

Abstract
Intriguing findings on genetic and environmental causation suggest a need to reframe the etiology of mental disorders. Molecular genetics shows that thousands of common and rare genetic variants contribute to mental illness. Epidemiological studies have identified dozens of environmental exposures that are associated with psychopathology. The effect of environment is likely conditional on genetic factors, resulting in gene‐environment interactions. The impact of environmental factors also depends on previous exposures, resulting in environment‐environment interactions. Most known genetic and environmental factors are shared across multiple mental disorders. Schizophrenia, bipolar disorder and major depressive disorder, in particular, are closely causally linked. Synthesis of findings from twin studies, molecular genetics and epidemiological research suggests that joint consideration of multiple genetic and environmental factors has much greater explanatory power than separate studies of genetic or environmental causation. Multi‐factorial gene‐environment interactions are likely to be a generic mechanism involved in the majority of cases of mental illness, which is only partially tapped by existing gene‐environment studies. Future research may cut across psychiatric disorders and address poly‐causation by considering multiple genetic and environmental measures across the life course with a specific focus on the first two decades of life. Integrative analyses of poly‐causation including gene‐environment and environment‐environment interactions can realize the potential for discovering causal types and mechanisms that are likely to generate new preventive and therapeutic tools.
Keywords: Psychiatric genetics, environmental risk factors, gene‐environment interactions, classification of mental disorders, life course research, schizophrenia, depression, bipolar disorder, autism
Major depressive disorder, schizophrenia, bipolar disorder and autism are among the most disabling and costly diseases1. They affect individuals from young age, and are associated with physical morbidity and early death2. The causal mechanisms underlying mental illness may hide keys to effective prevention and treatment, but remain poorly understood.
The last two decades have seen an expansion of knowledge punctuated by surprises that challenge previously held assumptions about mental illness. In this paper we provide a synthesis of current knowledge and direct further research to maximize the potential for meaningful discovery. While the focus is on generic principles underlying the causation of any mental illness, the majority of information comes from studies of schizophrenia, bipolar disorder, major depressive disorder and autism, on which most data have been accumulated.
We first review the genetic and environmental factors implicated in the etiology of mental illness, before adopting an integrative perspective that jointly considers genetic and environmental elements of causation. We conclude by outlining a framework for productive causal research.
GENETIC FACTORS IN THE CAUSATION OF MENTAL ILLNESS
All types of mental illness have a tendency to run in families, and the risk of developing an illness is associated with the degree of biological relatedness to the affected individual3, 4. This pattern of transmission strongly suggests genetic causation. Twin studies consistently show that monozygotic twins who share 100% of their nuclear DNA are more likely to be concordant on each disorder than dizygotic twins who share 50% of their genetic material5. This difference suggests that the causation of mental illness is to a large degree attributable to genetic factors.
There is a gradient of genetic contribution, with higher estimates of heritability for the more severe and less common disorders (autism, schizophrenia, bipolar disorder) and a lesser degree of heritability for more common and less severe disorders (anxiety, major depressive disorder)5.
The large heritability estimates promised an easy identification of the molecular genetic variants responsible for the causation of mental illness. Influential authorities estimated that severe mental illness, such as schizophrenia, was likely to be caused by several (2 to 9) genetic loci6, while others argued for a single gene causing most cases of schizophrenia7.
Three assumptions have shaped the field of genetic discovery: a) severe mental illness is caused by a small number of genes; b) there is a specific relationship between genotype and the type of mental illness and c) the genetic variants lead to mental illness through biological pathways independent of environment. Consequently, most genetic research has studied one mental disorder at a time by comparing cases with a specific diagnosis to controls, without accounting for environmental influences.
Over the last decade, the molecular genetic technology has offered the tools to study the genetic variants responsible for the transmission of liability for mental illness from parents to offspring. This decade of research has brought surprising findings that challenge the assumptions on which psychiatric genetics has been based. Genome‐wide association studies have identified more than a hundred variants associated with severe mental illness (Table 1)8‐11. Each of the variants has small effect and the number of associated variants keeps increasing with growing sample sizes8.
Table 1.
Genetic variants associated with mental illness
| Autism | Schizophrenia | Bipolar disorder | Depression | |
|---|---|---|---|---|
| Number of individuals in largest genetic sample to date | 13,088 cases with autism spectrum disorders and 16,664 controls | 36,989 cases with schizophrenia and 113,075 controls | 7,481 cases with bipolar disorder and 9,250 controls | 121,380 cases with depression and 338,101 controls |
| Number of genetic variants associated at genome‐wide level of statistical significance | 4 | 128 | 18 | 17 |
| Odds ratio of the most strongly associated genetic variant | 1.17 | 1.21 | 1.15 | 1.05 |
| Proportion of variance explained by common genetic variants across the genome | 14% | 23% | 25% | 5% |
Polygenic risk scores analyses consistently show that the prediction of mental illness improves by including more weakly associated genetic variants, suggesting that many thousands of genetic variants are involved in shaping the risk for most mental disorders12, 13. These involve both common single nucleotide polymorphisms and rare structural variants, such as deletions and insertions of stretches of DNA14.
Another consistent finding is that most common and rare genetic variants are non‐specifically associated with a range of mental disorders15, 16. Overall, approximately two thirds of genetic associations are common to schizophrenia, bipolar disorder and major depressive disorder15. There are also overlaps with genetic variants contributing to autism, attention‐deficit/hyperactivity disorder, and intellectual disabilities.
It has also become clear that the heritability estimates derived from twin studies do not translate into direct effects of molecular genetics variants15. The estimates based on case‐control studies with molecular genomic data suggest that genetic variants contribute only a fraction of the effect that was suggested by heritability estimates from twin studies (Figure 1). The most likely explanation for this “heritability gap” is that a large fraction of genetic effects are contingent on factors that are common to individuals growing up in the same family but not to unrelated individuals who participate in case‐control studies17, 18. A picture is emerging of a complex etiological mechanism, where genetic influence is thinly distributed across thousands of genetic variants of small effects that are contingent on environment and not specific to any single form of psychopathology.
Figure 1.
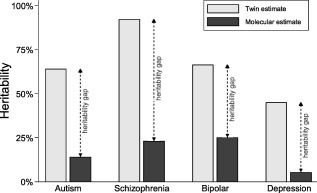
The heritability gap. Heritability (the proportion of causation attributable to genetic factors) has been estimated from differences of concordance between identical and fraternal twins (twin estimates) and from hundreds of thousands of single nucleotide polymorphisms across the human genome (molecular estimates). The large difference between the twin and molecular estimates is referred to as the “heritability gap”. Twin estimates are based on same‐sex twin pairs from a recent comprehensive meta‐analysis5. Molecular estimates are from large case‐control genome‐wide association studies8, 9, 10, 11.
ENVIRONMENTAL FACTORS IN THE CAUSATION OF MENTAL ILLNESS
The same twin studies which confirmed that mental illness is heritable have also demonstrated that environment matters. Concordance of genetically identical twins is far from perfect even for the most heritable types of mental illness, such as autism or schizophrenia5. While it is not possible to completely separate the effects of environment from errors in diagnosis, a realistic assessment suggests that environmental and genetic factors contribute equally to the causation of mental illness.
Since the 1960s, researchers have been identifying strong relationships between adverse social environment and mental illness. The bulk of the research on social causation has been based on the assumption that a single environmental factor may explain the causation of a specific diagnosis, irrespective of enduring characteristics of the exposed individual. Thus, social researchers tended to examine one aspect of environment and one mental disorder diagnosis at a time. The highlights of this research included identification of strong associations between severe adverse life events and depression19.
A number of studies of environmental factors have included longitudinal follow‐ups and documented both the long‐term effects of adversity in childhood and the close temporal relationship between severe life events and psychopathology onset in adulthood20, 21. With larger and more representative studies, additional and diverse environmental risk factors have been identified, including exposure to viral infections during gestation, vitamin D deficiency, growing up in urban environment, ethnic minority status, childhood maltreatment and bullying victimization (Table 2)22‐25.
Table 2.
Environmental factors associated with mental illness
| Autism | Schizophrenia | Bipolar disorder | Depression | |
|---|---|---|---|---|
| Pregnancy risk factors | ||||
| Infections | + | +++ | ++ | + |
| Malnutrition | +++ | ++ | ++ | |
| Heavy metals | +++ | ++ | ||
| Perinatal risk factors | ||||
| Preterm birth | ++ | ++ | ++ | ++ |
| Season of birth | ++ | +++ | ++ | + |
| Birth complications | +++ | +++ | 0 | |
| Childhood environment | ||||
| Urbanicity | +++ | +++ | + | + |
| Poverty | ++ | +++ | + | +++ |
| Maltreatment | N/A | ++ | ++ | +++ |
| Bullying | N/A | ++ | + | +++ |
| Drug use in adolescence | ||||
| Cannabis | N/A | +++ | ++ | + |
| Stimulants | N/A | +++ | ++ | 0 |
The number of + marks the strength of evidence (+ means some evidence of association/single report; ++ means moderate replicated evidence of association/multiple reports; +++ means strong evidence of association/multiple replications or good meta‐analysis). Evidence of no association is noted as 0. Empty cells reflect absence of evidence for or against association. No factor has been negatively associated with any of the disorders. Because of the early age at onset of autism, environmental factors occurring after age 3 cannot be reliably studied and are marked as not applicable (N/A).
Several general principles emerged. First, the same type of environmental exposure increases the risk of many different mental disorders. For example, urban environment was first identified as a risk factor for schizophrenia, but a systematic analysis showed that it is associated with increased risk of all types of mental disorders25. Second, many different types of environmental exposures contribute to the same disorder. For example, the risk of schizophrenia increases with maternal malnutrition, vitamin D deficiency and viral infections during pregnancy, low socio‐economic status, urban upbringing, minority status and childhood maltreatment, as well as exposure to stimulants, cannabis and tobacco26. Third, no constellation of adverse environmental exposures will result in psychopathology among all exposed individuals. Many individuals appear to be resilient and do not develop any mental disorder even if they are exposed to multiple adverse environmental factors27, 28.
Resilience appears to be related to a number of enduring personal characteristics that are partly heritable and partly shaped by previous environmental exposures28, 29. Experiences early in life may make a person more vulnerable or resilient to exposures later in development, resulting in a sequential environment‐environment interaction. For example, exposure to maltreatment in childhood may cause sensitization to the effects of specific types of stressful life events in adulthood30. The observation that unshared environment has greater influence on intellectual ability among twins growing up in families with low socio‐economic status also suggests a complex interplay between multiple environmental factors31.
A synthesis of current knowledge on environmental causation of mental illness suggests a complex picture with a multitude of social, physical and chemical exposures occurring at different stages of life, affecting the risk for a range of mental disorders. It is becoming increasingly unlikely that any given environmental factor could be a necessary and sufficient cause of any mental disorder. Instead of searching for single disorder‐specific environmental causes, researchers who want to explain or predict mental illness may need to jointly study a multitude of environmental influences across the life course, that may be summed up in cumulative poly‐environmental scores (E‐scores)32 or grouped in unique environment‐environment constellations31.
While the array of environmental factors that are known to be involved in the causation of mental illness is impressive, it may still only be the tip of an iceberg. Research designs to date have only been powered to detect environmental factors that are harmful for the vast majority of individuals. The types of environments that may good for some and bad for others are still waiting to be discovered.
GENE‐ENVIRONMENT CAUSATION OF MENTAL ILLNESS
No genetic variant and no environmental exposure on its own is a sufficient cause of mental illness. While it is possible that some cases of mental illness are caused by a combination of many genetic variants or a combination of multiple environmental exposures, the most likely scenario by far is that both genetic and environmental factors jointly contribute to the causation of mental illness. A causal mechanism where one or more genetic factors and one or more environmental factors are required to produce an outcome is gene‐environment interaction (GxE).
A ubiquitous role of GxE in the causation of mental illness is suggested by a contradiction between the results of epidemiological studies and twin studies that we call the shared environment paradox. Epidemiological research shows that a substantial proportion of cases of mental illness are attributable to environmental factors which are typically shared by whole families, such as socio‐economic class, poverty, urban environment, minority status, neighbourhood characteristics and childhood maltreatment22, 25, 33. Yet, twin studies allocate only a very small role to shared environmental factors5 (Figure 2). One explanation of the shared environment paradox is that the impact of the family‐wide environment depends on factors that are shared more between monozygotic than between dizygotic twins, i.e. genetic polymorphisms. If the effects of shared environment are conditional on genetic variants, the statistical models used in twin studies will fully attribute the joint effect to the genetic component, thus inflating heritability and reducing the estimate of shared environment34. In this way, GxE provide the most parsimonious explanation for both the shared environment paradox and the heritability gap.
Figure 2.
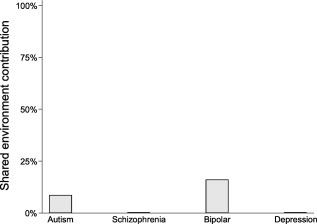
Shared environment paradox. Twin studies have consistently allocated little or no role in the causation of mental illness to environmental factors that are shared by members of the same family. The estimates plotted here are from a recent comprehensive meta‐analysis of twin studies5, based on same‐sex twin pairs. Estimates for schizophrenia and depression were actually negative, but since a negative contribution to variance is not possible, we plotted them at 0%.
In the last 15 years, researchers have started to identify specific genetic variants that may sensitize individuals to environmental factors. Like most molecular genetic research, the search for GxE started with tests of candidate polymorphisms in candidate genes. The success of such studies depends not just on picking the correct combination of a genetic variant and an environmental factor based on prior knowledge, but also on sampling and design that allows an approximation of a biological interaction with a statistical test.
Remarkably, some of these studies appear to have been successful in finding causal mechanisms. Some candidate gene GxE have been consistently replicated. For example, the interaction between low activity variants of the X‐chromosome‐linked monoamine oxidase A (MAO‐A) gene and childhood maltreatment leading to antisocial behavior in males has been replicated multiple times and confirmed in meta‐analyses35, 36. The interaction between brain derived neurotrophic factor (BDNF) gene variants and severe life events leading to depression has also been replicated and confirmed in a meta‐analysis37.
Other GxEs have proven less robust or more specific than originally reported. For example, the interaction between short variants of the serotonin transporter gene length polymorphism and adversity leading to depression has seen similar number of replications and non‐replications and it may be specific to childhood maltreatment leading to persistent depressive disorder38, 39, 40. Yet other reported GxE have proven unreplicable. For example, the interaction between catechol‐O‐methyltransferase (COMT) gene and cannabis use leading to psychotic symptoms has been reported, but not replicated in subsequent studies41.
More recent studies have screened a larger number of genes and polymorphisms to search for GxE. Such systematic search has led to the identification, among 152 polymorphisms in 42 genes related to cannabinoid signalling, of an interaction between a single nucleotide polymorphism in the serine/threonine kinase encoding gene AKT1 and cannabis use leading to psychosis42. This GxE has been replicated in independent samples43, 44, suggesting that this polymorphism sensitizes individuals to the psychosis inducing effects of tetrahydrocannabinol.
Finally, several genome‐wide environment interaction studies (GWEIS) have been completed to search for GxE without any pre‐existing hypothesis about the genetic variants involved. The first GWEIS concerned interactions of common genetic variants with prenatal exposure to cytomegalovirus and with stressful life events in the causation of schizophrenia45 and depression46, respectively. The existing GWEIS have limited statistical power, because most large genotyped samples are missing adequate measures of environment. At present, it is unclear whether the results of GWEIS will be more replicable than those of candidate GxE studies. An interim synthesis suggests that multiple GxE contribute to most types of mental illness, but no specific GxE explains a substantial proportion of cases.
Several studies suggest that multiple genetic variants shape the susceptibility to harmful and protective environmental factors. One study has shown that a score derived from over 2,800 schizophrenia‐associated variants in coding or regulatory genomic regions interacted with winter birth to increase the risk of schizophrenia47. In another study, a polygenic risk score of tens of thousands of common variants tapping the overall sensitivity to environment predicted the effects of negative parenting on emotional psychopathology as well as the effectiveness of intensive psychological treatment for anxiety48. As in studies of direct polygene‐disorder associations, the GxE increased in strength with more genetic variants being included in the polygenic risk score. The emerging pattern of findings suggests that sensitivity to environment is a highly polygenic trait with contributions from thousands of common genetic variants.
The examination of gene‐environment interplay is still in its infancy, and research available to date leaves many unanswered questions. The specificity of polygenic GxE to the type and timing of environmental exposures, the specificity or pleiotropy of GxE across types of mental disorders, and the role of rare structural variants in sensitivity to environment remain largely unexplored.
BOUNDARIES AND OVERLAP BETWEEN DISORDERS
The review of genetic and environmental factors above has concluded that most factors are associated with most types of mental illness. The apparent overlaps in causation has been generally ascribed either to pleiotropy, i.e. the same factors having the potential to cause multiple types of illness, or to the lack of validity of the diagnostic criteria for specific disorders.
Pleiotropy at the level of a single causal factor has been well documented: for example, the same variant (A‐allele of rs1006737) of the calcium channel gene CACNA1C has been associated with increased risk of bipolar disorder, schizophrenia, depression, anxiety and autism49, 50, 51. While inadequate validity of boundaries between diagnostic categories has also been amply demonstrated52, evidence supporting validity also exists, e.g. in the specificity of therapeutic response to lithium in bipolar disorder but not schizophrenia.
While both pleiotropy and inadequate validity of categorization are likely to be at play, the multifactorial causation also leaves the possibility of unique combinations of causal factors. For example: even if most risk factors are shared between bipolar disorder and schizophrenia, the loading and combination of factors that give rise to each of the two disorders may still be unique.
Since hundreds or thousands of environmental and genetic factors are likely involved in the causation of each disorder, the number of possible combinations is extremely large. The examination of these possible combinations has only just begun. One example involves both a mental disorder and a physical disorder: individuals with schizophrenia have less than half the risk of developing rheumatoid arthritis compared to the general population, even though schizophrenia and rheumatoid arthritis share environmental risk factors, including winter birth and tobacco smoking. Recently it has been shown that a polygenic risk associated with the immune system is associated with both increased risk of rheumatoid arthritis and reduced risk of schizophrenia. In addition, a polygenic risk score for schizophrenia interacts with winter season of birth to increase the likelihood of schizophrenia47. In this case, some environmental factors are shared, but genetic disposition distributed across thousands of variants may determine the relative risk of two competing outcomes.
Even if we are able to examine combinations of genetic and environmental factors, the question remains about the level of outcomes that is most likely to lead to success in etiological research. Most of the debate to date has focused on the distinction between categorical diagnoses and dimensional measures. This may have been a false focus. At present, it is unclear whether one of the approaches is more advantageous than the other. The experience with dimensional constructs introduced as part of the Research Domain Criteria framework over the past five years does not inspire hopes for major advances in etiological research. While dimensional measures may be more powerful for examining variation in common traits across the general population, the categorical diagnostic constructs remain more relevant to the severe types of mental illness that are most pertinent to psychiatry. When it comes to psychopathology, it is unlikely that the difference between complete absence of pathological symptoms and population average matters as much as the difference between average and severe psychopathology. Yet, the number of categories in the current classifications are too large and the boundaries between them lack validity52.
Because psychiatric research to date has been based on the now refuted assumption of diagnostic specificity, most studies are uninformative about the validity of specific diagnostic categories or dimensions52. The potential for discovery will likely be enhanced if researchers refocus on examining broad and heterogeneous samples of mental illness without exclusions based on diagnostic criteria and without constraining their measurement to consensus based constructs, categorical or dimensional. Shedding the constraints of diagnosis‐specific research does not require adopting another set of constraints and it does not necessitate transition from a categorical to a dimensional framework of inquiry. Examination of overlaps in etiological factors between disorders suggest that higher level broad categorical constructs may be more appropriate targets of etiological research than specific diagnostic categories. For example, the major overlap in both genetic and environmental contributors between major depressive disorder, bipolar disorder, schizophrenia and other types of psychotic illness suggests that a broad category of severe mental illness that encompasses major mood and psychotic disorders may be an appropriate unit of investigation.
DEVELOPMENTAL CONTEXT AND CLINICAL COURSE
Two major discoveries in psychiatry remain underrated and are not reflected in most etiological research. The first one is the continuity of pathology over the life course. From cohorts with complete and intensive long‐term follow up, it has become clear that most cases of mental illness start in childhood and adolescence. The early manifestations of psychopathology typically differ in kind from the eventual diagnoses in adulthood, yet are very strongly predictive of mental illness diagnosed across the life course.
Heterotypic continuity is the rule. For example, anxiety in childhood is a strong predictor of both major depressive disorder and bipolar disorder in adulthood. Oppositional defiant disorder in childhood predicts a broad range of psychopathology in adults, including depression, bipolar disorder, anxiety, substance use disorders and antisocial personality disorder. Yet, there is also a degree of specificity, with a systematic correspondence between the profile of childhood symptoms and the type of adult disorders53.
The fact that most individuals with mental illness will go through a number of diagnostic categories over their life course adds to the problems associated with diagnosis‐specific research52. It highlights the need to examine psychopathology broadly and in a developmental context. Since most mental illness starts in childhood and retrospective report is inaccurate, etiological research needs longitudinal designs that start in childhood, at birth or even earlier54.
The second discovery is that the course of mental illness varies between individuals and is only loosely related to the diagnostic category. Traditionally, some disorders have been conceptualized as episodic and other disorders as persistent, but longitudinal follow‐ups suggest that this conceptual distinction has limited validity. Mood disorders have been codified as episodic, yet on follow‐up they are marked by chronicity, with most individuals spending most of their time with depressive symptoms55, 56. Personality disorders have been conceptualized as persistent, but on follow‐up their symptoms show similar rates of remission and relapses as mood disorders do57.
Yet, within the same disorder, episodic and persistent cases may have distinct etiologies. For example, episodic cases of major depressive disorder are more strongly heritable58 and persistent cases are more strongly linked to childhood maltreatment59. The interaction between serotonin transporter gene length polymorphism and childhood maltreatment also appears to be specific to persistent depressive disorder39, 40. On the other hand, there is evidence that cycloid psychosis, a type of mental illness marked by a characteristic highly episodic course in spite of varied symptom content, may have distinct genetic underpinning60, 61. These examples suggest that time course of illness may be at least as important in etiological studies as the symptom profile.
The findings of longitudinal research outlined above highlight a massive caveat in prior etiological studies that grouped individuals into cases and controls based on symptom content in adulthood without reference to developmental context or time course of symptoms. Future etiological research will be improved by the systematic incorporation of a temporal dimension that has been conspicuously missing from both categorical and dimensional classifications used by most etiological studies to date.
LIMITS OF CURRENT APPROACHES
The last decade has seen a large amount of criticism of psychiatric research. It may be important to own up to both successes and failures and take a stock of what might have hindered the field from knowing more. Based on the review of etiological research in psychiatry outlined above, we conclude that four factors are limiting further progress.
One of the major limiting factors is assumed knowledge. Over the past five decades, psychiatry researchers have built their studies around the following assumptions: causes are diagnosis‐specific, disorders are caused by a small number of factors, and genetic factors have primacy over environmental influences. It is remarkable that some of the greatest discoveries in psychiatry occurred before these assumptions were established. For example, the discovery of lithium efficacy for bipolar disorder occurred thanks to investigation in an unselected group of patients62.
Another limiting factor lies in omissions. The diagnosis‐focused approach of the 20th century and the ensuing categories‐vs.‐dimensions discussion might have led to the neglect of the developmental context and the temporal dimension of psychopathology.
The final limitations we will discuss are related: statistical power and quality of measurement. Since many genetic and environmental factors contribute to most cases of mental illness, large representative samples with accurate measurements of genetic variation, environmental exposures and psychopathology over the life course are needed for etiological research. We have many studies with good measurement of environment, but they do not overlap much with studies with high standard of genetic measurement. We have some longitudinal studies with high quality measurements and we have some studies with a large number of individuals. Unfortunately, there has been little overlap between the two. The largest studies in psychiatry are either pulled together from many variably assessed samples or they suffer from large dropout rate and less accurate measurement. We may not get the answers about causation of mental illness unless experts in developmental psychopathology, environment and genetics join forces to work together on large longitudinal studies. Early examples of such collaborations are emerging and hopefully will be completed.
FRAMEWORK FOR DISCOVERY
To substantially advance our understanding of mental illness, the next generation of studies will need to embrace the complexity of poly‐gene‐environmental causation. The technology and methodological knowledge available today enables studies of multiple environmental and genetic factors without assumptions of independence. It is essential that the research studies are designed in a way that maximizes the potential for meaningful discovery by avoiding the pitfalls of assumptions, omissions, inadequate measurement and statistical power (Figure 3).
Figure 3.
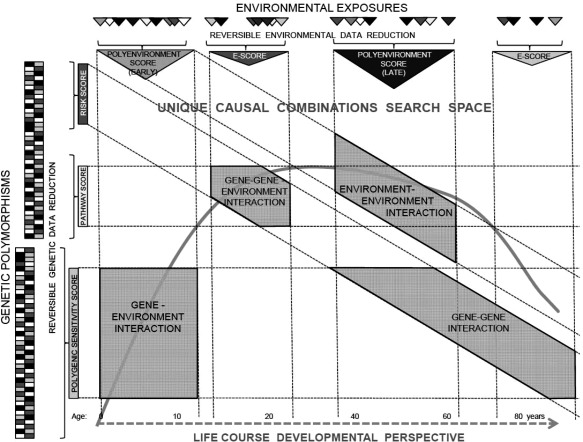
Framework for discovery. Life‐course developmental perspective and an open search space for unique combinations of genetic and environmental factors (including gene‐gene, gene‐environment, and environment‐environment interactions) are core elements that will enhance the potential for discovery in etiological research. Genetic and environmental data reduction – polygenic sensitivity scores, polygenic risk scores, genetic pathway scores, poly‐environmental scores (E‐scores) – may be a necessary intermediate step towards the discovery of broad poly‐gene‐environmental causal mechanisms, but the reduction process should be reversible to enable fine mapping of specific molecular and behavioral mechanisms.
Large longitudinal studies of samples that are not selected for a particular diagnosis are needed to enable new discovery. These studies should start in pregnancy, childhood or adolescence to capture the development of psychopathology and allow separation of cases from consequences. Repeated assessments of multiple aspects of environment during the individual's development should cover known environmental risk factors as well as key factors of environment that may be good for some and bad for others. Regular assessments of psychopathology across the life course are needed to establish true age at onset, track the course and record sequential comorbidity. Measurement of environment and psychopathology should use multiple independent sources of information to maximize objectivity and avoid common source bias (e.g., predictably high but uninformative correlations between questionnaires completed by the same individual at the same time). Instead of case‐control studies, genetic measurement should concentrate on samples with high‐quality longitudinal data on environment and psychopathology.
With broadly based assumption‐free designs, the onus will be on data analysis to make use of the resulting data in a way that can identify complete etiological mechanisms leading to mental illness. The key challenge for data analysts will be to embrace the complexity of causation while retaining the capacity to trace specific causal mechanisms. The data analysis may need to move from theory‐driven hypothesis testing focus to a theory‐free explanatory framework that aims to explain the causation of a large proportion of cases. It will be important to identify unique combinations of genetic and environmental variables that lead to mental illness, irrespective of whether the biological mechanism corresponds to the constrained concept of statistical interaction. The framework should be open to identify combinations of early and late environmental factors as well as of environmental and genetic factors.
Tools for such analyses are becoming widely available. For example, statistical learning offers a set of tools that are designed to maximize the use of rich datasets in the prediction and explanation of outcomes and at the same time provide understanding of how individual factors contribute to the prediction63. Methods are also being developed that make it possible to distinguish between causal heterogeneity and polyfactorial causation64. While the available methods potentially offer many ways of analyzing rich datasets, the model complexity will have to be kept proportional to available sample sizes. Given the vast number of potential factors to be considered, data analysis process will require a degree of data reduction in the initial stages. This may take the form of polygenic risk scores of disorder liability or environmental sensitivity48, genetic pathway scores47 and poly‐environmental risk scores32.
The degree of data reduction should not be excessive and the process may need to preserve developmental specificity: e.g., with separate procedures for childhood, adolescent and adulthood exposures. Data reduction also needs to be transparent, so that it is possible to follow a positive result back to the molecules and specific factors that drive the causal mechanisms. Specific constellations of molecular genetic and environmental factors will be needed to inform prevention and treatment65.
Eventually, the role of each genetic and environmental variable has to be understood in a way that enables independent replication and examination of the underlying biological mechanism. Embracing complexity in a transparent and assumption‐free framework will enable researchers to map complete causal mechanisms that explain why large groups of individuals develop mental illness. While this may be a bigger task than what previous generations of psychiatrists had envisioned, knowledge of complete causal mechanisms is necessary to meaningfully transform classification, prevention and treatment.
ACKNOWLEDGEMENTS
The work reported in this manuscript has been completed thanks to funding from the Canada Research Chairs Program, the Canadian Institutes for Health Research (grant nos. 124976, 142738 and 148394), Nova Scotia Health Research Foundation and the Dalhousie Medical Research Foundation.
REFERENCES
- 1. Whiteford HA, Degenhardt L, Rehm J et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet 2013;382:1575‐86. [DOI] [PubMed] [Google Scholar]
- 2. Walker ER, McGee RE, Druss BG. Mortality in mental disorders and global disease burden implications: a systematic review and meta‐analysis. JAMA Psychiatry 2015;72:334‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gottesman II, Laursen TM, Bertelsen A et al. Severe mental disorders in offspring with 2 psychiatrically ill parents. Arch Gen Psychiatry 2010;67:252‐7. [DOI] [PubMed] [Google Scholar]
- 4. Rasic D, Hajek T, Alda M et al. Risk of mental illness in offspring of parents with schizophrenia, bipolar disorder, and major depressive disorder: a meta‐analysis of family high‐risk studies. Schizophr Bull 2014;40:28‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Polderman TJ, Benyamin B, de Leeuw CA et al. Meta‐analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet 2015;47:702‐9. [DOI] [PubMed] [Google Scholar]
- 6. Gershon ES. Bipolar illness and schizophrenia as oligogenic diseases: implications for the future. Biol Psychiatry 2000;47:240‐4. [DOI] [PubMed] [Google Scholar]
- 7. Crow TJ. A continuum of psychosis, one human gene, and not much else – the case for homogeneity. Schizophr Res 1995;17:135‐45. [DOI] [PubMed] [Google Scholar]
- 8.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia‐associated genetic loci. Nature 2014;511:421‐7. [DOI] [PMC free article] [PubMed]
- 9. Hyde CL, Nagle MW, Tian C et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 2016;48:1031‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Psychiatric Genomics Consortium . Large‐scale genome‐wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011;43:977‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robinson EB, St Pourcain B, Anttila V et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat Genet 2016;48:552‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet 2013;9:e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wray NR, Lee SH, Mehta D et al. Research review: Polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry 2014;55:1068‐87. [DOI] [PubMed] [Google Scholar]
- 14.CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium. Contribution of copy number variants to schizophrenia from a genome‐wide study of 41,321 subjects. Nat Genet 2017;49:27‐35. [DOI] [PMC free article] [PubMed]
- 15. Cross‐Disorder Group of the Psychiatric Genomics Consortium . Identification of risk loci with shared effects on five major psychiatric disorders: a genome‐wide analysis. Lancet 2013;381:1371‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee SH, Ripke S, Neale BM et al. Genetic relationship between five psychiatric disorders estimated from genome‐wide SNPs. Nat Genet 2013;45:984‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uher R. Gene‐environment interactions in common mental disorders: an update and strategy for a genome‐wide search. Soc Psychiatry Psychiatr Epidemiol 2014;49:3‐14. [DOI] [PubMed] [Google Scholar]
- 18. Uher R. Gene‐environment interactions in severe mental illness. Front Psychiatry 2014;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown GW, Harris TO. Social origins of depression. A study of psychiatric disorder in women. London: Routledge, 1978.
- 20. Brown GW, Craig TK, Harris TO. Parental maltreatment and proximal risk factors using the Childhood Experience of Care & Abuse (CECA) instrument: a life‐course study of adult chronic depression ‐ 5. J Affect Disord 2008;110:222‐33. [DOI] [PubMed] [Google Scholar]
- 21. Arseneault L, Cannon M, Fisher HL et al. Childhood trauma and children's emerging psychotic symptoms: a genetically sensitive longitudinal cohort study. Am J Psychiatry 2011;168:65‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davis J, Eyre H, Jacka FN et al. A review of vulnerability and risks for schizophrenia: beyond the two hit hypothesis. Neurosci Biobehav Rev 2016;65:185‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arseneault L. The long‐term impact of bullying victimization on mental health. World Psychiatry 2017;16:27‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rai D, Lewis G, Lundberg M et al. Parental socioeconomic status and risk of offspring autism spectrum disorders in a Swedish population‐based study. J Am Acad Child Adolesc Psychiatry 2012;51:467‐76. [DOI] [PubMed] [Google Scholar]
- 25. Vassos E, Agerbo E, Mors O et al. Urban‐rural differences in incidence rates of psychiatric disorders in Denmark. Br J Psychiatry 2015;208:435‐40. [DOI] [PubMed] [Google Scholar]
- 26. Davis J, Eyre H, Jacka FN et al. A review of vulnerability and risks for schizophrenia: Beyond the two hit hypothesis. Neurosci Biobehav Rev 2016;65:185‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collishaw S, Pickles A, Messer J et al. Resilience to adult psychopathology following childhood maltreatment: evidence from a community sample. Child Abuse Negl 2007;31:211‐29. [DOI] [PubMed] [Google Scholar]
- 28. Cicchetti D. Resilience under conditions of extreme stress: a multilevel perspective. World Psychiatry 2010;9:145‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rutten BP, Hammels C, Geschwind N et al. Resilience in mental health: linking psychological and neurobiological perspectives. Acta Psychiatr Scand 2013;128:3‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Starr LR, Hammen C, Conway CC et al. Sensitizing effect of early adversity on depressive reactions to later proximal stress: moderation by polymorphisms in serotonin transporter and corticotropin releasing hormone receptor genes in a 20‐year longitudinal study. Dev Psychopathol 2014;26:1241‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hanscombe KB, Trzaskowski M, Haworth CM et al. Socioeconomic status (SES) and children's intelligence (IQ): in a UK‐representative sample SES moderates the environmental, not genetic, effect on IQ. PLoS One 2012;7:e30320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Padmanabhan JL, Shah JL, Tandon N et al. The “polyenviromic risk score”: aggregating environmental risk factors predicts conversion to psychosis in familial high‐risk subjects. Schizophr Res (in press). [DOI] [PMC free article] [PubMed]
- 33. Johnson SB, Riis JL, Noble KG. State of the art review: poverty and the developing brain. Pediatrics 2016;137:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taylor PJ. The unreliability of high human heritability estimates and small shared effects of growing up in the same family. Biol Theory 2008;2:387‐97. [Google Scholar]
- 35. Byrd AL, Manuck SB. MAOA, childhood maltreatment, and antisocial behavior: meta‐analysis of a gene‐environment interaction. Biol Psychiatry 2014;75:9‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taylor A, Kim‐Cohen J. Meta‐analysis of gene‐environment interactions in developmental psychopathology. Dev Psychopathol 2007;19:1029‐37. [DOI] [PubMed] [Google Scholar]
- 37. Hosang GM, Shiles C, Tansey KE et al. Interaction between stress and the BDNF Val66Met polymorphism in depression: a systematic review and meta‐analysis. BMC Med 2014;12:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karg K, Burmeister M, Shedden K et al. The serotonin transporter promoter variant (5‐HTTLPR), stress, and depression meta‐analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry 2011;68:444‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown GW, Ban M, Craig TK et al. Serotonin transporter length polymorphism, childhood maltreatment, and chronic depression: a specific gene‐environment interaction. Depress Anxiety 2013;30:5‐13. [DOI] [PubMed] [Google Scholar]
- 40. Uher R, Caspi A, Houts R et al. Serotonin transporter gene moderates childhood maltreatment's effects on persistent but not single‐episode depression: replications and implications for resolving inconsistent results. J Affect Disord 2011;135:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zammit S, Spurlock G, Williams H et al. Genotype effects of CHRNA7, CNR1 and COMT in schizophrenia: interactions with tobacco and cannabis use. Br J Psychiatry 2007;191:402‐7. [DOI] [PubMed] [Google Scholar]
- 42. van Winkel R. Family‐based analysis of genetic variation underlying psychosis‐inducing effects of cannabis: sibling analysis and proband follow‐up. Arch Gen Psychiatry 2011;68:148‐57. [DOI] [PubMed] [Google Scholar]
- 43. Di Forti M, Iyegbe C, Sallis H et al. Confirmation that the AKT1 (rs2494732) genotype influences the risk of psychosis in cannabis users. Biol Psychiatry 2012;72:811‐6. [DOI] [PubMed] [Google Scholar]
- 44. Morgan CJ, Freeman TP, Powell J et al. AKT1 genotype moderates the acute psychotomimetic effects of naturalistically smoked cannabis in young cannabis smokers. Transl Psychiatry 2016;6:e738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Borglum AD, Demontis D, Grove J et al. Genome‐wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol Psychiatry 2014;19:325‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dunn EC, Wiste A, Radmanesh F et al. Genome‐wide association study (GWAS) and genome‐wide by environment interaction study (GWEIS) of depressive symptoms in African American and Hispanic/Latina women. Depress Anxiety 2016;33:265‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee SH, Byrne EM, Hultman CM et al. New data and an old puzzle: the negative association between schizophrenia and rheumatoid arthritis. Int J Epidemiol 2015;44:1706‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Keers R, Coleman JR, Lester KJ et al. A genome‐wide test of the differential susceptibility hypothesis reveals a genetic predictor of differential response to psychological treatments for child anxiety disorders. Psychother Psychosom 2016;85:146‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Green EK, Grozeva D, Jones I et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry 2010;15:1016‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li J, Zhao L, You Y et al. Schizophrenia related variants in CACNA1C also confer risk of autism. PLoS One 2015;10:e0133247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pasparakis E, Koiliari E, Zouraraki C et al. The effects of the CACNA1C rs1006737 A/G on affective startle modulation in healthy males. Eur Psychiatry 2015;30:492‐8. [DOI] [PubMed] [Google Scholar]
- 52. Uher R, Rutter M. Basing psychiatric classification on scientific foundation: problems and prospects. Int Rev Psychiatry 2012;24:591‐605. [DOI] [PubMed] [Google Scholar]
- 53. Stringaris A, Goodman R. Longitudinal outcome of youth oppositionality: irritable, headstrong, and hurtful behaviors have distinctive predictions. J Am Acad Child Adolesc Psychiatry 2009;48:404‐12. [DOI] [PubMed] [Google Scholar]
- 54. Jaddoe VW, van Duijn CM, Franco OH et al. The Generation R Study: design and cohort update 2012. Eur J Epidemiol 2012;27:739‐56. [DOI] [PubMed] [Google Scholar]
- 55. Judd LL, Akiskal HS, Maser JD et al. A prospective 12‐year study of subsyndromal and syndromal depressive symptoms in unipolar major depressive disorders. Arch Gen Psychiatry 1998;55:694‐700. [DOI] [PubMed] [Google Scholar]
- 56. Judd LL, Akiskal HS, Schettler PJ et al. The long‐term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530‐7. [DOI] [PubMed] [Google Scholar]
- 57. Zanarini MC, Frankenburg FR, Reich DB et al. Attainment and stability of sustained symptomatic remission and recovery among patients with borderline personality disorder and axis II comparison subjects: a 16‐year prospective follow‐up study. Am J Psychiatry 2012;169:476‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McGuffin P, Katz R, Watkins S et al. A hospital‐based twin register of the heritability of DSM‐IV unipolar depression. Arch Gen Psychiatry 1996;53:129‐36. [DOI] [PubMed] [Google Scholar]
- 59. Nanni V, Uher R, Danese A. Childhood maltreatment predicts unfavorable course of illness and treatment outcome in depression: a meta‐analysis. Am J Psychiatry 2012;169:141‐51. [DOI] [PubMed] [Google Scholar]
- 60. Maj M. Cycloid psychotic disorder: validation of the concept by means of a follow‐up and a family study. Psychopathology 1990;23:196‐204. [DOI] [PubMed] [Google Scholar]
- 61. Pfuhlmann B, Jabs B, Althaus G et al. Cycloid psychoses are not part of a bipolar affective spectrum: results of a controlled family study. J Affect Disord 2004;83:11‐9. [DOI] [PubMed] [Google Scholar]
- 62. Cade JF. Lithium salts in the treatment of psychotic excitement. Med J Aust 1949;2:349‐52. [DOI] [PubMed] [Google Scholar]
- 63. Iniesta R, Stahl D, McGuffin P. Machine learning, statistical learning and the future of biological research in psychiatry. Psychol Med 2016;46:2455‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Han B, Pouget JG, Slowikowski K et al. A method to decipher pleiotropy by detecting underlying heterogeneity driven by hidden subgroups applied to autoimmune and neuropsychiatric diseases. Nat Genet 2016;48:803‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Uher R. The implications of gene‐environment interactions in depression: will cause inform cure? Mol Psychiatry 2008;13:1070‐8. [DOI] [PubMed] [Google Scholar]