, non-specific HO-1 induction with synthetic heme can confer significant resistance against renal IRI14, 15.
Myeloid cells (i.e. neutrophils, circulating monocyte, tissues macrophages and dendritic cells) can be important contributors to the pathogenesis of IRI-induced AKI9, 16, 17. During renal IRI, it has been shown that expression of HO-1 is an important trafficking regulator of myeloid cells9. Therefore, we hypothesized that the myeloid HO-1 might be critical in renal IRI. Herein, we compared IRI in transgenic mice with myeloid-restricted deletion of HO-1 (HO-1M-KO) to control mice. In addition, we used hemin to confer protection against IRI and to study the importance of a myeloid source of HO-1 in this preventive strategy.
Materials and Methods
Mice
HO-1M-KO mice were generated using the Cre/loxP system as described18. Two transgenic lines were used for the myeloid-specific deletion of HO-1: (1) C57BL/6 Hmox1 loxP/loxP mice in which Hmox1 allele was flanked by loxP sites, and (2) C57BL/6 LysM Cre/wt mice in which Cre recombinase is under the control of a myeloid-restricted promoter (LysM, lysozyme M). HO-1M-KO mice (i.e. LysM Cre/wt Hmox1 loxP/loxP) and littermate (LT) mice (i.e. LysM wt/wt Hmox1 loxP/loxP) were generated at the Institute for Medical Immunology by crossing C57BL/6 Hmox1 loxP/loxP mice with C57BL/6 LysM Cre/WT mice. The LysM Cre transgene mediates a specific deletion of loxP-flanked Hmox1 gene in myeloid cells. C57BL/6 wild-type (WT) mice were purchased from Harlan (Zeist, The Netherlands). LT or WT mice, when specified, were used as controls for HO-1M-KO mice. Eight- to twelve-week-old male animals were used for all experiments, and animals were bred in our specific pathogen-free animal facility. All experiments were conducted in compliance with the Principles of Laboratory Animal Care formulated by the National Institute of Health (Guide for the Care and Use of Laboratory Animals, Eighth Edition, National Research Council, 2010) and were approved by the local committee for animal welfare (Commission d’éthique du Biopole ULB Charleroi).
Renal IRI procedure
Mice were anesthetized with an intraperitoneal injection (340 μl/25 g) of a solution containing Dormicum® (1 mg/ml; Roche), Fentanyl® (78 μg/ml; Janssen-Cilag), and Haldol® (5 mg/ml; Janssen-Cilag). Body temperature was maintained at 37 °C throughout the procedure. Kidneys were exposed through midline incision, and both renal pedicles were clamped for 26 minutes using nontraumatic microsurgical clamps (S&T Microsurgical Instruments). Evidence of ischemia was confirmed by visualizing dark color of clamped kidneys. Restoration of blood flow was monitored before closing incision. Sham-operated mice underwent the same procedure except for clamping of the pedicles. Mice were sacrificed 24 hours or 7 days after reperfusion and samples were collected. When specified, mice received an intraperitoneal injection of hemin (5 mg/kg) or saline 24 h prior to surgery.
Preparation of hemin solution
Hemin (Ferriprotoporphyrin IX chloride, Sigma-Aldrich) was dissolved in 0.1 M NaOH, neutralized (to pH 7.2) with 1 M HCl and adjusted to concentration of 7.7 mM with distilled water. Aliquots were protected from light and stored at −80 °C until used. Hemin was then diluted in sterile saline (NaCl 0.9%) to appropriate concentration and filtered.
Generation of bone marrow-derived macrophages (BMDMs) and culture
Bone marrow cells were isolated from femurs and tibias of WT, LT and HO-1M-KO mice, and cultured in Petri dishes (Greiner Bio-one). Bone marrow cells were incubated at 37 °C in a 5% CO2 atmosphere. For generation of bone marrow-derived macrophages (BMDMs), bone marrow cells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with L-Glutamine, 4.5 g/l glucose (Lonza), 10% heat-inactivated fetal calf serum (FCS), nonessential amino acids, sodium pyruvate, penicillin/streptomycin, β-mercaptoethanol, and 20% supernatant derived from macrophage colony-stimulating factor (M-CSF)-producing L929 cells. At day 3 of culture, 5 ml of complete medium containing 60% L929 cells supernatant was added to each dish. BMDMs were allowed to grow until day 7 after isolation. The purity of BMDMs in culture was over 97% as confirmed by a Fluorescence-activated cell sorting (FACS) analysis of CD11b and F4/80 expression. BMDMs were then collected and cultured in resting and stimulated conditions either with lipopolysaccharide (LPS) 100 ng/ml (Sigma-Aldrich) or hemin 15 μM (Sigma-Aldrich) by using complete medium supplemented with 2% L929 cells supernatant for 24 hours at 37 °C in a 5% CO2 atmosphere.
RNA extraction and Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNA was extracted from BMDMs and kidney tissues using the MagnaPure LC RNA Isolation Kit High Performance and the MagnaPure LC RNA Isolation Kit III for tissue, respectively, according to manufacturer's instructions (Roche). Reverse transcription and real-time PCR were performed using LightCycler Multiplex RNA Virus Master (one-step procedure) on a LightCycler 480 apparatus (Roche). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a RNA loading control. The primers were custom ordered from Eurogentec as follows: GAPDH, forward, 5′-ATTGTCAGCAATGCATCCTG-3′, reverse, 5′-CCTTCCACAATGCCAAAGTT-3′ and probe, 5′-FAM-CCCTGGCCAAGGTCATCCATGA-TAMRA-3′; HO-1, forward, 5′-GCCGAGAATGCTGAGTTCAT-3′, reverse, 5′-AGGAAGCCATCACCAGCTTA-3′ and probe, 5′-FAM-AGAACTTTCAGAAGGGTCAGGTGTCCA-TAMRA-3′; IL-6, forward, 5′-GAGGATACCACTCCCAACAGACC-3′, reverse, 5′-AAGTGCATCATCGTTGTTCATACA-3′ and probe, 5′-FAM-CAGAATTGCCATTGCACAACTCTTTTCTCA-TAMRA-3′. Monocyte chemoattractant protein-1 (MCP-1), keratinocyte chemoattractant (KC), p53, and p21 primers were purchased as ready-made mix from Roche. BMDMs mRNA levels are expressed as 2−ΔΔCT in which CT represents “cycle of threshold”, ΔΔCT = ΔCTmouse of interest − ΔCTnaïve LT mouse, and ΔCT = CTgene of interest − CTGAPDH. Kidney tissue mRNA levels are expressed as 2−ΔΔCT in which CT represents “cycle of threshold”, ΔΔCT = ΔCTmouse of interest − ΔCTsham-operated WT C57BL/6 mouse, and ΔCT = CTgene of interest − CTGAPDH.
Western blot
Western blot analysis was performed as described19. BMDMs were lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) containing protease inhibitor cocktail (Sigma-Aldrich). Protein concentration was measured with the Micro BCA Protein Assay Kit (Thermo Scientific). Primary antibodies used were Actin pAb (Sigma-Aldrich) and HO-1 mAb (clone EP1391Y, Abcam). Secondary antibody used was donkey anti-rabbit IgG- horseradish peroxidase (HRP) (GE Healthcare).
Renal function assessment and histopathology
Renal function was evaluated by measuring creatinine in plasma samples as described20. Kidneys were fixed in 4% formaldehyde, embedded in paraffin, sectioned at 5-μm thickness, and stained with Periodic acid–Schiff–diastase and Masson Trichrome. Renal damage was assessed in a blinded manner by the Tubular Injury Score21. Briefly, the percentage of damaged tubules were assessed in the corticomedullary junction according to observation of necrosis, tubular dilatation, brush border loss, and cast deposition in 10 nonoverlapping fields for each sample (x400 magnification). A five-point scale was used: 0 = no damage, 1 = <10% of tubules injured, 2 = 10–25%, 3 = 25–50%, 4 = 50–75%, 5 = >75%. Interstitial fibrosis was quantified using NIH ImageJ software.
Immunohistochemistry
Paraffin-embedded 5-µm kidney sections were deparaffinized and rehydrated. Endogenous peroxidase activity was first quenched by H2O2 peroxidase blocking reagent (DakoCytomation). Macrophages and neutrophils were detected by using an anti-F4/80 antibody (1:50; eBioscience) and an anti–Ly-6G antibody (1:50; BD Biosciences), respectively; for 30 minutes at room temperature (RT). Sections were then washed and incubated with a secondary biotinylated goat anti-rabbit antibody (1:500; Jackson Immunoresearch) for 30 minutes at RT. Streptavidin-HRP was added and coloration was revealed using diaminobenzidine (DAB) with the substrate chromogen system from Dakocytomation. The number of F4/80+ and Ly-6G+ cells was counted in 10 non-overlapping fields (x400 magnification). Nitrotyrosine staining was performed using an anti-nitrotyrosine antibody (1:400; Abcam) and the OptiView DAB IHC Detection Kit (Ventana Medical Systems) according to manufacturer’s instructions. Nitrotyrosine intensity in the renal cortex was quantified using NIH ImageJ software.
Kidney homogenates preparation and Enzyme-Linked Immunosorbent Assay
Kidneys were diluted in lysis solution containing RIPA buffer (Sigma-Aldrich), protease inhibitor cocktail (Sigma-Aldrich), and phosphatase inhibitor cocktail PhosSTOP (Roche Life Science). Kidneys were homogenized with the MagNa Lyser (Roche Diagnostics). Homogenates were subsequently centrifuged at 12,000 rpm for 20 minutes at 4 °C, and the supernatants were stored at −80 °C until use. HO-1 and p62 enzyme-linked immunosorbent assay (ELISA) kits were purchased from Enzo Life Sciences. Interleukin-6 (IL-6), KC, and MCP-1 ELISA kits were purchased from R&D Systems. Renal cytokines were measured according to manufacturer's instructions. Values were corrected for the amount of renal proteins using Micro BCA Protein Assay Kit (Thermo Scientific).
Kidney tissue digestion protocol for flow cytometry
Kidneys were harvested and washed in RPMI 1640 medium supplemented with L-Glutamine and 25 mM Hepes (Lonza). Kidneys were dissociated in 5 ml digestion buffer containing collagenase IV (Worthington Biochemical) and DNase I (Roche) using the mouse lung dissociation program 1 on gentleMACS Dissociator (Miltenyi Biotec). Then, samples were incubated for 20 minutes at 37 °C and agitated every 15 minutes. Complete tissue dissociation was achieved using the mouse spleen dissociation program 4 on gentleMACS Dissociator (Miltenyi Biotec). Cell suspension was passed through a 70 μm cell strainer (BD Biosciences) and was adjusted to a 30 ml volume with Phosphate Buffer Saline (PBS)/Bovine Serum Albumin (BSA) 0.5%/Ethylenediaminetetraacetic acid (EDTA) 2 mM. Samples were centrifuged at 300 g for 5 minutes at 4 °C. Cell pellets were resuspended in 1 ml PBS-BSA 0.5%-EDTA 2 mM and suitable for flow cytometry staining protocol.
Flow cytometry
Kidneys were prepared as described above. HO-1 intracytoplasmic staining was performed through indirect labeling after cell surface marker staining using the Intracellular Fixation and Permeabilization Buffer Set (eBioscience) according to manufacturer’s instructions. PE-conjugated anti-mouse F4/80 (clone BM8), and anti-mouse CD16/CD32 (Fc block, clone 93) were purchased from eBioscience. FITC-conjugated anti-mouse IgG1 (clone A85-1), and APC-Cy7-conjugated anti-mouse CD11b (clone M1/70) were purchased from BD Biosciences. Unconjugated anti-mouse HO-1 (clone HO-1-1), and PE/Cy7-conjugated anti-mouse CD45 (clone 30-F11) were purchased from Abcam and BioLegend, respectively. Cytometric analysis was performed on a BD LSRFortessa cell analyzer (BD Biosciences) using FlowJo software (FlowJo LLC).
Statistical analysis
All data are expressed as mean ± standard error of the mean (SEM). A two-tailed nonparametric Mann-Whitney U test was used; P-values < 0.05 were considered to represent statistical significance. All graphs and statistical analyses were performed using GraphPad Prism 6.00 for Mac OS X (GraphPad Software, La Jolla California USA, www.graphpad.com).
Results
Renal myeloid cells upregulate HO-1 upon renal IRI
Based on CD11b and F4/80 expression by flow cytometric analysis, we defined three major myeloid cell sub-populations in the kidney (Fig. 1A). These populations were CD11b− F4/80+ (defined as P1), CD11b+ F4/80lo (defined as P2) and CD11bhi F4/80− cells (defined as P3). Most of the P3 cells were neutrophils according to their high Ly6G expression (Supplemental Fig. S1). Upon renal IRI, we noted a significant increase in the number of P3 cells, while P1 and P2 subsets remained unaffected (Fig. 1A,B). Interestingly, we observed a significant increase in HO-1 expression within both P1 and P2 populations 24 hours after reperfusion whereas P3 cells only displayed a slight induction of HO-1 (Fig. 1C,D).
Figure 1.
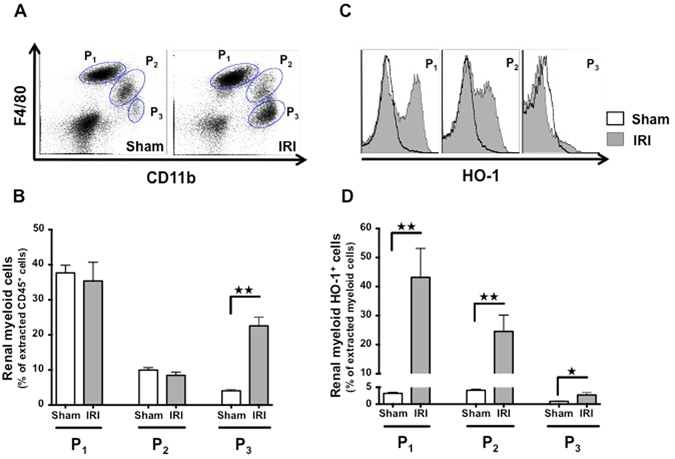
IRI induces HO-1 expression by kidney-infiltrating myeloid cells. Wild-type mice (WT) underwent either bilateral renal IRI for 26 minutes or sham surgery. Twenty-four hours later, mice were sacrificed and kidneys were harvested and processed for flow cytometry analysis. (A) Representative dot plots of sham-operated and IRI mice demonstrating three myeloid-cell type populations in the kidney. These distinct populations were CD11b− F4/80+ (P1), CD11b+ F4/80lo (P2), and CD11bhi F4/80− (P3). (B) Quantification of renal myeloid cells, presented as a proportion of the CD45+ cells extracted from the kidney after IRI (grey bars) or sham surgery (white bars). (C) Representative histograms depicting the level of intracellular HO-1 expression in the myeloid populations after IRI (grey filled area) or sham surgery (bold curve area). (D) Quantification of renal myeloid HO-1+ cells, presented as a proportion of the myeloid cells extracted from the kidney after IRI (grey bars) or sham surgery (white bars). Results are expressed as the mean ± SEM, ★p < 0.05; ★★p < 0.01. n = 5–7 per group.
Myeloid HO-1 protects kidney from IRI
HO-1M-KO mice were used to determine the role of myeloid HO-1 during IRI. Myeloid-restricted deletion of HO-1 was confirmed in both resting and LPS-stimulated BMDMs (Fig. 2A,B). We identified that HO-1M-KO mice exhibit increased susceptibility to renal IRI as demonstrated by the significant loss of renal function and more severe tubular injuries 24 hours after renal IRI in comparison to LT mice (Fig. 3A–C). Interestingly, after renal IRI, HO-1 expression within the whole kidney was identical between LT and HO-1M-KO mice (Fig. 4A,B), suggesting that, despite representing a minor source of HO-1 from a quantitative point of view, myeloid HO-1 mediates significant renal protection during IRI.
Figure 2.
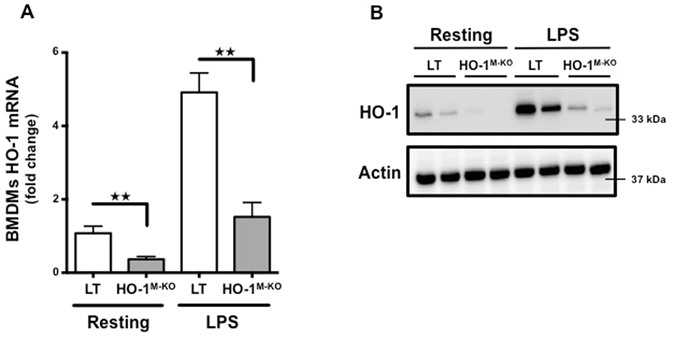
Myeloid-restricted deletion of HO-1 in HO-1M-KO mice. (A) qRT-PCR analysis of HO-1 mRNA levels and (B) representative images of western blot analysis for HO-1 in BMDMs generated from LT (white bars) and HO-1M-KO (grey bars) mice in resting and after LPS stimulation (100 ng/ml) for 24 hours. Results are expressed as the mean ± SEM, ★★p < 0.01 and representative of at least 3 independent experiments. n = 5 per group for HO-1 mRNA levels.
Figure 3.
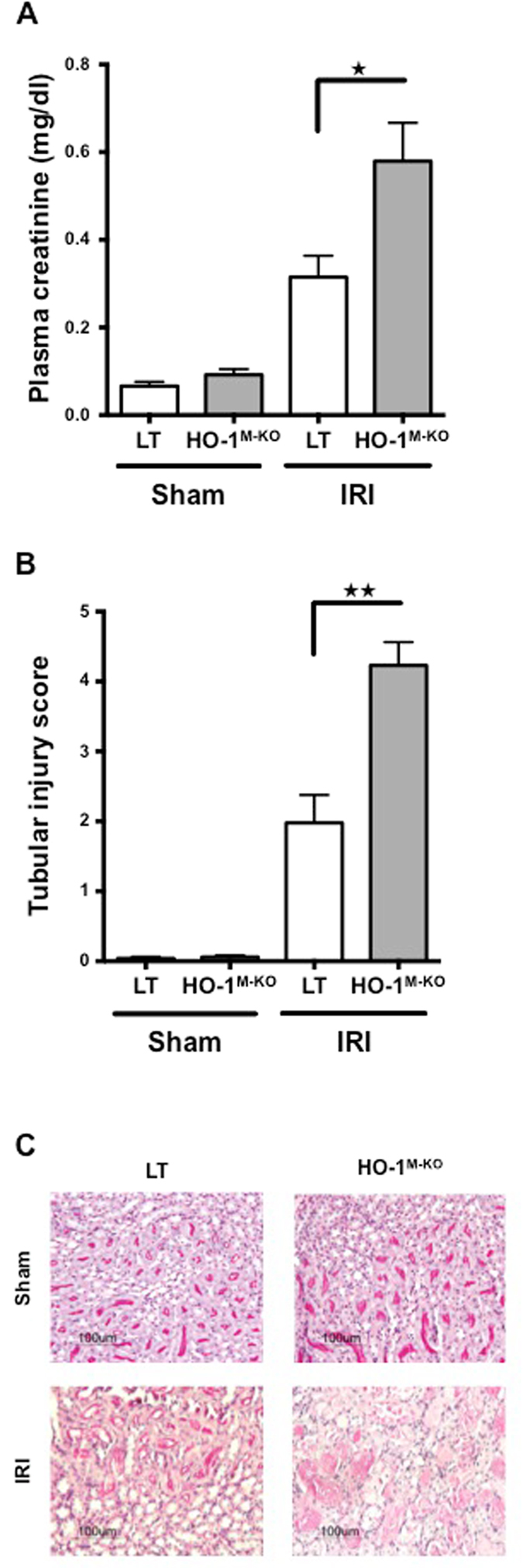
Myeloid-restricted ablation of HO-1 exacerbates renal IRI. (A) Plasma creatinine levels and (B) histological scoring for tubular damage in LT (white bars) and HO-1M-KO (grey bars) mice subjected to sham surgery or 24 hours of reperfusion after renal IRI. Results are expressed as the mean ± SEM, ★p < 0.05; ★★p < 0.01. n = 7–9 for IRI groups and n = 5 for sham groups. (C) Representative sections of corticomedullary junction from sham-operated and IRI after 1 day of reperfusion in LT and HO-1M-KO mice (PAS-D stained). Magnification, x200. Scale bar, 100 μm.
Figure 4.

Whole-tissue renal expression of HO-1 is similar between in LT and HO-1M-KO mice. (A) qRT-PCR analysis of HO-1 mRNA levels and (B) ELISA for HO-1 in LT (white bars) and HO-1M-KO (grey bars) mice subjected to sham surgery or 24 hours of reperfusion after renal IRI. Results are expressed as the mean ± SEM. n = 5–8 for IRI groups and n = 5 for sham groups.
Myeloid HO-1 mitigates both innate immune responses and oxidative stress upon renal IRI
The impact of myeloid HO-1 on renal inflammation 24 hours after IRI was analyzed. HO-1M-KO mice expressed increased levels of renal inflammatory mediators (i.e. IL-6, KC, and MCP-1) in comparison to LT (Fig. 5A). In addition, HO-1M-KO mice also exhibited increased neutrophil and macrophage infiltrates surrounding necrotic tubular cells in the corticomedullary junction (Fig. 5B–E). Finally, accumulation of tubular nitrotyrosine, a well-described marker of oxidative damage22, was exacerbated within the renal cortex of HO-1M-KO mice in comparison to LT (Fig. 5F,G).
Figure 5.
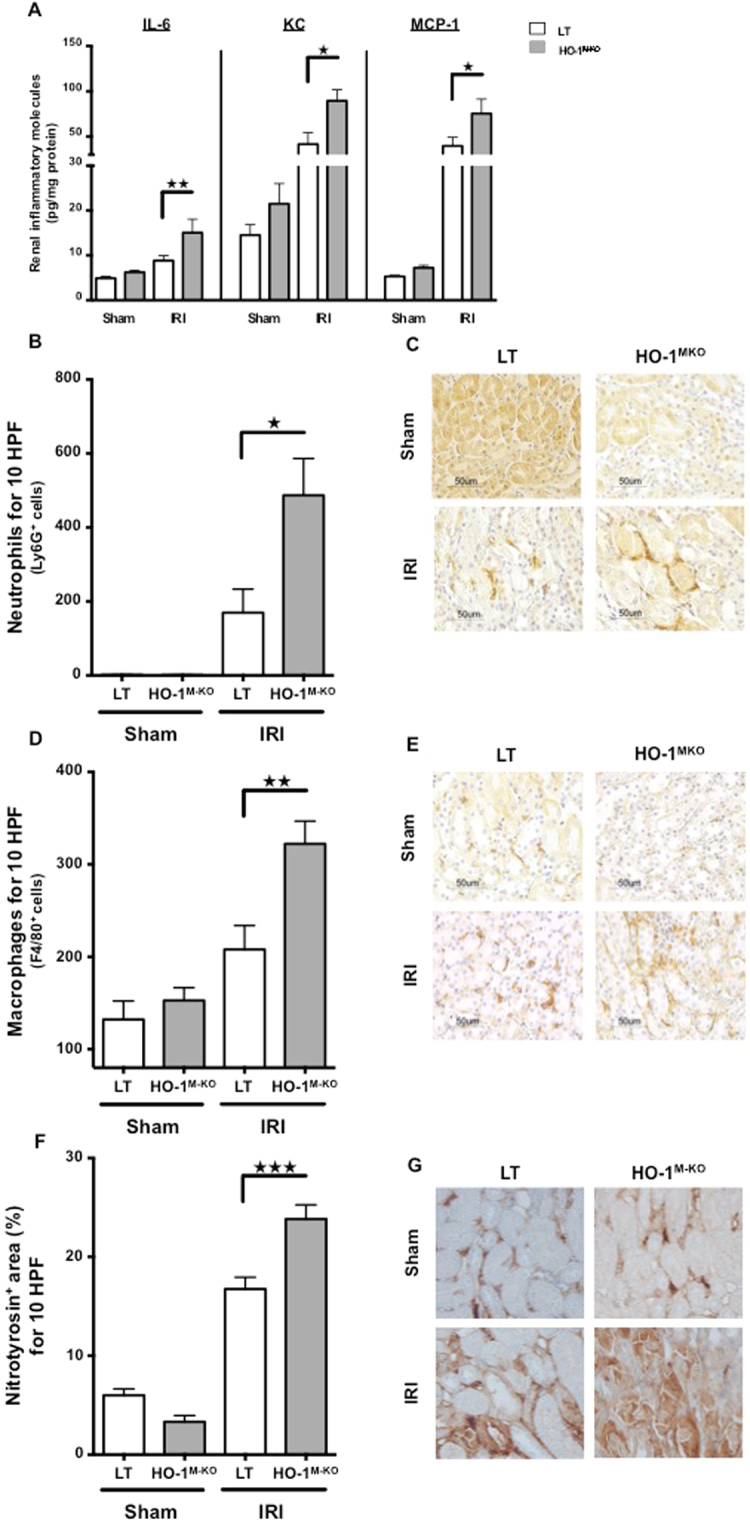
Myeloid HO-1 dampens innate immune responses and oxidative stress upon renal IRI. LT (white bars) and HO-1M-KO mice (grey bars) underwent sham surgery or 1 day of reperfusion after IRI. (A) Proinflammatory mediator levels in kidney. (B) Neutrophil and (D) macrophage influx counts. Representative renal sections stained for Ly6G (C) and F4/80 (E). Magnification, x400. Scale bar, 50 μm. (F) Percentage of nitrotyrosine-positive area in corticomedullary junction and (G) representative sections of nitrotyrosine immunostaining. Magnification, x400. Quantification for nitrotyrosine was done by using ImageJ software. Results are expressed as the mean ± SEM, ★p < 0.05; ★★p < 0.01; ★★★p < 0.001. n = 7–9 for IRI groups and n = 5 for sham groups.
Myeloid HO-1 deficiency leads to impaired tubular repair and renal interstitial fibrosis
Tubular cell-cycle arrest represents a major event that promotes interstitial fibrosis and finally chronic kidney disease (CKD) upon IRI5, 23. In order to investigate a possible role of myeloid HO-1 in the prevention of CKD, we analyzed the expression of the cell-cycle inhibitor p53 and its target p21. As compared to LT, HO-1M-KO mice exhibited a significant increase in both p53 and p21 mRNA expression 4 hours and 24 hours after IRI, respectively (Fig. 6A,B). In addition, we noted a significant accumulation of renal p62 24 hours post IRI in the HO-1M-KO animals as compared to LT, suggesting potential impaired autophagy, a phenomenon known to enhance interstitial fibrosis upon tubular stress24 (Fig. 6C). Finally, 7 days after reperfusion, HO-1M-KO mice exhibited more renal interstitial fibrosis in comparison to LT (Fig. 6D,E).
Figure 6.
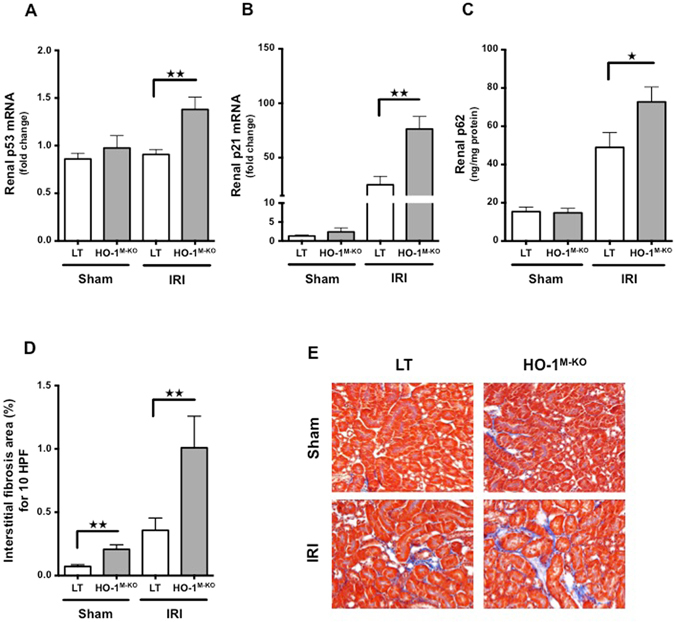
Myeloid HO-1 promotes renal repair and attenuates interstitial fibrosis induced by renal IRI. (A) p53 mRNA, (B) p21 mRNA and (C) p62 renal expression in LT (white bars) and HO-1M-KO (grey bars) mice subjected to sham surgery, 4 hours (p53 mRNA analysis) or 24 hours (p21 mRNA and p62 analysis) of reperfusion after renal IRI. Results are expressed as the mean ± SEM, ★p < 0.05; ★★p < 0.01. In p53/p21 experiments, n = 6–8 for IRI groups and n = 5–6 for sham groups. In p62 experiment, n = 9–10 for IRI groups and n = 6 for sham groups. (D) Percentage of fibrosis area and (E) representative renal sections of Masson Trichrome staining from sham-operated and IRI after 7 days of reperfusion in LT (white bars) and HO-1M-KO (grey bars) mice. Quantification was done using ImageJ software. Results are expressed as the mean ± SEM, ★★p < 0.01. n = 3–4 for IRI groups and n = 5 for sham groups. Magnification, x200.
Hemin-mediated protection against IRI depends on HO-1 upregulation by renal CD11b+F4/80lo myeloid cells
To determine whether the protection against IRI conferred by hemin pretreatment involved HO-1+ myeloid cells, we treated HO-1M-KO and WT mice either with hemin (5 mg/kg) or saline. Twenty-four hours after hemin administration, WT mice exhibited strong HO-1 expression within the renal P2 cells, while neither the P1 nor P3 subsets increased their HO-1 expression (Fig. 7A,B). Interestingly, hemin also induced HO-1 expression within the same subset of splenic myeloid cells (i.e. P2) (Supplemental Fig. S2). In contrast, no HO-1 induction in renal myeloid cells from HO-1M-KO mice was noted (Fig. 7C,D). Twenty-four hours after renal IRI, the proportion of HO-1+ P2 cells increased in the hemin- vs. the saline-treated WT mice while it remained unchanged in HO-1M-KO mice in both conditions (Fig. 8A–C). Concurrently, hemin-treated WT mice displayed renal resistance against IRI (Fig. 8D, Supplemental Fig. S3A–C) while hemin did not preserve renal function in HO-1M-KO mice (Fig. 8E). These results strongly suggest that CD11b+ F4/80lo cells (i.e. P2) are the main protective myeloid source of HO-1 within the kidney upon IRI. We also observed that, in comparison to WT mice, LT did not benefit from the protection against renal IRI when they were treated with hemin 24 hours before IRI (Supplemental Fig. S4A). This could be explained by a hypomorphic HO-1 allele in LT that was due to the modification of the Hmox1 allele with loxP sites (Supplemental Fig. S4B–D). However, the HO-1 hypomorphism was not sufficient to provoke particular susceptibility to renal IRI in LT as attested by similar creatinine levels compared to WT mice (Supplemental Fig. S5).
Figure 7.
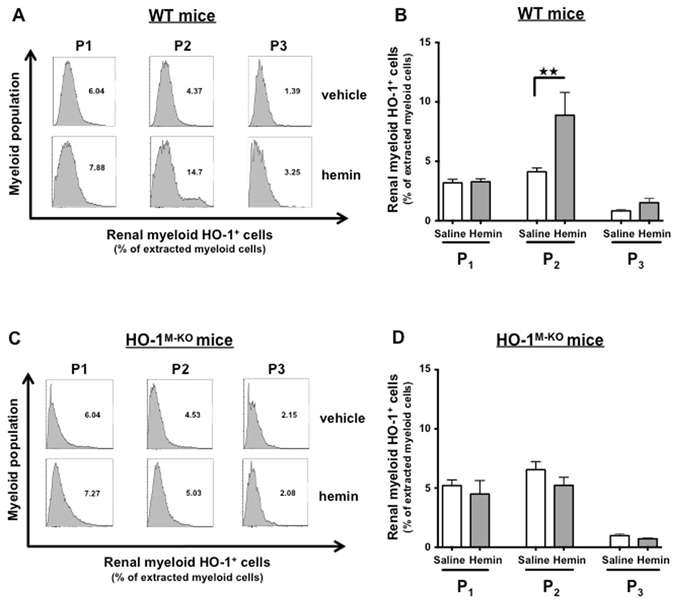
Hemin specifically induces HO-1 expression in renal CD11b+ F4/80lo (P2) myeloid cells. WT (A,B) and HO-1M-KO mice (C,D) were treated with hemin (5 mg/kg) or saline. Twenty-four hours after intraperitoneal injection, kidneys were harvested and homogenized for flow cytometry analysis. The renal myeloid cell populations were characterized according to the expression of CD11b and F4/80 surface markers (i.e., CD11b− F4/80+ (P1), CD11b+ F4/80lo (P2), and CD11bhi F4/80-(P3)). (A,C) Representative histograms depicting the level of intracellular HO-1 expression in the myeloid subpopulations after hemin administration or saline injection. (B,D) Quantification of renal myeloid HO-1+ cells, presented as a proportion of the myeloid cells extracted from the kidney after hemin (grey bars) or saline (white bars). Results are expressed as the mean ± SEM, ★★p < 0.01. n = 7–8 per group in WT mice and n = 5 per group in HO-1M-KO mice.
Figure 8.

Renal CD11b+ F4/80lo (P2) myeloid cells modulate myeloid HO-1-mediated protection against IRI. WT and HO-1M-KO mice were treated with hemin (5 mg/kg, grey bars) or saline (white bars) 24 hours before renal IRI. At day 1 of reperfusion, mice were sacrificed. (A) Representative dot plots of saline- and hemin-treated WT IRI mice showing the myeloid cell populations in the kidney according to the expression of CD11b and F4/80 surface markers (i.e., CD11b− F4/80+ (P1), CD11b+ F4/80lo (P2), and CD11bhi F4/80−(P3)). (B) Quantification of renal myeloid cells upon IRI, presented as a proportion of the renal CD45+ cells extracted from saline- and hemin-treated WT IRI mice. Results are expressed as the mean ± SEM, ★p < 0.05. n = 5–6 per group in WT mice. (C) Quantification of renal myeloid cells upon IRI, presented as a proportion of the renal CD45+ cells extracted from saline- and hemin-treated HO-1M-KO IRI mice. Results are expressed as the mean ± SEM. n = 3–5 per group in HO-1M-KO mice. Plasma creatinine levels in saline- and hemin-treated WT (D) and HO-1M-KO (E) mice subjected to sham surgery or 24 hours of reperfusion after renal IRI. Results are expressed as the mean ± SEM, ★★★p < 0.001. In WT mice, n = 16–20 for IRI groups and n = 6 for sham groups. In HO-1M-KO mice, n = 9 for IRI groups and n = 5 for sham groups.
Discussion
We have shown that myeloid HO-1 mediates protection against renal IRI and that its preemptive induction by hemin confers significant resistance against renal IRI. These findings contrast with the common belief that only epithelial (i.e. tubular cells) and endothelial cells are the critical source of HO-1 during renal IRI. This hypothesis was mainly supported by the intense susceptibility of fully HO-1-deficient to renal IRI10, 25, 26. However, Ferenbach DA et al. already demonstrated that genetically modified or hemin-induced HO-1+ macrophages, a subset of myeloid cells, mediate protection against renal IRI27, 28. Our results strongly support this observation. In addition to these previous studies, we have observed that, in response to IRI, naturally occurring myeloid HO-1 may already modulate the severity of AKI. Indeed, even if we showed that the global expression of HO-1 in the whole kidney was not affected, the absence of myeloid HO-1 was critical in the outcome of renal IRI. More recently, Hull et al. showed that HO-1 is a critical regulator of the trafficking of myeloid cells in AKI. However they did not report a difference between LT and HO-1M-KO mice in term of renal function (i.e. plasma creatinine) or in tubular damage 1 day after reperfusion9. In contrast, our results pointed out the myeloid HO-1 as a critical regulator of the earliest phases of IRI (i.e. lower plasma creatinine, tubular damage, and renal inflammation) that may mitigate the risk of severe AKI upon IRI. By inference, as severe IRI in a renal transplant also increases its immunogenicity, we may postulate that, by downsizing the early inflammatory response, the induction of myeloid HO-1 could be a regulatory mechanism decreasing transplant alloreactivity.
A cell-cycle arrest at the G2/M phase is associated with maladaptive repair and subsequent fibrosis in renal IRI5, 23. In our experiments, HO-1M-KO mice exhibited impaired renal repair upon IRI as suggested by the upregulation of cell-cycle regulatory proteins (i.e. p53, p21), potential disruption of autophagy (i.e. p62 accumulation) and early interstitial fibrosis, a central marker of CKD. The roles of cell-cycle inhibitors p53/p21 in the pathogenesis of AKI are complex and remain a matter of debate. For instance, if p53 expression by leukocytes has been identified as protective against AKI, its induction in tubular cells was associated with worsened AKI and subsequent interstitial fibrosis29–31. In line with a previous study32, the cell-cycle inhibitor p21 has been shown protective in AKI by mediating a cell-cycle arrest in G1 phase and therefore allowing repair of DNA-damage33. However, the authors observed that p21 was not involved in protection against renal fibrosis33. Herein, we focus on the link between p21 and the progression of AKI to fibrosis. Interestingly, even if p21 expression may confer early resistance to AKI, p21 expression by tubular cells also reflects their cellular senescence that is associated with limited regenerative ability of tubular cells and interstitial fibrosis following AKI suggesting an additional role for p2134, 35. Indeed, these different data are consistent with the recent concept that p21 has different effects during AKI or CKD/fibrosis progression36.
Altogether, these observations sustain the hypothesis that the upregulation of both p53 and p21 upon IRI in HO-1M-KO kidneys may encourage fibrotic processes in HO-1M-KO mice. Interestingly, fully HO-1-deficient mice exhibit more severe interstitial fibrosis upon AKI37 and renal fibrosis was also reported in HO-1M-KO mice 1week after IRI9. Further, our results suggest a link between myeloid HO-1 deficiency and renal fibrosis because of maladaptive repair.
Even if the role of myeloid HO-1 as a critical modulator of the late fibrotic processes remains a matter of debate, it has been shown that AKI could lead to severe CKD. Indeed, this is supported by the recent key concept of cell-cycle arrest and maladaptive repair of tubular cells that is induced by AKI itself5, 23. Briefly, a clear correlation between the severity of the renal lesions and the risk of maladaptive repair has been established in several experimental models, leading to interstitial fibrosis and subsequent CKD5, 23. Even if the loss of myeloid HO-1 expression before IRI was responsible for the observed kidney injury, a specific role for HO-1 during the later phases of renal repair after IRI is not excluded. For instance, HO-1 may decrease TGF-β expression at the tubular level, thereby preventing renal fibrosis38. HO-1 was also shown to modulate tubular autophagy, a major event involved in cell repair after insult39. Of interest, we have observed that HO-1M-KO mice exhibited p62 accumulation upon renal IRI which may be seen as a surrogate marker of impaired autophagy40.
Specific deletion of myeloid HO-1 was associated with sustained oxidative damage within the corticomedullary junction upon IRI. It is well known that HO-1 limits the heme- and hydrogen peroxide-mediated oxidative stress that heavily damages DNA and proteins and leads to cell death41. However, the fact that HO-1M-KO mice exhibit intense tubular damage due to excessive oxidative stress strongly suggests potential cross-talk between the tubular environment and the HO-1+ myeloid cells. Accordingly, the release of by-products of heme degradation by HO-1+ myeloid cells (i.e. carbon-monoxide, IL-10) may inhibit the apoptosis of the surrounding tubular cells, thereby promoting their survival42. Interestingly, HO-1M-KO mice exhibited sustained p53 expression in the whole kidney upon IRI. As tubular cells are major components of kidney extract, we may hypothesize that myeloid HO-1 may regulate some of their important functions upon IRI, decreasing the risk of both tubular apoptosis and cell-cycle arrest.
It has been shown that HO-1 expression was associated with CD11b+ F4/80lo macrophages that exhibit regulatory properties (i.e. “M2” macrophages)43. These M2 macrophages are potential modulators of inflammatory responses induced by renal IRI, thereby promoting renal repair after insult16. In this condition, HO-1+ myeloid cells may promote the development of a microenvironment dominated by regulatory macrophages that efficiently dampen early inflammatory responses and delay the onset of interstitial fibrosis44. However, the molecular mechanism of macrophage polarization mediated by HO-1 remains to be elucidated44. Interestingly, HO-1 modulates IL-10 expression45, 46 and HO-1+ macrophages express increased level of IL-1027 suggesting a positive-feed-forward loop between these two anti-inflammatory factors, favoring the acquisition of an IL-10-producing, M2-like phenotype. The macrophage-derived molecules that promote kidney repair are poorly known47. However, both the macrophage-secreted Wnt7b and chitinase-like protein BRP-39 have been shown to promote kidney regeneration after IRI48, 49. Accordingly, we may hypothesize that our HO-1+ CD11b+ F4/80lo anti-inflammatory macrophages protect the renal parenchyma upon IRI by secretion of some mediators which may interact with renal epithelial cells.
Even if we did not demonstrate it in our experiments, the intense inflammatory response that we observed in HO-1M-KO kidneys upon IRI might be explained through a phenotypic polarization toward “M1” macrophages. Indeed, in absence of HO-1, a lack of M2 macrophages was observed with an excess of inflammatory “M1” macrophages50. Then, these macrophages secrete pro-inflammatory mediators that amplify intrarenal inflammation and injury through interaction with kidney resident cells16, 47. Therefore, we assume that HO-1, in addition to its protective role against oxidative stress at the cellular level, also plays a critical role in the regulation of early innate immune responses induced by renal IRI.
The origin of HO-1+ myeloid cells that protect the injured kidney remains largely unknown. Theoretically, renoprotection might be provided by both resident and infiltrating HO-1+ myeloid cells in the kidney. In line with previous report28, we showed that, even in absence of IRI, hemin upregulated the HO-1 expression within CD11b+ F4/80lo myeloid cells in the kidney. This observation suggests that tissue-resident myeloid cells might be involved in the earliest phase of renal IRI. The recruitment of splenic macrophages that protect against IRI was already observed51, 52. We also noted that hemin induced HO-1 within spleen CD11b+ F4/80lo myeloid cells suggesting that extra-renal HO-1+ myeloid cells might represent a reservoir that can be recruited in the injured kidney after renal IRI. Indeed, one day after reperfusion, we observed a higher proportion of CD11b+ F4/80lo myeloid cells within the kidney of hemin-treated WT mice suggesting that these protective cells could have been recruited from extra-renal sites such as the spleen.
We identified that the intensity of myeloid HO-1 expression was an important determinant of efficient hemin-mediated protection against renal IRI. Indeed, we found that the floxation of the Hmox1 allele with loxP sites in LT significantly decreased both native and induced HO-1 expression in comparison to WT mice. This “hypomorphism” induced by allele floxation has been described in others Cre-LoxP knockout systems53, 54. Interestingly, even if the HO-1 hypomorphism was insufficient to cause particular susceptibility to renal IRI in LT compared to WT mice, it leaded to the loss of the hemin-mediated protection in LT. Therefore, we may postulate that myeloid HO-1 expression induced by hemin requires a critical level to be effective and that this minimal threshold in HO-1 expression was not reached in LT. By inference, the length polymorphism of guanosine thymidine dinucleotide (GT)n repeats in the promoter region of Hmox1 was associated with lower HO-1 activity55, mimicking the effect of a hypomorphic allele. Interestingly, longer (GT)n repeats in the Hmox1 promoter were associated with the occurrence of CKD and decreased renal function after cardiac surgery or kidney transplantation in humans11, 56–58. Also, it has been largely reported that hemin preconditioning significantly mitigates IRI-induced AKI14, 15. A recent study showed that hemin safely induces HO-1 in deceased donor renal transplant recipients but the benefit of this approach on the renal allograft survival remains still unknown59. Interestingly, myeloid HO-1 has been involved in the suppression of alloreactivity60–62. This suggests that myeloid HO-1 could have additional regulatory properties that may contribute to both better survival and tolerance of the allograft after IRI. However, further studies are needed to confirm the benefit of the HO-1 induction in renal transplantation.
In conclusion, our results strongly support the importance of HO-1+ myeloid cells for conferring resistance against renal IRI and are in line with other recent published works9, 27, 28. Myeloid HO-1 appears as a critical modulator of the interstitial inflammation and subsequent fibrosis that both follow renal IRI. Pharmacological HO-1 induction by hemin before renal IRI could be an efficient preventive strategy for limiting kidney damage in many situations such as renal transplantation.
Electronic supplementary material
Acknowledgements
We thank Eric Deprez (Laboratory of Experimental Nephrology, Université Libre de Bruxelles, Brussels, Belgium) for technical assistance. We wish to thank Céline Molle and Stanislas Goriely (Institute for Medical Immunology, Université Libre de Bruxelles, Gosselies, Belgium) for providing supernatant derived from M-CSF-producing L929 cells. We acknowledge the contribution of a medical writer, Sandy Field, PhD, for English language editing of this manuscript. This work was supported by grants from the “Fonds de la Recherche Scientifique Médicale” (F.R.S.M.) to M.R. and J.M.H., the “Fonds Erasme pour la Recherche Médicale” to J.M.H., and the “Société Belge d’Urologie” (S.B.U.) to M.R. This work was presented in abstract form at the 17th Congress of the European Society for Organ Transplantation (ESOT) in Brussels, Belgium (Brief Oral Presentation, BOS04 – Ischemia, Reperfusion, Metabolism and Aging, abstract N°BO33; 13–16 September 2015) and at the 16th Congress of the European Association of Urology (EAU) in Munich, Germany (Poster Session 48, Kidney Transplant: From Bench to clinical practice, abstract n°603; 11–15 March 2016).
Author Contributions
All the authors have made substantial contributions to the intellectual content of the manuscript. M.R., A.T., O.L., V.F., A.L. and J.M.H. participated in the design of the study and conceived the experiments. M.R., A.T., S.D., N.P. and J.M.H. performed the experiments and acquired the data. M.R., A.T., S.D., N.P., O.L., V.F., A.L. and J.M.H. analyzed and interpreted the data. M.R., A.L. and J.M.H. wrote the manuscript. A.T., S.D., N.P., M.P.S., T.R., O.L. and V.F. revised the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Alain Le Moine and Jean-Michel Hougardy jointly supervised this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-00220-w
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Maxime Rossi, Email: mrossi@ulb.ac.be.
Jean-Michel Hougardy, Email: Jean-Michel.Hougardy@erasme.ulb.ac.be.
References
- 1.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kruger B, et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci USA. 2009;106:3390–3395. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu WK, Famure O, Li Y, Kim SJ. Delayed graft function and the risk of acute rejection in the modern era of kidney transplantation. Kidney Int. 2015;88:851–858. doi: 10.1038/ki.2015.190. [DOI] [PubMed] [Google Scholar]
- 4.Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant. 2011;11:2279–2296. doi: 10.1111/j.1600-6143.2011.03754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11:264–276. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metzger RA, et al. Expanded criteria donors for kidney transplantation. Am J Transplant. 2003;3(Suppl 4):114–125. doi: 10.1034/j.1600-6143.3.s4.11.x. [DOI] [PubMed] [Google Scholar]
- 7.Matignon M, et al. Th-17 Alloimmune Responses in Renal Allograft Biopsies From Recipients of Kidney Transplants Using Extended Criteria Donors During Acute T Cell-Mediated Rejection. Am J Transplant. 2015;15:2718–2725. doi: 10.1111/ajt.13304. [DOI] [PubMed] [Google Scholar]
- 8.Ryter SW, Choi AM. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res. 2016;167:7–34. doi: 10.1016/j.trsl.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hull TD, et al. Heme Oxygenase-1 Regulates Myeloid Cell Trafficking in AKI. J Am Soc Nephrol. 2015;26:2139–2151. doi: 10.1681/ASN.2014080770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tracz MJ, et al. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int. 2007;72:1073–1080. doi: 10.1038/sj.ki.5002471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baan C, et al. Fundamental role for HO-1 in the self-protection of renal allografts. Am J Transplant. 2004;4:811–818. doi: 10.1111/j.1600-6143.2004.00420.x. [DOI] [PubMed] [Google Scholar]
- 12.Exner M, et al. Donor heme oxygenase-1 genotype is associated with renal allograft function. Transplantation. 2004;77:538–542. doi: 10.1097/01.TP.0000113467.36269.F8. [DOI] [PubMed] [Google Scholar]
- 13.Courtney AE, et al. Association of functional heme oxygenase-1 gene promoter polymorphism with renal transplantation outcomes. Am J Transplant. 2007;7:908–913. doi: 10.1111/j.1600-6143.2006.01726.x. [DOI] [PubMed] [Google Scholar]
- 14.Chok MK, et al. Renoprotective potency of heme oxygenase-1 induction in rat renal ischemia-reperfusion. Inflamm Allergy Drug Targets. 2009;8:252–259. doi: 10.2174/187152809789352186. [DOI] [PubMed] [Google Scholar]
- 15.Correa-Costa M, et al. Transcriptome analysis of renal ischemia/reperfusion injury and its modulation by ischemic pre-conditioning or hemin treatment. PLoS One. 2012;7:e49569. doi: 10.1371/journal.pone.0049569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22:317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, et al. Dendritic cells tolerized with adenosine A(2)AR agonist attenuate acute kidney injury. J Clin Invest. 2012;122:3931–3942. doi: 10.1172/JCI63170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzima S, Victoratos P, Kranidioti K, Alexiou M, Kollias G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J Exp Med. 2009;206:1167–1179. doi: 10.1084/jem.20081582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Preyat N, et al. Intracellular nicotinamide adenine dinucleotide promotes TNF-induced necroptosis in a sirtuin-dependent manner. Cell Death Differ. 2016;23:29–40. doi: 10.1038/cdd.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue GP, Fishlock RC, Snoswell AM. Determination of creatinine in whole blood, plasma, and urine by high-performance liquid chromatography. Anal Biochem. 1988;171:135–140. doi: 10.1016/0003-2697(88)90133-9. [DOI] [PubMed] [Google Scholar]
- 21.Leemans JC, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parajuli N, MacMillan-Crow LA. Role of reduced manganese superoxide dismutase in ischemia-reperfusion injury: a possible trigger for autophagy and mitochondrial biogenesis? Am J Physiol Renal Physiol. 2013;304:F257–267. doi: 10.1152/ajprenal.00435.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi M, et al. alphaKlotho Mitigates Progression of AKI to CKD through Activation of Autophagy. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2015060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovtunovych G, et al. Wild-type macrophages reverse disease in heme oxygenase 1-deficient mice. Blood. 2014;124:1522–1530. doi: 10.1182/blood-2014-02-554162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapturczak MH, et al. Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am J Pathol. 2004;165:1045–1053. doi: 10.1016/S0002-9440(10)63365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferenbach DA, et al. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol Ther. 2010;18:1706–1713. doi: 10.1038/mt.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferenbach DA, et al. The induction of macrophage hemeoxygenase-1 is protective during acute kidney injury in aging mice. Kidney Int. 2011;79:966–976. doi: 10.1038/ki.2010.535. [DOI] [PubMed] [Google Scholar]
- 29.Zhang D, et al. Tubular p53 regulates multiple genes to mediate AKI. J Am Soc Nephrol. 2014;25:2278–2289. doi: 10.1681/ASN.2013080902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutton TA, et al. p53 is renoprotective after ischemic kidney injury by reducing inflammation. J Am Soc Nephrol. 2013;24:113–124. doi: 10.1681/ASN.2012050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ying Y, Kim J, Westphal SN, Long KE, Padanilam BJ. Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. J Am Soc Nephrol. 2014;25:2707–2716. doi: 10.1681/ASN.2013121270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Megyesi J, Andrade L, Vieira JM, Jr., Safirstein RL, Price PM. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001;60:2164–2172. doi: 10.1046/j.1523-1755.2001.00044.x. [DOI] [PubMed] [Google Scholar]
- 33.Nishioka S, et al. The cyclin-dependent kinase inhibitor p21 is essential for the beneficial effects of renal ischemic preconditioning on renal ischemia/reperfusion injury in mice. Kidney Int. 2014;85:871–879. doi: 10.1038/ki.2013.496. [DOI] [PubMed] [Google Scholar]
- 34.Westhoff JH, et al. Telomere shortening reduces regenerative capacity after acute kidney injury. J Am Soc Nephrol. 2010;21:327–336. doi: 10.1681/ASN.2009010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Megyesi J, et al. Increased expression of p21WAF1/CIP1 in kidney proximal tubules mediates fibrosis. Am J Physiol Renal Physiol. 2015;308:F122–130. doi: 10.1152/ajprenal.00489.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canaud G, Bonventre JV. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol Dial Transplant. 2015;30:575–583. doi: 10.1093/ndt/gfu230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kie JH, Kapturczak MH, Traylor A, Agarwal A, Hill-Kapturczak N. Heme oxygenase-1 deficiency promotes epithelial-mesenchymal transition and renal fibrosis. J Am Soc Nephrol. 2008;19:1681–1691. doi: 10.1681/ASN.2007101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Correa-Costa M, et al. Induction of heme oxygenase-1 can halt and even reverse renal tubule-interstitial fibrosis. PLoS One. 2010;5:e14298. doi: 10.1371/journal.pone.0014298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Surolia R, et al. Heme oxygenase-1-mediated autophagy protects against pulmonary endothelial cell death and development of emphysema in cadmium-treated mice. Am J Physiol Lung Cell Mol Physiol. 2015;309:L280–292. doi: 10.1152/ajplung.00097.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolisetty S, et al. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J Am Soc Nephrol. 2010;21:1702–1712. doi: 10.1681/ASN.2010030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci USA. 1997;94:10925–10930. doi: 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brouard S, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med. 2000;192:1015–1026. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology. 2010;215:788–795. doi: 10.1016/j.imbio.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 44.Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys. 2014;564:83–88. doi: 10.1016/j.abb.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 46.Drechsler Y, et al. Heme oxygenase-1 mediates the anti-inflammatory effects of acute alcohol on IL-10 induction involving p38 MAPK activation in monocytes. J Immunol. 2006;177:2592–2600. doi: 10.4049/jimmunol.177.4.2592. [DOI] [PubMed] [Google Scholar]
- 47.Cao Q, Harris DC, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 2015;30:183–194. doi: 10.1152/physiol.00046.2014. [DOI] [PubMed] [Google Scholar]
- 48.Lin SL, et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci USA. 2010;107:4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt IM, et al. Chitinase-like protein Brp-39/YKL-40 modulates the renal response to ischemic injury and predicts delayed allograft function. J Am Soc Nephrol. 2013;24:309–319. doi: 10.1681/ASN.2012060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Devey L, et al. Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Mol Ther. 2009;17:65–72. doi: 10.1038/mt.2008.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gigliotti JC, et al. Ultrasound Modulates the Splenic Neuroimmune Axis in Attenuating AKI. J Am Soc Nephrol. 2015;26:2470–2481. doi: 10.1681/ASN.2014080769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gigliotti JC, et al. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J Am Soc Nephrol. 2013;24:1451–1460. doi: 10.1681/ASN.2013010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacLean HE, et al. A floxed allele of the androgen receptor gene causes hyperandrogenization in male mice. Physiol Genomics. 2008;33:133–137. doi: 10.1152/physiolgenomics.00260.2007. [DOI] [PubMed] [Google Scholar]
- 54.Taguchi K, et al. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol Cell Biol. 2010;30:3016–3026. doi: 10.1128/MCB.01591-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walther M, et al. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog. 2012;8:e1002579. doi: 10.1371/journal.ppat.1002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen YH, et al. Length polymorphism in heme oxygenase-1 and risk of CKD among patients with coronary artery disease. J Am Soc Nephrol. 2014;25:2669–2677. doi: 10.1681/ASN.2013111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leaf DE, et al. Length Polymorphisms in Heme Oxygenase-1 and AKI after Cardiac Surgery. J Am Soc Nephrol. 2016 doi: 10.1681/ASN.2016010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ozaki KS, et al. Improved renal function after kidney transplantation is associated with heme oxygenase-1 polymorphism. Clin Transplant. 2008;22:609–616. doi: 10.1111/j.1399-0012.2008.00832.x. [DOI] [PubMed] [Google Scholar]
- 59.Thomas RA, et al. Hemin Preconditioning Upregulates Heme Oxygenase-1 in Deceased Donor Renal Transplant Recipients: A Randomized, Controlled, Phase IIB Trial. Transplantation. 2016;100:176–183. doi: 10.1097/TP.0000000000000770. [DOI] [PubMed] [Google Scholar]
- 60.Martins PN, et al. Induction of heme oxygenase-1 in the donor reduces graft immunogenicity. Transplant Proc. 2005;37:384–386. doi: 10.1016/j.transproceed.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 61.De Wilde V, et al. Endotoxin-induced myeloid-derived suppressor cells inhibit alloimmune responses via heme oxygenase-1. Am J Transplant. 2009;9:2034–2047. doi: 10.1111/j.1600-6143.2009.02757.x. [DOI] [PubMed] [Google Scholar]
- 62.George JF, et al. Suppression by CD4+CD25+ regulatory T cells is dependent on expression of heme oxygenase-1 in antigen-presenting cells. Am J Pathol. 2008;173:154–160. doi: 10.2353/ajpath.2008.070963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.