

Abstract
I (R.P.M.) presented “The Year In G Protein-Coupled Receptor Research” at ENDO 2009. I first described the diversity of ligands and the five families into which the approximately 800 G protein-coupled receptors (GPCRs) are grouped, their basic structural architectures, their preeminent role in signaling, and the enormous scope for developing drugs targeted at GPCRs. I then spoke about some of the exciting breakthroughs in solving the atomic level structures of the active state of rhodopsin, β2-adrenergic, β1-adrenergic, and A2A-adenosine receptors. I also described studies on the structural changes accompanying the activation of the rhodopsin family of GPCRs. From these recent technical advances, we can anticipate that many more GPCR structures will emerge, which will afford us greater insight into their common and unique structural features and, particularly, the mechanisms underlying their activation. These insights will guide us in our understanding of how GPCRs operate, both in the normal and pathological situation. Although these crystal structures are highly informative, it is important to recognize that they represent static frozen conformations of a single GPCR state. New biophysical techniques are therefore being utilized to facilitate the dynamic monitoring of GPCR structural changes in relation to ligand activation. Solving of the crystal structures of GPCRs has also presented the real possibility of using the information of the ligand-binding pocket to allow in silico screening for novel small-molecule ligands. I then reviewed the concept of ligand-induced selective signaling of GPCRs, which is opening up new insights into more selective drug development. The assembly of GPCRs as homo- and heterooligomers and their phosphorylation and association with a vast array of trafficking and signal-modulating proteins are emerging as major mechanisms underlying the functioning of GPCRs. Differential expression and recruitment of these proteins provide a mechanism for subtle physiological regulation of cellular activity. Finally, I mentioned some of the GPCRs that have lately come to the fore as novel regulators in endocrinology. These included fatty acid-specific GPCRs expressed in pancreatic β-cells and novel neuroendocrine GPCRs regulating reproduction.
We would like to thank the Year In Endocrinology program organizers for having the vision to review the year in G protein-coupled receptors (GPCRs) and also the audacity to do so! There happen to have been more than 8000 publications on GPCRs this last year and even more than 850 reviews. Being unable to read either all of the reviews or the primary articles, we have necessarily been highly selective in singling out some of the exciting developments that have emerged from GPCR research during the last 12–18 months. We have also taken license to cover some developments from late 2007 as well as early this year and to describe some unpublished results, which colleagues have been kind enough to share with us.
First, we will highlight some of the really exciting discoveries of the atomic level structures of GPCRs. Next, we’ll review ligand and accessory protein modulation of signaling of GPCRs. The regulation of GPCR membrane expression will follow and, finally, we will mention some of the novel GPCRs that have lately come to the fore in endocrinology. I (R.P.M.) have asked my post-doc, Claire Newton, to assist in transforming my original talk to the written form and to coauthor the resulting article.
Background to GPCRs
GPCRs convey the majority (∼80%) of signal transduction across cell membranes. They are activated by diverse ligands, which vary from single photons through ions, odorants, amino acids, fatty acids, neurotransmitters, peptides/polypeptides, to proteolytic enzymes, which cleave off receptor fragments to generate an activating ligand and adhesion molecules. All GPCRs are located within the plasma membrane and have a common architecture consisting of seven-transmembrane (TM) domains, connected by extracellular (ECL) and intracellular (ICL) loops. One of the very special features of GPCRs is that they are highly druggable, to the extent that more than one third of all current therapeutics are directed at them. Despite this, so far only a small percentage of the approximately 800 known and verified GPCRs have been targeted for therapeutics. Hence, there remains enormous scope for researchers to delineate the numerous roles of GPCRs in basic physiology and pathophysiology and to thereby understand and develop new treatments for disease.
GPCRs may be classified as five major families. The largest family, and the one we’ll be focusing on for this article, is the Rhodopsin family, which comprises 672 family members, including 388 odorant receptors. Despite this large number, and the fact that among these only 63 are orphan receptors (i.e. ones for which ligands are unknown), relatively few have been identified as drug targets. Moreover, there are probably many more drug candidates in the family as odorant receptors, which have been assumed to be involved exclusively in the detection of odorants, are now emerging as the target of ligands in tissues other than the nasal neuroepithelium. For example, this year an odorant receptor, prostate-specific G protein-coupled receptor, was shown to be expressed in prostatic cancer cells, and researchers identified its ligand as a steroid hormone, androstenone, which has potent antiproliferative effects (1).
The four other, smaller, GPCR families are the Secretin (15 members), the Adhesion (33 members), the Glutamate (22 members), and the Frizzled/Taste (36 members). The family of GPCRs classified as adhesion molecules is particularly fascinating. They have a very extended N terminus thought to be involved in cell-cell contact. There are, as yet, no drugs targeting them, and the majority of them remain orphan receptors. But what is becoming evident from knockout studies in Caenorhabditis elegans and in rodents is that they are crucial in the early development of the embryo and also appear to have a number of physiological roles (2). There are no orphans among the 15 Secretin family and the 11 Frizzled GPCRs, but two thirds of the Glutamate and most of the Taste GPCRs remain as orphans. Only seven of all these GPCRs have been identified as drug targets.
New Atomic Level Structure of GPCRs
As previously mentioned, GPCRs have seven-TM domains connected by ICLs and ECLs. In the absence of structural knowledge, predictions of the atomic level structures of GPCRs were originally based on rather speculative molecular models, and later on low-resolution two-dimensional crystal structures of rhodopsin (3, 4). Nevertheless, this information provided insight on the relative positioning of the TM domains and inspiration for testing predictions of molecular interactions derived from the molecular models. We have used the two-dimensional GnRH receptor, which is the focus of our research and with which we are most familiar, to illustrate the salient functional features of Rhodopsin family GPCRs (Fig. 1). The GnRH receptor does have some unusual features that differ from the other members of this family (e.g. it lacks a C-terminal tail, and the TM2 asparagine (N87) and TM7 aspartate (D319) residues, which interact, are more frequently in reciprocal positions). Nevertheless, the GnRH receptor does have the major hallmarks of a rhodopsin family GPCR (see Fig. 1). By developing molecular models of the GnRH receptor based on the early rhodopsin-predicted structures, it was possible to mutate targeted residues that were proposed to configure the receptor, bind the ligand, or be involved in receptor activation or engagement of intracellular signaling proteins (see review in Ref. 5). For example, the putative interaction of TM residues mentioned above was tested by mutation and restoration of function by reciprocal mutation. Such established interactions are then firmly fixed in refining the molecular model. Residues conserved in all Rhodopsin family members (boxed in Fig. 1) were shown to be critical for function, as expected. Among these, the conserved DR sequence, located at the interface between TM3 and ICL2, (sometimes the D is substituted with another acidic residue, E) was thought to be involved in receptor activation and indeed, mutation of D to N, which would release R from the ionic bond between these residues, increases the coupling efficiency (6). Confirmation that the DR ionic bond is broken in the process of receptor activation has emerged from the recent crystal structures (see later). GnRH-binding sites were also identified (red residues in Fig. 1) and docking of the GnRH nuclear magnetic resonance (NMR) structure to engage these residues added further information for the constraints of the GnRH receptor structure and further refinement. Although considerable progress could be made in this way, in the absence of crystal structures, these physical structures were essential to transform interpretation into fact.
Fig. 1.

Two-dimensional representation of the human GnRH receptor. The seven-TM α-helical domains (boxed) are connected by three ECLs and three ICLs. Residues in squares are ones highly conserved throughout the rhodopsin family of GPCRs. Ligand binding residues (red) and residues thought to be important in receptor structure or binding pocket configuration (green) are shown. These include residues involved in disulfide bond formation (black lines) and a glycosylation site. Residues involved in receptor activation are shown in blue. Residues involved in coupling to G proteins are shown in orange. Putative protein kinase C (PKC) and protein kinase A (PKA) phosphorylation sites are indicated. The intermolecular interactions between GnRH I residues and the receptor are indicated with red lines. Residues shown to alter selectivity for relative binding affinities of GnRH I and GnRH II are shown in bold circles. [Modified with permission from R. P. Millar et al.: Front Neuroendocrinol 29:17, 2008 (23 ). ©Elsevier.]
Scientists faced a big challenge of obtaining accurate information on the three-dimensional structure of GPCRs to reveal the TM helical clusters surrounding the binding pocket. Crucial to an understanding of how any protein carries out its functions is the knowledge of the protein’s three-dimensional structure. Such structural information provides the necessary framework for determining mechanisms of action of proteins. In 2000, Palczewski et al. (7), solved the structure of rhodopsin, which had a major impact in confirming and refuting previous predictions on the structure and function of rhodopsin. The structure also provided more accurate information for sequence alignment and modeling of other GPCRs. This has been followed over the last 18 months by the solving of the structures of the β2-adrenergic, β1-adrenergic, and the A2A-adenosine receptors, as well as the structure of the unliganded active opsin (Ops) and the active opsin bound to an 11-amino acid C-terminal sequence of Gα transducin (Ops*) (8, 9, 10, 11, 12, 13), all of which has been key to our understanding of the molecular mechanisms underlying GPCR activation for transmission of signals into cells. Aside from the important biomedical implications of solving the structures of these receptors, the findings provide insight into understanding how proteins in general can take up different conformations, into their associations with other molecules and into the signaling/activation process.
The first crystal structures that emerged after that of rhodopsin were from Brian Kobilka’s laboratory. Solving the β2-adrenergic crystal structure presented an enormous challenge in producing sufficient pure protein, stabilizing flexible domains such as ICL3, defining appropriate detergent/lipidic environments and crystallization conditions, and developing microdiffraction technology to obtain x-ray data from very small crystals. Kobilka and co-workers (10) first used an antibody to stabilize the ICL3 (which is highly mobile and compromises crystallization) and achieved a 3.4–3.7Å-resolution, which revealed the TM domains very clearly. This was followed up with another crystal structure in which ICL3 was stabilized by substitution with lysozyme, a highly structured molecule, which improved the resolution to 2.4Å so that ECL structure could be visualized as well as elements of the ICLs (13). The structure also shows membrane cholesterol interaction with TM domains 2, 3, and 4, which is suggested to be the site at which small molecules can act allosterically at GPCRs to potentially activate or inactivate them (14) (Fig. 2).
Fig. 2.
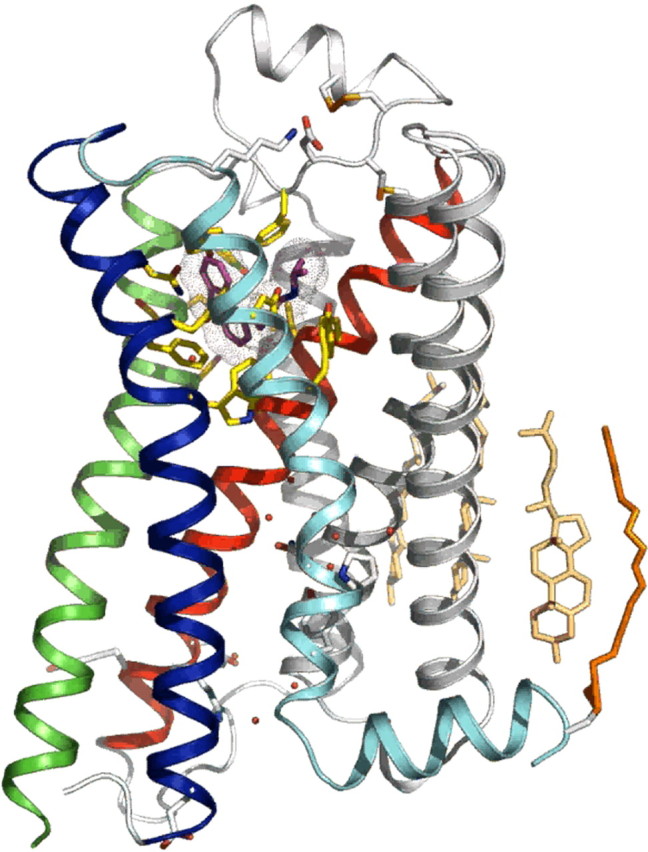
Three-dimensional structure of the β2-adrenergic receptor. The three-dimensional structure of the β2-adrenergic receptor stabilized by lysozyme substitution of ICL3, with a resolution of 2.4Å. The inverse agonist carazolol cocrystallized with the receptor is shown in purple, bound to cognate residues (yellow). ECL2 is shown as a helical structure above the TM helices. Molecules of water are shown in red, membrane cholesterol molecules interacting with the receptor are shown in beige, and an ICL, made up of the carboxyl terminus tethered to the membrane by an palmitoyl residue, is shown in orange. [Figure kindly supplied by Brian Kobilka, Department of Molecular and Cellular Physiology, Stanford University School of Medicine, Stanford, CA.]
Comparison of the four solved GPCR structures reveal two important features. The first is what we call “structural convergence” (i.e. the similarities in structure). The second is “structural divergence” (i.e. the features that differ). Figure 3A shows solved structures of four GPCRs. It is evident from superimposition of the TM domains that these are strikingly similar. The docked ligands (shown in orange) are also fairly similar in occupying much the same space, although adenosine docks somewhat more superficially (15). Thus, GPCRs tend to accommodate these small molecules with similar spatial arrangement, but the particular interactions with amino acid side chains are quite different. The structures of the ECL2, however, are quite divergent. In rhodopsin, for example, Fig. 3B shows that the top of the TM cluster seems to be occluded by the N terminus and ECL2 domains, whereas the top of the TM cluster appears to be open in the other receptors with ECL2 positioned to one side (15). The occlusion of the TM cluster pocket by ECL2 presented a considerable conundrum when the structure of rhodopsin was first solved because ligands would not have access to the binding pocket. In forming this highly structured closure at the top of rhodopsin, retinol is retained within an environment where it would not be easily hydrolyzed. Thus, the new solved structures revealing that extracellular domains in the β2-adrenergic and adenosine A2A receptors are located to one side of the TM cluster provided a mechanism for small ligand access.
Fig. 3.

Structures of GPCRs solved to date. Comparison of the three-dimensional structures of rhodopsin/opsin, β-adrenergic, and adenosine receptors. All structures are shown with the same orientation. TM helices are shown in purple, intracellular regions are shown in blue and extracellular regions are shown in brown. The ligands are shown in orange, and bound lipids are shown in yellow. A, Transmembrane view of the structures of rhodopsin/opsin, β2-adrenergic, β1-adrenergic, and adenosine A2A receptors showing similar relative positions of the TM domains and location of the ligand-docking sites with the adenosine analog occupying a slightly more superficial position. B, Extracellular view of rhodopsin, β-adrenergic, and adenosine receptor structures showing occlusion of the binding pocket by the extracellular domains for rhodopsin but accessibility of the ligand-binding pockets for the β-adrenergic and adenosine receptors. [Both panels reproduced with permission from M. A. Hanson and R. C. Stevens: Structure 17:8, 2009 (15 ). ©Cell Press.]
GPCR activation
One of the big questions in GPCR function is how does receptor activation occur? There have been many postulates over the years and a plethora of data and speculations presented on the basis of modeling, biophysical data, and mutagenesis studies. Thus, the three-dimensional structures solved during the past year have contributed enormously to advancing our understanding of the molecular mechanisms involved in receptor activation. Figure 4A beautifully demonstrates what changes occur in the rhodopsin activation process. Opsin, which reflects the active state of rhodopsin, is depicted in yellow helices (9); active state opsin bound to the C-terminal peptide of the G protein (transducin) in orange helices (11); and inactive rhodopsin, in mauve (16). TM6, which is predicted to move, does so around 3 or 4Å in the active opsin crystal structures. This movement allows access for the G protein transducin to interact with helices (TMs) 6 and 5 and induction of an α-helical-type structure as it associates with helices 7, 6, and 5. These molecular changes are brought about by the breaking of the ionic lock in rhodopsin comprising ionic interactions in the conserved E/DR motif between Arg135 and the adjacent Glu134, and with a second Glu247 in TM6 (11). The disruption of the Arg ionic bond with its adjacent Asp acidic residue in TM3 had been implicated by Ballesteros and colleagues (6) in the activation of the GnRH receptor a decade ago. The recent crystal structures of active opsin and inactive rhodopsin clearly show that activation is accompanied by the disruption of the Arg135/Glu134 ionic bond (Fig. 4B). This results in the formation of a new interaction of Arg135 with Tyr223 in TM5 (11).
Fig. 4.
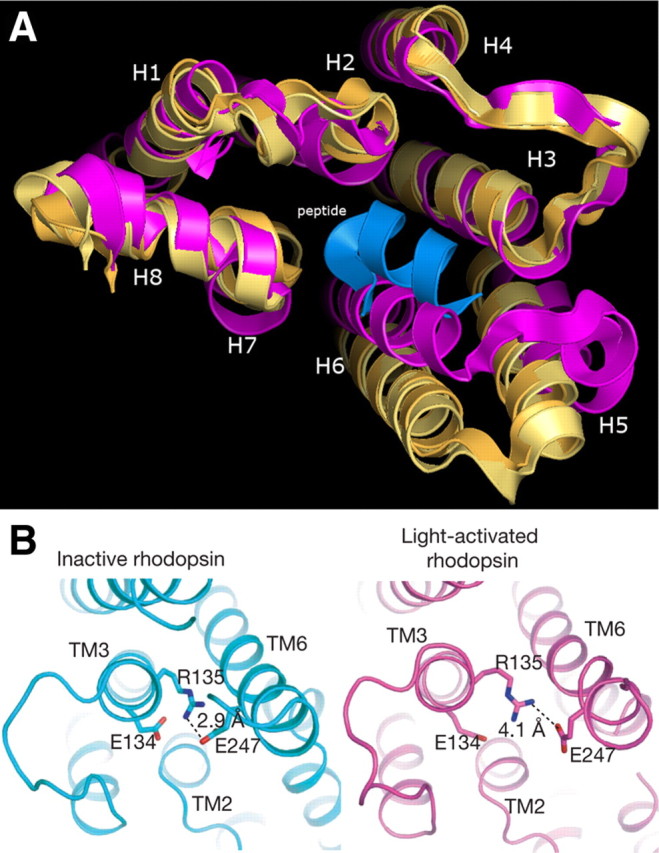
Comparison of inactive and active rhodopsin conformations. A, Comparison of the structures of active opsin (Ops), active opsin bound to a C-terminal fragment of Gα transducin (Ops*) (both yellow) and rhodopsin (purple), depicting the change in position of TM6 (H6) upon activation. [Figure kindly supplied by Richard Henderson, MRC Laboratory of Molecular Biology, Cambridge, UK.] B, Presence of the ionic lock between Arg135 and the adjacent Glu134 in TM3 and Glu247 in TM6. In the light-activated structure the Arg135/Glu134 ionic bond is broken, and the distance of Glu247 from Arg135 has increased. [Reproduced with permission from S. G. Rasmussen et al.: Nature 450:383, 2007 (10 ). ©Macmillan Publishers Ltd.]
Limitations of GPCR crystal structures: alternative biophysical approaches
Although crystal structures of GPCRs have revolutionized our understanding of the operation of these receptors, they do have limitations. First, it is extremely difficult and labor intensive to produce three-dimensional x-ray crystals; therefore, the degree to which producing crystals of the large number of GPCRs is achievable is uncertain and will depend upon new improved methodology. Second, these structures represent a single conformational state, yet GPCRs appear to be highly dynamic and assume different conformational states and go through various transitions in their interactions with ligands and intracellular signaling and modulating proteins. Most of the current crystal structures (with the exception of opsin) appear to represent an inactive or partially active conformation, possibly because the stability of this state is greater and it is a preferred conformation for crystallization. Therefore, other biophysical techniques are needed to inform us about the dynamic changes in GPCR structures upon their activation.
One exciting development from Wayne Hubbell’s laboratory is site-directed spin labeling of rhodopsin. This technique involves specifically labeling the receptor with spin labels (organic molecules able to interact with other molecules and which contain an unpaired electron) before analysis by electron paramagnetic resonance spectroscopy. This dynamic new technology allowed them to look at the real-time structural movements in rhodopsin and demonstrated that TM6 moved several angstroms on activation (17).
Another approach, used by Brian Kobilka’s group, involved substituting putative interacting amino acids of the β2-adrenergic receptor with cysteine and tryptophan. The cysteines were then labeled with the fluorophore bimane, and the quenching of the fluorescent signal by the tryptophan was used to monitor the distance between the two residues when the receptor is in the active (agonist-bound) or inactive (inverse agonist-bound) states. When activated with an agonist, an increase in signal occurred, which reflected the movement of a fluorescent cysteine residue in TM6 away from a tryptophan TM3 so that quenching no longer occurred (18). This technique has now been used with other residues to start to build up an overall picture of the movements and conformational changes in GPCRs on activation.
Yet another elegant approach, used by Brian Kobilka and colleagues to study changes in conformation of the β2-adrenergic receptor, employed two-dimensional nuclear magnetic resonance (NMR) spectroscopy after systematically labeling lysine residues with 13C. This enabled the identification of signals from specific lysines. The lysines at the intracellular face (distal TM regions and adjacent loops) were solvent exposed and mobile, whereas K235 in ECL3 was highly constrained in accordance with it forming a salt bridge with an aspartate residue in ECL2. The movement of this residue was then demonstrated by the spectral change after agonist, but not inverse agonist, binding (65). 13C NMR spectroscopy has also been used to show the ECL2 is displaced from the retinal binding site upon activation of rhodopsin (19).
These fascinating insights into the molecular functioning of GPCRs represent a superb intellectual tour de force. But do these fundamental discoveries have relevance to solving medical problems? Do they, for example, provide the means to develop new drugs? In a recent study from Kolb et al. (20), 972,608 molecules were screened for their ability to occupy the carazolol-binding site in the β2-adrenergic receptor crystal structure. Because carazolol is an inverse agonist, the receptor is thought to be in the inactive conformation. From this screen, 25 of the best fitting molecules were selected and characterized pharmacologically. Six of them had binding affinities of less than 4 μm [one with an inhibition constant (Ki) of 9 nm], and five were inverse agonists. The finding is encouraging because it demonstrates the potential of in silico screening based on crystal structure. In fact, the best selected compound is among the most effective β2-adrenergic receptor inverse agonists. It is also intriguing that unprecedented new classes of molecules emerged from the screen, which were not expected to be ones that would occupy the binding site. This work is significant because it demonstrates the proof of the concept that GPCR crystal structures can be used for structure-based discovery of new ligands.
What’s New in GPCR Signaling?
Moving on to GPCR signaling, we’re going to highlight some selected areas that we feel are likely to impact significantly on our understanding of the diversity of GPCR effects on physiology and pathophysiology and how this is going to influence drug development.
Ligand-induced selective signaling (LiSS)
The first is a concept we call LiSS, whereby different ligands selectively recruit different intracellular signaling proteins to produce different phenotypic effects in cells (21). The classical view of how GPCRs operate is conventionally viewed as the receptor occurring in two states: an inactive state and an active state, which is stabilized by agonist binding. In this active state the receptor is able to co-opt the intracellular signaling machinery and activate a cascade of events culminating in the physiological function of the cell. This simple bimodal switch model has been under scrutiny for sometime. What has recently emerged is that receptors can assume many conformations. Each of these conformations can potentially interact with a ligand in a highly selective manner. In turn, this specific receptor conformation selectively interacts with a specific intracellular signaling complex. By the same token the preponderance of a specific signaling complex in a particular cell will tend to stabilize a certain receptor conformation, thereby inducing selectivity for a certain ligand (Fig. 5A).
Fig. 5.
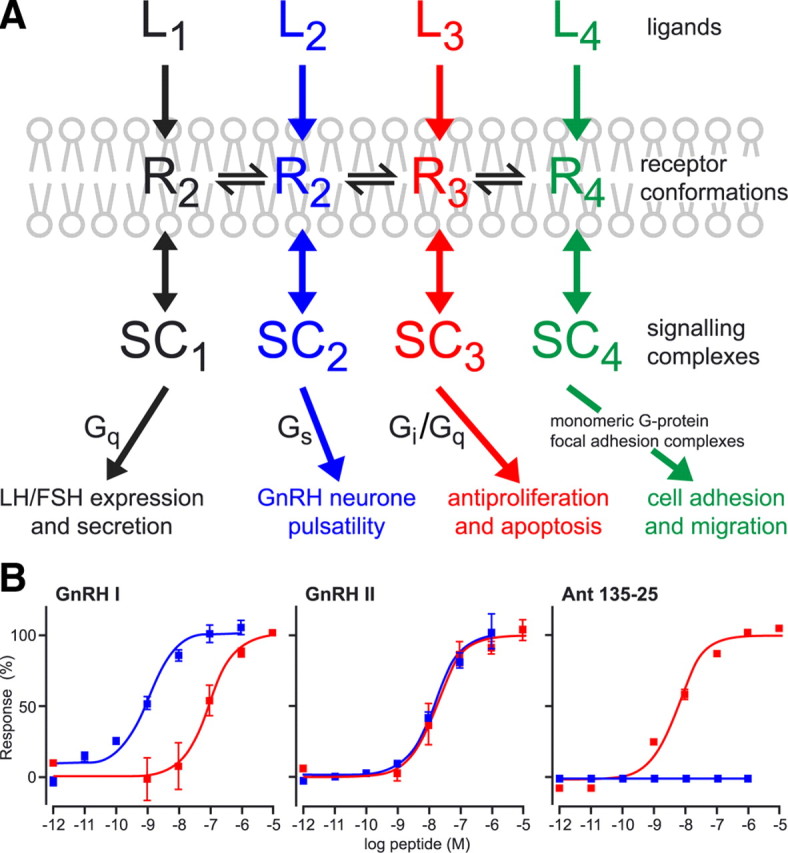
GnRH receptor ligand-induced selective signaling. A, Schematic depicting the concept of multiple active states (R1–R4) of a single GPCR (GnRH receptor) that are selective for different agonist ligands (L1–L4). The different active receptor states are selectively coupled to different signaling complexes (SC1–SC4) that give rise to different effects in cells. [Modified with permission from R. P. Millar et al.: Front Neuroendocrinol 29:17, 2008 (23 ). ©Elsevier.] B, Effects of GnRH I, GnRH II, and GnRH antagonist (135-25) on stimulation of Gαq G proteins (inositol phosphate production) (blue) and inhibition of proliferation (red) in HEK 293 cells stably transfected with the rat GnRH receptor. [Reprinted with permission from R. P. Millar et al.: Front Neuroendocrinol 29:17, 2008 (23 ). ©Elsevier.]
The LiSS concept was originally promulgated by Terry Kenakin (22) and is rapidly becoming a generic theme for GPCRs. This phenomenon is referred to by different groups using a variety of terms such as: “functional selectivity,” “biased agonism,” “ligand-selective agonism,” “agonist-directed trafficking of signaling,” or “agonist-receptor trafficking.” It has important implications in specific drug development and in minimizing side effects. One example, of many that have emerged in recent years, comes from our own studies on the effects of the two naturally occurring GnRHs, GnRH I and GnRH II, operating through the single GnRH type I receptor. GnRH I is much more potent in generating inositol phosphate than in its antiproliferative effects on certain cells, whereas GnRH II does not show much difference between these two effects. An extreme example is a GnRH antagonist (135-25), which has no intrinsic stimulation of inositol phosphate generation but has potent antiproliferative activity (23) (Fig. 5B). We have gone on to show that the Tyr8 of GnRH II is the main determinant of selective antiproliferative effects (24) and identified residues in the TM domains (25) and ECLs (26) of the GnRH receptor that determine selectivity for ligand binding.
The LiSS concept has now been demonstrated for many GPCRs (e.g. see Refs. 27, 28, 29, 30, 31) and is creating a new level of sophistication, which challenges the dogma that ligand engagement of a GPCR consistently elicits a specific intracellular signal. Instead, it has become increasingly clear that the nature of the ligand and the dynamically changing intracellular environment alter the flavor of the signaling. Indeed, it appears that there is a new era of drug discovery on our doorstep, in which screening for novel ligands will not simply involve receptor binding and/or the most convenient high-throughput functional signal output but instead will screen for the appropriate intracellular signal, which reflects the desired phenotypic response of a cell for a disease state or pathophysiology. Equally, appropriate cells will have to be used to ensure an appropriate intracellular context. Although these challenges are substantial, we believe they will bear fruit in the longer term efforts of GPCR drug discovery in the spin-off benefits of reduced failure in the clinic through lack of specificity and off-target effects.
GPCR signaling independent of G proteins
Another area of development over the last few years is GPCR signaling through proteins other than G proteins. It is becoming increasingly apparent that there are many ways in which GPCRs can signal independently of G proteins. So much so, that a case has been made for abandoning the term “G protein-coupled receptors” and referring to them as “seven-transmembrane receptors.” The first convincing evidence for the existence of GPCR-independent signaling came from the laboratory of Lefkowitz and associates (32) and has since been extensively reviewed (e.g. see Ref. 33). An example is angiotensin II at its AT1 receptor activating both β-arrestin and G proteins. When antagonists such as angiotensin II-receptor blockers (losartan and valsartan) engage the binding site, neither signal is propagated. However, another type of antagonist (SII) does not activate the G protein pathway but exclusively recruits β-arrestin and activates ERK (34). Thus, it is imperative when developing drugs to utilize the signaling outfit appropriate for the clinical condition when screening small-molecule libraries. Furthermore, the use of generic high-throughput assays, such as monitoring β-arrestin recruitment, have limitations in small molecule-screening programs.
Other Mechanisms of Modulating GPCR Function
As discussed earlier, ligand-induced selective signaling can be accomplished through single GPCRs recruiting different G proteins and also non-G protein-signaling molecules (i.e. G protein-independent signaling). The dynamic regulation of the relative preponderance of these cell membrane and intracellular molecules will therefore modulate the flavor of the signaling. Similarly, variations in cellular proteins that influence the expression of GPCRs, their translation and trafficking to the cell membrane, and their internalization will have marked effects on ligand stimulation of cells. As summarized in Fig. 6, the first levels of GPCR regulation are gene transcription, translation, and posttranslational processing, which may be regulated by the ligand itself and by other hormones and factors. The synthesized receptor may then be regulated by other sets of proteins in its trafficking to the membrane of the cell. On arrival at the cell membrane, the GPCR may further associate with numerous membrane and intracellular proteins that will potentially alter ligand affinity, ligand selectivity, signaling, cytoskeletal and extracellular matrix interactions, and receptor internalization. GPCRs may further undergo homo- or heterooligomerization to induce transactivation of other receptors or lead to signal modification. GPCR phosphorylation, acetylation, palmityolation, ubiquitinylation, and myristoylation also modify receptor functional properties. Clearly, the integrated effects of all these possibilities are vast. New developments in the effects of phosphorylation or oligomerization have been selected for description in this section, and regulators of membrane trafficking will be touched on in the next section.
Fig. 6.

Potential mechanisms for regulation of GPCRs. Schematic describing how GPCRs can be regulated at many levels: from their biosynthesis (gene transcription, translation, and posttranslational processing) through their trafficking to the cell membrane and, once at the cell surface, through oligomerization and interactions with various other nonreceptor proteins or through modifications such as differential phosphorylation.
Differential receptor phosphorylation
The effects of differential receptor phosphorylation on signaling events has been reviewed by Tobin et al. (35). Using the M3 muscarinic receptor as an example, they demonstrated characteristic fingerprints of receptor phosphorylation in different cells, each with its own spectrum of kinases (Fig. 7A). Each fingerprint imparts both a different flavor of signaling and a different phenotype in cells. The M3 receptor has also been mutated so that certain types of phosphorylation cannot take place, and knock-in mice have been created with associated differential phenotypes, demonstrating the importance of differential phosphorylation in regulation of cellular responses to GPCR activation (35). This represents an interesting level of regulation and opens up many possibilities for differential phosphorylation of GPCRs, which will be affected by expression, activation, deactivation, and recruitment of kinases and phosphatases, all of which influence their conformation and their ability to recruit and activate intracellular signaling pathways (Fig. 7B).
Fig. 7.
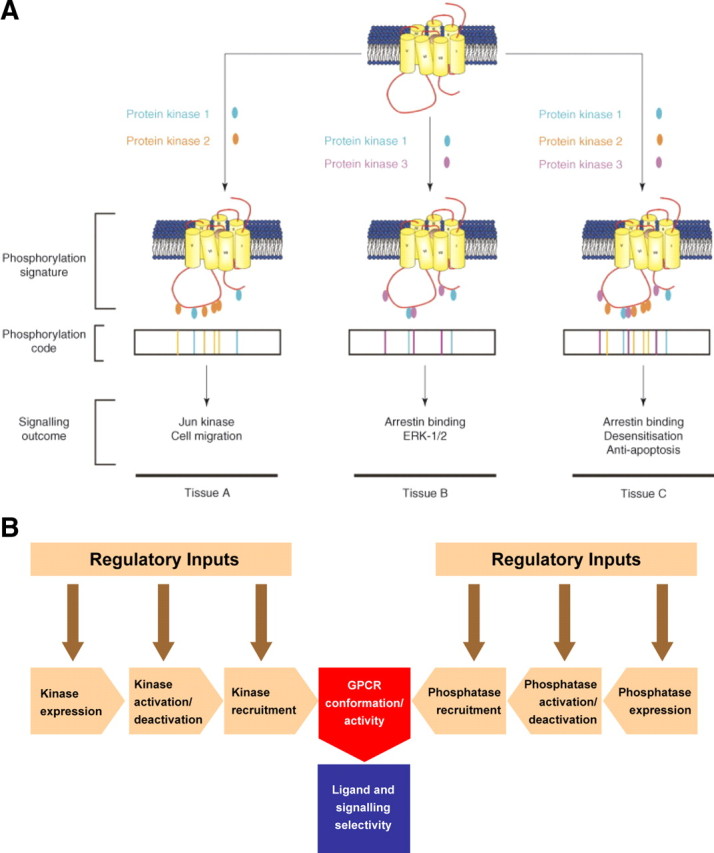
Differential GPCR phosphorylation. A, Schematic describing how differential receptor phosphorylation can lead to differential signaling outcomes. A single GPCR subtype that is able to be phosphorylated by three different protein kinases at distinct sites on the C-terminal tail and ICL3 is shown. These hypothetical protein kinases are employed in different combinations resulting in different phosphorylation profiles. This translates to a code that directs the signaling outcome of the receptor. Thus, the same receptor subtype expressed in three different tissues (tissues A–C) can show different signaling outcomes based on differential receptor phosphorylation. [Reprinted with permission from A. B. Tobin et al.: Trends Pharmacol Sci 29:413, 2008 (35 ). ©Elsevier.] B, Schematic describing potential mechanisms mediating differential GPCR phosphorylation. The phosphorylation of receptors within different cell types can potentially be regulated by several factors, including the expression, activation/deactivation, and recruitment of different kinases and phosphatases. Each level of regulation will itself be under the control of different regulatory inputs. All these factors may result in differential phosphorylation of receptors, leading to an alteration in their conformational activity and subsequent differential ligand/signaling selectivity.
Receptor oligomerization
Receptor oligomerization has attracted a great deal of attention in the last few years. In a recent publication by Ferré et al. (36), a conceptual framework was laid out to classify the various types of oligomerization and their resultant effects on receptor function. As summarized in Fig. 8, they suggest that receptors that are inactive in binding or signaling as monomers, but which become active as oligomers, should be known as “homomeric/heteromeric receptors,” whereas receptors that are intrinsically active as monomers but have new activities as oligomers should be designated as “receptor homomers/ heteromers.”
Fig. 8.

Receptor oligomers: definitions and conceptual framework. Ferré et al. (36 ) have proposed the following revised definitions for oligomeric receptor nomenclature: homomeric/heteromeric receptors are oligomers comprised of two or more receptor molecules that are nonfunctional as monomers but are active as oligomers; receptor homomers/heteromers are oligomers comprised of two or more (functional) receptor molecules, which results in novel biochemical properties distinct from those of the individual monomers. X, Inactive; √, active.
Oligomerization of GPCRs can influence their signaling in several ways. For example, as shown in Fig. 9, dopamine D1 and D2 receptors, as monomers, signal through Gαs and Gαi, respectively, but D1/ D2 heteromers recruit a different signaling pathway, Gαq (37). Interestingly, dopamine signaling in the brain is predominantly through Gαq, suggesting that D1/D2 heteromers predominate in vivo (38).
Fig. 9.

Signal switching by dopamine receptor heteromers. Lee et al. (37 ) have demonstrated that, when individually expressed, dopamine D1 and D2 receptors are activated by the agonist SKF 83959 they signal through coupling to Gαs and Gαi, respectively. However, SKF 83959-activated D1/D2 receptor heteromers couple to Gαq, which equates to in vivo signaling.
Another way in which receptor oligomerization can affect receptor function is through a process known as “transactivation” in which oligomerization of two defective receptors is able to restore receptor functionality. For example, it has been shown that coexpression of two mutant LH receptors in cells (one which cannot signal but can bind LH and a second that can signal but cannot bind LH) allows the receptors to compliment each other so that LH is bound and receptor signaling is restored (39). These findings have now been taken further by Rivero-Müller et al., who have provided the only direct evidence of GPCR oligomerization in vivo by creating knock-in mice with each of these mutant receptors, both of which show highly regressed reproductive tracts (Rivero-Müller, A., personal communication). However, crossing the two knock-in strains to produce offspring with one allele of each mutant receptor restores reproductive activity. This is a most elegant and singular demonstration that receptor oligomers are formed in vivo.
What are the potential future prospects and implications for research in GPCR oligomerization? All indications are that receptor oligomerization will become an important area of study for understanding the biology and pathophysiology of disease, and that the phenomenon offers novel therapeutic possibilities. A good example is that of rimonabant, a cannabinoid receptor 1 antagonist/inverse agonist, which was originally developed as an antagonist of nicotine addiction and therefore an aid to stop smoking. The drug was found to be very effective in diminishing the urge to smoke, but a desirable side effect of treatment was observed in appetite suppression. Research revealed that cannabinoid receptor 1 dimerizes with the appetite-stimulating orexin-1 receptor and that rimonabant antagonizes orexin A stimulation of ERK 1/2 through the orexin-1 receptor (40). Another example is δ-opioid/μ-opioid receptor heteromers. δ- And μ-opioid receptor interactions have been implicated in μ-opioid receptor-mediated morphine tolerance and physical dependence. A hybrid bivalent molecule consisting of μ-opioid receptor agonist coupled with a δ-opioid receptor antagonist, which targets these heteromers, has the potential to convey analgesia without generating these unwanted side effects (41).
These are just a few examples of GPCR heteromers. The formation of heteromers between GPCRs and other receptors or nonreceptor proteins suggests that these interactions create a huge spectrum of additional subtleties in the way that GPCR activities may be modulated and how they may be involved in cross talk with other regulatory systems.
Regulation of GPCR Membrane Expression
Understanding the mechanisms and identifying the proteins involved in receptor internalization, down-regulation, and uncoupling from signaling proteins has been an area of intensive research for many years. An emerging area of considerable interest is the identification and delineation of the roles of proteins involved in escorting GPCRs through the various cellular compartments to the cell surface. GPCRs are dynamically regulated to adapt responses of cells to cognate ligands by regulating receptor trafficking to the cell surface or internalization and degradation/recycling of receptors. These processes therefore represent novel means of physiological regulation and targets for potential therapeutic intervention (42). It is not possible to describe all these proteins here but of particular interest is a group of proteins, listed in Fig. 10, called “nonclassical private GPCR chaperones,” which interact with GPCRs in intracellular compartments to facilitate their cell-surface expression (43). One such example, melanocortin 2-receptor accessory protein (MRAP), is an excellent example of the importance of these proteins. Genetically inherited hypocortisolism in patients with normal ACTH and melanocortin-2 (MC2) receptor genes presented a clinical conundrum until mutations in MRAP were identified that ablated its ability to shuttle the MC2 receptor to the surface of the cell (44, 45, 46). Given the large number of intracellular proteins involved in regulating GPCR trafficking to the cell surface, it is evident that there are many opportunities for understanding how cell surface of expression of GPCRs is regulated and how this may be modulated for therapeutic interventions. For example, inhibiting MRAP function could potentially be used as an adjunct therapy for Cushing’s disease, because the overproduction of ACTH could be counteracted by the absence of MC2 receptor expression on the cell surface. The importance of MRAP, and its paralog MRAP2, in the regulation of melanocortin receptors has been recently reviewed, in detail, by Webb and Clark (47).
Fig. 10.

Nonclassical private GPCR chaperones. A list of nonclassical chaperones and escorts that regulate GPCR translocation to the plasma membrane (43 ).
Pharmacochaperones: rescue of mutant and underexpressing GPCRs
A relatively new area of GPCR pharmacology is that of pharmacochaperones. These are hydrophobic small molecules that can penetrate the cell membrane, bind to the nascent GPCR, and rescue or shuttle mutant and underexpressing GPCRs to the cell membrane. Conn and Janovick (48) have pioneered the use of cell-permeant small molecule GnRH receptor antagonists to rescue poorly expressed GnRH mutant receptors. In our own laboratory, we routinely employ such antagonists to facilitate the characterization of GnRH receptor mutations directed at delineating receptor function. As an example, a mutant human GnRH receptor, which exhibits no inositol phosphate response to GnRH, exhibits a robust response to GnRH after preincubation with a small molecule GnRH antagonist from Neurocrine Bioscience (NBI42902) (Tello, J. A., unpublished data) (Fig. 11A). Similar results have been observed using a mutant human LH receptor, which was identified in patients with impaired fertility and has been shown to be retained intracellularly (49). When exposed to a cell-permeant small molecule agonist the mutant receptors were recruited to the cell surface (Fig. 11B). In addition, treating the mutant receptor with LH elicits a very poor response compared with the wild type, whereas treatment with the small molecule agonist restores functionality (Newton, C. L., unpublished data). There are now quite a few examples of the rescue of underexpressing mutant GPCR endocrine receptors (50).
Fig. 11.
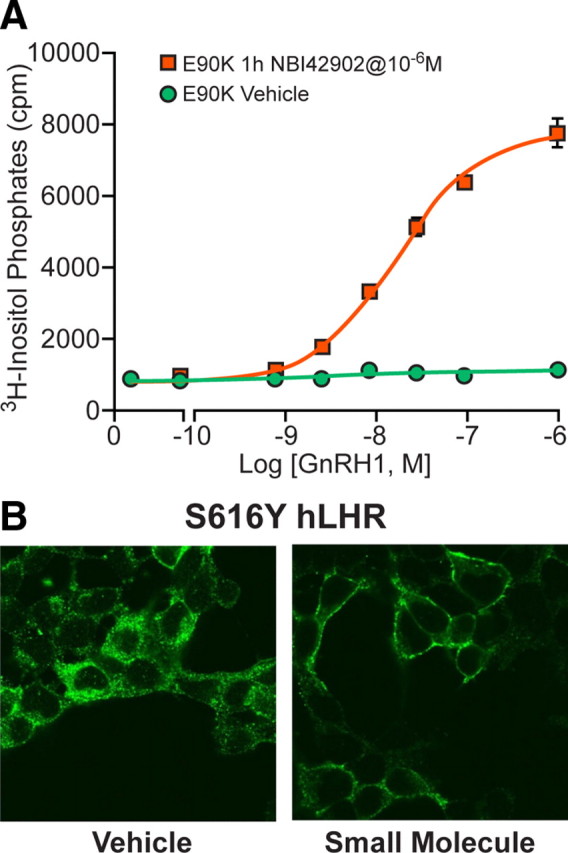
Rescue of mutant GPCRs using pharmacochaperones. A, Rescue of mutant GnRH receptors by the small-molecule pharmacochaperone NBI42902. Cells expressing mutant (E90K) human GnRH R1 were preincubated in the presence/absence of NBI42902 (1 μm) for 1 h before measurement of inositol phosphate accumulation in response to stimulation with GnRH I. In the absence of the small molecule no inositol phosphate production in measured, but a robust response occurs after preincubation with the pharmacochaperone. [Figure kindly provided by Javier Tello, MRC Human Reproductive Sciences Unit, Edinburgh, UK (unpublished data)]. B, Rescue of mutant LH receptors by a small-molecule pharmacochaperone. Cells expressing mutant (S616Y) human LH receptor (kindly supplied by Deborah Segaloff, Department of Molecular Physiology and Biophysics, The University of Iowa, Iowa City, IA) were preincubated in the presence/absence of a small-molecule agonist for 24 h, before permeabilization of the cells, labeling of the receptors with fluorescent (green) antibodies, and visualization by confocal microscopy. In the absence of the small-molecule receptor, localization is largely intracellular. However, preincubation with the pharmacochaperone causes translocation of the receptor to the cell surface as is seen in untreated wild-type human LH receptors (Newton, C. L., unpublished data).
Novel GPCRS and Ligands in Endocrinology
A number of new roles for GPCRs and their ligands have been identified over the past 18 months. We have selected examples of novel regulators of pancreatic β-cell function and novel GPCRs regulating gonadotropic hormones.
Novel pancreatic β-cell GPCRs
About 20 GPCRs have now been shown to be expressed in pancreatic β-cells, all of which can potentially stimulate or inhibit insulin secretion. The glucagon-like peptide 1 (GLP1) receptor is one of these, and GLP1 receptor activators are already being used clinically for the treatment of type 2 diabetes (51). Insulin secretion is stimulated by glucose transport through the glucose transporter 2 into the β-cell. Glucose metabolism generates ATP which leads to the closing of potassium channels, depolarization, and opening of voltage-gated calcium channels. Calcium entry into the cell elicits the exocytosis of insulin. Activation of GPCRs such as GLP1 can enhance the amount of intracellular calcium, for example through activation of Gαq/11 and subsequent generation of inositol triphosphate and release of calcium from intracellular stores, thereby potentiating glucose stimulation of insulin secretion. Among the other GPCRs identified in β-cells are the newly discovered free fatty acid receptors, GPR40, 43, and 41. GPR40 couples to Gαq/11, so we might predict that free fatty acid would enhance the calcium response of the β-cell to glucose and increase insulin secretion. And indeed, this appears to be the case. Insulin responses to glucose are improved in mutant mice overexpressing the GPR40 receptor and in normal rats treated with GRP40 agonists (52, 53, 54). Intriguingly, Milligan and colleagues (55) have shown that thiazolidinedione compounds (glitazones) used in the treatment of type 2 diabetes, are able to activate GPR40 through the recruitment of Gαq/11 leading to ERK 1/2 MAPK activation. The various glitazones tested revealed different effective doses (ED50) and time frames of ERK1/2 activation (Fig. 12). These observations suggest that part of the action of glitazones may be through GPR40 and that this might underlie variations in the clinical effectiveness of different glitazones.
Fig. 12.

Differential thiazolidinedione activation of GPR40. Different thiazolidinedione drugs provide distinct kinetics and efficacy of ERK1/2 MAPK phosphorylation. Cells expressing GPR40 were treated with a range of thiazolidinedione drugs [rosiglitazone (100 μm), ciglitazone (100 μm), troglitazone (10 μm), and pioglitazone (100 μm)], and the phosphorylation of ERK1/2 MAPK was measured over a 3-h period. [Adapted with permission from N. J. Smith et al.: J Biol Chem 284:17527, 2009 (55 ). ©American Society for Biochemistry and Molecular Biology.]
Novel neuroendocrine GPCRs regulating reproduction
There have been a number of breakthroughs in neuroendocrinology in the last year. Two of these are briefly touched on here. After the seminal discovery that kisspeptin/GPR54 acts as a major whole-body sensor mediating diverse effects on the GnRH neuron (56, 57, 58), Topaloglu et al. (59) described mutations in neurokinin B (NKB) and its receptor (TACR3), which give rise to hypogonadotropic hypogonadism and pubertal failure in four Turkish families. How does this relate to the kisspeptin/GPR54 system? Interestingly, NKB colocalizes to kisspeptin neurons, along with Dynorphin A, in ewes (60), so we now have an enrapturing challenge to understand the interplay of these three neuropeptides in modulating the GnRH neuron through their cognate GPCRs. Are they being cosecreted and interacting at the level of the GnRH neuron or are they working in an autocrine feedback type of modality by modulating each others secretion in kisspeptin neurons, or possibly a combination of both (Fig. 13)? Their relative biosynthesis, processing, and secretion may also be differentially regulated by various neuronal inputs, providing a further level for subtlety of regulation.
Fig. 13.

NKB and GnIH: novel regulators of gonadotropin in man. Schematic describing the potential mechanisms for control of gonadotropin secretion by NKB and GnIH. Kisspeptin (Kiss) released from kisspeptin neurons within the hypothalamus is known to control secretion of GnRH from GnRH neurons through interaction with its cognate receptor (KissR/GPR54). Secretion of GnRH from these neurons then leads to secretion of gonadotropins (LH/FSH) from pituitary gonadotropes. NKB is colocalized in kisspeptin neurons and is postulated to be a novel regulator of GnRH secretion either through direct interaction with GnRH neurons or through autocrine interactions with kisspeptin neurons. GnIH is a potent inhibitor of GnRH stimulation of gonadotropin secretion from cultured gonadotropes but may also operate by inhibiting the activity of GnRH neurons.
Another discovery, that of a gonadotropin-inhibitory hormone (GnIH) in birds some years ago, has gathered momentum recently with studies in mammals. In the laboratory of Clarke et al. (61), GnIH (like kisspeptin, an RF-amide neuropeptide) was found to be a very potent inhibitor of GnRH stimulation of gonadotropin secretion, in cultured ovine gonadotropes and in vivo. GnIH may also operate by directly or indirectly inhibiting the GnRH neurons themselves (62, 63, 64).
The discovery of NKB, dynorphin A, and GnIH as neuroendocrine regulators has provided new opportunities for research on novel GPCRs in fine tuning the hypothalamic-pituitary-gonadal axis and provides new pathways in which to interrogate feedback mechanisms and metabolic, photoperiod, and behavioral influences on the reproductive system (Fig. 13).
Conclusion: What Lies Ahead?
The pioneering technical advances recently accomplished are promising to facilitate the crystallization of additional GPCRs and the solving of their structures. Nevertheless, the challenges remain formidable, and the structural determination of substantial numbers of GPCRs is not very likely in the near future. Solving additional GPCR structures, and especially those of receptors from other GPCR families, and receptors for other classes of ligands, will provide insights into common and unique structural features involved in ligand binding, receptor activation, and interactions with signaling proteins. This information, in turn, will assist in guiding molecular modeling of unsolved GPCR structures more accurately.
The limitations of crystal structures in representing a singular nondynamic state or conformation of GPCRs may defy satisfactory resolution of the full repertoire of events underlying GPCR function. The development of novel biophysical techniques for monitoring these structural changes is encouraging and has considerable promise for future studies.
The insights into GPCR structure and function also appear to be bringing the holy grail of in silico discovery and development of small-molecule analogs closer to reality. Further finessing of the approach and translation into success in drug discovery is now a very reasonable expectation.
The discovery of a multitude of intracellular protein interactions with GPCRs points to almost limitless potential for modulation of their expression, ligand selectivity, and signaling to produce an array of cellular and physiological phenotypes. A particular challenge will be the determination of the actual in vivo functioning of GPCRs and the development of specific therapeutics targeting specific phenotypes. However, the recognition of the diversity of cell context-driven function provides the first steps toward more specific drug development.
We can also anticipate that the generation of knockout and hypomorph animals for every GPCR, and the discovery of GPCR ligand analogs and modulators of GPCR function, will provide early preclinical tools for delineating physiological function and regulation.
Deorphanization of GPCRs is advancing at pace, and the relatively under-explored large group of odorant receptors and smaller GPCR families offer exciting possibilities to discover new regulators of the endocrine system.
Acknowledgments
We thank Graeme Milligan, Brian Kobilka, Craig McArdle, Richard Henderson, Scott Struthers, Zhi-liang Lu, Adam Pawson, Antonia Roseweir, and Javier Tello for helpful input into the presentation.
Footnotes
Disclosure Summary: The authors have nothing to disclose.
First Published Online December 17, 2009
Abbreviations: ECL, Extracellular loop; GLP1, glucagon-like peptide 1; GnIH, gonadotropin-inhibitory hormone; GPCR, G protein-coupled receptor; ICL, intracellular loop; LiSS, ligand-induced selective signaling; NKB, neurokinin B; NMR, nuclear magnetic resonance; TM, transmembrane.
References
- 1.Neuhaus EM, Zhang W, Gelis L, Deng Y, Noldus J, Hatt H2009. Activation of an olfactory receptor inhibits proliferation of prostate cancer cells. J Biol Chem 284:16218–16225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yona S, Lin HH, Siu WO, Gordon S, Stacey M2008. Adhesion-GPCRs: emerging roles for novel receptors. Trends Biochem Sci 33:491–500 [DOI] [PubMed] [Google Scholar]
- 3.Krebs A, Villa C, Edwards PC, Schertler GF1998. Characterisation of an improved two-dimensional p22121 crystal from bovine rhodopsin. J Mol Biol 282:991–1003 [DOI] [PubMed] [Google Scholar]
- 4.Schertler GF1998. Structure of rhodopsin. Eye (Lond) 12 :504–510 [DOI] [PubMed]
- 5.Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR2004. Gonadotropin-releasing hormone receptors. Endocr Rev 25:235–275 [DOI] [PubMed] [Google Scholar]
- 6.Ballesteros J, Kitanovic S, Guarnieri F, Davies P, Fromme BJ, Konvicka K, Chi L, Millar RP, Davidson JS, Weinstein H, Sealfon SC1998. Functional microdomains in G-protein-coupled receptors. The conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J Biol Chem 273:10445–10453 [DOI] [PubMed] [Google Scholar]
- 7.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M2000. Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289:739–745 [DOI] [PubMed] [Google Scholar]
- 8.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC2008. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP2008. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454:183–187 [DOI] [PubMed] [Google Scholar]
- 10.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK2007. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450:383–387 [DOI] [PubMed] [Google Scholar]
- 11.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP2008. Crystal structure of opsin in its G-protein-interacting conformation. Nature 455:497–502 [DOI] [PubMed] [Google Scholar]
- 12.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF2008. Structure of a β1-adrenergic G-protein-coupled receptor. Nature 454:486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC2007. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318:1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weis WI, Kobilka BK2008. Structural insights into G-protein-coupled receptor activation. Curr Opin Struct Biol 18:734–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanson MA, Stevens RC2009. Discovery of new GPCR biology: one receptor structure at a time. Structure 17:8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF2004. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol 343:1409–1438 [DOI] [PubMed] [Google Scholar]
- 17.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL2008. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci USA 105:7439–7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao X, Parnot C, Deupi X, Ratnala VR, Swaminath G, Farrens D, Kobilka B2006. Coupling ligand structure to specific conformational switches in the β2-adrenoceptor. Nat Chem Biol 2:417–422 [DOI] [PubMed] [Google Scholar]
- 19.Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, Ziliox M, Sakmar TP, Sheves M, Reeves PJ, Smith SO, Eilers M2009. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat Struct Mol Biol 16:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK2009. Structure-based discovery of β2-adrenergic receptor ligands. Proc Natl Acad Sci USA 106:6843–6848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Millar RP, Pawson AJ2004. Outside-in and inside-out signaling: the new concept that selectivity of ligand binding at the gonadotropin-releasing hormone receptor is modulated by the intracellular environment. Endocrinology 145:3590–3593 [DOI] [PubMed] [Google Scholar]
- 22.Kenakin T1995. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci 16:232–238 [DOI] [PubMed] [Google Scholar]
- 23.Millar RP, Pawson AJ, Morgan K, Rissman EF, Lu ZL2008. Diversity of actions of GnRHs mediated by ligand-induced selective signaling. Front Neuroendocrinol 29:17–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.López de Maturana R, Pawson AJ, Lu ZL, Davidson L, Maudsley S, Morgan K, Langdon SP, Millar RP2008. Gonadotropin-releasing hormone analog structural determinants of selectivity for inhibition of cell growth: support for the concept of ligand-induced selective signaling. Mol Endocrinol 22:1711–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu ZL, Coetsee M, White CD, Millar RP2007. Structural determinants for ligand-receptor conformational selection in a peptide G protein-coupled receptor. J Biol Chem 282:17921–17929 [DOI] [PubMed] [Google Scholar]
- 26.Pfleger KD, Pawson AJ, Millar RP2008. Changes to gonadotropin-releasing hormone (GnRH) receptor extracellular loops differentially affect GnRH analog binding and activation: evidence for distinct ligand-stabilized receptor conformations. Endocrinology 149:3118–3129 [DOI] [PubMed] [Google Scholar]
- 27.Leduc M, Breton B, Galés C, Le Gouill C, Bouvier M, Chemtob S, Heveker N2009. Functional selectivity of natural and synthetic prostaglandin EP4 receptor ligands. J Pharmacol Exp Ther 331:297–307 [DOI] [PubMed] [Google Scholar]
- 28.Cussac D, Boutet-Robinet E, Ailhaud MC, Newman-Tancredi A, Martel JC, Danty N, Rauly-Lestienne I2008. Agonist-directed trafficking of signalling at serotonin 5-HT2A, 5-HT2B and 5-HT2C-VSV receptors mediated Gq/11 activation and calcium mobilisation in CHO cells. Eur J Pharmacol 594:32–38 [DOI] [PubMed] [Google Scholar]
- 29.Woo AY, Wang TB, Zeng X, Zhu W, Abernethy DR, Wainer IW, Xiao RP2009. Stereochemistry of an agonist determines coupling preference of β2-adrenoceptor to different G proteins in cardiomyocytes. Mol Pharmacol 75:158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Georgieva T, Devanathan S, Stropova D, Park CK, Salamon Z, Tollin G, Hruby VJ, Roeske WR, Yamamura HI, Varga E2008. Unique agonist-bound cannabinoid CB1 receptor conformations indicate agonist specificity in signaling. Eur J Pharmacol 581:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jongsma M, van Unen J, van Loenen PB, Michel MC, Peters SL, Alewijnse AE2009. Different response patterns of several ligands at the sphingosine-1-phosphate receptor subtype 3 (S1P(3)). Br J Pharmacol 156:1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ2003. Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA 100:10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajagopal K, Lefkowitz RJ, Rockman HA2005. When 7 transmembrane receptors are not G protein-coupled receptors. J Clin Invest 115:2971–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Violin JD, Lefkowitz RJ2007. β-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci 28:416–422 [DOI] [PubMed] [Google Scholar]
- 35.Tobin AB, Butcher AJ, Kong KC2008. Location, location, location.site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci 29:413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KD, Pin JP, Volkow ND, Waldhoer M, Woods AS, Franco R2009. Building a new conceptual framework for receptor heteromers. Nat Chem Biol 5:131–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lançca AJ, O'Dowd BF, George SR2004. Dopamine D1 and D2 receptor Co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem 279:35671–35678 [DOI] [PubMed] [Google Scholar]
- 38.Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, O'Dowd BF, George SR2007. D1–D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci USA 104:654–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee C, Ji I, Ryu K, Song Y, Conn PM, Ji TH2002. Two defective heterozygous luteinizing hormone receptors can rescue hormone action. J Biol Chem 277:15795–15800 [DOI] [PubMed] [Google Scholar]
- 40.Ellis J, Pediani JD, Canals M, Milasta S, Milligan G2006. Orexin-1 receptor-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J Biol Chem 281:38812–38824 [DOI] [PubMed] [Google Scholar]
- 41.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS2005. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci USA 102:19208–19213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooray SN, Chan L, Webb TR, Metherell L, Clark AJ2009. Accessory proteins are vital for the functional expression of certain G protein-coupled receptors. Mol Cell Endocrinol 300:17–24 [DOI] [PubMed] [Google Scholar]
- 43.Achour L, Labbé-Jullié C, Scott MG, Marullo S2008. An escort for GPCRs: implications for regulation of receptor density at the cell surface. Trends Pharmacol Sci 29:528–535 [DOI] [PubMed] [Google Scholar]
- 44.Chung TT, Webb TR, Chan LF, Cooray SN, Metherell LA, King PJ, Chapple JP, Clark AJ2008. The majority of adrenocorticotropin receptor (melanocortin 2 receptor) mutations found in familial glucocorticoid deficiency type 1 lead to defective trafficking of the receptor to the cell surface. J Clin Endocrinol Metab 93:4948–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooray SN, Almiro Do Vale I, Leung KY, Webb TR, Chapple JP, Egertová M, Cheetham ME, Elphick MR, Clark AJ2008. The melanocortin 2 receptor accessory protein exists as a homodimer and is essential for the function of the melanocortin 2 receptor in the mouse y1 cell line. Endocrinology 149:1935–1941 [DOI] [PubMed] [Google Scholar]
- 46.Metherell LA, Chapple JP, Cooray S, David A, Becker C, Rüschendorf F, Naville D, Begeot M, Khoo B, Nürnberg P, Huebner A, Cheetham ME, Clark AJ2005. Mutations in MRAP, encoding a new interacting partner of the ACTH receptor, cause familial glucocorticoid deficiency type 2. Nat Genet 37:166–170 [DOI] [PubMed] [Google Scholar]
- 47.Webb TR, Clark AJ 23 October 2009. Minireview: The melanocortin 2 receptor accessory proteins. Mol Endocrinol 10.1210/me. 2009-0283 [DOI] [PMC free article] [PubMed]
- 48.Conn PM, Janovick JA2009. Trafficking and quality control of the gonadotropin releasing hormone receptor in health and disease. Mol Cell Endocrinol 299:137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mizrachi D, Segaloff DL2004. Intracellularly located misfolded glycoprotein hormone receptors associate with different chaperone proteins than their cognate wild-type receptors. Mol Endocrinol 18:1768–1777 [DOI] [PubMed] [Google Scholar]
- 50.Dunham JH, Hall RA2009. Enhancement of the surface expression of G protein-coupled receptors. Trends Biotechnol 27:541–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahrén B2009. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov 8:369–385 [DOI] [PubMed] [Google Scholar]
- 52.Brownlie R, Mayers RM, Pierce JA, Marley AE, Smith DM2008. The long-chain fatty acid receptor, GPR40, and glucolipotoxicity: investigations using GPR40-knockout mice. Biochem Soc Trans 36:950–954 [DOI] [PubMed] [Google Scholar]
- 53.Doshi LS, Brahma MK, Sayyed SG, Dixit AV, Chandak PG, Pamidiboina V, Motiwala HF, Sharma SD, Nemmani KV2009. Acute administration of GPR40 receptor agonist potentiates glucose-stimulated insulin secretion in vivo in the rat. Metabolism 58:333–343 [DOI] [PubMed] [Google Scholar]
- 54.Nagasumi K, Esaki R, Iwachidow K, Yasuhara Y, Ogi K, Tanaka H, Nakata M, Yano T, Shimakawa K, Taketomi S, Takeuchi K, Odaka H, Kaisho Y2009. Overexpression of GPR40 in pancreatic β-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes 58:1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith NJ, Stoddart LA, Devine NM, Jenkins L, Milligan G2009. The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. J Biol Chem 284:17527–17539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roseweir AK, Millar RP2009. The role of kisspeptin in the control of gonadotrophin secretion. Hum Reprod Update 15:203–212 [DOI] [PubMed] [Google Scholar]
- 57.Oakley AE, Clifton DK, Steiner RA2009. Kisspeptin signaling in the brain. Endocr Rev 30:713–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tena-Sempere M2006. KiSS-1 and reproduction: focus on its role in the metabolic regulation of fertility. Neuroendocrinology 83:275–281 [DOI] [PubMed] [Google Scholar]
- 59.Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Ozbek MN, Imamoglu S, Akalin NS, Yuksel B, O'Rahilly S, Semple RK2009. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the central control of reproduction. Nat Genet 41:354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goodman RL, Lehman MN, Smith JT, Coolen LM, de Oliveira CV, Jafarzadehshirazi MR, Pereira A, Iqbal J, Caraty A, Ciofi P, Clarke IJ2007. Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology 148:5752–5760 [DOI] [PubMed] [Google Scholar]
- 61.Clarke IJ, Sari IP, Qi Y, Smith JT, Parkington HC, Ubuka T, Iqbal J, Li Q, Tilbrook A, Morgan K, Pawson AJ, Tsutsui K, Millar RP, Bentley GE2008. Potent action of RFamide-related peptide-3 on pituitary gonadotropes indicative of a hypophysiotropic role in the negative regulation of gonadotropin secretion. Endocrinology 149:5811–5821 [DOI] [PubMed] [Google Scholar]
- 62.Wu M, Dumalska I, Morozova E, van den Pol AN, Alreja M2009. Gonadotropin inhibitory hormone inhibits basal forebrain vGluT2-gonadotropin-releasing hormone neurons via a direct postsynaptic mechanism. J Physiol 587:1401–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ducret E, Anderson GM, Herbison AE2009. RFamide-related peptide-3, a mammalian gonadotropin-inhibitory hormone ortholog, regulates gonadotropin-releasing hormone neuron firing in the mouse. Endocrinology 150:2799–2804 [DOI] [PubMed] [Google Scholar]
- 64.Anderson GM, Relf HL, Rizwan MZ, Evans JJ2009. Central and peripheral effects of RFamide-related peptide-3 on luteinizing hormone and prolactin secretion in rats. Endocrinology 150:1834–1840 [DOI] [PubMed] [Google Scholar]
- 65.Kobilka B, Structure and dynamics of the human β2 adrenoceptor. Proc British Pharmalogical Society 3rd Focused Meeting, Cell Signaling, The University of Leicester, Leicester, UK, 2009