

Abstract
T-cell and natural killer–cell lymphomas are a relatively rare and heterogeneous group of diseases that are difficult to treat and usually have poor outcomes. To date, therapeutic interventions are of limited efficacy and there is a pressing need to find better treatments. In recent years, advances in molecular biology have helped to elucidate the underlying genetic complexity of this group of diseases and to identify mutations and signaling pathways involved in lymphomagenesis. In this review, we highlight the unique biological characteristics of some of the different subtypes and discuss how these may be targeted to provide more individualized and effective treatment approaches.
Keywords: T-cell lymphoma, classification, treatment
Background and Etiology
T-cell and natural killer (NK)–cell lymphomas account for less than 20% of non-Hodgkin lymphoma (NHL) overall but show a striking geographical and racial variation in incidence.1,2 For example, T-cell lymphomas in general, and certain subtypes, such as adult T-cell leukemia/lymphoma (ATLL) or some aggressive Epstein-Barr virus (EBV)–associated T-cell and NK-cell NHLs, are much more commonly found in Asian populations. By contrast, enteropathy-associated T-cell lymphomas occur much more frequently in individuals of Welsh or Irish descent who share a common genetic predisposition to gluten-sensitive enteropathy.1,3 Figure 1 depicts the major lymphoma subtypes by geographic regions.2
Figure 1.
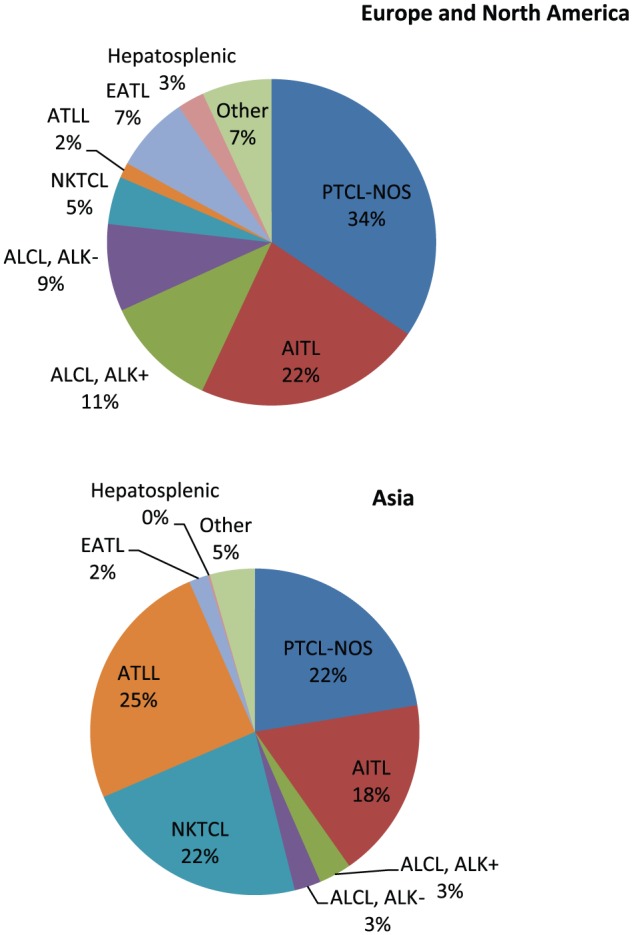
Distribution of major T and NK neoplasms by geographic region. AITL indicates angioimmunoblastic T-cell lymphoma; ALCL, ALK+, anaplastic large cell lymphoma, anaplastic lymphoma kinase positive; ALCL, ALK−, anaplastic large cell lymphoma, anaplastic lymphoma kinase negative; ATLL, adult T-cell leukemia/lymphoma; EATL, enteropathy-associated T-cell lymphoma; NKTCL, natural killer/T-cell lymphoma; PTCL-NOS, peripheral T-cell, not otherwise specified. Adapted from Vose et al.2
Although little is known about other potential environmental exposures contributing to their etiology, viral infectious agents have been implicated in the development of certain types of T-cell and NK-cell NHLs: seropositivity for the human T-lymphotropic virus type I (HTLV-1), for example, predisposes individuals to the development of ATLL, whereas EBV has been associated with a number of T-cell and NK-cell lymphomas both in children and adults.1
Classification and Diagnosis
Because NK cells show some immunophenotypic and functional similarities to T cells, lymphomas derived from both cell types are generally considered together; tumors are thought to arise from cells at various stages of differentiation, and as with B-cell lymphomas, both indolent and aggressive forms of the disease are recognized. Table 1 presents the current World Health Organization (WHO) classification of mature T-cell and NK-cell neoplasms; disease entities are classified by morphology, immunophenotype, and genetic characteristics.1,4 However, 1 large international study found that diagnostic accuracy and consensus diagnosis by a panel of expert hematopathologists vary considerably depending on the type of lymphoma and the availability of useful markers such as expression of anaplastic lymphoma kinase (ALK). Although the agreement of diagnosis in ALK-positive anaplastic large cell lymphoma (ALCL) was found to be 97% in this study, the consensus diagnosis of other subtypes, such as ALK-negative ALCL and of the most common subtype, peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS) was much lower at 74% and 75%, respectively.2
Table 1.
WHO classification of mature T and NK neoplasms.4
| Usually indolent | |
|---|---|
| T-cell large granular lymphocytic leukemia | |
| Hydroa vacciniforme-like lymphoproliferative disorder | |
| Indolent T-cell lymphoproliferative disorder of the gastrointestinal tract | |
| Subcutaneous panniculitis-like T-cell lymphoma | |
| Mycosis fungoides | |
| Primary cutaneous CD30-positive T-cell lymphoproliferative disorders Lymphomatoid papulosis Primary cutaneous anaplastic large cell lymphoma Primary cutaneous acral CD8-positive T-cell lymphoma Primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder | |
| Breast implant–associated anaplastic large cell lymphoma | |
| Usually aggressive | |
| Nodal | Extranodal |
| Systemic EBV-positive T-cell lymphoma of childhood | Extranodal NK/T-cell lymphoma, nasal type |
| Adult T-cell leukemia/lymphoma | Enteropathy-associated T-cell lymphoma |
| Peripheral T-cell lymphoma, not otherwise specified | Monomorphic epitheliotropic intestinal T-cell lymphoma |
| Angioimmunoblastic T-cell lymphoma | Hepatosplenic T-cell lymphoma |
| Follicular T-cell lymphoma | Sézary syndrome |
| Nodal peripheral T-cell lymphoma with TFH phenotype | Primary cutaneous gamma-delta T-cell lymphoma |
| Anaplastic large cell lymphoma Anaplastic lymphoma kinase positive Anaplastic lymphoma kinase negative |
Primary cutaneous CD 8–positive aggressive epidermotropic cytotoxic T-cell lymphoma |
| Typically leukemic presentation | |
| T-cell prolymphocytic leukemia | |
| Chronic lymphoproliferative disorder of NK cells | |
| Aggressive NK-cell leukemia | |
Abbreviations: EBV, Epstein-Barr virus; NK, natural killer; TFH, follicular helper T; WHO, World Health Organization.
Recent advances in molecular biology and genetics have led to the identification of new mutations and gene expression profiles (GEPs) that should lead to a more accurate subclassification of T-cell NHL in the future. To date, the only frequently recurring chromosomal translocation with a demonstrable prognostic impact occurs in ALK-positive ALCL (t(2;5) (p23;q35)). By contrast, ALK-negative ALCL previously was only recognized as a provisional entity in the WHO classification, until results from GEP studies, which revealed distinct GEP signatures, led to improved criteria to distinguish it from CD30+ PTCL. Similarly, the discovery that both ALK-positive and ALK-negative ALCLs show constitutive activation of JAK/STAT signaling pathways has provided a unifying genetic basis for the morphologic and phenotypic similarities shared by both tumor types.4
Other genetic abnormalities, including mutations in epigenetic regulators such as TET2, IDH, and DNMT3A may be useful in dissecting the complexity of the genetic landscape of nodal PTCL further, allowing a clearer distinction between angioimmunoblastic T-cell lymphoma (AITL) and PTCL-NOS. Gene expression profile may also provide key insights into the heterogeneity of the largest subgroup, PTCL-NOS.
Current Standard of Care
In comparison with the treatment of B-cell lymphomas, where the addition of immunotherapeutic agents to standard combination chemotherapy regimens has led to improved outcomes for patients, treatment options for T-cell and NK-cell lymphomas are limited and rarely curative.2,5,6 Traditionally, despite their differences in pathology and clinical presentation, the most common entities, PTCL-NOS, AITL, ALCL (both ALK positive and ALK negative), are treated in a similar fashion, with treatment regimens extrapolated from the treatment of aggressive B-cell malignancies.5–7 Thus, the CHOP chemotherapy regimen, comprising cyclophosphamide, doxorubicin, vincristine, and prednisolone, has been the most widely studied,6 and a retrospective meta-analysis of patients treated with CHOP or CHOP-like regimens reported a 5-year overall survival (OS) of 38.5%.6,8Another large, multicenter retrospective analysis of 1314 cases of PTCL-NOS and extranodal NK/T-cell lymphoma (ENKTCL) treated with anthracycline-containing regimens found that outcomes vary depending on histologic subtype: 14% 5-year OS in ATLL, 32% in PTCL-NOS, 32% in AITL, 70% in ALK-positive ALCL, and 49% in ALK-negative ALCL.2
Attempts to improve this CHOP induction chemotherapy have had limited success. Adding etoposide to the existing CHOP regimen was found to improve 3-year event-free survival in a group of 343 patients treated by the German High-Grade Non-Hodgkin Lymphoma Study Group from 51% to 75.4%, but only if patients were less than 60 years of age and had a normal lactate dehydrogenase.9 In addition, the subtype that appeared to benefit most was ALK-positive ALCL, which already enjoys the best outcomes. The role for etoposide is therefore unclear.
Shortening the time between the administrations of chemotherapy cycles from 3 to 2 weeks (CHOP-21 versus CHOP-14) proved beneficial for older patients, whereas adding etoposide to the biweekly regimen (CHOEP [cyclophosphamide, doxorubicin, etoposide, vincristine, and prednisone]) improved outcomes for younger patients in the same trial.10,11 However, these trials were for aggressive NHLs and therefore dominated by B-cell subtypes.
In younger patients, the French Groupe d’Etude des Lymphomes de l’Adultes showed that dose-intensified doxorubicin, cyclophosphamide, vindesine, bleomycin, and prednisone (ACVBP) was superior to abbreviated chemotherapy consolidated by early high-dose chemotherapy and autologous stem cell transplant (ASCT) again in a mixed population of aggressive B-cell and T-cell NHLs.12 Anecdotal reports have discussed the use of a hyper-CVAD (fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) regimen in advanced cases of ENKTCL.13
Phase 2 trials have shown a benefit to ASCT in first remission with 3- to 5-year progression-free survival (PFS) of 24% to 53%.14,15 However, most of the patients remain refractory to primary induction therapy or are not medically fit enough for ASCT. For these patients, median OS is less than 6 months.16
As optimal therapy remains undefined, new approaches to manage T-cell and NK-cell lymphomas are clearly needed. It is hoped that therapeutic targeting of driver oncogenic pathways and genetic mutations elucidated from GEP and mutational analysis will result in better clinical outcomes in the future.
PTCL Subtypes With Distinct Biological Features
Adult T-cell leukemia/lymphoma
Adult T-cell leukemia/lymphoma is a rare T-cell malignancy that occurs exclusively in patients infected with human T-lymphotropic virus type I (HTLV-1). The virus is primarily transmitted through breastfeeding, although blood borne transmission can also occur.17 HTLV-1 infection alone is insufficient to cause ATLL, with only approximately 3% to 5% of seropositive patients progressing to ATLL, often after a latency period of many years. It is recognized that in addition to HTLV-1 infection, further oncogenic mutations are required for the development of ATLL.18
Four distinct subtypes of ATLL are recognized, as per the Shimoyama classification, which are important when considering appropriate treatments.19–21 Smoldering and chronic forms, which are relatively indolent at diagnosis, can often be managed with a “watch and wait” approach. Lymphomatous and acute (leukemic) forms, which are more aggressive at presentation, typically carry a very poor prognosis and require treatment on diagnosis. Progression of indolent forms to the more aggressive types frequently occurs, although this can take several years.
There is no clear consensus regarding the optimal treatment of ATLL. It is recognized that the lymphomatous and acute forms are aggressive with short median OS in the absence of treatment. Even with treatment, refractory disease is common and remission duration is often short.19,21 Treatment strategies have been designed around multiagent chemotherapy with a clear role for consolidative stem cell allograft in appropriately selected patients where a reasonable response following induction treatment can be attained. In addition, strategies incorporating antiviral therapy have also been tested. More recently, molecular approaches, including the anti-C-C chemokine receptor type 4 (CCR4) antibody mogamulizumab, have been shown to have antitumor activity in the relapsed/refractory (r/r) setting.
Indolent disease
Patients with smoldering and chronic favorable presentations of ATLL often do not require multiagent chemotherapy at diagnosis. This is based on data which show that median OS is around 4 years post diagnosis.22 More recently, a retrospective series has shown a 100% 5-year OS in this group of patients with antiviral therapy alone and this is described in further detail below.23
Aggressive disease
Patients with lymphomatous and acute presentations of ATLL are typically unwell at diagnosis (hypercalcemia, B symptoms, and organ infiltration) and have been treated with multiagent chemotherapy up front. The efficacy of CHOP-like regimes for this group of patients has been demonstrated in a number of retrospective analyses of PTCL/ATLL studies as well as control arms of randomized trials.24–26
More recently, a Japanese trial has demonstrated the efficacy of VCAP-AMP-VECP (vincristine, cyclophosphamide, doxorubicin, and prednisolone; doxorubicin, ranimustine, and prednisolone; vindesine, etoposide, carboplatin, and prednisolone chemotherapy in this group of patients, albeit at the expense of considerably increased treatment toxicity.26
Mogamulizumab
C-C chemokine receptor type 4 is widely expressed on ATLL neoplastic cells as well as other helper T cell–derived lymphomas. The defucosylated, anti-CCR4 humanized monoclonal antibody mogamulizumab has been approved in Japan for the treatment of r/r ATLL, cutaneous T-cell lymphoma, and PTCL-NOS on the basis of phase 1/2 studies which demonstrated efficacy and acceptable toxicity.27 Removal of fucosyl groups provides the antibody with greater antibody-dependent cellular cytotoxicity. In the largest randomized study of mogamulizumab so far conducted, it has demonstrated efficacy in r/r ATLL (overall response rate [ORR] 23.4% by independent assessor (11/47), with PFS of 3 months, compared with 0/24 for investigator choice treatment).28 An earlier nonrandomized phase 2 study demonstrated 50% ORR in the r/r setting in a smaller group (28) of patients.29 This provides a clear example where an understanding of disease biology (in this case, CCR4 expression) has led to advances in treatment.
Antiviral therapy
Antiviral agents have been widely used as part of treatment regimens for ATLL, although without a plausible biological explanation for their efficacy. HTLV-1 infection in ATLL is believed to be in a latent phase, and it is unclear, therefore, how such an approach may potentially improve outcomes. Typically, the antiviral agent zidovudine plus interferon alfa have been used.30
The most comprehensive evaluation of antiviral agents in ATLL has been a retrospective meta-analysis involving 245 patients in France, United Kingdom, United States, and Martinique.23 This study included patients with smoldering, chronic, lymphomatous, and acute ATLL treated with antiviral therapy alone and antiviral therapy plus chemotherapy. For patients with indolent presentations of ATLL, antiviral therapy demonstrated apparent efficacy with 100% 5-year OS compared with 42% in patients treated with chemotherapy or chemotherapy plus antiviral therapy. Similarly, patients with acute (leukemic) ATLL antiviral therapy alone improved 5-year OS from 10% to 28% over treatments including chemotherapy. This benefit was not, however, noted for patients with lymphomatous ATLL where antiviral therapy alone resulted in considerably poorer 5-year OS compared with chemotherapy-based treatments (0% vs 18%). These data, while interesting, are derived from a retrospective study and should be translated cautiously into clinical practice, preferably in the context of prospective studies.
ENKTCL nasal type
Extranodal NK/T-cell lymphoma nasal type is an aggressive T-cell lymphoma, which, although rare, is markedly more prevalent in Asia and Central and South America than in Western countries.31 It shows a 2:1 male:female predominance and in most cases is derived from NK cells, although may occasionally be T-cell derived—this can be confirmed by gene rearrangement studies. Typically, it presents with extranodal limited-stage disease affecting the oropharyngeal tissues and nasal/paranasal sites, although more disseminated presentations, particularly affecting gastrointestinal tract, skin, spleen, adrenal gland, and testes, do occur. Infection of malignant cells by EBV can frequently be demonstrated.32 It is an aggressive disease with a poor 5-year OS.
Extranodal NK/T-cell lymphoma frequently demonstrates chemoresistance, and refractory disease or early relapses are common. This has led to the search for alternative treatment strategies that demonstrate greater efficacy in this setting. One approach has been the use of l-asparaginase to augment multiagent chemotherapy.
Nasal disease
The treatment of ENKTCL is dependent on the stage of the disease at presentation. Stage I or contiguous stage II disease can be treated with radiotherapy +/− multiagent chemotherapy with curative intent. More advanced disease carries a markedly shorter median duration of remission and is generally treated with multiagent chemotherapy. For limited-stage disease, adjuvant chemotherapy postradiotherapy probably does lead to improved OS. This has been demonstrated in retrospective studies. For example, a retrospective series of 136 cases of ENKTCL showed survival benefit for nasal cases receiving radiotherapy in addition to chemotherapy.33 Combined modality therapy has therefore become standard of care in this setting.34
Extranasal disease
For disseminated disease, there is no consensus as to the most effective form of multiagent chemotherapy and no relevant randomized trials exist. Extranodal NK/T-cell lymphoma frequently expresses high levels of P-glycoprotein that gives it a multidrug resistance phenotype.35 This may explain frequent resistance to anthracycline-based chemotherapy, although this remains unclear.36
Most recent studies have focused on the role of l-asparaginase, a drug which has activity that is not affected by P-glycoprotein expression due to its extracellular mode of action. It has demonstrated efficacy in newly diagnosed and relapsed disease.37,38 This drug was incorporated into the LVP (l-asparaginase, vincristine, and prednisolone) regimen with radiotherapy where it produced an 81% complete response (CR) in newly diagnosed disease.39 The 5-year survival data from this study have now been published and show an OS and PFS both of 64%.40 Similarly, GELOX (gemcitabine, l-asparaginase, and oxaliplatin) elicited a 74.1% CR in a prospective phase I/II study with a 2-year PFS of 86%.41 A subsequent retrospective study comparing GELOX with EPOCH (etoposide, vincristine, doxorubicin, cyclophosphamide, and prednisone), which lacks asparaginase, showed a superior CR, ORR, and PFS for GELOX.42
A more intense asparaginase-containing chemotherapy regimen SMILE (dexamethasone, methotrexate, ifosfamide, l-asparaginase, and etoposide) has also been assessed in newly diagnosed and r/r disease giving an OR response rate of 78%, with 47% PFS at a median follow-up of 31 months, which was similar for newly diagnosed and r/r disease, albeit at the expense of significant toxicities.43
Most recently, a Chinese randomized controlled study including 42 patients has compared safety and efficacy of DDGP (dexamethasone, cisplatin, gemcitabine, and polyethylene glycol-l-asparaginase) and SMILE regimens for newly diagnosed advanced NK/T-cell lymphoma. This was a randomized controlled, multicenter, and open-label clinical trial.44 This study demonstrated significant improvements in PFS, OS (74% compared with 45% for 2-year OS), and tolerability from DDGP compared with SMILE.
Anaplastic large cell lymphoma
In common with many other types of T-cell NHL, the prognosis of ALCL is often poor. This is, however, dependent on whether malignant cells express ALK (described above), which is recognized to confer a significantly better prognosis. In general, ALK-positive tumors affect children and younger adults, and this may account for the improved prognosis in this group. In keeping with this, advanced-stage ALK-positive disease, particularly when diagnosed in older patients, carries a prognosis that is comparable with ALK-negative disease.
Multiagent chemotherapy, typically anthracycline based, is the mainstay of treatment of ALCL and this is recognized in national guidelines.34,45 CHOP is most frequently used. For limited-stage disease, abbreviated chemotherapy plus involved field radiotherapy can be used. For more advanced-stage disease, 6 cycles of CHOP are preferred. Typically, consolidation, usually with autologous stem cell transplant, is recommended for appropriately selected patients with high-risk disease.
In addition to the above, 2 specific treatments for ALCL are being investigated, based on the biology of ALCL—brentuximab vedotin, which is specific for CD30 commonly expressed on ALCL malignant cells and ALK inhibitors used in ALK-positive ALCL.
Brentuximab vedotin
Brentuximab vedotin is an antibody-drug conjugate (ADC) with specificity for CD30. The chemotherapy moiety is monomethyl auristatin E which is an antitubulin agent. It has demonstrated efficacy in classical Hodgkin lymphoma as well as ALCL. The original phase 2 study in r/r ALCL showed that it could induce CRs in more than half of the patients when used as monotherapy. The ORR rate was 86%. The median durations of ORR and CR were 12.6 and 13.2 months, respectively.46 ECHELON-2, a study comparing brentuximab vedotin + CHP with CHOP in newly diagnosed ALCL remains ongoing. Brentuximab is currently used in r/r disease, often as a bridge to consolidation with stem cell transplant.
ALK inhibitors
Crizotinib and ceritinib are inhibitors of ALK and have also been used in a limited fashion, in ALK-positive ALCL. Crizotinib has been used as monotherapy in a small series of patients with relapsed ALK-positive ALCL who were refractory to chemotherapy. 10 of 11 (90.9%) of patients responded by entering at least a partial remission, and there is evidence that these are durable remissions with 63.7% PFS at 2 years.47
In a phase 1 ceritinib trial for patients with advanced ALK-positive tumors, 3 of 304 patients had ALK-positive ALCL. All 3 patients achieved a response (2 CRs, 1 PR) with ongoing remissions of at least 20 months.48 Clearly, these data relate to very small groups of patients, but they do demonstrate potential therapies for the future.
Angioimmunoblastic T-cell lymphoma
Angioimmunoblastic T-cell lymphoma comprises approximately 19% of cases of T-cell NHL.49 Originally, it was defined on the basis of typical clinical, morphological, and immunophenotypic characteristics. More recently, the recognition of recurrent genetic mutations and a characteristic GEP has improved diagnostic accuracy. It has also led to a broadening and redefinition of this subgroup to include some cases that share these genetic abnormalities but that would previously have been classified as PTCL-NOS. Furthermore, it has highlighted key pathways that may be involved in the pathogenesis of AITL and are potential therapeutic targets.
Clinical features
Patients with AITL usually present with acute onset systemic symptoms, although, more rarely, they present with asymptomatic lymphadenopathy. About 90% of patients present with advanced (stage III-IV) disease. B symptoms, rash, ascites, and pleural effusions and autoimmune phenomena such as polyarthritis and positive direct antiglobulin test are common.
Morphological features
Involved lymph nodes show complete or partial effacement of the normal architecture by a paracortical infiltrate. This infiltrate consists of malignant T cells and numerous inflammatory cells, including plasma cells, histiocytes, eosinophils, B-immunoblasts, which are often positive for EBV. There is prominent neovascularization with high endothelial venules with thickened or hyalinized walls. Expanded networks of follicular dendritic cells (FDCs), best appreciated with immunostains for dendritic cell markers (eg, CD21, CD23, and CD35), surround these abnormal blood vessels.
Immunophenotypic characteristics
Angioimmunoblastic T-cell lymphoma cells express pan T-cell antigens (CD4+, CD3+, CD2+ with variable expression of CD5 and CD7) as well as cell surface markers that are usually expressed on follicular helper T (TFH) cells: CD10, BCl6, PD-1, CXCL13, and ICOS.
Diagnostic uncertainty may still arise, however, because of the variable expression of these cell surface markers on other subtypes of PTCL and the similarity of the background milieu to an inflammatory state. T-cell receptor (TCR) rearrangement studies may clarify this to some extent but are hampered by the relatively low burden of neoplastic cells within the malignant lesion.
Gene expression profiling and TFH derivation
Gene expression profile studies provide further diagnostic information and have confirmed the TFH cell as the likely cell of origin of AITL. Gene expression profile analyses of an array of T-cell NHL samples and cell lines have shown that AITL cases have a signature that is distinct from other T-cell NHL subtypes.50,51 The AITL signature has a strong imprint from the tumor microenvironment (overexpression of B-cell–related and FDC-related genes, chemokines, and genes implicated in angiogenesis), as well as overexpression of genes characteristic of normal TFH cells, for example, CXC13, BCL6, PD-1, CD40L, and NFATC1. Oncogenic pathways including nuclear factor κB (NF-κB), interleukin (IL)-6, and transforming growth factor β were also identified.
Normal TFH cells migrate to B-cell follicles and provide help to B cells for high-affinity antibody production.52 The close interaction between TFH and B cells may explain some of the clinical characteristics of AITL. For example, clonal IGHV rearrangements are seen in approximately 15% of AITL cases, and hypergammaglobulinemia and autoimmune phenomena are relatively common. There is also a recognized increased risk of developing diffuse large B-cell lymphoma.53 Epstein-Barr virus–positive polyclonal B cells are highly prevalent in AITL (95% cases are EBV-encoded small RNA positive). Most of the malignant T cells are, however, negative. This suggests against a primary role for EBV in AITL tumorigenesis and instead indicates that EBV-infected B cells may take advantage of the permissive TFH-driven interactions.54
A proportion of cases that have been reported morphologically as PTCL-NOS, and lack some of the phenotypic hallmarks of AITL, share this TFH—such as GEP signature, 20% in 1 case series.51 This has led to their unification in the latest WHO classification system under a common subtype of PTCL with a TFH phenotype.4 This group comprises AITL, TFH lymphoma, and nodal PTCL with TFH phenotype. The WHO diagnostic criteria specify that they should express at least 2 to 3 TFH-related markers, including PD-1, CD10, BCL6, CXCl13, ICOS, SAP, and CCR5. However, although AITL, TFH lymphoma, and nodal PTCL with TFH phenotype share a common GEP, they are maintained as separate disease entities because of their distinct clinical and pathological features.
Recurrent genetic mutations
In addition to this characteristic GEP profile, AITL and PTCLs with a TFH phenotype share unifying recurrent genetic mutations. Using exome and transcriptome sequencing of AITL tumor samples, a specific RHOA mutation, encoding a p.Gly17Val substitution, was identified in 55.3% of AITL cases.55 It was also present in a lower proportion of PTCL-NOS (7.7%) and NK/T-cell lymphoma (15%) but absent in B-cell lymphomas. RHOA encodes a small guanosine triphosphatase protein in the RAS family: the p.Gly17Val mutation results in a loss of function. The group hypothesizes that this mutation is key to AITL tumorigenesis. It was shown to increase proliferation in cell lines, possibly by altering AKT kinase phosphorylation and signaling, downstream of the TCR.
IDH2 mutations are another potential driver mutation and have been identified in 20% to 45% of AITL cases.56 They were absent in other B-cell and T-cell lymphomas. IDH2 mutations are thought to alter enzymatic function, leading to accumulation of oncometabolites that affect hypoxia signaling and histone and DNA methylation.
TET2 mutations are also frequently identified in AITL (47%) as well as PTCL-NOS, particularly those expressing TFH cell markers (58%).57 These are mainly frameshift and nonsense mutations similar to those seen in myeloid malignancies. However, unlike in myeloid malignancies in AITL, IDH2 and TET2 mutations may coincide. DNMT3A mutations have also been detected in 33% of patients and seem to coincide with TET2 mutations.58 DNMT3A and TET2 are involved in DNA and histone methylation, suggesting potential epigenetic pathways for tumorigenesis in AITL.
Several other less frequent but recurrent mutations identified in this group, including mutations in genes involved in DNA damage response (ATM), escape from immune surveillance mechanisms (B2M and CD58) and TCR signaling through SRC kinases (FYN).59
Peripheral T-cell lymphoma, not otherwise specified
Peripheral T-cell lymphoma, not otherwise specified is a heterogeneous group of predominantly nodal T-cell lymphomas that do not meet the immunophenotypic, morphological, or molecular criteria for other subgroups. Currently, this group comprises approximately 30% to 50% of new PTCL diagnoses.60 It is likely that this group comprises a conglomerate of further subtypes that have yet to be defined. Gene expression profile is beginning to lend evidence to this theory.
Clinical features
Approximately, 38% of patients have nodal disease alone, 49% have nodal and extranodal disease, and 13% have extranodal disease only. Circulating lymphoma cells may be seen, but leukemic presentation is rare. Some cases are associated with eosinophilia or hemophagocytosis.
Morphological features
Morphological heterogeneity within PTCL-NOS has long been recognized: diffuse small cleaved, mixed, large cell, immunoblastic subtypes are recognized.61 There is usually effacement of the normal architecture by sheets of atypical lymphoid cells in a diffuse or paracortical distribution. There are typically a variety of admixed cells including eosinophils, plasma cells, B cells, and histiocytes.
Immunophenotypic characteristics
There is no characteristic immunophenotype for PTCL-NOS. T-cell–associated antigens, including CD2, CD3, CD5, and CD7, are variably expressed. Most of the cases express CD4 only but some tumors may be CD4−/CD8−, CD4−/CD8+, or CD4+/CD8+. T-cell receptor gene rearrangements may commonly be detected, although this has limitations as a diagnostic tool, as discussed previously.
GEP of PTCL-NOS
One large study that performed GEP on 372 cases of T cell-NHL was able to reclassify 37% of PTCL-NOS into other subgroups, including AITL, ALCL, and NK-cell NHLs.60 This still left 32% of cases in the PTCL-NOS group. These same GEP studies identified 2 meaningful subtypes within this PTCL-NOS group, with prognostic, and potentially therapeutic, relevance. About 33% of cases where categorized by high expression levels of GATA 3 and its target genes, CCR4, CXCR7, and IL18RA. There was enrichment of gene proliferation signals such as MYC and MTOR. This subtype was associated with worse outcomes, 19% OS at 5 years. The other subtype showed high expression of TBX21 and EOMES and their target genes, CXCR3, IL2RB, CCL3, and interferon gamma. This subtype showed enrichment of interferon gamma and NF-κB–induced signatures and was associated with a more favorable clinical prognosis, 38% 5-year OS. The 2 subtypes could be distinguished with 80% confidence according to these differences in GEP. No significant differences regarding immunophenotypic characteristics, specifically CD4 or CD8 positivity, or clinical characteristics could be identified.
New therapies in clinical trials
A number of new agents have been used in T-cell NHL in clinical trials with some promising initial results. These include conventional chemotherapy agents, epigenetic modifying agents, immune modulators, kinase inhibitors, and monoclonal antibodies.
Chemotherapy agents
In the phase 2 PROPEL (Pralatrexate in Patients With Relapsed or Refractory Peripheral T-cell Lymphoma) trial, which included 115 patients with r/r T-cell NHL who had received ≥1 line of chemotherapy, the antifolate drug, pralatrexate, had a 29% ORR and 11% CR rate, with median duration of response of 10.1 months.62 Cell of origin may be important even in response to conventional chemotherapy agents: response rates in AITL were 8% versus 31% in PTCL-NOS. In the phase 2 BENTLY (Bendamustine in Patients With Refractory or Relapsed T-cell Lymphoma) trial, which included 60 patients with r/r PTCL treated with bendamustine, ORR was 50% with a CR of 28%. Progression-free survival was relatively short at 3.6 months.63
Monoclonal antibodies and antibody conjugates
Brentuximab vedotin has shown benefit in CD30+ ALCL: ORR 86% with 57% CR and median PFS of 12.6 months.46 It was also effective, although slightly less so, in CD30+ PTCL-NOS and AITL: ORR 43%, PFS: 6.7 months.64 Interestingly, response appeared to be independent of CD30 expression levels. Similarly, response rate appeared to be independent of CD25 (IL2 receptor) expression level in a trial of the IL-2-diphtheria toxin fusion protein, denileukin.65 Overall response rate was 61.5% in CD25-positive tumors and 45.5% in CD25-negative tumors, with a median PFS of 6 months. The anti-CD52 monoclonal antibody, alemtuzumab, has previously been tested in r/r PTCL and had a 36% ORR but with short lived responses and significant toxicity, in terms of infective complications.66
Immune modulators
Lenalidomide is an immune modulator and has antiangiogenic activity. It has some single agent activity in r/r T-cell NHL: 26% ORR, median PFS: 4 months.67 Response rates were slightly higher in PTCL-NOS (43%) and AITL (33%), which might fit with the importance of interactions with the microenvironment in T-cell NHL subtypes with a TFH phenotype. The immune checkpoint inhibitors are being used with impressive results across an array of solid organ malignancies and B-cell lymphomas. They have not specifically tested in T-cell NHL to date. However, the expression of PD-1 on TFH cells is associated with high-affinity antibody responses.52 Abrogation of this cross talk by an immune checkpoint inhibitor may have therapeutic value.
Histone deacetylase inhibitors
The high frequency of TET2 mutations in PTCL-NOS and AITL suggests that aberrant epigenetic modifications may be important in pathogenesis. Histone deacetylase inhibitors, such as romidepsin, are therefore an exciting area of investigation. In particular, patients who respond to these agents tend to have more durable responses than to other investigational agents. A phase 2 clinical trial of romidepsin in r/r PTCL led to romidepsin’s approval by the Food and Drug Administration (it remains unlicensed in the United Kingdom and Europe). Overall response rate was 25%, including 15% of patients with CR.68 Median duration of response was 17 months.
Drugs targeting oncogenic pathways
Gene expression profile studies have shown increased activation of oncogenic pathways in association with specific T-cell NHL subtypes. These oncogenic pathways are also potential drug targets. For example, increased activity of NF-κB has been demonstrated in AITL and some PTCL-NOS subtypes. The NF-κB inhibitor bortezomib may therefore be of therapeutic benefit. In a small phase 2 trial of 15 patients with mycosis fungoides or PTCL with limited skin involvement, ORR was 67% with response duration of 7 to 14 months.69 Gene expression profile on ENKTCL samples demonstrated consistent enrichment of several distinct gene signatures, including upregulation of the NOTCH and AURKA signaling pathways. AURKA is involved in several well-characterized oncogenic pathways, including upregulation of C-MYC and inhibition of TP53. Molecular inhibitors of the NOTCH and AURKA pathways induced cell cycle arrest and apoptosis in ENKTCL lines, identifying these as promising candidates for therapeutic trials in ENKTCL lymphoma, a subtype that has historically been associated with very poor outcomes.70
Kinase inhibitors
Activating mutations of kinases can be seen in various T-cell NHL subtypes. Small-molecule inhibitors of these kinases, many of which are already available for other malignancies, may therefore be effective. The therapeutic potential of crizotinib in ALK-positive r/r ALCL has been discussed above. There is a high frequency of STAT3 mutations in hepatosplenic T-cell lymphoma49 and STAT5 B mutations in T-cell prolymphocytic leukemia,71 which may be targetable by existing JAK2 inhibitors. Platelet-derived growth factor receptor α is overexpressed in PTCL-NOS, raising the possibility of efficacy for imatinib.72 Dasatinib has been demonstrated to impair the growth of PTCL-NOS cell lines expressing the FYN kinase gene mutation, which is present in some PTCL-NOS tumors.59
Conclusions
Much work has been done in recent years to classify T-cell NHL into biologically and clinically relevant subtypes. An increased understanding of the biology of ATLL has led to specific therapies such as antiviral agents and mogamulizumab. l-asparaginase has improved the treatment of ENKTCL and owes its introduction in part to an understanding of mechanisms of drug resistance in this condition. The development of an anti-CD30 ADC has revolutionized the outcome of patients with r/r ACLCL and ALK inhibitors appear active in the small number of cases in which they have been used. Most of the PTCL cases in North America and Europe, however, fall into the bracket of PTCL-NOS and AITL. Emerging evidence suggests that a more informative way of thinking about these subtypes is to reclassify them into TFH-derived and non–TFH-derived lymphomas, as reflected by the revised WHO classification system. Targeting driver mutations in these different groups offers hope of more personalized and effective treatments. Existing medication, such as histone deacetylase inhibitors, may be more effective in the TFH-derived subgroup. The challenge to the international community is to develop biologically driven trials in subtypes of patients with T-cell NHL using a sufficient number of participants to show statistically meaningful results.
Acknowledgments
G.P.C. acknowledges support from the Blood Theme of the NIHR Oxford Biomedical Research Centre.
Footnotes
Peer review:Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 166 words, excluding any confidential comments to the academic editor.
Funding:The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: CATH, SS, JABB, and GPC wrote the manuscript. GPC planned the overview of the manuscript. CATH and GPC reviewed and edited the manuscript. All authors reviewed and approved the final manuscript.
Disclosures and Ethics: As a requirement of publication, the authors have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
- 1. Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Geneva, Switzerland: World Health Organization; 2008. [Google Scholar]
- 2. Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124–4130. [DOI] [PubMed] [Google Scholar]
- 3. Armitage JO. The aggressive peripheral T-cell lymphomas: 2015. Am J Hematol. 2015;90:665–673. [DOI] [PubMed] [Google Scholar]
- 4. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abramson JS, Feldman T, Kroll-Desrosiers AR, et al. Peripheral T-cell lymphomas in a large US multicenter cohort: prognostication in the modern era including impact of frontline therapy. Ann Oncol. 2014;25:2211–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117:6756–6767. [DOI] [PubMed] [Google Scholar]
- 7. Moskowitz AJ, Lunning MA, Horwitz SM. How I treat the peripheral T-cell lymphomas. Blood. 2014;123:2636–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abouyabis AN, Shenoy PJ, Sinha R, Flowers CR, Lechowicz MJ. A systematic review and meta-analysis of front-line anthracycline-based chemotherapy regimens for peripheral T-cell lymphoma. ISRN Hematol. 2011;2011:623924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmitz N, Trumper L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116:3418–3425. [DOI] [PubMed] [Google Scholar]
- 10. Pfreundschuh M, Trumper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of elderly patients with aggressive lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood. 2004;104:634–641. [DOI] [PubMed] [Google Scholar]
- 11. Pfreundschuh M, Trumper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of young patients with good-prognosis (normal LDH) aggressive lymphomas: results of the NHL-B1 trial of the DSHNHL. Blood. 2004;104:626–633. [DOI] [PubMed] [Google Scholar]
- 12. Gisselbrecht C, Lepage E, Molina T, et al. Shortened first-line high-dose chemotherapy for patients with poor-prognosis aggressive lymphoma. J Clin Oncol. 2002;20:2472–2479. [DOI] [PubMed] [Google Scholar]
- 13. Shapiro M, Wasik MA, Junkins-Hopkins JM, et al. Complete remission in advanced blastic NK-cell lymphoma/leukemia in elderly patients using the hyper-CVAD regimen. Am J Hematol. 2003;74:46–51. [DOI] [PubMed] [Google Scholar]
- 14. d’Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30:3093–3099. [DOI] [PubMed] [Google Scholar]
- 15. Kewalramani T, Zelenetz AD, Teruya-Feldstein J, et al. Autologous transplantation for relapsed or primary refractory peripheral T-cell lymphoma. Br J Haematol. 2006;134:202–207. [DOI] [PubMed] [Google Scholar]
- 16. Mak V, Hamm J, Chhanabhai M, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J Clin Oncol. 2013;31:1970–1976. [DOI] [PubMed] [Google Scholar]
- 17. Mahieux R, Gessain A. Adult T-cell leukemia/lymphoma and HTLV-1. Curr Hematol Malig Rep. 2007;2:257–264. [DOI] [PubMed] [Google Scholar]
- 18. Ohshima K. Molecular pathology of adult T-cell leukemia/lymphoma. Oncology. 2015;89:7–15. [DOI] [PubMed] [Google Scholar]
- 19. Kato K, Akashi K. Recent advances in therapeutic approaches for adult T-cell leukemia/lymphoma. Viruses. 2015;7:6604–6612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–87). Br J Haematol. 1991;79:428–437. [DOI] [PubMed] [Google Scholar]
- 21. Yared JA, Kimball AS. Optimizing management of patients with adult T cell leukemia-lymphoma. Cancers (Basel). 2015;7:2318–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takasaki Y, Iwanaga M, Imaizumi Y, et al. Long-term study of indolent adult T-cell leukemia-lymphoma. Blood. 2010;115:4337–4343. [DOI] [PubMed] [Google Scholar]
- 23. Bazarbachi A, Plumelle Y, Carlos Ramos J, et al. Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol. 2010;28:4177–4183. [DOI] [PubMed] [Google Scholar]
- 24. Kawano N, Yoshida S, Kuriyama T, et al. Clinical features and treatment outcomes of 81 patients with aggressive type adult T-cell leukemia-lymphoma at a single institution over a 7-year period (2006–2012). Intern Med. 2015;54:1489–1498. [DOI] [PubMed] [Google Scholar]
- 25. Nishi Y, Fukushima T, Nomura S, et al. Characterization of patients with aggressive adult T-cell leukemia-lymphoma in Okinawa, Japan: a retrospective analysis of a large cohort. Int J Hematol. 2016;104:468–475. [DOI] [PubMed] [Google Scholar]
- 26. Tsukasaki K, Utsunomiya A, Fukuda H, et al. VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J Clin Oncol. 2007;25:5458–5464. [DOI] [PubMed] [Google Scholar]
- 27. Remer M, Al-Shamkhani A, Glennie M, et al. Mogamulizumab and the treatment of CCR4-positive T-cell lymphomas. Immunotherapy. 2014;6:1187–1206. [DOI] [PubMed] [Google Scholar]
- 28. Phillips A, Fields P, Hermine O, et al. A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator’s choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T-cell leukemia-lymphoma (ATL). In: 2016 ASCO Annual Meeting Oral Abstracts http://meetinglibrary.asco.org/content/167138-176. Published 2016. [Google Scholar]
- 29. Ishida T, Joh T, Uike N, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30:837–842. [DOI] [PubMed] [Google Scholar]
- 30. Fields PA, Taylor GP. “Antivirals” in the treatment of adult T cell leukaemia- lymphoma (ATLL). Curr Hematol Malig Rep. 2012;7:267–275. [DOI] [PubMed] [Google Scholar]
- 31. Haverkos BM, Pan Z, Gru AA, et al. Extranodal NK/T cell lymphoma, nasal type (ENKTL-NT): an update on epidemiology, clinical presentation, and natural history in North American and European cases. Curr Hematol Malig Rep. 2016;11:514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gru AA, Haverkos BH, Freud AG, et al. The Epstein-Barr virus (EBV) in T cell and NK cell lymphomas: time for a reassessment. Curr Hematol Malig Rep. 2015;10:456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Au WY, Weisenburger DD, Intragumtornchai T, et al. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood. 2009;113:3931–3937. [DOI] [PubMed] [Google Scholar]
- 34. Dearden CE, Johnson R, Pettengell R, et al. Guidelines for the management of mature T-cell and NK-cell neoplasms (excluding cutaneous T-cell lymphoma). Br J Haematol. 2011;153:451–485. [DOI] [PubMed] [Google Scholar]
- 35. Huang WT, Huang CC, Weng SW, Eng HL. Expression of the multidrug resistance protein MRP and the lung-resistance protein LRP in nasal NK/T cell lymphoma: further exploring the role of P53 and WT1 gene. Pathology. 2009;41:127–132. [DOI] [PubMed] [Google Scholar]
- 36. Kim GE, Yang WI, Lee SW, et al. Lack of correlation between P-glycoprotein and chemotherapy resistance in nasal NK/T-cell lymphomas. Leuk Lymphoma. 2004;45:1857–1864. [DOI] [PubMed] [Google Scholar]
- 37. Yong W. Clinical study of l-asparaginase in the treatment of extranodal NK/T-cell lymphoma, nasal type. Hematol Oncol. 2016;34:61–68. [DOI] [PubMed] [Google Scholar]
- 38. Yong W, Zheng W, Zhu J, et al. L-asparaginase in the treatment of refractory and relapsed extranodal NK/T-cell lymphoma, nasal type. Ann Hematol. 2009;88:647–652. [DOI] [PubMed] [Google Scholar]
- 39. Jiang M, Zhang H, Jiang Y, et al. Phase 2 trial of “sandwich” L-asparaginase, vincristine, and prednisone chemotherapy with radiotherapy in newly diagnosed, stage IE to IIE, nasal type, extranodal natural killer/T-cell lymphoma. Cancer. 2012;118:3294–3301. [DOI] [PubMed] [Google Scholar]
- 40. Zhang L, Jiang M, Xie L, et al. Five-year analysis from phase 2 trial of “sandwich” chemoradiotherapy in newly diagnosed, stage IE to IIE, nasal type, extranodal natural killer/T-cell lymphoma. Cancer Med. 2016;5:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang L, Wang ZH, Chen XQ, et al. First-line combination of gemcitabine, oxaliplatin, and L-asparaginase (GELOX) followed by involved-field radiation therapy for patients with stage IE/IIE extranodal natural killer/T-cell lymphoma. Cancer. 2013;119:348–355. [DOI] [PubMed] [Google Scholar]
- 42. Wang H, Wuxiao ZJ, Zhu J, et al. Comparison of gemcitabine, oxaliplatin and L-asparaginase and etoposide, vincristine, doxorubicin, cyclophosphamide and prednisone as first-line chemotherapy in patients with stage IE to IIE extranodal natural killer/T-cell lymphoma: a multicenter retrospective study. Leuk Lymphoma. 2015;56:971–977. [DOI] [PubMed] [Google Scholar]
- 43. Kwong YL, Kim WS, Lim ST, et al. SMILE for natural killer/T-cell lymphoma: analysis of safety and efficacy from the Asia Lymphoma Study Group. Blood. 2012;120:2973–2980. [DOI] [PubMed] [Google Scholar]
- 44. Li X, Cui Y, Sun Z, et al. DDGP versus SMILE in newly diagnosed advanced natural killer/T-cell lymphoma: a randomized controlled, multicenter, open-label study in China. Clin Cancer Res. 2016;22:5223–5228. [DOI] [PubMed] [Google Scholar]
- 45. Horwitz SM, Zelenetz AD, Gordon LI, et al. NCCN guidelines insights: non-Hodgkin’s lymphomas, version 3.2016. J Natl Compr Canc Netw. 2016;14:1067–1079. [DOI] [PubMed] [Google Scholar]
- 46. Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30:2190–2196. [DOI] [PubMed] [Google Scholar]
- 47. Gambacorti Passerini C, Farina F, Stasia A, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Inst. 2014;106:djt378. [DOI] [PubMed] [Google Scholar]
- 48. Richly H, Kim TM, Schuler M, et al. Ceritinib in patients with advanced anaplastic lymphoma kinase–rearranged anaplastic large-cell lymphoma. Blood. 2015;126:1257–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iqbal J, Wilcox R, Naushad H, et al. Genomic signatures in T-cell lymphoma: how can these improve precision in diagnosis and inform prognosis? Blood Rev. 2016;30:89–100. [DOI] [PubMed] [Google Scholar]
- 50. de Leval L, Rickman DS, Thielen C, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109:4952–4963. [DOI] [PubMed] [Google Scholar]
- 51. Iqbal J, Weisenburger DD, Greiner TC, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood. 2010;115:1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ahearne MJ, Allchin RL, Fox CP, Wagner SD. Follicular helper T-cells: expanding roles in T-cell lymphoma and targets for treatment. Br J Haematol. 2014;166:326–335. [DOI] [PubMed] [Google Scholar]
- 53. Smith JL, Hodges E, Quin CT, McCarthy KP, Wright DH. Frequent T and B cell oligoclones in histologically and immunophenotypically characterized angioimmunoblastic lymphadenopathy. Am J Pathol. 2000;156:661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bräuninger A, Spieker T, Willenbrock K, et al. Survival and clonal expansion of mutating “forbidden” (immunoglobulin receptor-deficient) Epstein-Barr virus-infected b cells in angioimmunoblastic T cell lymphoma. J Exp Med. 2001;194:927–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yoo HY, Sung MK, Lee SH, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:371–375. [DOI] [PubMed] [Google Scholar]
- 56. Cairns RA, Iqbal J, Lemonnier F, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lemonnier F, Couronne L, Parrens M, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120:1466–1469. [DOI] [PubMed] [Google Scholar]
- 58. Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Palomero T, Couronne L, Khiabanian H, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123:2915–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rudiger T, Weisenburger DD, Anderson JR, et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkin’s Lymphoma Classification Project. Ann Oncol. 2002;13:140–149. [DOI] [PubMed] [Google Scholar]
- 62. O’Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29:1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Damaj G, Gressin R, Bouabdallah K, et al. Results from a prospective, open-label, phase II trial of bendamustine in refractory or relapsed T-cell lymphomas: the BENTLY trial. J Clin Oncol. 2013;31:104–110. [DOI] [PubMed] [Google Scholar]
- 64. Horwitz SM, Advani RH, Bartlett NL, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood. 2014;123:3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dang NH, Pro B, Hagemeister FB, et al. Phase II trial of denileukin diftitox for relapsed/refractory T-cell non-Hodgkin lymphoma. Br J Haematol. 2007;136:439–447. [DOI] [PubMed] [Google Scholar]
- 66. Enblad G, Hagberg H, Erlanson M, et al. A pilot study of alemtuzumab (anti-CD52 monoclonal antibody) therapy for patients with relapsed or chemotherapy-refractory peripheral T-cell lymphomas. Blood. 2004;103:2920–2924. [DOI] [PubMed] [Google Scholar]
- 67. Toumishey E, Prasad A, Dueck G, et al. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T-cell lymphoma. Cancer. 2015;121:716–723. [DOI] [PubMed] [Google Scholar]
- 68. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30:631–636. [DOI] [PubMed] [Google Scholar]
- 69. Zinzani PL, Musuraca G, Tani M, et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:4293–4297. [DOI] [PubMed] [Google Scholar]
- 70. Iqbal J, Weisenburger DD, Chowdhury A, et al. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic gammadelta T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro. Leukemia. 2011;25:348–358. [DOI] [PubMed] [Google Scholar]
- 71. Kiel MJ, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124:1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Piccaluga PP, Rossi M, Agostinelli C, et al. Platelet-derived growth factor alpha mediates the proliferation of peripheral T-cell lymphoma cells via an autocrine regulatory pathway. Leukemia. 2014;28:1687–1697. [DOI] [PubMed] [Google Scholar]