

Abstract
Estrogen-related receptor α (ERRα) is an orphan nuclear receptor highly expressed in the kidney, an organ playing a central role in blood pressure regulation through electrolyte homeostasis and the renin-angiotensin system. Physiological analysis revealed that, relative to wild-type mice, ERRα null mice are hypotensive despite significant hypernatremia, hypokalemia, and slight hyperreninemia. Using a combination of genome-wide location analysis and expression profiling, we demonstrate that ERRα regulates the expression of channels involved in renal Na+ and K+ handling (Scnn1a, Atp1a1, Atp1b1) and altered in Bartter syndrome (Bsnd, Kcnq1). In addition, ERRα regulates the expression of receptors implicated in the systemic regulation of blood pressure (Ghr, Gcgr, Lepr, Npy1r) and of genes within the renin-angiotensin pathway (Ren1, Agt, Ace2). Our study thus identifies ERRα as a pleiotropic regulator of renal control of blood pressure, renal Na+/K+ homeostasis, and renin-angiotensin pathway and suggests that modulation of ERRα activity could represent a potential avenue for the management of hypertension.
This physiological genomics study identifies the orphan nuclear receptor ERRα as a regulator of blood pressure, Na+/K+ homeostasis and the renin-angiotensin pathway.
The kidneys play a central role in blood pressure regulation through the production of renin and maintenance of electrolyte homeostasis (1). Defects in the expression or activity of renal ion channels and electrolyte transporters induce a panel of physiological conditions and symptoms, notably Bartter syndrome, a genetically inherited disorder of renal electrolyte transport caused by the loss-of-function mutations in various ion channels (2). Although considerable progress has been made in understanding ion channel function and associated channelopathies of the kidney, knowledge about the molecular mechanisms underlying their coordinated expression in this tissue is still lacking.
Estrogen-related receptor α (ERRα; NR3B1) is an orphan member of the nuclear receptor superfamily (3). In the absence of a cognate ligand, ERRα is a constitutive transcription factor and its activity is dependent on the presence of coregulatory proteins and posttranslational modifications generated in response to external cues (4, 5, 6, 7, 8, 9, 10, 11). ERRα is ubiquitously expressed, but its highest expression levels are detected in tissues with elevated energy demands such as the heart, brown adipose tissue, and the kidneys (12, 13, 14). The expression of ERRα is also known to be under circadian regulation in the liver, skeletal muscle, bone, uterus, and adipose tissues (15, 16). Whether the expression of ERRα is subjected to the same circadian regulation in the kidney is still unknown.
Acting as a transcription factor, ERRα has been shown to control the expression of genes involved in all aspects of bioenergetics including fat and glucose metabolism and energy production by the mitochondria as well as intracellular fuel sensing (17). In agreement with these findings, phenotypic analyses of mice lacking ERRα have shown that, whereas ERRα is dispensable for normal development, growth, and reproduction (18), ERRα-deficient mice are unable to respond to physiological challenges requiring energy demands above levels necessary to maintain basic physiological functions (17, 19).
Recently, the combination of chromatin immunoprecipitation (ChIP) and genomic DNA arrays (ChIP-on-chip) allowed the identification of nuclear receptor-binding sites in both promoters and more distal enhancers of genes as well as the construction of extensive regulatory networks (20, 21). For example, the combination of ChIP-on-chip, microarray gene expression profiling, and phenotypic analysis of genetically modified mice has led to the discovery of comprehensive ERRα-dependent transcriptional programs in the heart, macrophages, and breast cancer cells that were associated with biological processes specific to each tissue (17, 22, 23, 24, 25, 26). In this work, we used the powerful mix of a physiological genomics-based approach together with phenotypic studies of ERRα null mice to explore the role of ERRα in the kidneys. Taken together, our results revealed that ERRα null mice display a phenotype reminiscent of a Bartter-like syndrome and identify ERRα as an important transcriptional regulator of two important components of blood pressure control by the kidney, namely Na+/K+ homeostasis and the renin-angiotensin pathway.
Results
Hypernatremia and hypokalemia in the ERRα knockout mice
To test the role of ERRα in the kidneys, we initially compared the overall renal function and electrolyte handling in wild-type vs. ERRα null mice by measuring the urine and blood constituents (Table 1). ERRα null mice tend to drink slightly less, although not in a statistically significant manner, but are excreting significantly less urine volume than their wild-type siblings. The urinary measures retained the same differences and level of significance when corrected for body weight which, as previously observed, is slightly smaller due to a lower fat content (Ref. 18 and data not shown). The urinary excretion of creatinine was found to be significantly decreased in the ERRα null mice. However, the blood concentrations of creatinine were similar between the wild-type and the ERRα null mice, and the glomerular filtration rate was slightly reduced but not significantly. The blood Na+ concentrations were in the normal range for both groups but were significantly higher in the ERRα null mice, resulting in a mild hypernatremia relative to the wild-type animals. Also, the blood concentrations of K+ in the ERRα null mice, although still in the normal range, were significantly lower resulting in a relative hypokalemia compared with the wild-type mice. No significant difference was observed with regard to blood calcium and chloride content. ERRα null mice also excreted the same amount of chloride and Ca2+ as the wild-type mice, suggesting a normal chloride and Ca2+ handling under basal physiological conditions. Taken together, these results suggest that the ERRα null mice possess a relatively normal renal filtration capacity, but also indicate a deficiency in Na+/K+ handling with a mechanism favoring Na+ retention.
Table 1.
Blood and urinary parameters in wild type and ERRα null mice
| WT | ERRα KO | P Value | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | sem | Mean | sem | ||||||
| Body weight (g) | 24.5 | 0.3 | 22.2 | 0.6 | 0.002 | ||||
| Water intake (ml) | 4.6 | 0.7 | 3.4 | 0.1 | 0.100 | ||||
| Urine volume (ml) | 1.07 | 0.12 | 0.54 | 0.12 | 0.008 | ||||
| GFR (ml/d) | 1.00 | 0.11 | 0.75 | 0.12 | 0.129 | ||||
| Urinary parameters (mmol/d) | |||||||||
| Creatinine | 4.0 | 0.3 | 2.4 | 0.4 | 0.025 | ||||
| Na+ | 166.7 | 24.4 | 94.6 | 14.5 | 0.035 | ||||
| K+ | 323.5 | 38.3 | 206.1 | 40.6 | 0.069 | ||||
| Na/K ratio | 0.51 | 0.04 | 0.49 | 0.05 | 0.816 | ||||
| Chloride | 270.1 | 46.2 | 160.8 | 28.6 | 0.097 | ||||
| Ca2+ | 2.1 | 0.5 | 1.9 | 0.5 | 0.971 | ||||
| Blood parameters | |||||||||
| Urea | 8.32 | 0.53 | 8.23 | 0.30 | 0.885 | ||||
| Creatinine | 18.21 | 1.87 | 17.40 | 1.95 | 0.760 | ||||
| Na+ | 140.79 | 0.90 | 144.80 | 1.00 | 0.005 | ||||
| K+ | 8.29 | 0.21 | 6.73 | 0.15 | <0.001 | ||||
| Na+/K+ ratio | 16.04 | 0.65 | 20.30 | 0.62 | <0.001 | ||||
| Chloride | 107.95 | 0.63 | 108.60 | 0.63 | 0.455 | ||||
| Ca2+ | 2.17 | 0.07 | 2.22 | 0.03 | 0.462 | ||||
Values are presented as mean ± sem Unpaired Student t test was used for statistical analysis. Measurements were made at ZT4 on five metabolic cages each housing three to five mice per cage. A total of 19 mice per genotype were used for the analysis. GFR, Glomerular filtration rate; KO, knockout; WT, wild type.
ERRα knockout mice are hypotensive
Because electrolyte homeostasis can affect blood pressure, we used telemetry to determine whether the ERRα null mice showed differences in blood pressure parameters. Continuous measurements over a 72-h period are presented in Fig. 1, A and B. Telemetry analysis revealed that the ERRα null mice display a significantly lower diastolic and systolic blood pressure between zeitgeber time (ZT) 12 and ZT24, the activity period of nocturnal animals (Fig. 1, A and B, respectively). Heart rate was identical between the ERRα null and wild-type mice during this period (Fig. 1C). However, during the resting period (between ZT24 and ZT12), the heart rate of the ERRα null mice tends to remain slightly higher (Fig. 1C). In addition, the ERRα null mice showed a decreased activity state compared with the wild type during the nocturnal period (Fig. 1D). As noted above, the expression of ERRα has been shown to cycle according to a circadian rhythm in a tissue-specific manner in the liver, skeletal muscle, and brown and white adipose tissues as well as in estrogen-responsive tissues (15, 16). Therefore, we tested whether there existed a circadian regulation of ERRα expression in the kidneys. We found that the level of expression of renal ERRα mRNA was indeed cycling according to the circadian rhythm (Fig. 1E) and that the cycling of ERRα closely paralleled that of blood pressure. These results indicate that the presence of ERRα is required to sustain elevated blood pressure of mice during the period of nocturnal activity.
Fig. 1.

Blood pressure parameters and heart rate in ERRα null mice. A, Telemetry analysis of diastolic blood pressure over a 72-h period. B, Telemetry analysis of systolic blood pressure over a 72-h period. C, Heart rate of wild-type and ERRα null mice over a 72-h period. Gray shading, Nighttime periods (ZT12–ZT24); gray line, wild-type mice; black line, ERRα null mice; bpm, beats per min. D, Activity levels of wild-type and ERRα null mice over a 72-h period. E, Analysis of the temporal expression of ERRα in the kidney over a 24-h period. *, P < 0.05.
Identification of ERRα target genes in the kidney by ChIP-on-chip
To determine whether the transcriptional program governed by ERRα in the kidneys could provide a molecular basis for the phenotypic differences observed in the ERRα null animals, we performed a ChIP-on-chip experiment using tiled genomic DNA arrays covering the extended promoter regions (∼−5.5 to ∼+2.5 kb from transcriptional start sites) of approximately 17,000 mouse genes and chromatin isolated from wild-type mouse kidneys. Analysis of the ChIP-on-chip dataset identified 1391 high-confidence ERRα-binding sites mapping to 1366 different genes (Table S1 published as supplemental data on The Endocrine Society’s Journals Online web site at http://endojournals.mend.org). The ERRα target gene list was then analyzed using FatiGO. In agreement with the known function of ERRα in energy homeostasis (17), genes involved in the oxidative phosphorylation process [as reflected in the gene ontology (GO) term oxidoreductase (0016491), mainly containing mitochondrial enzymes] were enriched in the analysis (Fig. 2A). Other categories of interest enriched for ERRα targets included transcriptional repressors (Ncor2, Sin3A, Hdac5) and activators (Ncoa1, PPARa) and enzymes with transferase activity (Got1, Pdk4, Stk11). In addition and consistent with the differences observed in electrolyte handling by the ERRα null mice, the analysis also revealed that the ion transporter activity category (GO:0015075) was significantly enriched among the ERRα targets (Fig. 2A). We further dissected the ion specificity within the ion transporter activity category and found that a great proportion of target genes within this category were implicated in Na+ and K+ handling (Fig. 2B).
Fig. 2.
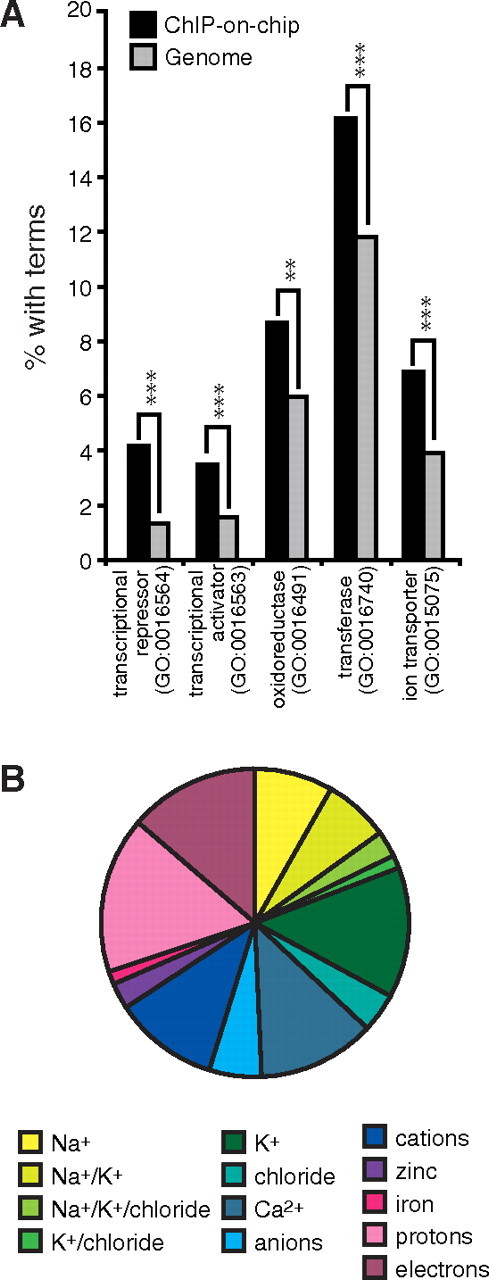
ERRα target genes are enriched for specific biological functions. A, Comparison between the functionally enriched biological functions associated with ERRα targets and the overall genomes. (**, P < 0.01; ***, P < 0.001). B, Subdivision of ERRα target genes within the ion transporter category shows a large proportion of genes encoding proteins implicated in Na+ and K+ handling. GO, Gene Ontology.
The binding profiles of a set of representative ERRα targets of the ion transporter activity category (GO: 0015075) are presented in Fig. 3A, and details on the identified bindings sites of the genes of interest are listed in Table 2. We observed that genes crucial for Na+ handling and, more precisely, Na+ reabsorption were target genes of ERRα. Indeed, ERRα was bound to the regulatory regions of the Na+/K+ ATPase α, β, and γ subunits (Atp1a1, Atp1a3, Atp1b1, Atp1b2, Fxyd2) and the key epithelial Na+ channel ENaC (Scnn1a) (27). ERRα was also found to be bound to the regulatory region of the K+ channels KvLQT1 (Kcnq1) and Kv5.1 (Kcnj16), Bartter syndrome-related K+/chloride channel β-subunit Barttin (Bsnd) and the chloride/carbonate anion exchanger 1 (AE1; Slc4a1) as well as the K+/chloride cotransporters KCC3 (Slc12a6) and KCC4 (Slc12a7). We also noticed that genes implicated in Na+ handling, without being ion transporters themselves, were also ERRα target genes. The serum/glucocorticoid-regulated kinase 2 (Sgk2) plays a crucial role in activating and prolonging the surface expression of K+ and Na+ channels, notably ENaC (Scnn1a) (35). Also, the genes encoding the transcription factor NFAT5 (also known as TonEBP) and the receptor for GH (Ghr) were also identified as ERRα targets (Fig. 3B). NFAT5 is crucial for the response to osmotic stress and GH, through activation of its receptor Ghr, affects blood pressure through regulation of peripheral vascular tone in addition to its direct antinatriuretic effects on tubular Na+ and water reabsorption (28). Finally, ERRα target genes other than ion transporters, but known to play a role in electrolyte handling or showing an association to blood pressure regulation or hypertension, include Agt, Add3, Aldoa, Anxa5, ApoA4, ApoC3, Fgf1, Fgf9, Gclc, and Lnpep (supplemental Table S1).
Fig. 3.

Genome-wide identification of ERRα-bound segments within extended promoter regions in mouse kidneys. A, Enrichment ratio profiles for ERRα at extended promoter regions of genes encoding ion channels. B, Enrichment ratio profiles for ERRα at extended promoter regions of genes encoding diverse proteins implicated in blood pressure regulation. Arrows indicate the transcriptional start sites for each gene. Black boxes, Exons. Chr, Chromosome.
Table 2.
ERRα ChIP-on-chip target genes associated with Na+ and/or K+ transport
| Gene | Common name | Transport | Peak probe | Location | Bindingsite | ||||
|---|---|---|---|---|---|---|---|---|---|
| Chr | Start | End | |||||||
| Atp1a1 | NaK-ATPase α1 | Na+/K+ | 3 | 101732164 | 101732220 | Intron | TCAAGGTCA | ||
| Atp1a3 | NaK-ATPase α3 | Na+/K+ | 7 | 24718010 | 24718054 | Promoter | TGAAGGTCA | ||
| Atp1b1 | NaK-ATPase β1 | Na+/K+ | 1 | 166292531 | 166292588 | Intron | cCAAGGgCA | ||
| 1 | 166293675 | 166293734 | Intron | TTAAGGTCA | |||||
| 1 | 166295022 | 166295066 | Promoter | AGGTCA | |||||
| 1 | 166295318 | 166298801 | Promoter | TCAAGGTCA | |||||
| 1 | 166297763 | 166297820 | Promoter | gGAAGGTCA | |||||
| Atp1b2 | NaK-ATPase β2 | Na+/K+ | 11 | 69420940 | 69422229 | Intron | AGGTCA | ||
| Bsnd | Barttin | Cl−/K+ | 4 | 105994809 | 105994867 | Promoter | TCAAGGTCA | ||
| Fxyd2 | NaK-ATPase γ | Na+/K+ | 9 | 45149761 | 45151240 | Promoter | cCAAGGTCA | ||
| 9 | 45150607 | 45150651 | Promoter | TCAAGGTCA | |||||
| 9 | 45152194 | 45152248 | Promoter | cTAAGGTCA | |||||
| 9 | 45157922 | 45157971 | Intron | TCAAGGTCA | |||||
| Hcn3 | — | K+/Na+ | 3 | 89245442 | 89246567 | Intron | — | ||
| Kcnc3 | Kv3.3 | K+ | 7 | 44458383 | 44459841 | Promoter | — | ||
| Kcnh2 | Kv11.1 | K+ | 5 | 23861900 | 23861944 | Promoter | TAAAGGTCc | ||
| Kcnj11 | Kir6.2 | K+ | 7 | 45967896 | 45969085 | Intron | AGGTCA | ||
| Kcnj16 | Kir5.1 | K+ | 11 | 110783890 | 110783949 | Promoter | TGAAGGTCA | ||
| Kcnq1 | Kv7.1/KvLQT1 | K+ | 7 | 142917727 | 142917784 | Intron | TCAAGGgCA | ||
| Kcnq3 | Kv7.3/KvLQT3 | K+ | 15 | 66116269 | 66116313 | Promoter | — | ||
| Kctd15 | — | K+ | 7 | 34362551 | 34363715 | Promoter | — | ||
| Kctd8 | — | K+ | 5 | 69619598 | 69620713 | Intron | TGAAGGaCA | ||
| Scnn1a | αENaC | Na+ | 6 | 125283488 | 125283545 | Promoter | TCAAGGTCA | ||
| 6 | 125286119 | 125286172 | Promoter | TCAAGGTCA | |||||
| 6 | 125287189 | 125287233 | Promoter | TAcAGGTCA | |||||
| Slc12a6 | KCC3 | Cl−/K+/Na+ | 2 | 112065842 | 112067213 | Promoter | AGGTCA | ||
| Slc12a7 | KCC4 | Cl−/K+/Na+ | 13 | 74230441 | 74230485 | Intron | cCAAGGTCA | ||
| 13 | 74230735 | 74230787 | Intron | TCAAGGTgA | |||||
ATPase, Adenosine triphosphatase; Chr, Chromosome; —, none. Lower case letters represent mismatch from consensus sequence, TNAAGGTCA.
Identification of differentially expressed genes in the ERRα null kidney
We next wanted to further explore the interconnection between the observed phenotype and the transcriptional program in the ERRα null kidney. We found 652 differentially expressed genes in the kidneys of the ERRα null mice compared with wild-type mice (276 up-regulated genes, 428 down-regulated genes; fold change (log2) cut-off of ±0.3 and a P value cut-off of <0.05) (GSE16623). Manual inspection of those genes revealed that several of them, involved in different biological processes, have been implicated in blood pressure regulation (Fig. 4A). Of interest, several ion transporters and channels known to play a role in electrolyte homeostasis are differentially expressed in the kidney of the ERRα null mice vs. wild-type mice (Fig. 4A). As can be expected, genes identified by the microarray analysis include several primary ERRα target genes identified by our ChIP-on-chip analysis (e.g. Bsnd, Kcnq1, Slc4a1, Aldoa, Fgf1, Fgf9, and Gclc). The differential expression of Avpr1a, Ptgds, Atp4a, Slc2a5, Kcnq1, and Kcnj1 (ROMK) observed from the microarray was validated by quantitative real-time RT-PCR (qRT-PCR) (Fig. 4B). In addition, the expression levels of Scnn1a and Atp1a1, two key genes responsible for Na+ reabsorption, identified by the ChIP-on-chip analysis as ERRα target genes but not by the microarray analysis, were assessed by qRT-PCR (Fig. 4C). The levels of expression of both genes were found significantly up-regulated in the kidneys of ERRα null mice.
Fig. 4.

Gene expression profiling in ERRα null kidneys vs. wild-type kidneys. A, Schematic representation of a subset of differentially regulated genes in ERRα null kidneys that are involved in blood pressure regulation. Data are presented as relative fold change (log2) vs. wild-type kidneys. B, qRT-PCR validation performed on RNA isolated from adult mouse kidneys used in the microarray analysis. Relative fold (log2) expression levels between ERRα null and wild-type kidneys were normalized to Arbp levels, and bars represent mean (±sem). C, Relative expression of Scnn1a and Atp1a1 in kidneys of wild-type and ERRα mice by qRT-PCR (*, P < 0.05). KO, Knockout; WT, wild type.
ERRα regulates the expression of the renin-angiotensin-aldosterone system (RAAS) genes in the kidney
The RAAS is intertwined with Na+/K+ homeostasis in the regulation of blood pressure (29). We first measured the plasma renin concentration (PRC) and plasma aldosterone concentration (supplemental Fig. S1). The PRC was slightly increased in the ERRα null mice, and no difference was observed in the plasma aldosterone concentration. Aldosterone is a regulator of sodium and potassium homeostasis via its activating effect on the mineralocorticoid receptor (MR). The expression levels of the MR and of the hydroxysteroid dehydrogenases-11β1 and -11β2 enzymes responsible for the conversion of cortisone to cortisol that can bind the MR, were also unchanged in the ERRα null kidneys at ZT15 (supplemental Fig. S1).
Considering that the angiotensinogen gene (Agt) was identified as an ERRα target gene in the kidney by ChIP-on-chip analysis (supplemental Table S1), we then investigated whether ERRα was regulating other components of the renin-angiotensin system at the transcriptional level. Because the region corresponding to the main regulatory element of Ren1, an approximately 240-bp region located 2.9 kb upstream of the transcriptional start site and referred to as the “renin kidney enhancer” (30), was not included on the array used in our ChIP-on-chip analysis, we probed this region by standard RT-PCR ChIP for possible ERRα binding. As shown in Fig. 5A, the Ren1 enhancer was enriched with the ERRα antiserum. In the same manner, we identified ERRα-bound segments within the promoters of both Ace2 and Agt (Fig. 5A). In agreement with ERRα binding, we observed that the expression of both Ace2 and Agt (Fig. 5B) as well as Ren1 (Fig. 5C) was up-regulated at the mRNA level in the kidneys of ERRα null mice, suggesting a repressive effect of ERRα on the expression of these genes. Also, the difference in Ren1 mRNA levels between wild-type and ERRα null mice was exacerbated when the mice were fed a Na+-deficient diet. In addition, the repression of renin expression usually induced by high salt was not as efficient in the ERRα null mice (Fig. 5C). Finally, to confirm the direct repressive effect of ERRα on renin expression suggested by the differences in the effect of Na+ diets on renin mRNA levels in vivo, we introduced three different small interfering RNA (siRNA) duplexes targeting mouse ERRα mRNA in the immortalized juxtaglomerular As4.1 cell line (Fig. 5E). The As4.1 cells retain renin expression in culture and are thus recognized as a good model to study renin expression (31). All three duplexes induced a 2.5- to 3-fold increase in renin mRNA levels (Fig. 5D) as well as renin protein levels (Fig. 5F). This is consistent with a complex regulatory role of ERRα on Na+ channels expression and/or activity and suggests a repressive role for ERRα on renin expression.
Fig. 5.

Regulation of genes involved in the RAAS by ERRα. A, Standard ChIP validation of ERRα binding to targets not identified in the original ChIP-on-chip experiment and involved in the RAAS pathway. Results shown are of one experiment based on two independent immunoprecipitations prepared from a pool of 85 mouse kidneys. B, Ace2 and Agt are up-regulated in the kidneys of ERRα null mice as assessed by qRT-PCR. C, Relative expression by qRT-PCR of Ren1 in kidneys of wild-type and ERRα null mice fed a low sodium (LS) or high high sodium (HS) diet as compared with mice fed standard chow (STD). The kidneys were collected at the ZT12 time point. D, Knockdown of Esrra in As4.1 cells using three different siRNA duplexes increases Ren1 expression as assessed by qRT-PCR. E, Western blot shows the level of ERRα in As4.1 cells transfected with the three siRNA duplexes. F, Western blot shows the level of renin in As4.1 cells transfected with the three siRNA duplexes. (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Equal loading was assessed by Ponceau red staining of the corresponding membrane. KO, Knockout; WT, wild type; siERRα, ERRα siRNA duplex.
Discussion
The kidney is involved in the regulation of blood pressure via Na+ and K+ handling as well as the production of renin, the rate-limiting step of the renin-angiotensin pathway. Here, using a combination of genome-wide location analysis, expression profiling, and physiological studies, we showed that the blood pressure of ERRα null mice is abnormal and that ERRα controls genes involved in the regulation of this function in the kidney. The ERRα-dependent transcriptional program in the kidney comprises several genes involved directly in K+ and Na+ homeostasis (Bsnd, Kcnq1, Kcnj16, Kcnj11, Atp1a1, Atp1a3, Atp1b1, Atp1b2, Scnn1a, Slc12a6, Slc12a7) or indirectly through regulation of the activity of these channels (Sgk2), or through impaired response to systemic cues regulating Na+ handling (Ghr, Gcgr, Lepr, Npy1r) as well as targets of the renin-angiotensin pathway (Agt, Ace2, Ren1).
The ERRα null mice are viable and show no obvious defects other than their smaller size, previously associated with a reduced fat mass (18). Indeed, the basal phenotype of the ERRα null mice is inconspicuous in many aspects, and the application of a physiological stressor is often necessary to uncover the role of ERRα in vivo (17, 19). Despite their adapted phenotype, the ERRα null mice showed significant alterations in electrolyte handling at basal level, suggesting a role of ERRα in this pathway. Indeed, the renal transcriptional program of ERRα correlates with the observed phenotype because the deregulated expression of several genes involved in controlling electrolyte balance are ERRα targets in the kidney.
Aldosterone is known to increase the expression and/or activity of ionic channels (32, 33, 34). The hormone can also trigger Na+ retention through the stimulation of ENaC expression. Aldosterone also activates the kinase Sgk2, which, in turn, influences the activity and surface residency time of ENaC and other ion channels (35). In the ERRα null mice, aldosterone levels, measured at ZT4, were not different from wild-type animals although variation of aldosterone concentrations at a later time point cannot be excluded. The global physiological effects observed in the ERRα null mice seems to imply that ERRα plays a role in the cardiorenal axis. Indeed, the dissection of the renal-specific mechanisms involved in the regulation of natremia and blood pressure by ERRα is excluded in our total knockout mouse model because a systemic effect arising from the cardiac (or other organ) defect cannot be ruled out in the current setting. However, the global regulation of genes involved in these processes, demonstrated by the renal transcriptional program and microarray analysis of the ERRα null kidneys, validates our hypothesis of a general role of ERRα in the control of natremia and blood pressure and awaits further studies concerning protein expression and activity of the various channels identified by our ChIP-on-chip. From a general point of view, ERRα could affect natremia and blood pressure in a direct manner by favoring Na+ excretion as well as by participating in the repression of the renin-angiotensin pathway genes. In addition, ERRα directly regulates the expression of Sgk2, Fxyd2 (γ-subunit of the NaK-ATPase) and indirectly regulates the expression of Tsc22d (also known as GILZ), thus potentially affecting in a broader manner the activity and stability of Na+ and K+ channels (35). Also, the protective role of ERRα could also involve a better response to the natriuretic effects of glucagon, leptin, and neuropeptide Y considering that Gcgr, Lepr, and Npy1r are down-regulated in the ERRα null kidney. The response to vasopressin is also likely to be impaired in the ERRα null mice considering vasopressin receptors, Avpr1a and Avpr2, are down-regulated in the ERRα null kidneys. Therefore, the hypernatremia of the ERRα null mice cannot be attributed to the effects of vasopressin. In light of our current model, the hypernatremia does not seem attributable to aldosterone effects either, because the expression of the mineralocorticoid receptor (MR) and of the enzymes regulating the levels of cortisol, which could activate the MR, are unchanged in the ERRα null kidneys at nighttime (supplemental Fig. S1). Indeed, the in-depth study of these potential renal mechanisms, and of how the various players of electrolyte balance regulated by ERRα interplay together in regulating natremia and blood pressure via ERRα, will require the generation of kidney-specific ERRα null mice. Furthermore, genes involved in vascular reactivity, such as the prostaglandin pathway (Ptgds, Hpgd) and others (Guca2a), were found to be down-regulated. Given these findings, together with the previous report of the up-regulation of the endothelial nitric oxide synthase gene by ERRα (36), our study constitutes a proper rationale for future studies on the role of ERRα in vasculature. From a cardiac point of view, ERRα could ameliorate overall blood pressure homeostasis via an improvement of cardiac function. It was recently shown that the ERRα null mice have a slightly decreased cardiac output and contractile force correlating with deregulated expression of genes regulating cardiac calcium handling and contractility (23, 24). This latter study, however, did not identify the differences in blood pressure most likely because measurements were performed during the day. In addition, the ERRα null mice were also demonstrating an impaired cardiac remodeling response to pressure overload induced by transaortic constriction (24). Given the global picture brought by the published cardiac phenotype of the ERRα null mice with our renal and blood pressure phenotype, we can speculate that ERRα impinges on the cardiorenal axis for homeostatic maintenance of blood pressure.
Taken together, the renal transcriptional program and the expression profile in the ERRα null kidneys suggest a role for ERRα in many aspects of electrolyte handling. Despite the absence of salt-wasting and metabolic alkalosis under standard housing conditions, ERRα null mice display a Bartter-like phenotype, principally owing to the simultaneous presence of a lower blood pressure along with hypokalemia, an elevated plasma level of renin, and to the down-regulation of channels associated with the etiology of Bartter syndrome (Bsnd, Kcnj1, Slc12a1 encoding, respectively, Barttin, ROMK, and NKCC2). In individuals affected by Bartter syndrome, the symptoms are thought to derive from a cascade of adaptive physiological reactions in response to the salt wasting caused by the loss of function of one or more ion channels. In the ERRα null mice, however, the final phenotype is likely caused by an accumulation of minor deregulations of multiple genes within the same pathway. Because ERRα also regulates the expression of Agt and Ren1, the adaptive cascades are likely to be impaired by the absence of ERRα. The fact that the removal of ERRα by siRNA in As4.1 cells induces a more pronounced increase in renin mRNA levels than that observed in vivo suggests that other mechanisms come into play and potentially counteract the effect of the absence of ERRα on renin transcription in vivo. Indeed, the ERRα null mice are hypernatremic, and this condition itself could diminish the increase of renin expression in the ERRα null kidney. This explanation could also apply to the plasma renin concentration. Further studies using tissue-specific ERRα null mice would be helpful to distinguish the causes from the consequences for the observed differences in blood pressure and to precisely assess the contribution of the kidneys in the observed phenotype.
ERRβ, an ERR isoform closely related to ERRα (3), has recently been implicated in the development of the endolymph-producing cells of the inner ear and the regulation of inner ear fluid homeostasis (37). Several differentially expressed genes in the stria vascularis of the ERRβ null mice (Kcne1, Atp1b2, and Kcnq1 among others) are modulated by ERRα in the kidney. Also the ERRγ null mice die at birth from heart defects that have been associated with a shorter depolarization phase and decreased expression of voltage-dependent cardiac Na+ channels (22). The ERRγ null mice displayed a prolonged QT interval on ECG. QT prolongation is typically associated to a longer repolarization phase of the cardiac action potential, which rely principally on rectifying K+ currents, suggesting that ERRγ could also be involved in K+ channels regulation in the heart. Indeed, we have previously shown that ERRα and ERRγ can bind as heterodimers and share the same target genes in the heart (23). Taken together with these studies, our results substantiate the implication of all members of the NR3B subgroup as regulators of the electrolyte handling transcriptional program.
In conclusion, this study connects ERRα with long-term blood pressure regulation in part via a global regulation of renal electrolyte-handling program and the renin-angiotensin pathway suggesting that ERRα, and potentially other isoforms of the ERR family, could be targeted for pharmacological regulation of blood pressure disorders. In addition, taken together with the previously shown cardiac phenotypic studies of the ERRα null mice, our study suggests a role of ERRα in the systemic homeostasis of blood pressure through the cardiorenal axis.
Materials and Methods
Mice and diets
The ERRα null mice (C57/BL6 background) were previously described (18). The animals were housed on a 12-h light, 12-h dark cycle (lights off at 1900 h). Male mice (2–3 months old) were used throughout the study. For the salt response analysis the mice were fed ad libitum with a standard diet containing 0.49% NaCl (TD.90208, Harlan Laboratories, Madison, WI) or a Na+-deficient diet (TD.90228, Harlan Laboratories) for 5 d. At ZT12, the mice were euthanized, and the tissues were removed, snap frozen in liquid nitrogen, and stored at −80 C until subsequent analysis. All the manipulations were performed in accordance with the McGill Animal Care Committee guidelines.
Telemetry monitoring
The telemetry transmitters were surgically implanted and mice (three per genotype) were allowed 7 d of postsurgery recovery before telemetry data acquisition. Data were collected for 72 consecutive hours. Statistical significance was reached for the nighttime period only (two-way ANOVA with Bonferroni posttest, P < 0.05) for all parameters except heart rate and activity state (n.s.).
Urine and blood constituents analysis
Briefly, mice were housed in metabolic cages (five cages of three to five mice per cage for a total of 19 mice per genotype) and were fed standard diet and water ad libitum. Mice were allowed an adaptation period of 1 wk before urine collection over a 24-h period. For plasma aldosterone measurements, nonstressed blood samples were collected at ZT4. The plasma was separated by centrifugation. Plasma aldosterone concentration was measured by RIA. The rest of the blood was collected by cardiac puncture at ZT4 for the electrolytes, creatinine, and blood urea nitrogen measurements. For the plasma renin concentration determination, mice were housed in standard conditions. Nonstressed blood samples were collected before and after a low-salt diet-feeding period. The plasma was separated by centrifugation, and PRC was measured by noncommercial RIA (n = 7–13 per group).
ChIP assays and ChIP-on-chip on extended promoter arrays
Chromatin was prepared from C57/Bl6 from a pool of kidneys taken around ZT4 from a total of 85 mice. ChIP labeling was performed as previously described (38) on chromatin immunoprecipitated with an antiserum against ERRα and nonimmunoprecipitated as control. The labeled samples were hybridized according to the Agilent Mammalian ChIP-on-chip Protocol (version 9.2; Agilent Technologies, Palo Alto, CA) in duplicate on mouse extended promoter microarrays (Agilent 2X 244 K array format) covering approximately 8 kb around the transcriptional start sites of approximately 17,000 genes. The slides were air dried and scanned immediately using the Agilent scanner (G2565BA) according to the protocol provided [Agilent Mammalian ChIP-on-chip Protocol (version 9.2)]. The data were then extracted using the Agilent Feature Extraction Software (version 9.5.1) using the grid template for mm8 mouse genome assembly, according to the protocol provided. The extracted data were subsequently analyzed with the ChIP Analytics Software (version 1.3.1, Agilent). The data were normalized using the Lowess normalization method, and significant binding peaks were detected using the Whitehead Peak Detection method. Complete datasets and bed files are available upon request. For standard ChIP assays, quantification of DNA enrichment was achieved by qRT-PCR with specific primers designed to amplify the regions of interest. Quantification by qRT-PCR were carried out using the LightCycler 2.0 (Roche Canada, Laval, Quebec, Canada).
Microarray analysis
Triplicate microarray analyses of wild-type and ERRα-null kidneys (collected at ZT4) were conducted at the McGill University and Génome Québec Innovation Centre using 10 μg of total RNA and hybridized to Affymetrix MOE430 2.0 GeneChIP arrays (Affymetrix, Santa Clara, CA). In this study, the mean of the expression data obtained from the individual wild-type and ERRα-null mice was determined to identify genes with altered levels of expression in the knockout mice vs. their wild-type littermates obtained using the Genespring X software (Agilent). A P value cutoff of <0.05 and a relative fold change (log2) cutoff of ±0.3 were used, and genes were classified by biological function based on Gene Ontology annotation (http://fatigo.org/), National Center for Biotechnology Information gene descriptions, and UniprotKB annotations.
siRNA knockdown assays
As4.1 cells were cultured in DMEM (Invitrogen, Burlington, Ontario, Canada) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were plated in 6-cm dishes 16–18 h before transfection with HiPerfect (QIAGEN, Mississauga, Ontario, Canada) according to the manufacturer’s instructions. Briefly, the cells were transfected with 30 nm of a scrambled siRNA in parallel with three different siRNA duplexes targeting Esrra (Integrated DNA Technologies, Coralville, IA). After 48 h, the cells were collected and frozen until subsequent analysis.
RNA isolation, reverse transcription, and qRT-PCR
Before RNA isolation, the frozen kidneys were pulverized using a frozen mortar and pestle. Liquid nitrogen was added regularly to the powder during the pulverization procedure to prevent thawing. Total RNA was isolated from frozen cell pellets or ground mouse kidneys using the RNeasy miniprep or midiprep kit (QIAGEN) according to the manufacturer’s instructions. For each sample, 2 μg of total RNA was reverse transcribed to cDNA using the SuperScript First-Strand Synthesis system (Invitrogen) according to the manufacturer’s protocol. The abundance of mRNA transcripts was assessed by LightCycler 480 (Roche) and LightCycler 480 SYBR Green I Master mix (Roche) according to the manufacturer’s instructions. Values were normalized to Arbp control gene.
Protein extraction and Western blotting
As4.1 cells were lysed in modified radioimmunoprecipitation assay buffer containing mini protease inhibitor tablet (Roche) for 30 min at 4 C, after which the cellular debris were pelleted at 10,000 rpm for 10 min at 4 C. Protein concentration was determined with Bradford Ultra reagent (Expedeon) according to the manufacturer’s instructions. Total protein extracts (20 or 100 μg) were resolved by SDS-PAGE and subjected to Western blot analysis using an in-house rabbit polyclonal anti-ERRα antibody (7) or goat antirenin antibody (R&D Biosciences, Minneapolis, MN) or rabbit anti-MR (SC-11412; Santa Cruz Biotechnology, Santa Cruz, CA). Equal loading was assessed by Ponceau red staining of the corresponding membrane.
Acknowledgments
We thank Carlo Ouellet (Rosalind and Morris Goodman Cancer Centre, Montreal, Quebec, Canada) and Chantale Mercure (Rosalind and Morris Goodman Cancer Centre) for skillful technical assistance and Dr. Christian Deschepper (Laboratory of Molecular Biochemistry of Hypertension, Clinical Research Institute of Montreal, Montreal, Quebec, Canada) for the As4.1 cell line.
NURSA Molecule Pages:
Nuclear Receptors: ERR-α.
Footnotes
This work was supported by grants from the Canadian Institutes for Health Research (CIHR). A.M.T. was supported by a Canada Graduate Scholarship doctoral award from CIHR.
Disclosure Summary: The authors have nothing to disclose.
First Published Online November 9, 2009
Abbreviations: Agt, Angiotensinogen; ChIP, chromatin immunoprecipitation; ChIP-on chip, combination of ChIP and genomic DNA arrays; ERRα, estrogen-related receptor α; MR, mineralocorticoid receptor; PRC, plasma renin concentration; qRT-PCR, quantitative RT-PCR; RAAS, renin-angiotensin-aldosterone system; siRNA, small interfering RNA; ZT, zeitgeber time.
References
- 1.Suzuki H, Saruta T2004. An overview of blood pressure regulation associated with the kidney. Contrib Nephrol 143:1–15 [DOI] [PubMed] [Google Scholar]
- 2.Jentsch TJ, Hübner CA, Fuhrmann JC2004. Ion channels: function unravelled by dysfunction. Nat Cell Biol 6:1039–1047 [DOI] [PubMed] [Google Scholar]
- 3.Giguère V, Yang N, Segui P, Evans RM1988. Identification of a new class of steroid hormone receptors. Nature 331:91–94 [DOI] [PubMed] [Google Scholar]
- 4.Huss JM, Kopp RP, Kelly DP2002. Peroxisome proliferator-activated receptor coactivator-1α (PGC-1α) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-α and -γ. Identification of novel leucine-rich interaction motif within PGC-1α. J Biol Chem 277:40265–40274 [DOI] [PubMed] [Google Scholar]
- 5.Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman IG, Heyman RA, Lander ES, Spiegelman BM2004. ERRα and GABPAα/β specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci USA 101:6570–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A2003. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor ERRα. J Biol Chem 278:9013–9018 [DOI] [PubMed] [Google Scholar]
- 7.Laganière J, Tremblay GB, Dufour CR, Giroux S, Rousseau F, Giguère V2004. A polymorphic autoregulatory hormone response element in the human estrogen related receptor α (ERRα) promoter dictates PGC-1α control of ERRα expression. J Biol Chem 279:18504–18510 [DOI] [PubMed] [Google Scholar]
- 8.Kamei Y, Ohizumi H, Fujitani Y, Nemoto T, Tanaka T, Takahashi N, Kawada T, Miyoshi M, Ezaki O, Kakizuka A2003. PPARγ coactivator 1β/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proc Natl Acad Sci USA 100:12378–12383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barry JB, Giguère V2005. Epidermal growth factor-induced signaling in breast cancer cells results in selective target gene activation by orphan nuclear receptor estrogen-related receptor α. Cancer Res 65:6120–6129 [DOI] [PubMed] [Google Scholar]
- 10.Tremblay AM, Wilson BJ, Yang XJ, Giguère V2008. Phosphorylation-dependent sumoylation regulates ERRα and γ transcriptional activity through a synergy control motif. Mol Endocrinol 22:570–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ariazi EA, Kraus RJ, Farrell ML, Jordan VC, Mertz JE2007. Estrogen-related receptor α1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol Cancer Res 5:71–85 [DOI] [PubMed] [Google Scholar]
- 12.Sladek R, Bader JA, Giguère V1997. The orphan nuclear receptor estrogen-related receptor α is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol Cell Biol 17:5400–5409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villena JA, Hock MB, Chang WY, Barcas JE, Giguère V, Kralli A2007. Orphan nuclear receptor ERRα is essential for adaptive thermogenesis. Proc Natl Acad Sci USA 104:1418–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ2006. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 126:789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, Mangelsdorf DJ, Evans RM2006. Nuclear receptor expression links the circadian clock to metabolism. Cell 126:801–810 [DOI] [PubMed] [Google Scholar]
- 16.Horard B, Rayet B, Triqueneaux G, Laudet V, Delaunay F, Vanacker JM2004. Expression of the orphan nuclear receptor ERRα is under circadian regulation in estrogen-responsive tissues. J Mol Endocrinol 33:87–97 [DOI] [PubMed] [Google Scholar]
- 17.Giguère V2008. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev 29:677–696 [DOI] [PubMed] [Google Scholar]
- 18.Luo J, Sladek R, Carrier J, Bader JA, Richard D, Giguère V2003. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor α. Mol Cell Biol 23:7947–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villena JA, Kralli A2008. ERRα: a metabolic function for the oldest orphan. Trends Endocrinol Metab 19:269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deblois G, Giguère V2008. Nuclear receptor location analyses in mammalian genomes: from gene regulation to regulatory networks. Mol Endocrinol 22:1999–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carroll JS, Brown M2006. Estrogen receptor target gene: an evolving concept. Mol Endocrinol 20:1707–1714 [DOI] [PubMed] [Google Scholar]
- 22.Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguère V, Evans RM2007. ERRγ directs and maintains the transition to oxidative metabolism in the post-natal heart. Cell Metab 6:13–24 [DOI] [PubMed] [Google Scholar]
- 23.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguère V2007. Genone-wide orchestration of cardiac functions by orphan nucler receptors ERRα and γ. Cell Metab 5:345–356 [DOI] [PubMed] [Google Scholar]
- 24.Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguère V, Murphy E, Kelly DP2007. The nuclear receptor ERRα is required for the bioenergetic and functional adaption to cardiac pressure overload. Cell Metab 6:25–37 [DOI] [PubMed] [Google Scholar]
- 25.Sonoda J, Laganière J, Mehl IR, Barish GD, Chong LW, Li X, Scheffler IE, Mock DC, Bataille AR, Robert F, Lee CH, Giguère V, Evans RM2007. Nuclear receptor ERRα and coactivator PGC-1β are effectors of IFN-γ induced host defense. Genes Dev 21:1909–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deblois G, Hall JA, Perry MC, Laganière J, Ghahremani M, Park M, Hallett M, Giguère V2009. Genome-wide identification of direct target genes implicates estrogen-related receptor α as a determinant of breast cancer heterogeneity. Cancer Res 69:6149–6157 [DOI] [PubMed] [Google Scholar]
- 27.Zacchia M, Trepiccione F, Morelli F, Pani A, Capasso G2008. Nephrotic syndrome: new concepts in the pathophysiology of sodium retention. J Nephrol 21:836–842 [PubMed] [Google Scholar]
- 28.Kamenicky P, Viengchareun S, Blanchard A, Meduri G, Zizzari P, Imbert-Teboul M, Doucet A, Chanson P, Lombès M2008. Epithelial sodium channel is a key mediator of growth hormone-induced sodium retention in acromegaly. Endocrinology 149:3294–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carey RM, Siragy HM2003. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev 24:261–271 [DOI] [PubMed] [Google Scholar]
- 30.Petrovic N, Black TA, Fabian JR, Kane C, Jones CA, Loudon JA, Abonia JP, Sigmund CD, Gross KW1996. Role of proximal promoter elements in regulation of renin gene transcription. J Biol Chem 271:22499–22505 [DOI] [PubMed] [Google Scholar]
- 31.Sigmund CD, Okuyama K, Ingelfinger J, Jones CA, Mullins JJ, Kane C, Kim U, Wu CZ, Kenny L, Rustum Y, Dzau, VJ, Gross, KW1990. Isolation and characterization of renin-expressing cell lines from transgenic mice containing a renin-promoter viral oncogene fusion construct. J Biol Chem 265:19916–19922 [PubMed] [Google Scholar]
- 32.Loffing J, Zecevic M, Féraille E, Kaissling B, Asher C, Rossier BC, Firestone GL, Pearce D, Verrey F2001. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol 280:F675–F682 [DOI] [PubMed]
- 33.Shigaev A, Asher C, Latter H, Garty H, Reuveny E2000. Regulation of sgk by aldosterone and its effects on the epithelial Na(+) channel. Am J Physiol Renal Physiol 278:F613–F619 [DOI] [PubMed]
- 34.Musch MW, Lucioni A, Chang EB2008. Aldosterone regulation of intestinal Na absorption involves SGK-mediated changes in NHE3 and Na+ pump activity. Am J Physiol Gastrointest Liver Physiol 295:G909–G919 [DOI] [PMC free article] [PubMed]
- 35.Friedrich B, Feng Y, Cohen P, Risler T, Vandewalle A, Bröer S, Wang J, Pearce D, Lang F2003. The serine/threonine kinases SGK2 and SGK3 are potent stimulators of the epithelial Na+ channel α,β,γ-ENaC. Pflugers Arch 445:693–696 [DOI] [PubMed] [Google Scholar]
- 36.Sumi D, Ignarro LJ2003. Estrogen-related receptor a 1 up-regulates endothelial nitric oxide synthase expression. Proc Natl Acad Sci USA 100:14451–14456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J, Nathans J2007. Estrogen-related receptor β/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Dev Cell 13:325–337 [DOI] [PubMed] [Google Scholar]
- 38.Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ2002. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev 16:235–244 [DOI] [PMC free article] [PubMed] [Google Scholar]