

Abstract
Oxalicumone A (POA), a novel dihydrothiophene-condensed chromone, was isolated from the marine-derived fungus Penicillium oxalicum. Previous reports demonstrated that POA exhibits strong activity against human carcinoma cells, thus it has been suggested as a bioactive anticancer agent. To research the toxic effect of POA on cultured normal epithelial human kidney-2 (HK-2) cells and evaluate its clinical safety, cell survival was evaluated by the Cell Counting Kit-8 assay and apoptosis was evaluated by Hoechst 33258 staining, flow cytometry, caspase-3 activity assay and western blotting. 2′,7′-Dichlorofluorescin diacetate and JC-1 dye staining was used to evaluate reactive oxygen species (ROS) production and mitochondrial membrane potential (MMP), respectively. The results indicated that POA inhibited HK-2 cell growth and promoted apoptosis, by increasing levels of Fas cell surface cell receptor and the B-cell lymphoma 2 associated protein X apoptosis regulator (Bax)/B-cell lymphoma 2 apoptosis regulator (Bcl-2) ratio. POA treatment also induced release of ROS and loss of MMP in HK-2 cells. Compared with untreated control, a significant decrease was also demonstrated in superoxide dismutase activity and glutathione content with POA treatment, accompanied by enhanced release of N-acetyl-β-D-glucosaminidase, increased leakage of lactate dehydrogenase, increased malondialdehyde formation and increased release of nitric oxide. In conclusion, the present in vitro study revealed that POA exhibits antiproliferation activity on HK-2 cells, through stimulation of apoptosis and oxidative stress injury, which may be relevant to its clinical application. The present study may, therefore, offer valuable new information regarding the use of POA as a candidate novel antitumor drug for clinical use.
Keywords: Penicillium oxalicum, oxalicumone A, human kidney-2 cells, cytotoxicity, apoptosis, mitochondria
Introduction
The fungus Penicillium oxalicum (Trichocomaceae) is known for its capacity to produce novel acetogenins, sesquiterpenoids, diterpenoids, prostanoids and steroids that exhibit bioactivities including antitumor, antituberculosis, anti-inflammation, and antioxidant effects (1,2). Penicillium oxalicum SCSGAF 0023 was isolated from the South China Sea gorgonian Muricella flexuosa by Zhang et al (3). Previous studies on the dihydrothiophene-condensed chromone oxalicumone A (POA; chemical structure in Fig. 1), isolated from a culture broth of Penicillium oxalicum SCSGAF 0023 (4), demonstrated that it exhibits significant antitumor activity against several carcinoma cell lines, including A375, SW-620 and HeLa, with IC50 values of 8.9, 7.8 and 18.4 µM, respectively (5). This suggests that POA might serve as a candidate for a novel antitumor drug. However, whether POA is toxic to normal cells, in vivo or in vitro, has not been reported to date, and this would be a major limitation to the clinical application of POA. In the drug discovery and development pipeline, toxicity information is important in guiding pharmaceutical application and optimization. Thus, investigations of the possible toxicity and its mechanisms are vital for POA clinical development, in order to determine drug safety.
Figure 1.
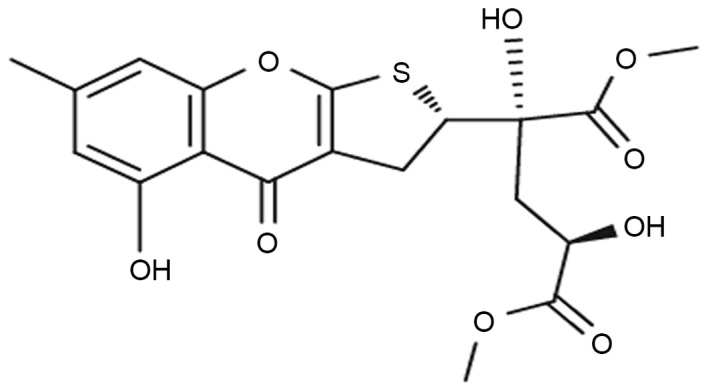
Chemical structure of oxalicumone A.
The function of renal proximal tubules is to concentrate the glomerular filtrate by reabsorption of essential molecules, and hence they are easily injured by drugs and chemicals that are eliminated through the kidney (6). Since the proximal tubule is one of the most common sites of injury by nephrotoxic drugs, screening and understanding the toxicity potential of drug candidates on renal proximal tubule cells is important as a first step to drug discovery.
In the present study, several biological endpoints were examined to assess the possible toxic effects of POA on human kidney-2 (HK-2) cells in vitro and the underlying mechanism.
Materials and methods
Materials
D/F12 medium and fetal bovine serum (FBS) were purchased from Hyclone; GE Healthcare Life Sciences (Logan, UT, USA) and Biological Industries (Kibbutz Beit-Haemek, Israel), respectively. The Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). Trypsin, dimethyl sulfoxide (DMSO), and Hoechst 33258 were purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). The Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining kit, DNA content quantitation assay kit, 5,5,6,6-tetra-chloro-1,1,3,3-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) dye and caspase-3 activity assay kit were purchased from Nanjing KeyGen Biotech. Co., Ltd. (Nanjing, China). Glutathione (GSH; cat. no. CEA294Ge) and N-acetyl-β-D-Glucosaminidase (NAG; cat. no. CSB-E07444 m) ELISA kits were purchased from Uscn Life Science, Inc. (Wuhan, China) and CUSABIO Biotech. Co., Ltd. (Wuhan, China), respectively. Radioimmunoprecipitation assay (RIPA) lysis buffer and enhanced chemiluminescence (ECL) kit were purchased from Biomiga, Inc. (San Diego, CA, USA) and Beyotime Institute of Biotechnology (Haimen, China), respectively. The bicinchoninic acid (BCA) protein assay kit was purchased from BioTeke Corporation (Beijing, China). Fas cell surface death receptor (Fas; dilution, 1:4,000; cat. no. ab133619), B-cell lymphoma 2 apoptosis regulator (Bcl-2; dilution, 1:4,000; cat. no. ab182858), Bcl-2 associated protein X apoptosis regulator (Bax; dilution, 1:4,000; cat. no. ab32503) and β-actin (dilution, 1:4,000; cat. no. ab16039) antibodies were purchased from Abcam (Cambridge, UK). Horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (dilution, 1:80,000; cat. no. IH-0011) was obtained from Boster Systems, Inc. Pleasanton. CA, USA. All other chemicals were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). POA was provided by the South China Sea Institute of Oceanology (Guangzhou, China). The structure of POA was determined by infrared, nuclear magnetic resonance and mass spectrometry and its purity of >98% was determined by high performance liquid chromatography. POA was dissolved in DMSO and phosphate buffer saline (PBS) to obtain stock solutions (40 mM), which were stored at −20°C. Prior to use in an experiment, the stock solution was diluted to the indicated concentrations with culture medium. During the experiments, the DMSO content in the medium never exceeded 0.5% (v/v).
Cell culture
HK-2 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and were grown in D/F12 supplemented with 10% FBS in a humidified incubator at 37°C in the presence of 5% CO2. The culture medium was changed every 2 days. Cells for assays were detached by a solution of 0.25% trypsin and 0.02% EDTA.
CCK-8 cell viability assay
HK-2 cell viability was evaluated by the CCK-8 assay. Briefly, HK-2 cells (1×104cells/well) were seeded in 96-well microplates and then cultured in D/F12 growth medium for 24 h. Subsequently, the medium was replaced with D/F12 growth medium containing 10, 20, 30, 40, 50, 60, 70, 80, 90 or 100 µM POA. Cells containing equal volumes of cell culture medium but no POA (0 µM), were used as a control in each experiment throughout the study. Following exposure to POA for 24, 48 or 72 h, 10 µl of the CCK-8 assay solution was added into each well, followed by incubation of the microplates at 37°C in 5% CO2/95% air for 2 h. Finally, absorption was measured at 450 nm using a microplate reader (PerkinElmer, Inc., Waltham, MA, USA), with a reference wavelength of 650 nm (7). Three different experiments were performed and the average value was calculated.
Morphological changes in the cell and nucleus
Morphological changes in the HK-2 cells were evaluated by phase contrast optical microscopy (Leica Microsystems Gmbh, Wetzlar, Germany). Morphological changes of the cell nuclei were evaluated by fluorescent visualization with Hoechst 33258 staining. Briefly, cells (4×104 cells/well) cultured on slides were treated with 0, 20 or 40 µM POA for 24 h. Following treatment, cells were washed with PBS, fixed with 4% paraformaldehyde for 10 min and then incubated for 5 min with 5 mg/ml Hoechst 33258 fluorescent dye. The cells were then washed, dried, and photographed using a fluorescence microscope.
Annexin V/PI staining assay
The early apoptosis rate was measured using Annexin V-FITC/PI double staining and a Accuri™ C6 FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) with BD CFlow Software v.264.15 (BD Biosciences). Following treatment with 0, 20 or 40 µM POA for 24 h, 5×105 HK-2 cells were harvested by centrifugation at 800 × g for 5 min at 4°C, washed twice with ice-cold PBS and resuspended in 500 µl binding buffer, followed by the addition of 5 µl Annexin V-FITC conjugate and 5 µl PI buffer, according to the manufacturer's protocol. Following incubation in the dark for 15 min at room temperature, the cells were analyzed by flow cytometry. Each determination is based on the acquisition of 10,000 events (8).
Cell cycle phase analysis
Following treatment with 0, 20 or 40 µM POA for 24 h, 5×105 cells were collected by centrifugation at 800 × g for 5 min at 4°C, washed twice with PBS, and then fixed with 70% chilled ethanol for 12 h. Following fixation, cells were washed twice with PBS and incubated in PBS containing 50 mg/ml PI, 1 mg/ml RNase A and Triton X-100 (0.5%) at 4°C for 30 min in the dark. The fluorescence emitted from the PI-DNA complex was measured using Accuri™ C6 FACScan flow cytometry (BD Biosciences) with BD CFlow Software v.264.15 (BD Biosciences). The cells with nuclei with sub-G1 content were considered apoptotic cells (9).
Activation of caspase 3
A caspase 3 activity assay kit was used to measure caspase 3 activity, as previously described (10). In brief, 1×106 cells were treated with 0, 20, 40, or 80 µM POA for 24 h, then cells were harvested by centrifugation at 800 × g for 5 min at 4°C, washed twice with ice-cold PBS, resuspended in lysis buffer and left on ice for 60 min. The lysate was centrifuged at 12,000 × g at 4°C for 5 min. The cell supernatant was incubated with the enzyme specific colorimetric substrate acetyl-Asp-Gla-Val-Asp-phosphorylated nitroanilide (Ac-DEVD-pNA) in assay buffer for 2 h at 37°C. The concentration of pNA from the Ac-DEVD-pNA substrate was determined by the optical absorbance at 405 nm using a microplate reader (PerkinElmer, Inc., Waltham, MA, USA).
Assays of antioxidant status
For assays of GSH, superoxide dismutase (SOD), malondialdehyde (MDA), nitric oxide (NO), N-acetyl-β-D-glucosaminidase (NAG), lactate dehydrogenase (LDH) and reactive oxygen species (ROS), HK-2 cells (1×106 cells/well) were seeded in 6-well plates and then cultured in D/F12 growth medium for 24 h. Subsequently, the medium was replaced with D/F12 growth medium containing 0, 20, 40, or 80 µM POA and incubated for 24 h at 37°C. Following treatment, 1×106 cells were harvested by centrifugation at 800 × g for 5 min at 4°C, then lysed on ice for 30 min using lysis buffer (50 mmol Tris-HCl, 1.0 mmol/l EDTA, 150 mmol/l NaCl and 0.1% SDS), and centrifuged once more at 5,000 × g for 10 min at 4°C. The supernatant was used for the enzymatic assays. For assessment of extracellularly released molecules, the culture medium was analyzed following the 24 h POA treatment.
GSH content
The antioxidant enzyme GSH was measured using a GSH ELISA kit, as per the manufacturer's instructions. GSH concentration was determined by measurement of the absorbance at 450 nm using an ELISA Reader (PerkinElmer, Inc., Waltham, MA, USA).
SOD activity
SOD is a scavenger of superoxide. SOD activity was detected using the xanthine/xanthine oxidase method based on the production of O2− anions (11). HK-2 cells and lysates were prepared as described above, but in 25 cm2 culture flasks at 1×106 cells/flask. The SOD activity in the cell lysates was determined using a formula calculation based on absorbance values at 550 nm (11). Activity of SOD is expressed as units per mg of cellular protein (U/mg prot).
Determination of lipid peroxidation
The concentration of MDA, an end product generated from lipid peroxidation, was measured using an MDA detection kit according to the manufacturer's instructions (12). Briefly, MDA reacts with thiobarbituric acid (TBA) at 95–100°C in acidic conditions and the reaction produces a pink MDA-TBA conjugate, which can be measured using an EnSpire multi-mode microplate reader (PerkinElmer, Inc., Waltham, MA, USA) at 532 nm. The cellular MDA concentration was expressed as nmol/mg of cellular protein (13).
Assessment of NO
NO, a potentially toxic molecule, is a labile, diffusible product of mammalian cells. It serves as a short-lived messenger molecule involved in diverse biological phenomena such as cytotoxicity (14). NO levels were estimated by measuring the accumulation of nitrite in the culture medium, which is the resulting byproduct of NO metabolites. The culture medium samples were tested with a NO detection kit, according to the manufacturer's instruction. Absorbance at 550 nm was measured with a microtiter plate reader (PerkinElmer, Inc., Waltham, MA, USA). A range of sodium nitrite concentrations were used to generate the standard curve. The cellular NO content was expressed as µmol/g of cellular protein.
NAG content
NAG is a lysosomal enzyme that is present in proximal tubular cells. The activity of NAG is generally low in HK-2 cells and increases as a consequence of the breakdown of renal tubular cells (15). The culture medium samples were tested for NAG by ELISA (16), according to the manufacturer's instructions. Absorbance at 450 nm was measured with a microtiter plate reader, and NAG concentration was determined based on a standard curve.
LDH leakage assay
LDH is a cytoplasmic oxidoreductase, that is important in maintaining cell membrane integrity. When cells are damaged, LDH leaks into the culture medium. LDH leakage was measured by testing the culture medium samples with an LDH ELISA, according to the manufacturer's instructions. Absorbance at 450 nm was measured with a microtiter plate reader and the concentration of LDH was determined based on a standard curve.
Measurement of ROS production
ROS are oxygenic free radicals which can alter the balance of endogenous protective systems, such as glutathione and enzymatic antioxidant defense systems (17). ROS production in cells was detected by a standard, cell permeable, fluorescent dye method, 2,7-dichlorofluorescin diacetate (DCFH-DA). Intracellular esterases hydrolyze DCFH-DA to 2,7-dichlorofluorescin (DCFH), which is then oxidized by ROS to dichlorofluorescein (DCF), and therefore amount of fluorescence reflects ROS production in cells. Fluorescence intensity was measured by Accuri™ C6 flow cytometry (BD Biosciences) with BD CFlow Software v.264.15 (BD Biosciences) at excitation and emission wavelengths of 485 nm and 530 nm, respectively (18).
Mitochondrial membrane potential (MMP)
Flow cytometry following JC-1 staining was used to evaluate loss of MMP (19,20). HK-2 cells (5×105 cells/well) in 6-well culture plates were treated with 0, 20, 40 or 80 µM POA for 24 h. Cells were trypsinized, washed twice with ice-cold PBS, resuspended in PBS and labeled with 5 µg/ml JC-1 cationic dye for 30 min in the dark at 37°C. Following labeling, cells were washed twice with PBS, resuspended in PBS at a concentration of 1 to 5×106 cells/ml and analyzed by Accuri™ C6 flow cytometry (BD Biosciences) with BD CFlow Software v.264.15 (BD Biosciences).
Western blot analysis
HK-2 cells cultured at a density of 1×106 cells/well in 6-well microplates were treated with 0, 20 or 40 µM POA for 24 h. The treated cells were lysed in RIPA lysis buffer containing 1% phenylmethane sulfonyl fluoride and incubated on ice for 30 min. Lysates were centrifuged at 8,000 × g for 8 min at 4°C, and the protein concentration in the supernatant was determined by BCA assay. A total of 60 µg/lane protein was separated by 12% SDS-PAGE and then electrotransferred to a polyvinylidene fluoride membrane. The membrane was incubated in 5% skim milk in TBST (0.1% Tween-20) buffer for 2 h, followed by incubation with the primary antibody (diluted in TBST with 5% skim milk) overnight at 4°C. The incubated membrane was washed 3 times with TBST prior to incubation for 1 h at 25°C with the secondary antibody which was diluted in TBST with 5% skim milk. Following 3 washes with TBST, the immune complexes were detected using an ECL kit.
Statistical analysis
Data are presented as the mean ± standard deviation of at least 3 independent experiments. Statistical differences between means among multiple groups were analyzed with one way analysis of variance followed by either the Bonferroni or Tamhane's T2 post hoc test when appropriate. Statistical analysis was performed using SPSS v.16.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Cytotoxic effect of POA on HK-2 cells
CCK-8 assays were used to determine the effect of POA on the growth of HK-2 cells. As presented in Fig. 2, cell growth became markedly inhibited from dose concentrations of 70, 50 and 50 µM POA at 24, 48, or 72 h, respectively. Increased cytotoxicity was observed as time and POA concentration increased (Fig. 2). DMSO, which was the vehicle control for the treatments, did not exhibit any inhibitory effect on the cell viability (0 µM POA; Fig. 2).
Figure 2.
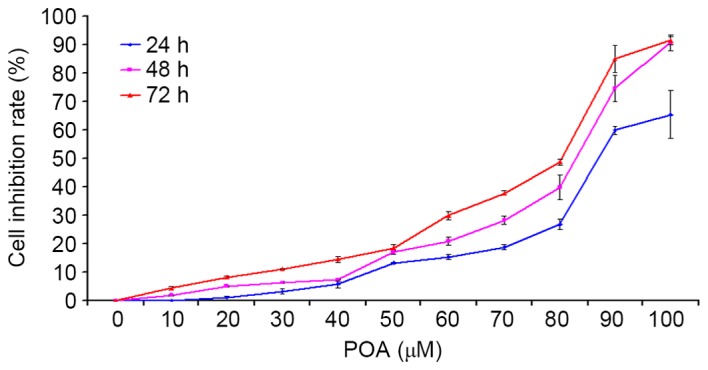
Effect of POA on HK-2 cell viability. HK-2 cells were treated with 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, or 100 µM POA for 24, 48 or 72 h, and cell viability was measured by the Cell Counting Kit-8 assay. Data are presented as the mean ± standard deviation of 3 independent experiments. POA, oxalicumone A.
Effect of POA on morphological changes in HK-2 cells and their nuclei
Using a phase contrast microscope, the untreated HK-2 cells appeared to assume a common spindle cell shape with intact nuclei (Fig. 3A). In the presence of 20 µM POA for 24 h, there were slight morphological alterations when compared with the untreated HK-2 cells, however, in the presence of 40 µM POA for 24 h, the HK-2 cells contracted and became rounded and detached from the substrate (Fig. 3A). HK-2 cells were stained with the nuclear dye Hoechst 33258, which also serves as marker of apoptosis. Control nuclei did not exhibit distinct changes (Fig. 3B). In the presence of 40 µM POA, the cells' nuclei revealed brighter Hoechst 33258 staining, karyopyknosis and fragmented chromatin, indicating that they might be undergoing apoptosis. By contrast, there were no obvious alterations in the nuclei with the 20 µM POA treatment (Fig. 3B).
Figure 3.
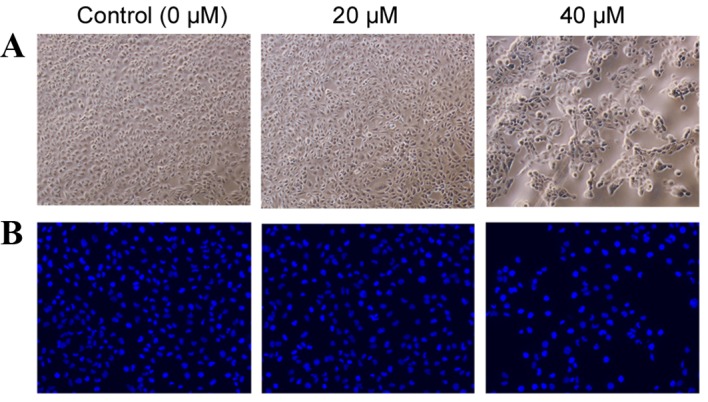
Morphological changes in HK-2 cells and their nuclei in response to POA treatment. (A) Phase contrast images of HK-2 cells following treatment with 0, 20 or 40 µM POA for 24 h (original magnification, ×40). (B) Fluorescence photomicrograph of HK-2 cells stained with Hoechst 33258 following treatment with 0, 20 or 40 µM POA for 24 h (original magnification, ×40). POA, oxalicumone A.
Effect of POA on HK-2 cell apoptosis
The apoptotic effect of POA on HK-2 cells was further investigated by flow cytometric analysis. Annexin V-FITC/PI double-labeling was used for the detection of phosphatidylserine (PS) externalization, a characteristic of early apoptosis (21,22). As demonstrated in Fig. 4A, a significant increase in early apoptotic cells was observed in cells treated with 20, 40 or 80 µM POA for 24 h (P<0.05), when compared with untreated cells (Annexin V+/PI− cells, as measured in the Q4 quadrant of the plots; Fig. 4A). To gain insights into the mechanism of the growth inhibition activity of POA in HK-2 cells, its effect on cell cycle distribution was examined by flow cytometry. The number of apoptotic cells in each treatment group was determined by observing the SubG1 apoptotic peaks, which are attributed to the reduced DNA content. As demonstrated in Fig. 4B, treatment with POA for 24 h resulted in a significant, dose-dependent, increase in the number of apoptotic cells and a significant increase in the proportion of cells in the S and G2/M phases compared with untreated cells. By contrast, a significant reduction of the proportion of cells in the G1 phase was observed in POA-treated cells compared with untreated cells (Fig. 4B). In conclusion, the results from the Annexin V-FITC/PI double-labeling assay and the cell-cycle analysis indicated that POA-mediated growth inhibition was accompanied by a dose-dependent increase in apoptosis.
Figure 4.

Effect of POA on cell apoptosis and cell cycle arrest. HK-2 cells were treated with 0, 20, 40 or 80 µM POA for 24 h. (A) The percentage of apoptotic cells was measured by flow cytometric analysis following Annexin V-FITC/PI double staining. Early apoptotic cells are quantified in the Q4 quadrant of the plots. (B) Cell cycle analysis was performed by PI staining and flow cytometry following exposure to 0, 20, 40 or 80 µM POA for 24 h. Data are presented as the mean ± standard deviation of 3 independent experiments. *P<0.05, **P<0.01 and ***P<0.001 vs. control. POA, oxalicumone A; FITC, fluorescein isothiocyanate; PI, propidium iodide.
Effect of POA on caspase 3 activity
Caspase 3 is a key regulator of the terminal phase of apoptosis (23). As demonstrated in Fig. 5, a significant, dose-dependent increase in caspase 3 activity was observed in HK-2 cells treated with 20, 40 or 80 µM POA for 24 h when compared with control untreated cells (P<0.001).
Figure 5.

Effect of POA on caspase 3 activity. HK-2 cells were treated with 0, 20, 40 or 80 µM POA for 24 h and caspase 3 activity was measured by ELISA. Data are presented relative to untreated control cells and as the mean ± standard deviation of 3 independent experiments. ***P<0.001 vs. control. POA, oxalicumone A.
GSH content
The content of the crucial, cellular, non-enzymatic antioxidant GSH was determined by ELISA. Treatment with 40 and 80 µM POA induced a significant decrease in cellular GSH content (Fig. 6A). The level of GSH in the control cells was 3.85 µg/ml, and this was reduced to 3.71 (P>0.05 vs. control), 3.19 (P<0.01 vs. control) and 2.81 (P<0.01 vs. control) µg/ml with 20, 40 and 80 µM POA treatment, respectively (Fig. 6A).
Figure 6.

Effect of POA on the antioxidant status of HK-2 cells. HK-2 cells were treated with 0, 20, 40 or 80 µM POA for 24 h and the cell lysates (for endogenous molecules assessment) or the culture medium (for assessment of molecules released extracellularly) were tested by ELISA or specific dye kits. (A) GSH content. (B) SOD activity. (C) MDA content. (D) NO content. (E) NAG content. (F) LDH release. Data are presented as the mean ± standard deviation of 3 independent experiments. *P<0.05, **P<0.01 and ***P<0.001 vs. control. POA, oxalicumone A; GSH, glutathione; SOD, superoxide dismutase; MDA, malondialdehyde; NO, nitric oxide; NAG, N-acetyl-β-D-Glucosaminidase; LDH, lactate dehydrogenase.
SOD activity
HK-2 cells treated with 40 or 80 µM POA demonstrated a significant decrease in SOD activity compared with control (Fig. 6B). The SOD activity of the control cells was 10.93 U/ml, which was reduced to 10.55 (P>0.05 vs. control), 9.47 (P<0.001 vs. control) and 9.12 (P<0.001 vs. control) U/ml in cells treated with 20, 40 and 80 µM POA, respectively (Fig. 6B).
MDA content
MDA is a well-studied intermediate of oxidative stress (24). HK-2 cells treated with 20, 40 and 80 µM POA exhibited a significant increase in MDA levels (Fig. 6C). MDA levels in control cells were 0.40 nmol/mg of total protein, and these levels were increased to 0.72 (P<0.01 vs. control), 1.22 (P<0.001 vs. control) and 1.65 nmol/mg (P<0.001 vs. control) with 20, 40 and 80 µM POA, respectively (Fig. 6C).
NO release
A significant effect of POA was observed on NO production in HK-2 cells: In control cells, the concentration of NO released in the culture medium was 11.42 µmol/l, and this was significantly decreased to 10.20, 7.25 and 4.42 µmol/l with 20, 40 and 80 µM POA treatment, respectively (P<0.001 vs. control; Fig. 6D).
NAG release
NAG is commonly regarded as a marker of proximal tubular cell integrity, and measurement of its release is used as an indicator of early kidney cells injury (25). As demonstrated in Fig. 6E, the NAG release level in HK-2 cells was significantly increased to 1.19-(P<0.01 vs. control), 1.40-(P<0.001 vs. control) and 1.70-fold (P<0.001 vs. control) relative to the control cells, with 20, 40 and 80 µM POA, respectively.
LDH leakage
Cellular LDH leakage in the culture medium was measured as a key signal of damaged cells. LDH activity was significantly increased to 1.48-(P<0.01 vs. control), 2.02-(P<0.01 vs. control) and 3.15-fold (P<0.001 vs. control) relative to control cells with 20, 40 and 80 µM POA, respectively (Fig. 6F).
Effect of POA on ROS production
ROS is a critical regulator of cellular homeostasis, and overproduction of ROS induces apoptosis and cell death (26). The results presented in Fig. 7 indicated that POA treatment significantly promoted ROS accumulation in HK-2 cells, in a dose-dependent manner. Treatment of HK-2 cells with 20, 40 or 80 µM POA for 24 h increased the % of cells that exhibit ROS accumulation to 3.8, 6.2 and 15.2% of total, respectively, compared with 0.9% of total in the control (Fig. 7).
Figure 7.
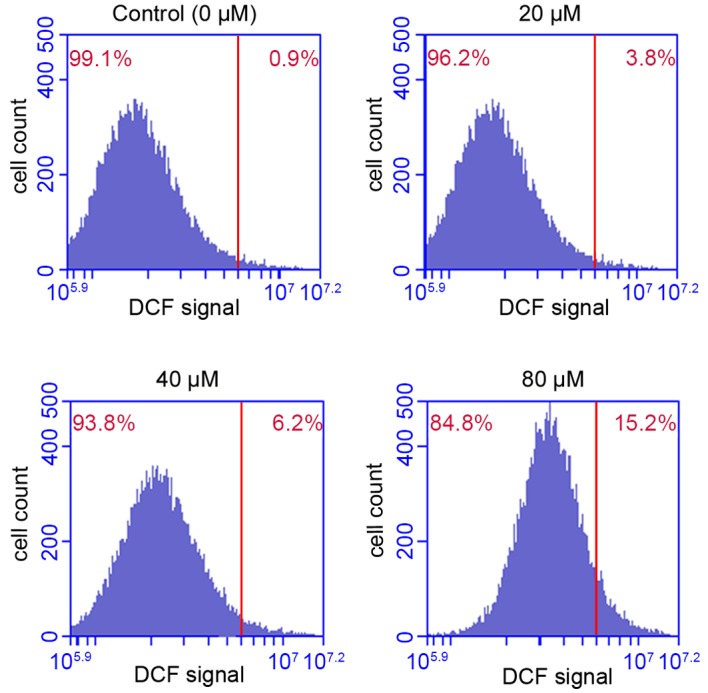
Effect of POA treatment on ROS production in HK-2 cells. HK-2 cells were treated with 0, 20, 40 or 80 µM POA for 24 h, then ROS production was measured by DCFH-DA dye staining and flow cytometry analysis. The % on the upper right corner of the plots denotes the proportion of the total cells analyzed that exhibited positive DCF staining. Data are presented as the mean ± standard deviation of 3 independent experiments. POA, oxalicumone A; ROS, reactive oxygen species; DCFH-DA, 2,7-dichlorofluorescin diacetate; DCF, 2,7-dichlorofluorescin.
Effect of POA on MMP
Disruption of mitochondrial integrity is one of the early events leading to apoptosis (27). Accumulation of the cationic dye JC-1 was assessed by flow cytometry to evaluate the effect of POA on MMP. Following exposure of HK-2 cells to 20, 40 or 80 µM POA for 24 h, MMP disruption was detected in 14.8, 16.2 and 26.5% of total cells, respectively, compared with 12.3% of total cells in the untreated control group (Fig. 8).
Figure 8.

Effect of POA on MMP depolarization in HK-2 cells. Cells were treated with 0, 20, 40 or 80 µM POA for 24 h, and MMP depolarization was measured by JC-1 staining and flow cytometry analysis. Data are presented as the mean ± standard deviation of 3 independent experiments. POA, oxalicumone A; MMP, mitochondrial membrane potential; JC-1, 5,5,6,6-tetra-chloro-1,1,3,3-tetraethylbenzimidazolyl- carbocyanine iodide.
Effect of POA on expression of apoptotic markers
Bcl-2 is a Bcl-2 family anti-apoptotic protein, which helps cells to prevent apoptosis. Bax is a Bcl-2 family pro-apoptotic protein, that translocates from the internal to the outer mitochondrial membrane, inducing the release of pro-apoptotic factors, and resulting in apoptosis. The expression levels of Fas, Bax and Bcl-2 were analyzed by western blot in HK-2 cells treated with 0, 20 or 40 µM POA for 24 h (Fig. 9). POA treatment significantly increased protein expression levels of Fas and Bax compared with control (Fig. 9). By contrast, the expression of Bcl-2 was significantly decreased with POA treatment, compared with control (Fig. 9). These results indicated that POA treatment resulted in an upregulation of the pro-apoptotic proteins Fas and Bax and a downregulation of the anti-apoptotic protein Bcl-2.
Figure 9.

Effect of POA on protein expression levels of Fas, Bax and Bcl-2 in HK-2 cells. HK-2 cells were treated with 0, 20, or 40 µM POA for 24 h, and cell lysates were analyzed by western blot for protein expression of apoptotic markers Fas, Bax and Bcl-2. β-actin was used as an internal control. (A) Representative images. (B) Quantification of density signals from the western blot images. Results are reported as fold ratio of control untreated cells for each protein. Data are presented as the mean ± standard devation of ≥3 indepndent experiments. **P<0.01 and ***P<0.001 vs. control. POA, oxalicumone A; Fas, Fas cell surface death receptor; Bax, Bcl-2 associated protein X apoptosis regulator; Bcl-2, B-cell lymphoma 2 apoptosis regulator.
Discussion
Previous studies have demonstrated that POA exhibits significant cytotoxicity against several carcinoma cell lines with IC50≤10 µM (3,5) however, the pharmacological mechanism remains unknown. The present study was designed to assess for the first time the toxic effect of POA on proximal tubular cells. Using a cell viability assay, it was demonstrated that POA treatment inhibited proliferation of HK-2 cells in a dose-dependent manner (Fig. 2), suggesting that POA treatment can induce cytotoxicity in human renal epithelial cells.
Apoptosis, a highly structured and ordered process, is a fundamental form of cell death, and its purpose is to eliminate superfluous, harmful, and metabolically perturbed cells (28,29). In the present in vitro study, cell nuclei stained by Hoechst 33258 displayed karyopyknosis, deepened staining and karyorrhexis following POA treatment, which is consistent with cellular shrinkage, chromatin condensation, and nuclear fragmentation (Fig. 3). These morphological characteristics are typical of apoptosis. Apoptosis was further confirmed by flow cytometry experiments, where POA was demonstrated to significantly increase the proportion of cells undergoing early apoptosis in a dose-dependent manner, compared with control (Fig. 4A). In addition, cell cycle analysis revealed that POA treatment resulted in an increase of the sub-G1 cell population, which is a hallmark of apoptosis (30), as well as an increase in the S-phase cell population, compared with control (Fig. 4B). These results indicate that POA treatment accelerated the cell cycle, caused abnormal proliferation and induced apoptosis in the HK-2 cells. Furthermore, an increase in POA-induced caspase 3 activity was demonstrated (Fig. 5), suggesting that the mechanism of POA-mediated apoptosis on HK-2 cells was associated with the activation of this key executor of apoptosis.
To examine whether the toxicity effect of POA on HK-2 cells involves the mitochondria, the expression levels of proteins that regulate apoptosis via the Fas, mitochondrial, and endoplasmic reticulum signaling pathways were analyzed. It has been reported that Fas is important in the initiation of the cell death signaling pathway (31). As demonstrated in Fig. 9, POA treatment increased the expression of Fas, which suggests that POA-induced apoptosis in HK-2 may be regulated via the mitochondria-mediated intrinsic apoptotic pathway.
It is well-established that the mitochondrial death pathway is regulated by members of the Bcl-2 family. This protein family can be divided into either anti-apoptotic [such as Bcl-2 and Bcl-2-like 1 protein extra-large (Bcl-XL)] or pro-apoptotic [such as Bax, BH3 interacting domain death agonist (Bid) and Bcl-2-like protein 11 (Bim)] members (32,33). The Bax protein translocates to the outer membrane of mitochondria, induces the release of pro-apoptotic factors and induces apoptosis (34), while the Bcl-2 proteins sequester in mitochondria, inhibit the release of pro-apoptotic proteins and factors from liposomes, and suppress apoptosis (35). The ratio of Bax/Bcl-2 proteins thus determines whether the cell will survive or undergo apoptosis (36). In the present study, POA treatment induced expression of the pro-apoptotic protein Bax and reduced expression of the anti-apoptotic protein Bcl-2 (Fig. 9). This results in a significant increase of the Bax/Bcl-2 ratio, which indicates that POA alters the balance between anti- and pro-apoptotic protein members in the cell, and eventually induces apoptosis via the Bcl-2 mitochondrial signaling pathway.
Since POA treatment induced apoptosis, its effect on ROS production was analyzed further. Excessive generation of ROS compromises cellular function and integrity (37). As demonstrated in Fig. 7, POA treatment was demonstrated to induce a significant and dose-dependent increase in ROS production compared with control, which is in agreement with POA inducing apoptosis and cytotoxicity. POA treatment also induced disruption of MMP compared with control cells (Fig. 8). It has been reported that alterations in the mitochondrial functions through increased ROS generation and the disruption of MMP can lead to cell death (38). Thus, the increased ROS generation and the depolarization of MMP observed following POA treatment further confirms its pro-apoptotic role via the mitochondrial pathway.
Cellular redox balance is maintained by various enzymatic and nonenzymatic antioxidant systems. The disruption of this balance by exogenous substances results in cell damage (39). The antioxidant systems include antioxidant enzymes, including SOD and aldo-keto reductase, and nonenzymatic molecules, including GSH, carotenoid and coenzyme (40). The main physiological function of GSH is scavenging free radicals and antioxidants (41). In the present study, POA treatment decreased the content of GSH in cells, compared with control (Fig. 6A). SOD is a common component of the cellular antioxidant systems, blocking cell damage resulting from oxygen free radicals, and promptly repairing the damaged cells. In the present study, POA treatment significantly decreased SOD activity (Fig. 6B), which is in agreement with the effect of POA on GSH. These results further confirmed that POA induced oxidative stress on HK-2 cells.
It has been reported that membrane lipids are major targets of free radicals (42). An increase in the levels of lipid peroxidation products, such as MDA, is an indication of membrane lipid damage (43). As demonstrated in Fig. 6C, POA treatment elevated MDA levels compared with control, which suggests that POA induces lipid peroxidation injury. These results confirm a POA-mediated oxidative stress induction, consistent with the results from the SOD and GSH measurements.
NO is a free radical that is a key participator in both physiological and pathological processes. Similar to other free radical species, small amounts of NO in the cell exhibit a protective effect, while excessive amounts of NO induce cell damage (44). LDH is a cytoplasmic, glycolytic enzyme, that is highly expressed in kidney cells. When cells are damaged, LDH leaks from the cell cytoplasm to the extracellular space (45). The present study revealed that NO and LDH levels were increased following POA treatment compared with control, which indicated that POA induced damage in HK-2 cells (Fig. 6D and F). NAG is generally considered a reliable and sensitive enzyme marker of tubular epithelia injury (46,47). POA treatment resulted in increased NAG release (Fig. 6E), which further confirmed the cytotoxicity effect of POA on HK-2 cells.
In conclusion, using a wide variety of experimental assays, the present study demonstrated that POA significantly induced cytotoxicity and apoptosis in human renal epithelial cells via the mitochondrial pathway. Analysis of antioxidant systems activity indicated that POA induced damage to cellular antioxidant enzymes, and changed the cellular antioxidant balance towards cell toxicity. It remains unclear whether the toxicological mechanism identified in the present study is the same as the pharmacological mechanism, and this needs to be further investigated. The present study evaluated for the first time (to the best of our knowledge) POA-mediated toxicity and its mechanism in vitro, which is valuable information for novel drug discovery. HK-2 cells are suitable for toxicity studies in vitro, because the proximal tubule cells of the kidney are the most common site of injury by nephrotoxic drugs. Eventually, the present study may contribute towards the potential use of POA in clinical application.
Acknowledgements
This work was financed by the National Marine Public Welfare Research Project of China (grant no. 201305017), National Natural Science Foundation of China (grant no. 81573638) and Xinhuo Planning Project of Guangzhou University of Chinese Medicine (grant no. XH20150107).
References
- 1.Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat Prod Rep. 2012;29:144–222. doi: 10.1039/C2NP00090C. [DOI] [PubMed] [Google Scholar]
- 2.Qi SH. Bioactive compounds from marine gorgonian corals. In: Rahman A, editor. Studies In Natural Products Chemistry. 1st. Vol. 38. Elsevier; Oxford: 2012. pp. 325–352. [DOI] [Google Scholar]
- 3.Zhang XY, Bao J, Wang GH, He F, Xu XY, Qi SH. Diversity and antimicrobial activity of culturable fungi isolated from six species of the South China Sea gorgonians. Microb Ecol. 2012;64:617–627. doi: 10.1007/s00248-012-0050-x. [DOI] [PubMed] [Google Scholar]
- 4.Sun YL, He F, Liu KS, Zhang XY, Bao J, Wang YF, Nong HX, Xu XY, Qi SH. Cytotoxic dihydrothiophene-condensed chromones from marine-derived fungus Penicillium oxalicum. Planta Med. 2012;78:1957–1961. doi: 10.1055/s-0033-1350805. [DOI] [PubMed] [Google Scholar]
- 5.Sun YL, Bao J, Liu KS, Zhang XY, He F, Wang YF, Nong XH, Qi SH. Cytotoxic dihydrothiophene-condensed chromones from the marine-derived fungus Penicillium oxalicum. Planta Med. 2013;79:1474–1479. doi: 10.1055/s-0033-1350805. [DOI] [PubMed] [Google Scholar]
- 6.Wu Y, Connors D, Barber L, Jayachandra S, Hanumegowda UM, Adams SP. Multiplexed assay panel of cytotoxicity in HK-2 cells for detection of renal proximal tubule injury potential of compounds. Toxicol In Vitro. 2009;23:1170–1178. doi: 10.1016/j.tiv.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Jia L, Kuang Z, Wu T, Hong Y, Chen X, Leung WK, Xia J, Cheng B. The in vitro and in vivo antitumor effects of clotrimazole on oral squamous cell carcinoma. PLoS One. 2014;9:e98885. doi: 10.1371/journal.pone.0098885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tripathi M, Singh BK, Mishra C, Raisuddin S, Kakkar P. Involvement of mitochondria mediated pathways in hepatoprotection conferred by Fumaria parviflora Lam. extract against nimesulide induced apoptosis in vitro. Toxicol In Vitro. 2010;24:495–508. doi: 10.1016/j.tiv.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Nigam N, George J, Srivastava S, Roy P, Bhui K, Singh M, Shukla Y. Induction of apoptosis by (6)-gingerol associated with the modulation of p53 and involvement of mitochondrial signaling pathway in B(a)P-induced mouse skin tumorigenesis. Cancer Chemother Pharmacol. 2010;65:687–696. doi: 10.1007/s00280-009-1074-x. [DOI] [PubMed] [Google Scholar]
- 10.Terada Y, Inoshita S, Hanada S, Shimamura H, Kuwahara M, Ogawa W, Kasuga M, Sasaki S, Marumo F. Hyperosmolality activates Akt and regulates apoptosis in renal tubular cells. Kidney Int. 2001;60:553–567. doi: 10.1046/j.1523-1755.2001.060002553.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhang JQ, Shen M, Zhu CC, Yu FX, Liu ZQ, Ally N, Sun SC, Li K, Liu HL. 3-Nitropropionic acid induces ovarian oxidative stress and impairs follicle in mouse. PLoS One. 2014;9:e86589. doi: 10.1371/journal.pone.0086589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lund AK, Knuckles TL, Akata Obot C, Shohet R, McDonald JD, Gigliotti A, Seagrave JC, Campen MJ. Gasoline exhaust emissions induce vascular remodeling pathways involved in atherosclerosis. Toxicol Sci. 2007;95:485–494. doi: 10.1093/toxsci/kfl145. [DOI] [PubMed] [Google Scholar]
- 13.Zhu CP, Hu W, Wu H, Hu X. No evident dose-response relationship between cellular ROS level and its cytotoxicity-a paradoxical issue in ROS-based cancer therapy. Sci Rep. 2014;4:5029. doi: 10.1038/srep05029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuste JE, Tarragon E, Campuzano CM, RosBernal F. Implications of glial nitric oxide in neurodegenerative diseases. Front Cell Neurosci. 2015;9:322. doi: 10.3389/fncel.2015.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiu JS. Models used to assess renal function. Drug Dev Res. 1994;32:247–255. doi: 10.1002/ddr.430320409. [DOI] [Google Scholar]
- 16.Ali RJ, Al-Obaidi FH, Arif HS. The role of urinary N-acetyl Beta-D-glucosaminidase in children with urological problems. Oman Med J. 2014;29:285–288. doi: 10.5001/omj.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prasad NR, Menon VP, Vasudev V, Pugalendi KV. Radioprotective effect of sesamol on gamma-radiation induced DNA damage, lipid peroxidation and antioxidants levels in cultured human lymphocytes. Toxicology. 2005;209:225–235. doi: 10.1016/j.tox.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Sharikabad MN, Ostbye KM, Lyberg T, Brørs O. Effect of extracellular Mg(2+) on ROS and Ca(2+) accumulation during reoxygenation of rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2001;280:H344–H353. doi: 10.1152/ajpheart.2001.280.1.H344. [DOI] [PubMed] [Google Scholar]
- 19.Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) Biochem Biophys Res Commun. 1993;197:40–45. doi: 10.1006/bbrc.1993.2438. [DOI] [PubMed] [Google Scholar]
- 20.Yao J, Jiang Z, Duan W, Huang J, Zhang L, Hu L, He L, Li F, Xiao Y, Shu B, Liu C. Involvement of mitochondrial pathway in triptolide-induced cytotoxicity in human normal liver L-02 cells. Biol Pharm Bull. 2008;31:592–597. doi: 10.1248/bpb.31.592. [DOI] [PubMed] [Google Scholar]
- 21.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 22.Zamai L, Falcieri E, Marhefka G, Vitale M. Supravital exposure to propidium iodide identifies apoptotic cells in the absence of nucleosomal DNA fragmentation. Cytometry. 1996;23:303–311. doi: 10.1002/(SICI)1097-0320(19960401)23:4<303::AID-CYTO6>3.3.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 23.Wilson MR. Apoptotic signal transduction: Emerging pathways. Biochem Cell Biol. 1998;76:573–582. doi: 10.1139/o98-056. [DOI] [PubMed] [Google Scholar]
- 24.Bonnes-Taourel D, Guérin MC, Torreilles J. Is malonaldehyde a valuable indicator of lipid peroxidation. Biochem Pharmacol. 1992;44:985–988. doi: 10.1016/0006-2952(92)90132-3. [DOI] [PubMed] [Google Scholar]
- 25.Yamashita T, Doi K, Hamasaki Y, Matsubara T, Ishii T, Yahagi N, Nangaku M, Noiri E. Evaluation of urinary tissue inhibitor of metalloproteinase-2 in acute kidney injury: A prospective observational study. Crit Care. 2014;18:716. doi: 10.1186/s13054-014-0716-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: The mitochondria-dependent andmitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–1180. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]
- 27.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi (m)) in apoptosis; an update. Apoptosis. 2003;8:115–128. doi: 10.1023/A:1022945107762. [DOI] [PubMed] [Google Scholar]
- 28.Jacobson MD, Weil M, Raff MC. Programmed cell death in the animal development. Cell. 1997;88:347–354. doi: 10.1016/S0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 29.Scaffidi C, Kirchhoff S, Krammer PH, Peter ME. Apoptosis signaling in lymphocytes. Curr Opin Immunol. 1999;11:277–285. doi: 10.1016/S0952-7915(99)80045-4. [DOI] [PubMed] [Google Scholar]
- 30.Pirocanac EC, Nassirpour R, Yang M, Wang J, Nardin SR, Gu J, Fang B, Moossa AR, Hoffman RM, Bouvet M. Bax-induction gene therapy of pancreatic cancer. J Surg Res. 2002;106:346–351. doi: 10.1006/jsre.2002.6473. [DOI] [PubMed] [Google Scholar]
- 31.Waring P, Müllbacher A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol Cell Biol. 1999;77:312–317. doi: 10.1046/j.1440-1711.1999.00837.x. [DOI] [PubMed] [Google Scholar]
- 32.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 33.Nakazawa Y, Kamijo T, Koike K, Noda T. ARF tumor suppressor induces mitochondria-dependent apoptosis by modulation of mitochondrial Bcl-2 family proteins. J Biol Chem. 2003;278:27888–27895. doi: 10.1074/jbc.M300510200. [DOI] [PubMed] [Google Scholar]
- 34.Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, et al. Inhibition of Bax channel- forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 35.Borner C. The Bcl-2 protein family: Sensors and checkpoints for life-or-death decisions. Mol Immunol. 2003;39:615–647. doi: 10.1016/S0161-5890(02)00252-3. [DOI] [PubMed] [Google Scholar]
- 36.Lanave C, Santamaria M, Saccone C. Comparative genomics: The evolutionary history of the Bcl-2 family. Gene. 2004;333:71–79. doi: 10.1016/j.gene.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 37.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urra FA, Cordova-Delgado M, Pessoa-Mahana H, Ramírez-Rodriguez O, Weiss-Lopez B, Ferreira J, Araya-Maturana R. Mitochondria: A promising target for anticancer alkaloids. Curr Top Med Chem. 2013;13:2171–2183. doi: 10.2174/15680266113139990150. [DOI] [PubMed] [Google Scholar]
- 39.Rosas HD, Doros G, Bhasin S, Thomas B, Gevorkian S, Malarick K, Matson W, Hersch SM. A systems-level ‘misunderstanding’: The plasma metabolome in Huntington's disease. Ann Clin Transl Neurol. 2015;2:756–768. doi: 10.1002/acn3.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brzović-Šarić V, Landeka I, Šarić B, Barberić M, Andrijašević L, Cerovski B, Oršolić N, Đikić D. Levels of selected oxidative stress markers in the vitreous and serum of diabetic retinopathy patients. Mol Vis. 2015;21:649–664. [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitt B, Vicenzi M, Garrel C, Denis FM. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study. Redox Biol. 2015;6:198–205. doi: 10.1016/j.redox.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valko M, Jomova K, Rhodes CJ, Kuca K, Musílek K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. 2016;90:1–37. doi: 10.1007/s00204-015-1579-5. [DOI] [PubMed] [Google Scholar]
- 43.Dubner D, Gisone P, Jaitovich I, Perez M. Free radicals production and estimation of oxidative stress related to gamma irradiation. Biol Trace Elem Res. 1995;47:265–270. doi: 10.1007/BF02790126. [DOI] [PubMed] [Google Scholar]
- 44.Förstermann U, Sessa WC. Nitric oxide synthases: Regulation and function. Eur Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, Sukhatme VP, Seth P. LDH-A Inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer (HLRCC) Mol Cancer Ther. 2009;8:626–635. doi: 10.1158/1535-7163.MCT-08-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laina A, Schwartz D. Renal tubular and molecular events in acute renal failure. Nephron. 1994;68:413–418. doi: 10.1159/000188316. [DOI] [PubMed] [Google Scholar]
- 47.Jongman RM, van Klarenbosch J, Molema G, Zijlstra JG, de Vries AJ, van Meurs M. Angiopoietin/Tie2 dysbalance is associated with acute kidney injury after cardiac surgery assisted by cardiopulmonary bypass. PLoS One. 2015;10:e0136205. doi: 10.1371/journal.pone.0136205. [DOI] [PMC free article] [PubMed] [Google Scholar]