

Abstract
Adiponectin is an important antiatherogenic adipocytokine that inhibits inflammation, insulin resistance, and oxide stress. Inflammation in the vascular adventitia is a crucial factor in the pathogenesis of atherosclerosis. Adventitial fibroblasts (AFs) can proliferate, divide into myofibroblasts, and migrate to the intima to become a new component of atherosclerotic plaque under inflammation and atherosclerosis. We investigated whether adiponectin might prevent AFs from proliferating, migrating, and transforming into myofibroblasts. Cultured AFs were stimulated with lipopolysaccharide (LPS) in the presence or absence of adiponectin. Methyl thiazolyl tetrazolium assay and migration and scratch-wound assays demonstrated that adiponectin reduced the AF proliferation and migration induced by LPS, respectively, whereas treatment with AdipoR1 small interfering (si) RNA (siAdipoR1), AMP-activated protein kinase (AMPK) siRNA (siAMPK), and an AMPK inhibitor reversed the effect. Immunocytochemistry and Western blot revealed that adiponectin reduced the transition of AFs to myofibroblasts, and treatment with siAdipoR1, siAMPK, and the AMPK inhibitor increased the transition. RT-PCR, Western blotting, and nitric oxide (NO) assay showed that adiponectin reduces induced NO synthase (iNOS) and nitrotyrosine expression and NO and ONOO− production induced by LPS. Treatment with siAdipoR1, siAMPK, and the AMPK inhibitor significantly attenuated adiponectin-induced phosphorylation of AMPK and its downstream target acetyl-coenzyme A carboxylase and up-regulated iNOS mRNA and protein expression, which resulted in a marked increase of NO and ONOO− production. In apolipoprotein E-deficient mice, immunohistochemistry of treated vascular adventitia showed that both iNOS expression and ONOO− production could be reversed with an adenovirus-adiponectin vector. Taken together, these results suggest that adiponectin reduces LPS-induced NO production and nitrosative stress and prevents AFs from proliferating, transforming to myoflbroblasts, and migrating to the intima, thus worsening atherosclerosis, by inhibiting the AdipoR1-AMPK-iNOS pathway in AFs.
Adiponectin can inhibit migration and the transition of AFs induced by LPS. Adiponectin secreted by adipose tissue around adventitia may be a new target in atherosclerosis.
Atherosclerosis has been recognized as an inflammatory disease. Oxidant stress, production of •O2− and its derived oxidants, such as peroxynitrite (ONOO−), can contribute to the onset of atherosclerosis (1). Because arterial injury, in general, is initiated at the interface with circulating blood, most studies performed to unravel the mechanisms involved in injury-induced arterial responses have focused on the innermost layer (intima) rather than on the outermost adventitial layer. However, increasing evidence suggests that the adventitia is a mediator of atherosclerosis and vascular dysfunction (2, 3, 4).
As the main cell types in adventitia, adventitial fibroblasts (AFs) can differentiate into myofibroblasts (MFs), migrate, proliferate and secrete cytokines, and play a critical role in the adventitial response to injury. It is noteworthy that the aortic adventitia is a potential source of nitric oxide (NO) (5), and adventitial inflammation can stimulate the formation of radical oxygen species (6). However, the physiological or pathophysiological role of nitric stress induced by adventitial inflammation remains largely unknown, and its relation to cardiovascular disease is unclear.
Adiponectin is an adipocytokine secreted from adipose tissue (7). Adiponectin plays a role as an antiinflammatory factor, and it is also related to the development of atherosclerosis, hypertension, and coronary heart disease (8, 9, 10, 11). The overexpression of adiponectin can ameliorate atherosclerosis through attenuating endothelial inflammatory response in apolipoprotein E-deficient (ApoE−/−) mice (11). Our previous study showed that adiponectin treatment in adventitia can also reduce the size of atherosclerotic plaques (12). We recently reported that adiponectin receptors are expressed in adventitial tissues and AFs, which implies that adiponectin can have a biologic effect via adventitia. However, the mechanisms by which adiponectin exerts its antiatherosclerosis effects via vascular adventitia remain unknown.
We aimed to determine whether atherosclerosis is amplified in oxidant and nitric stress induced by adventitial inflammation, and whether the enhanced oxidant and nitrosative stress can be rescued by adventitial administration of adiponectin. We also aimed to delineate the mechanisms by which adiponectin may confer its antiinflammatory effects via the adventitia under atherosclerosis and inflammation.
Results
Adiponectin (APN) inhibited lipopolysaccharide (LPS)-induced proliferation and migration of AFs
Compared with AFs of LPS group, methyl thiazolyl tetrazolium (MTT) assay showed that the 490 nm OD value in AFs of the APN + LPS group was decreased markedly (Fig. 1A). The 10 μg/ml LPS-induced increased migration of AFs was significantly reduced with APN (10 μg/ml) (42.83 ± 2.14 vs. 15.67 ± 1.58, P < 0.01) (Fig. 1B). To further determine the effect of APN on AF migration, scratch-wound assay was conducted to examine cell migrating across the wound edge into the scratch area (Fig. 1B). APN reduced AFs migration induced by LPS into the scratch area than those treated with LPS alone. These suggest a significant contribution of APN to lessening the LPS-mediated AF proliferation and migration.
Fig. 1.

The effect of APN (10 μg/ml) on proliferation and transformation to MFs and migration of LPS-treated AFs. A, Effects of APN and LPS on AF proliferation by MTT assay. B, Representative graphs of AF migration across the wound edges 96 h after scratch wounding of cells. C, Immunofluorescent staining of AF transformation to MFs. Green, FIFC staining; blue, DAPI staining. D, α-SM-actin protein expression. Expression of β-tubulin band was used as a loading control. APN, AFs were incubated with APN (10 μg/ml) for 24 h; LPS, AFs were incubated with LPS (10 μg/ml) for 24 h; APN + LPS, AFs were incubated with APN (10 μg/ml) for 2 h before LPS (10 μg/ml) treatment for 24 h; con (control group), AFs were treated with PBS. **, P < 0.01 vs. con; ##, P < 0.01 vs. LPS.
APN decreased LPS-induced transformation from AFs to MFs
Immunocytochemistry revealed that, compared with AFs treated with LPS, those treated with APN and LPS showed decreased transformation to MFs (Fig. 1C). In addition, similar to the immunocytochemistry results, Western blotting showed that the protein expression of anti-α-smooth muscle (SM) actin in the APN + LPS group was higher than that of the LPS group (Fig. 1D).
APN inhibited LPS-induced NO synthase (iNOS) expression and NO production in AFs
To evaluate the effect of APN on iNOS expression and NO production in AFs induced by LPS, we detected the expression of iNOS mRNA and protein and NO production. AFs were pretreated with APN in different doses (3, 10, and 30 μg/ml) for 2 h and 10 μg/ml APN for different times (1/2, 1, 2, 6, 12, 24, and 48 h). AFs were then treated with LPS (10 μg/ml) for 24 h. The data showed that APN decreased the LPS-induced iNOS mRNA in a dose-dependent manner, with maximal effect at 10 μg/ml. iNOS mRNA and protein levels were lower in APN (3, 10, 30 μg/ml)-treated cells than in AFs treated with LPS alone (all P < 0.01). APN also decreased the LPS-increased iNOS mRNA expression in a time-dependent manner. Compared with control, iNOS mRNA level was significantly decreased at 1/2, 1, 2, 6, and 12 h (all P < 0.01), with a peak effect at 2 h (Fig. 2A). LPS-increased iNOS protein level was significantly decreased at 2 h, with a peak effect at 6 h (P < 0.001). Consistent with the iNOS expression, LPS-increased NO production in culture media was also decreased in APN-treated AFs in a dose- and time-dependent manner (all P < 0.01) (Fig. 2C).
Fig. 2.

Effect of APN on LPS-mediated AF iNOS mRNA and protein expression and NO production in cultured medium. A, iNOS mRNA expression. B, iNOS protein expression. Expression of β-actin band was used as a loading control. C, NO production in cultured medium. **, P < 0.01 vs. con; ##, P < 0.01 vs. LPS. Values are means ± sem. con, Control group.
siRNA of AdipoR1 can inhibit the expression of AdipoR1 in AFs
To determine the function of APN in AFs, we first investigated whether the receptor of APN, AdipoR1, is expressed in AFs. RT-PCR and Western blot analysis showed that AdipoR1 mRNA and protein are expressed in AFs, approximately to the same level as in skeletal muscle cells (Fig. 3, A and B). Furthermore, immunofluorescent staining confirmed that AdipoR1 protein is expressed in mouse AFs (Fig. 3C). We next examine whether the expression of AdipoR1 can be inhibited by AdipoR1 siRNA (siAdipoR1).Western blot analysis revealed that treatment with 50 pmol/liter siAdipoR1 blocked the expression of AdipoR1 protein, whereas as a control, a treatment with siβ-actin blocked the expression of β-actin protein, indicating that the knockdown effect of siAdipoR1 was specific (Fig. 3, D and E). The effect of knockdown by siAdipoR1 gradually increased with increasing dose, and showed a maximal effect at 250 pmol/liter (Fig. 3D).
Fig. 3.
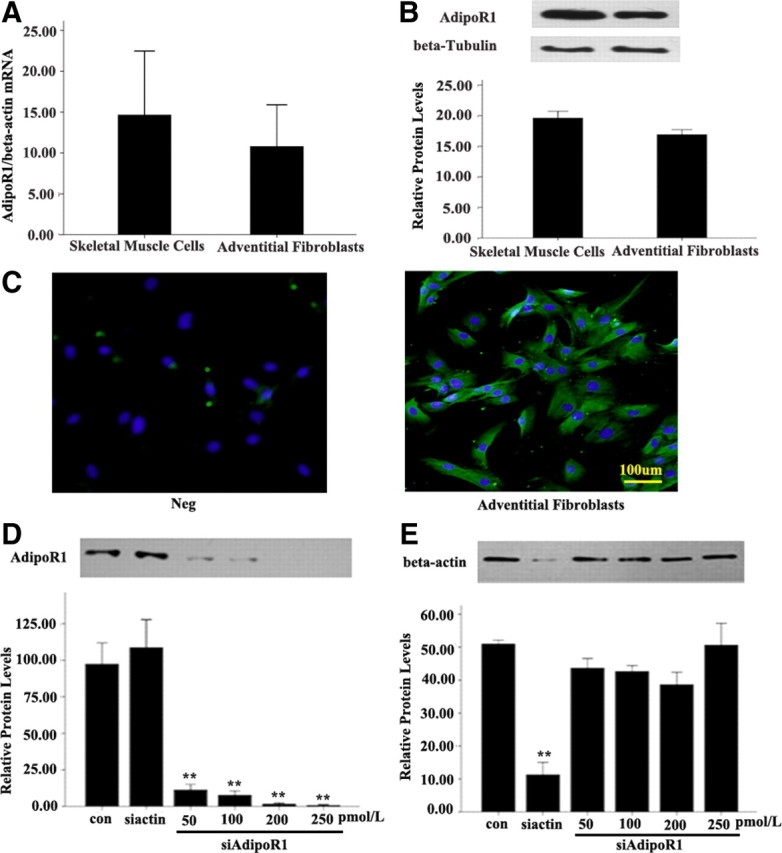
Expression of AdipoR1 in normal AFs and skeletal muscle cells and effect of siAdipoR1. A, AdipoR1 mRNA expression levels normalized to that of β-actin mRNA expression. P > 0.05 vs. skeletal muscle cells. B, AdipoR1 protein expression. Expression of β-tubulin band was used as a loading control. P > 0.05 vs. skeletal muscle cells. C, Immunofluorescent staining of mouse AFs with antibody to AdipoR1. Neg, negative control; Green, FIFC staining; Blue, DAPI staining. D, The siRNA restrained only siAdipoR1, which shows that its knockdown effect was specific, and the knockdown effect of siAdipoR1 was increased in a dose-dependent manner. N, transfection of irrelevant siRNA; siβ-actin, transfection of siRNA of β-actin (100 pm); siAdipoR1, transfection of siRNA of AdipoR1 in different doses. **, P < 0.01 vs. control (con). Values are means ± sem.
Treatment of siRNA of AdipoR1 and AMPK inhibitor compound C reversed the APN-reduced expression of iNOS and nitrotyrosine and production of NO and ONOO− induced by LPS
To elucidate the signaling pathways involved, the regulation of iNOS and nitrotyrosine, and NO and ONOO− production by APN were examined. AFs were pretreated with si-AdipoR1 for 48 h and the indicated kinase inhibitors for 30 min and then after incubated with 10 μg/ml APN for an additional 2 h and 10 μg/ml LPS for 24 h. Treatment with siAdipoR1 (100 pmol/liter) and the AMPK inhibitor compound C (20 μmol/liter) effectively reversed APN-reduced iNOS induced by LPS expression by 47.7% (P < 0.01) and 41.7% and (all P < 0.001), respectively, and nitrotyrosine expression by 60.2% and 55.5%, respectively (both P < 0.01) (Fig. 4, A and B). Treatment with siAdipoR1 (100 pmol/liter) and the AMPK inhibitor compound C (20 μmol/liter) also enhanced APN-reduced NO and ONOO− production in APN stimulated conditions (Fig. 4, C and D).
Fig. 4.

Effects of siRNA of AdipoR1 and AMPK inhibitor compound C on APN-reduced expression of iNOS and nitrotyrosine and production of NO and ONOO−. A, Expression of iNOS and nitrotyrosine. Expression of β-actin was a loading control. B, Production of NO. C, Production of ONOO−. **, P < 0.01 vs. control group (con); ##, P < 0.01 vs. LPS; $$, P < 0.01 vs. APN + LPS. Values are means ± sem.
Effects of APN, AdipoR1 siRNA, and AMPK inhibitor compound C on AMPK signaling pathway
To identify the molecular sources of the APN-regulated iNOS expression and NO production, protein phosphorylation of AMPK and acetyl-coenzyme A carboxylase (ACC) was determined (Fig. 5). AMPK and ACC protein phosphorylation was increased in AFs treated with APN and LPS (all P < 0.01). However, siAdipoR1 (100 pmol/liter) and the AMPK inhibitor compound C (20 μmol/liter) inhibited this effect (all P < 0.01).
Fig. 5.

Effect of APN, siAdipoR1, and AMPK inhibitor compound C on AMPK (p-AMPK) and ACC protein phosphorylation (p-ACC). Expression of β-actin was a loading control. **, P < 0.01 vs. control group (con); ##, P < 0.01 vs. LPS; $$, P < 0.01 vs. APN + LPS. Values are means ± sem.
Treatment of siRNA of AMPK reversed the APN-reduced expression of iNOS and nitrotyrosine and production of NO and ONOO− induced by LPS
To elucidate the signaling pathways involved again, the regulation of iNOS and nitrotyrosine, and NO and ONOO− production by siAMPK were examined. AFs were pretreated with siAMPK (10 μm) for 48 h and then incubated with 10 μg/ml APN for an additional 2 h and 10 μg/ml LPS for 24 h. Treatment with siAMPK (10 μm) effectively reversed APN-reduced iNOS induced by LPS and nitrotyrosine expression, respectively (both P < 0.01) (Fig. 6, A and B). Treatment with siAMPK (10 μm) also enhanced APN-reduced NO and ONOO− production in APN-stimulated conditions (Fig. 6, D and E), which was similar to the effect of compound C.
Fig. 6.

Effects of siRNA of AMPK on APN-reduced expression of iNOS and nitrotyrosine and production of NO and ONOO−. A, Expression of iNOS. B, Expression of nitrotyrosine. C, Expression of AMPK on treatment with siAMPK (10 μm). D, Production of NO. E, Production of ONOO−. **, P < 0.01 vs. control group (con); ##, P < 0.01 vs. LPS; $$, P < 0.01 vs. APN + LPS. Values are means ± sem.
Effect of siRNA of AdipoR1, siAMPK, and AMPK inhibitor compound C on proliferation, migration, and transformation of AFs
We also determined whether siAdipoR1, siAMPK, and AMPK inhibitor compound C affect the proliferation, migration, and transformation of AFs to MFs under APN-stimulated conditions. AFs treated with siAdipoR1 (100 pmol/liter), siAMPK (10 μm), and the AMPK inhibitor compound C (20 μmol/liter), underwent more proliferation and migration than those treated with APN + LPS on MTT assay (Fig. 7D) and scratch-wound assay (Fig. 7A). Furthermore, siAdipoR1 (100 pmol/liter), siAMPK (10 μm), and the AMPK inhibitor compound C (20 μmol/liter) significantly increased the number of migrating AFs under APN-stimulated conditions (all P < 0.01) (Fig. 7B), and increased the transformation to MFs (Fig. 7C).
Fig. 7.
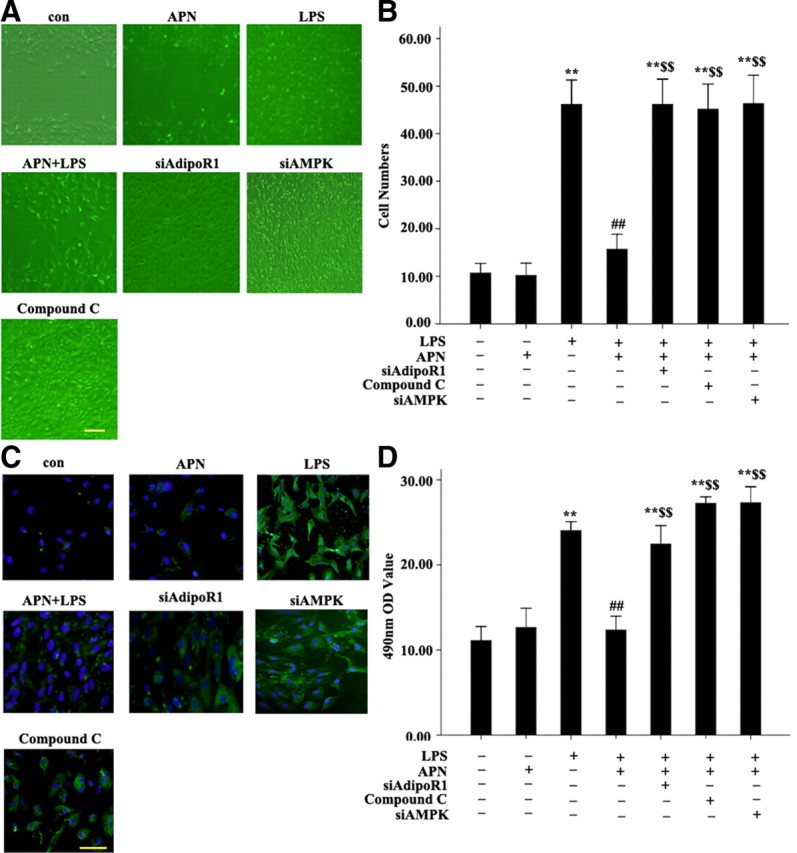
Effect of siAdipoR1, siAMPK, and AMPK inhibitor compound C on APN-reduced proliferation, migration and transformation of AFs. A, Scratch-wound assay results. B, Migration assay results. C, Immunofluorescent staining results of α-SM-actin. LPS, cells treated with LPS (10μg/ml) for 24 h; APN + LPS, cells pretreated with APN (10 μg/ml) for 2 h, then LPS (10 μg/ml) for 24 h; siAdipoR1 + APN + LPS, AFs treated with siAdipoR1, then with APN (10 μg/ml) for 2 h, then LPS (10 μg/ml) for 24 h; siAMPK + APN + LPS, cells treated with siAMPK (10 μm), then with APN (10 μg/ml) for 2 h, then LPS (10 μg/ml) for 24 h; compound C + APN + LPS, cells treated with compound C (20 μm) for 30 min, then with APN (10 μg/ml) for 2 h, then LPS (10 μg/ml) for 24 h. D, MTT assay results. **, P < 0.01 vs. control group (con); ##, P < 0.01 vs. LPS; $$, P < 0.01 vs. APN + LPS. Values are means ± sem.
Effect of APN gene transfection on the expression of iNOS and nitrotyrosine in ApoE−/− mice
Because we found production of NO and expression of iNOS and nitrotyrosine in cultured AFs, we examined whether APN treatment through adventitia may decrease this production animal model. Immunohistochemical staining of ApoE−/− carotid arteries showed iNOS and nitrotyrosine increased, and treatment with Ad-APN through adventitia markedly attenuated iNOS and nitrotyrosine formation (Fig. 8).
Fig. 8.
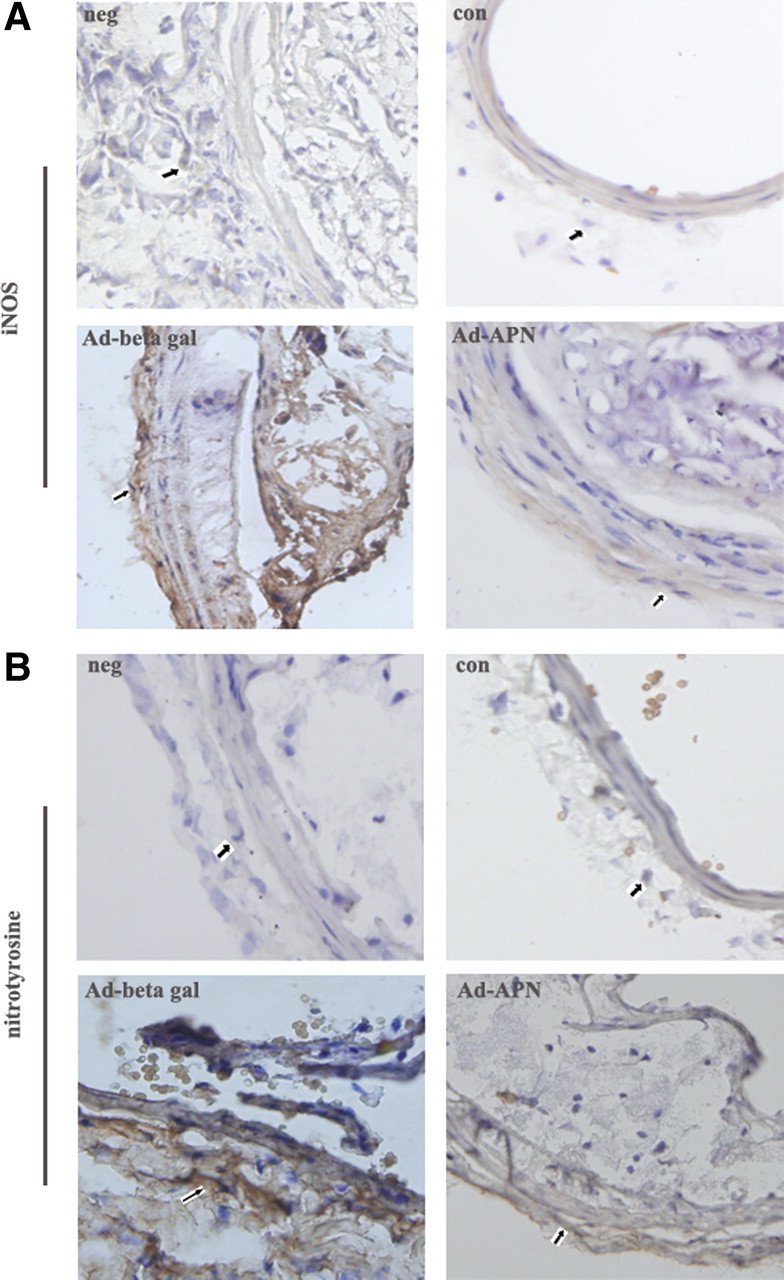
Effect of APN gene transfection on the expression of iNOS and nitrotyrosine in ApoE−/− mice. A, Expression of iNOS. B, Expression of nitrotyrosine. Neg, negative control; con, control group; Ad-β gal, Ad-β gal group; Ad-APN, Ad-APN group. The array showed the positive cell.
Discussion
In this study, we aimed to investigate nitrosative stress induced by adventitial inflammation, the possible rescue by adventitial administration of APN and the mechanisms involved in isolated mouse AFs and ApoE−/− mice. First, we determined that AFs can express AdipoR1 at both mRNA and protein levels. Second, with use of recombinant mouse APN, we showed that APN can inhibit the LPS-induced proliferation, migration of AFs, and decrease the LPS-mediated transformation of AFs to MFs in atherosclerosis. Third, we showed that APN operates in LPS-mediated inflammation at least in part by activation of an AdipoR1-AMPK-iNOS signaling pathway.
The arterial wall represents a highly plastic three-dimensional structure with a unique adaptive capacity to face changes in blood pressure, flow, and shear stress taking place during development and in some vascular pathological conditions (13, 14, 15, 16). The role of adventitia in providing cells and molecules with the capacity to influence neointima formation and vascular remodeling has recently received considerable attention (17, 18, 19, 20, 21). During the response to injury, resident adventitia cells can be reprogrammed to have different structural and functional behavior. AFs in experimental models of severe and mild balloon injury (17, 18, 19, 20) can be phenotypically converted into smooth muscle-like cells, the MFs. The first evidence for adventitial cell migration to the subendothelial space was obtained by pulse-labeled experiments using bromodeoxyuridine incorporated in proliferating cells (17, 18, 19). We found that LPS, an infective stimulus, can increase the proliferation and migration of AFs and promote the transformation of AFs to MFs. Our previous study showed that APN treatment in adventitia can also reduce the size of atherosclerotic plaques (12). However, whether APN can reverse the proliferation, migration, and transformation of AFs induced by LPS in vitro is unknown. In the study, we found that APN could inhibit the proliferation and migration of AFs and decrease the transformation of AFs to MFs with adventitial inflammation induced by LPS, which implies a role for APN in vessel protection.
Nitrosative stress initiates an inflammatory cascade that includes acute-phase protein synthesis, up-regulation of inflammatory adhesion molecules, and proinflammatory cytokine release (22, 23). iNOS is implicated in chronic metabolic disorders such as atherosclerosis (24, 25) and obesity-linked diabetes (26, 27) in which an inflammatory condition also is believed to play a pathogenic role. Adventitia has an independent iNOS system and can produce plenty of NO under inflammatory stimulation. The relationship between APN and iNOS in AFs stimulated by LPS is unclear. Our results showed that LPS stimulation produces a significant up-regulation of iNOS, which then up-regulated iNOS-mediated NO production and nitrotyrosine expression. Administration of APN markedly down-regulated the increased iNOS and nitrotyrosine expression and excessive NO production. These data indicate that preconditioning with APN was capable of effectively inhibiting the inflammatory response and nitrosative stress in LPS-activated AFs. In addition, we showed that iNOS and nitrotyrosine expression were markedly increased in areas where perivascular constrictive silica collars were placed in ApoE−/− mice fed a high-fat diet. We also showed that administration of Ad-APN to adventitia reversed iNOS and nitrotyrosine expression. These results imply that APN is a natural molecule that inhibits atherosclerosis-induced nitrosative stress.
AMPK is a heterotrimeric signaling kinase and a critical energy-sensing pathway with important functions in stimulating glucose uptake. A vascular protective role of AMPK has been demonstrated in several cell types (28, 29, 30). Our present study demonstrated AMPK activity down-regulated by LPS and up-regulated by APN in AFs. Pharmacological activation of AMPK by 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) and an antidiabetic drug inhibits iNOS induction in cells and tissues exposed to inflammatory mediators (31). Gene-silencing experiments confirmed that AMPK-activating agents blunt iNOS-mediated NO production, at least in part, via activation of AMPK (32). APN down-regulates NAPDH oxidase Nox4 expression (33) and protects the heart against ischemia-reperfusion injury by inhibiting iNOS and the resulting oxidative/nitrative stress (34). In the present study, we demonstrated that iNOS and nitrotyrosine expression and NO production were markedly increased under siAdipoR1 transfection and treatment with the AMPK inhibitor compound C but decreased with APN. These results provide the first direct evidence to demonstrate that APN inhibits iNOS expression in LPS-mediated AFs and thus attenuates nitrosative stress-induced migration of AFs and the transformation of AFs to MFs, in part, by an AdipoR1-AMPK pathway.
Physiological or pharmacological concentrations of NO exert significant cardioprotectiive effects, whereas the reaction product between NO and •O2−, ONOO−, is extremely cytotoxic. Our results showed that APN inhibits inflammation-induced iNOS expression and blocks ONOO− formation in vitro and in vivo and reduces the LPS-induced migration of AFs and the transition of AFs to MFs, in part, via an AdipoR1-AMPK-iNOS pathway. These novel findings suggest that reduced APN concentration, like that observed in type 2 diabetes and coronary heart disease, may aggravate atherosclerotic development, in part. APN secreted by adipose tissue around adventitia may be a new target in the treatment of atherosclerotic disease.
Materials and Methods
Materials
Cell culture medium and supplements were purchased from GIBCO-BRL (Rockville, MD). Recombinant mouse APN, which is produced in Escherichia coli, LPS, and an AMPK inhibitor compound C were purchased from Biovendor (Heidelberg, Germany), Sigma-Aldrich (St. Louis, MO), and Merck (Darmstadt, Germany), respectively. For Western blot analysis, rabbit antiphosphorylated-AMP kinase (AMPK), rabbit anti-AMPK, rabbit anti-AMPK α1, rabbit anti-AMPKα2, rabbit antiphosphorylated ACC, rabbit anti-ACC, and rabbit anti-α- SM-actin antibodies were purchased from Cell Signaling Technology (Beverly, MA). Rabbit anti-iNOS antibody and rabbit antinitrotyrosine antibody was purchased from Upstate (Chicago, IL). A secondary antibody (goat antirabbit IgG) for immunoblotting, MTT, and 4,6-diamidino-2-phenylindole hydrochloride (DAPI) were obtained from Sigma-Aldrich (St. Louis, MO). The enhanced chemiluminescence system for Western blot analysis was purchased from Pierce (Rockford, IL). All other chemicals were of the highest grade available commercially.
Cell culture
Male C57BL/6J mice, 16–18 wk old, were obtained from Shandong University. The mice were anesthetized with inhaled isoflurane and then killed by vertebral dislocation. Thoracic aortas were removed and cleaned under sterile conditions. The medium was separated from the adventitia. The adventitia was rinsed three times with culture medium. The isolated adventitia was then cut into 1- to 2-mm2 flat segments and placed on 0.1% gelatin-coated dishes. Fresh medium consisting of DMEM supplemented with 10% fetal bovine serum (FBS; GIBCO, Grand Island, NY), 2 mm l-glutamine, 50 UI/ml penicillin, and 50 μg/ml streptomycin (Sigma, Oakville, Ontario, Canada) was added into each well. The explants were incubated in a humidified incubator at 37 C in a 95% air-5% CO2 atmosphere until AFs reached confluence, typically 7–12 d. From the fourth day onward, the culture medium was removed and replaced with fresh medium every 48 h. Confluent cells were subsequently harvested for passage with a solution of trypsin (0.05%) and EDTA (0.02%) (Sigma). When cells from passage 1 or 2 had reached 80% confluence, they were frozen and kept at −70 C until further use.
Experimental cell grouping
The cells were serum starved and synchronized for 24 h. To observe the effects of APN on AF proliferation, migration, and transformation to MF, AFs were divided into groups for treatment: AFs (untreated), AFs-APN, AFs-LPS, AFs-APN + LPS. In the experiment of 10 μg/ml APN treated for 2 h, AFs were divided into groups for treatment: AFs (untreated), AFs + sicontrol (irrelevant siRNA), AFs-APN, AFs-LPS, AFs-APN + LPS, AFs-APN + small interfering RNA of AdipoR1 (siAdipoR1) + LPS, AFs-APN + compound C + LPS and AFs-APN + small interfering RNA of AMPK (siAMPK) + LPS groups. In culture medium, the final concentration of APN, LPS, compound C, sicontrol, siAdipoR1, and siAMPK duplexes was 10 μg/ml, 10 μg/ml, 20 μm, 200 pmol/liter, 200 pmol/liter, and 10 μm, respectively.
siRNA preparation and transfection
RNA interference was used to down-regulate the expression of AdipoR1 in AFs. SMARTpool siRNA and SMARTpool reagents for AdipoR1, β-actin, and nonspecific control siRNA duplexes were designed and synthesized by Shanghai Genephama CO (China). The small interfering RNA (siRNAs) sequences used for targeted silencing of mouse α1/α2 AMPK isoforms (sc-45312) was supplied by Santa Cruz Biotechnology (Santa Cruz, CA). siRNA A (sc-37007) was used as negative control for experiments using α1/α2 AMPK-targeted siRNA transfection. For gene knockdown experiments, AFs were plated in 6-cm dishes and cultured for 48 h in DMEM containing 10% FBS. Then, after 24 h of incubation in medium without FBS and antibiotics, cells were transfected with specific siRNAs with use of transfection reagent according to the manufacturer’s instructions. After another 48 h of culture, cells were recultured in DMEM containing 10% FBS.
MTT assay
The growing cell density were adjusted to 5 × 104 cells/ml. The cells were placed in 96-well dishes, incubated for 24 h under routine conditions, then serum starved for 24 h in 0.5% DMEM, transfected with reagents, and continuously incubated for 20 h. The supernatant fluid was discarded from the wells, then 5 mg/ml MTT was added to every well, respectively. The cells culture was continued for 4 h; 150 μl of DMSO was added, and the culture was agitated for 10 min. The OD of cells in well was quantitated at 490 nm by an enzyme-linked immunometric meter.
Migration assay
AFs were serum starved for 24 h in DMEM without FBS, then detached with use of trypsin-EDTA, and 3.0 × 104 cells/ml were seeded into Transwell plates (12-μm filter pores, 12-mm diameter, Costar). On the basolateral side of the Transwell chamber, medium containing 2% FBS (negative control) or 2% FBS with 10 μg/ml LPS was added. After 4 h of incubation at 37 C and 5% CO2, AFs were fixed and stained with viola crystallina. The number of cells that migrated to the basolateral side of the chamber was scored manually.
Scratch-wound assay
AFs harvested by trypsinization were seeded in 12-well plates at 5000 cells/well and grown until reaching confluence. A wound was gently introduced in the center of the cell monolayers with use of a sterile 200-μl pipette tip. To remove the cell debris, the wells were washed twice with PBS, and the cells were incubated with or without siAdipoR1 or compound C for 48 h and 0.5 h, respectively. Then after the incubation with or without APN for 2 h, cells were incubated with LPS for 24 h. Phase-contrast images of regions marked along the wound area were obtained with use of an inverted microscope (Axiovert; Carl Zeiss Meditec GmbH) immediately after creating the wound and at the end of 96 h.
Determination of NO generation
NO is unstable, but it forms stable end products, namely, nitrite and nitrate. Hence, the content of nitrite and nitrate is a suitable index of NO generation. The AF culture medium was harvested and the level of nitrite was measured with the Griess Reagent Kit (Nitric Oxide Assay Kit, Beyotime, Shanghai China) according to the manufacturer’s instructions.
Measurement of ONOO−
ONOO−-dependent oxidation of dihydrorhodamine 123 to rhodamine 123 was estimated based on the method described by Kooy et al. (35) Samples were added to the rhodamine buffer (pH 7.4) containing 6.25 μm dihydrorhodamine 123 and 125 μm diethylenetriaminepentaacetic acid and incubated 5 min at 37 C. The absorbance was measured at 500 nm, which is the absorbance of rhodamine 123.
RT-PCR
After stimulation of culture AFs for 6 h, total RNA was extracted by use of Trizol (Roche, Basel, Swiss) and Moloney murine leukemia virus. The mRNA levels were quantitated by quantitative real-time PCR (qPCR). Primer of iNOS was designed by standard PCR reaction chemistry with the addition of the fluorescent DNA-binding dye SYBR Green I (Roche, Basel, Swiss). Serial dilutions of an external standard with a predefined known concentration were used to create a standard curve. Results from qPCR were analyzed by use of LightCycler 3.5 (Roche, Basel, Switzerland). The determination of unknown sample concentrations involved determination of the crossing point value, which correlated inversely with the log of the initial template concentration. Results were controlled for real-time efficiency and normalized with those of endogenous control (β-actin).
Western blot analysis
After treatment, AFs were washed three times with ice-cold PBS and then were lysed in buffer containing protease inhibitors, and total protein was extracted and detected by Western blot analysis (36). Protein concentration in cell lysates was measured by use of a Bio-Rad DC Protein Assay Kit (Pierce, Rockford, IL). Protein (50 μg) underwent treatment with 10% polyacrylamide gel, was separated at 120 V for 2 h and transferred with 200 mA for 2 h. Membranes were blocked with 5% milk for 60 min at 37 C. Protein was incubated with primary antibodies at 4 C overnight, and secondary antibodies were incubated at room temperature for 2 h. After a washing with TBS-Tween (10 min, three times), membranes underwent detection by the ECL Western Blot detection system (Amersham Pharmacia, Deisenhofen, Germany). Sample loadings were normalized by Western blot analysis with an anti-β-actin polyclonal antibody (Sigma-Aldrich, St. Louis, MO).
Immunocytochemistry
Expression and localization of AdipoR1 in AFs were examined on indirect immunofluorescent staining. Cultured AFs in chamber slides were fixed in 4% formalin buffer for 30 min at 4 C. After serial washing with PBS and blocking in 5% goat serum in PBS for 1 h, the same primary antibody for adipoR1 used for immunofluorescent staining was diluted in PBS and applied to the slides and incubated overnight at 4 C. Secondary anti-IgG antibody of the specific species of the primary antibodies, conjugated with Fluorescein Isothiocyante (FIFC) and DAPI, were used. AFs treated with normal goat IgG served as a negative control. The immunofluorescent staining of AFs was then examined on a fluorescent microscopy (Olympus, Tokyo, Japan).
Adenoviral vectors and animal surgery
Adenovirus producing the full-length APN was constructed with use of the Adenovirus Expression Vector Kit (TaKaRa, Kyoto, Japan) as described previously (37). Based on the effects of different titers of Ad-APN on serum APN concentration in mice by ELISA, the optimal concentration of Ad-APN we used was 300 multiplicity of infection.
Male ApoE−/− mice (10–12 wk old; Beijing University, Beijing, China) were housed at a constant temperature (24 C) under a 12-h dark/12-h light cycle and received a high-fat diet (0.25% cholesterol and 15% cocoa butter) for 12 wk. Carotid atherosclerotic lesions were induced during surgery by use of perivascular constrictive silica collars placed on the left common carotid arteries (12). Eight weeks after surgery, all mice were divided to groups: the Ad-APN group (n = 30), the Ad-βgal group (n = 30), and the control group (n = 30). The left common carotid arteries were dissociated. The silica collars on the left common carotid arteries were removed carefully. Ad-APN and Ad-βgal were trickled into the silica collar parts of common carotid arteries of mice in the Ad-APN and Ad-βgal groups, respectively. In contrast, saline was trickled into the common carotid arteries of control mice. The experiment conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised 1985). Mice were euthanized, and carotid arteries were excised.
Immunohistochemistry
Frozen cross-sections (3 μm thick) of carotid arteries embedded in optimal cutting temperature (OCT) compound after overnight fixation in 10% formalin were mounted on slides. Sections were immunohistochemically stained for the presence of iNOS and nitrotyrosine with rabbit iNOS and nitrotyrosine antibodies and IgG (negative control), dilution 1:100. Goat antirabbit biotin (1:100, Sigma Diagnostics) was used as a secondary antibody, and ABC-horseradish-conjugated peroxidase was used for visualization.
Statistical analysis
Results are presented as mean ± sem. Comparisons were analyzed by Student’s t test or one-factor ANOVA. A P < 0.05 was considered statistically significant. All experiments were repeated at least three times.
Acknowledgments
We thank Drs. Wang Xuping, Jinbo Feng, Jiang Hong, Chunxi Liu, and Rong Wang for technical assistance.
Footnotes
This work was supported by The National Basic Research Program of China (973 Program, 2006CB503803), the HI-TECH Technique and Development Program of China (863 Program, 2007AA02Z448), the National Natural Science Foundation of China (No. 30728025), and Shandong Province (Grant 1020).
X.-j.C., L.C., and L.L. contributed equally to this study.
Disclosure Summary: The authors have nothing to disclose.
First Published Online November 4, 2009
Abbreviations: ACC, Acetyl-coenzyme A carboxylase; AF, adventitial fibroblasts; AMPK, AMP-activated protein kinase; APN, adiponectin; ApoE−/−, apolipoprotein E-deficient; DAPI, 4,6-diamidino-2-phenylindole hydrochloride; FBS, fetal bovine serum; iNOS, induced NO synthase; LPS, lipopolysaccharide; MF, myofibroblast; MTT, methyl thiazolyl tetrazolium; NO, nitric oxide; qPCR, quantitative real-time PCR; si, small interfering; siAdipoR1, siRNA of AdipoR1; SM, smooth muscle.
References
- 1.Muscoli C, Cuzzocrea S, Riley DP, Zweier JL, Thiemermann C, Wang ZQ, Salvemini D2003. On the selectivity of superoxide dismutase mimetics and its importance in pharmacological studies. Br J Pharmacol 140:445–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilcox JN, Scott NA1996. Potential role of the adventitia in arteritis and atherosclerosis. Int J Cardiol 54(Suppl):S21–S35 [DOI] [PubMed]
- 3.Piñeiro R, Iglesias MJ, Gallego R, Raghay K, Eiras S, Rubio J, Diéguez C, Gualillo O, González-Juanatey JR, Lago F2005. Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Lett 579:5163–5169 [DOI] [PubMed] [Google Scholar]
- 4.Tan KC, Xu A, Chow WS, Lam MC, Ai VH, Tam SC, Lam KS2004. Hypoadiponectinemia is associated with impaired endothelium-dependent vasodilation. J Clin Endocrinol Metab 89:765–769 [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Du Y, Cohen RA, Chobanian AV, Brecher P1999. Adventitia as a source of inducible nitric oxide synthase in the rat aorta. Am J Hypertens 12:467–475 [DOI] [PubMed] [Google Scholar]
- 6.Kawai J, Ando K, Tojo A, Shimosawa T, Takahashi K, Onozato ML, Yamasaki M, Ogita T, Nakaoka T, Fujita T2004. Endogenous adrenomedullin protects against vascular response to injury in mice. Circulation 109:1147–1153 [DOI] [PubMed] [Google Scholar]
- 7.Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K1996. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (adipose most abundant gene transcript 1). Biochem Biophys Res Commun 221:286–289 [DOI] [PubMed] [Google Scholar]
- 8.Kumada M, Kihara S, Sumitsuji S, Kawamoto T, Matsumoto S, Ouchi N, Arita Y, Okamoto Y, Shimomura I, Hiraoka H, Nakamura T, Funahashi T, Matsuzawa Y, Osaka CAD Study Group2003. Coronary artery disease. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol 23:85–89 [DOI] [PubMed] [Google Scholar]
- 9.Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, Hotta K, Nishida M, Takahashi M, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y1999. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation 100:2473–2476 [DOI] [PubMed] [Google Scholar]
- 10.Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, Ohashi K, Sakai N, Shimomura I, Kobayashi H, Terasaka N, Inaba T, Funahashi T, Matsuzawa Y2002. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 106:2767–2770 [DOI] [PubMed] [Google Scholar]
- 11.Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB2004. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 291:1730–1737 [DOI] [PubMed] [Google Scholar]
- 12.Li CJ, Sun HW, Zhu FL, Chen L, Rong YY, Zhang Y, Zhang M2007. Local adiponectin treatment reduces atherosclerotic plaque size in rabbits. J Endocrinol 193:137–145 [DOI] [PubMed] [Google Scholar]
- 13.Owens GK1995. Regulation and differentiation of vascular smooth muscle cells. Physiol Rev 75:487–517 [DOI] [PubMed] [Google Scholar]
- 14.Stenmark KR, Mecham RP1997. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu Rev Physiol 59:89–144 [DOI] [PubMed] [Google Scholar]
- 15.Hungerford JE, Little CD1999. Developmental biology of the vascular smooth muscle cell: building a multilayered vessel wall. J Vasc Res 36:2–27 [DOI] [PubMed] [Google Scholar]
- 16.Sartore S, Franch R, Roelofs M, Chiavegato A1999. Molecular and cellular phenotypes and their regulation in smooth muscle. Rev Physiol Biochem Pharmacol 134:235–320 [DOI] [PubMed] [Google Scholar]
- 17.Wilcox JN, Scott NA1997. Potential role of the adventitia in arteritis and atherosclerosis. Int J Cardiol 54(Suppl):S21–S35 [DOI] [PubMed]
- 18.Zalewski A, Shi Y1997. Vascular myofibroblasts: lessons from coronary repair and remodeling. Arterioscler Thromb Vasc Biol 17:41. [DOI] [PubMed] [Google Scholar]
- 19.Faggin E, Puato M, Zardo L, Franch R, Millino C, Sarinella F, Pauletto P, Sartore, Chiavegato A1999. Smooth muscle-specific SM22 protein is expressed in the adventitial cells of the balloon-injured rabbit carotid artery. Arterioscler Thromb Vasc Biol 19:1393–1404 [DOI] [PubMed] [Google Scholar]
- 20.Li G, Chen SJ, Oparil S, Chen YF, Thompson JA2000. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid artery. Circulation 101:1362–1365 [DOI] [PubMed] [Google Scholar]
- 21.Gutterman DD1999. Adventitia-dependent influences on vascular function. Am J Physiol 277:H1265–H1272 [DOI] [PubMed]
- 22.Szabó C, Dawson VL1998. Role of poly (ADP-ribose) synthetase in inflammation and ischemia-reperfusion. Trends Pharmacol Sci 19:287–298 [DOI] [PubMed] [Google Scholar]
- 23.Willy C, Dahouk S, Starck C, Kaffenberger W, Gerngross H, Plappert UG2000. DNA damage in humane leucocytes after ischemia/reperfusion injury. Free Radical Biol Med 28:1–12 [DOI] [PubMed] [Google Scholar]
- 24.Cromheeke KM, Kockx MM, De Meyer GR, Bosmans JM, Bult H, Beelaerts WJ, Vrints CJ, Herman AG1999. Inducible nitric oxide synthase colocalizes with signs of lipid oxidation/peroxidation in human atherosclerotic plaques. Cardiovasc Res 43:744–754 [DOI] [PubMed] [Google Scholar]
- 25.Behr-Roussel D, Rupin A, Simonet S, Bonhomme E, Coumailleau S, Cordi A, Serkiz B, Fabiani JN, Verbeuren T2000. J Effect of chronic treatment with the inducible nitric oxide synthase inhibitor N-iminoethyl-l-lysine or with l-arginine on progression of coronary and aortic atherosclerosis in hypercholesterolemic rabbits. Circulation 102:1033–1038 [DOI] [PubMed] [Google Scholar]
- 26.Perreault M, Marette A2001. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 7:1138–1143 [DOI] [PubMed] [Google Scholar]
- 27.Shimabukuro M, Ohneda M, Lee Y, Unger RH1997. Role of nitric oxide in obesity-induced β cell disease. J Clin Invest 100:290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouedraogo R, Wu X, Xu SQ, Fuchsel L, Motoshima H, Mahadev K, Hough K, Scalia R, Goldstein BJ2006. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: evidence for involvement of a cAMP signaling pathway. Diabetes 55:1840–1846 [DOI] [PubMed] [Google Scholar]
- 29.Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K2004. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem 279:1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X, Mahadev K, Fuchsel L, Ouedraogo R, Xu SQ, Goldstein BJ2007. Adiponectin suppresses IκB kinase activation induced by tumor necrosis factor-α or high glucose in endothelial cells: role of cAMP and AMP kinase signaling. Am J Physiol Endocrinol Metab 293:E1836–E1844 [DOI] [PubMed]
- 31.Kuo CL, Ho FM, Chang MY, Prakash E, Lin WW2008. Feb 15 Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J Cell Biochem 103:931–940 [DOI] [PubMed] [Google Scholar]
- 32.Pilon G, Dallaire P, Marette A2004. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem 279:20767–20774 [DOI] [PubMed] [Google Scholar]
- 33.Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, Goldstein BJ2008. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118:1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL2007. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 115:1408–1416 [DOI] [PubMed] [Google Scholar]
- 35.Kooy NW, Royall JA, Ischiropoulos H, Beckman JS1994. Peroxynitrite-mediated oxidation of dihydrorhodamine 123. Free Rad Biol Med 16:149–156 [DOI] [PubMed] [Google Scholar]
- 36.Khalil N, Xu YD, O'Connor R, Duronio V2005. Proliferation of pulmonar interstitial fibroblasts is mediated by transforming growth factor-β1 induced release of extracellular fibroblast growth factor-2 and phosphorylation of p38MAPK and JNK. J Biol Chem 280:43000–43009 [DOI] [PubMed] [Google Scholar]
- 37.Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y2002. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 8:731–737 [DOI] [PubMed] [Google Scholar]