

Abstract
Mutations in Fused in Sarcoma/Translocated in Liposarcoma (FUS) cause familial forms of amyotrophic lateral sclerosis (ALS), a neurodegenerative disease characterized by progressive axonal degeneration mainly affecting motor neurons. Evidence from transgenic mouse models suggests mutant forms of FUS exert an unknown gain-of-toxic function in motor neurons, but mechanisms underlying this effect remain unknown. Towards this end, we studied the effect of wild type FUS (FUS WT) and three ALS-linked variants (G230C, R521G and R495X) on fast axonal transport (FAT), a cellular process critical for appropriate maintenance of axonal connectivity. All ALS-FUS variants impaired anterograde and retrograde FAT in squid axoplasm, whereas FUS WT had no effect. Misfolding of mutant FUS is implicated in this process, as the molecular chaperone Hsp110 mitigated these toxic effects. Interestingly, mutant FUS-induced impairment of FAT in squid axoplasm and of axonal outgrowth in mammalian primary motor neurons involved aberrant activation of the p38 MAPK pathway, as also reported for ALS-linked forms of Cu, Zn superoxide dismutase (SOD1). Accordingly, increased levels of active p38 MAPK were detected in post-mortem human ALS-FUS brain tissues. These data provide evidence for a novel gain-of-toxic function for ALS-linked FUS involving p38 MAPK activation.
Introduction
Mutations in Fused in Sarcoma/Translocated in Liposarcoma (FUS/TLS or FUS) are causative for amyotrophic lateral sclerosis (ALS)1, 2, a progressive and fatal neurodegenerative disease characterized by progressive dying back degeneration of both upper and lower motor neurons3. Analysis of pathogenic mechanisms associated with mutant FUS in mammalian cells is complicated because FUS is a multifunctional protein engaged in numerous aspects of RNA and DNA processing, and overexpression of FUS WT proved cytotoxic3. Recent reports describing a series of transgenic FUS animal models provide strong evidence for mutant FUS exerting a gain-of-toxic function in the cytoplasm that results in motor neuron degeneration4–6. However, specific toxic function(s) associated with FUS mutations have not been elucidated, and could involve defects in RNA-related processes3, cytoskeletal transport7 and/or the proteostasis network8.
Deficits in fast axonal transport (FAT) and abnormal activation of protein kinases are early pathogenic events in both SOD1-related and sporadic forms of ALS9–15, but whether ALS-linked FUS affects FAT has not been evaluated. To address this issue, recombinant FUS proteins were perfused into isolated squid axoplasm and the effect on FAT examined. Although FUS WT had no discernable effect, ALS-FUS variants robustly inhibited anterograde (conventional kinesin-dependent) and modestly inhibited retrograde (cytoplasmic dynein-dependent) FAT when perfused into squid axoplasm. Irrespective of whether the disease-causing mutation affects the nuclear localization signal (e.g., R521G or R495X) or the glycine-rich domain (e.g., G230C) of FUS, all disease variants tested similarly inhibited FAT. Addition of mutant FUS with the molecular chaperone Hsp110 mitigated this impairment, suggesting that a misfolded conformation of mutant FUS underlies this gain-of-toxic function.
Pharmacological experiments further indicated that impairments in FAT in squid axoplasm and in axon outgrowth in murine primary motor neurons by mutant FUS both require activation of the p38 MAPK pathway, which has been broadly implicated in various neurodegenerative disorders including Alzheimer’s Disease and ALS16–19. Indeed, aberrant activation of the p38 MAPK pathway by ALS-related forms of Cu, Zn superoxide dismutase (SOD1) was previously demonstrated in squid axoplasm10, 12, 20 and transgenic mouse models expressing ALS-linked mutant SOD112, 21–23. We detected elevated levels of phosphorylated p38 MAPK in post-mortem CNS tissues derived from individuals harboring FUS mutations, providing support that the phenotypes observed in vitro in this report are relevant to the human disease. Although SOD1 and FUS have no known overlapping physiological functions, mutant and misfolded forms of these proteins appear to converge on a common pathogenic pathway that involves p38 MAP kinases. Together these results identify p38 MAPK activation as a novel gain of toxic function of mutant FUS that results in reductions in FAT and strengthens the association between the p38 MAPK axis and neurodegeneration in ALS.
Results
ALS-FUS inhibits fast axonal transport
Cytoskeletal abnormalities, including impaired axonal transport, are implicated as early events in the pathogenesis of ALS and other neurodegenerative disorders24. We have demonstrated the utility of isolated squid axoplasm as a powerful ex vivo system to study alterations in FAT induced by disease-related neuropathogenic proteins10, 12, 20, 24–27, which were confirmed in mammalian disease models12 and in human post-mortem central nervous system (CNS) tissues10, 28, 29. Here, we sought to determine whether ALS-linked mutant FUS proteins also affect FAT in the squid axoplasm preparation. To this end, recombinant wild-type (WT) and three distinct ALS-linked FUS variants (G230C, R521G and R495X) were expressed and purified as glutathione-S-transferase (GST)-tagged recombinant proteins (Fig. 1A). Mutations R495X and R521G are located within a nuclear localization signal (NLS) that induce FUS translocation from the nucleus to the cytoplasm30, with R521 representing a “hot-spot” for ALS-linked FUS mutations31. The R495X truncation mutation leads to an omission of the entire NLS and a more aggressive disease phenotype (i.e., earlier age of onset and shorter disease duration) in affected patients30, 32. Conversely, mutations in the Gly-rich region of FUS, such as G230C, do not affect the cellular localization of exogenously expressed FUS in mammalian cells33. Because untagged FUS is intrinsically prone to aggregation34 and undergoes liquid-liquid phase separation35–38, the GST tag was maintained throughout this study to enhance the solubility of FUS proteins. Recombinant GST-FUS has been employed in various functional assays in vitro, and therefore the tag is not expected to disrupt the fold or function of this protein34, 39, 40. Moreover, the GST protein itself does not have an effect on FAT25.
Figure 1.

ALS-linked mutant FUS proteins impair anterograde and retrograde FAT. (A) ALS-linked mutations (G230C, R495X and R521G) investigated in this study are mapped onto the domain structure of GST-FUS. RRM = RNA recognition motif, RGG = arginine-glycine-glycine-rich, ZFD = zinc-finger domain and NLS = nuclear localization signal. (B–G) FUS proteins (2.5 μM) were perfused into isolated squid axoplasm and fast axonal transport (FAT) rates (μm/s) of membrane bounded-organelles measured as a function of time (minutes). For (B,C), slopes of the linear best fits for velocities obtained for each axoplasm (D–G) were averaged and plotted as bar graphs in Graphpad Prism with the standard error of the mean (SEM) for anterograde (blue bars; A) and retrograde (red bars; B) velocities. Those conditions with statistical significance (p < 0.05) relative to FUS WT are denoted by *, determined by one-way Anova and Tukey post-hoc test for multiple comparisons. (D–G) Motility plots comprised of the raw data for every axoplasm (‘n’ denotes the number of axoplasms analyzed for each condition) are shown, where linear best fits of the compiled data are shown for anterograde (blue arrowheads; linear fit shown as a solid blue line) and retrograde (red arrowheads; linear fit shown as a solid red line) directions. Protein obtained from at least two independent protein expression and purifications were included for each FUS variant.
Next, FUS proteins were perfused into squid axoplasm and their effect on FAT determined using video-enhanced contrast differential interference (DIC) microscopy27. The use of DIC microscopy in the axoplasm preparation allows for quantitation of anterograde (conventional kinesin-dependent) and retrograde (cytoplasmic dynein-dependent) transport of membrane-bounded organelles (MBOs) in real time27, 41. Because the plasma membrane is removed from the axon in a manner that leaves intact the FAT machinery and polarity of the axonal cytoskeleton, recombinant FUS proteins could be directly perfused into the axoplasm and their effect on FAT quantitatively measured27. Recombinant FUS proteins were perfused into squid axoplasm at 2.5 µM, a physiologically relevant concentration in mammalian cells38. Perfusion of FUS WT (2.5 µM) had no effect on either anterograde or retrograde FAT over the entire time course of the assay (50 min; Fig. 1B–D). In striking contrast, perfusions with either FUS G230C, R495X or R521G significantly inhibited anterograde FAT and, to a lesser extent, retrograde FAT (Fig. 1B,C,E,F,G). Together, these data reveal FAT inhibition as a novel, toxic gain of function for mutant FUS. Given the similar effect of all three variants tested, we focused our next experiments on FUS R521G, a mutation with high frequency in the ALS-FUS population31.
Inhibition of FAT by ALS-FUS is mediated by p38 MAPK
The inhibitory effect of mutant FUS proteins on FAT was independent from nucleus-dependent activities (i.e., transcription) because neuronal cell bodies are absent from the axoplasm preparation41. Endogenous axoplasmic kinases and phosphatases are plausible candidates, as kinases mediate the inhibitory effect of other neuropathogenic proteins on FAT by promoting abnormal phosphorylation of conventional kinesin and cytoplasmic dynein, major molecular motors responsible for FAT in mature neurons. For example, activation of cJun N-terminal kinases (JNK) was found to mediate the inhibitory effect of pathogenic androgen receptor and huntingtin on FAT25, 26. Based on these precedents, we co-perfused the JNK-specific kinase inhibitor SP600125 with FUS R521G. However, inhibition of JNK did not rescue the inhibitory effect of mutant-FUS on FAT (Fig. 2A–C).
Figure 2.

The inhibitory effect of mutant-FUS on FAT is mediated by activated p38 MAPK. FUS R521G was co-perfused with the indicated pharmacological inhibitor or Hsp110 into squid axoplasm and fast axonal transport evaluated as described in Fig. 1. (A–C) Co-perfusion of the pharmacological JNK kinase inhibitor SP600125 (0.5 μM) with R521G failed to rescue FAT inhibition. In contrast, inhibition of the p38 MAPK pathway by co-perfusion of either SB203850 (5 μM) or NQDI-1 (20 μM) prevented the toxic effects of FUS R521G on FAT (A,B,D,E). (F) Similarly, co-perfusion of Hsp110 (0.6 μM) and R521G ameliorated the inhibition of FAT by mutant FUS. Those conditions with statistical significance (p < 0.05) relative to FUS R521G are denoted by *, determined by one-way Anova and Tukey post-hoc test for multiple comparisons.
The beta-isoform of p38 mitogen-activated protein kinase (MAPK) also inhibits both anterograde and retrograde FAT in isolated squid axoplasm12, and ALS-related forms of ALS have been shown to inhibit FAT through a mechanism involving p38 activation10, 12, 20, prompting us to evaluate the potential role of this kinase. Strikingly, co-perfusion of FUS R521G with the p38 MAPK pharmacological inhibitor SB203580 completely prevented the adverse effects of this protein on FAT, restoring FAT rate values to that of control axoplasms (Fig. 2A,B,D). p38 kinases are typically activated by sequential activation of upstream MAPK kinases (MKKs) and MAPK kinase kinases (MKKKs). An example of the latter is apoptosis signal-regulating kinases (ASKs), which mediates the inhibitory effects of pathogenic SOD1 variants on FAT20. Therefore, we assessed the effect of a pharmacological ASK1 inhibitor (NQDI-1) on FUS-mediated inhibition of FAT. Significantly, NQDI-1 also prevented the adverse effects of FUS R521G on FAT (Fig. 2A,B,E). Taken together, results from vesicle motility assays indicate that pathogenic forms of two seemingly different ALS-linked proteins (FUS and SOD1) converge on a common pathway that involves aberrant activation of p38-specific MAP kinase pathways. There are, however, mechanistic differences between SOD1- and FUS-induced inhibition of FAT. Whereas both anterograde and retrograde FAT are impaired by mutant FUS (Figs 1 and 2), only anterograde FAT was significantly inhibited by misfolded SOD110, 12, 20. We previously detected phosphorylated p38 MAPK in squid axoplasm perfused with misfolded SOD1 by immunoblot analysis12. However, the same mammalian anti-p38 MAPK antibody used previously failed to detect activated p38 MAPK in squid perfused with mutant FUS (data not shown). These results suggest that different p38 MAPK isoforms may be activated by mutant FUS compared to misfolded SOD1, and that these different isoforms may not be detected by our available anti-phosphorylated p38 antibodies.
The molecular chaperone Hsp110 mitigates the effect of mutant FUS on FAT
Protein misfolding is a hallmark feature of neurodegenerative diseases, including ALS8. Indeed, FUS aggregates are detected within neurons and glia of human post-mortem ALS-FUS tissue2, 42, in cultured cells1, and in vitro 34. To address the possibility that a misfolded form of mutant FUS activates the p38 MAPK pathway, the molecular chaperone Hsp110 was co-perfused with mutant FUS protein. Hsp110 has been shown to stabilize unfolded proteins and synergize with other chaperones such as Hsp(c)70 and Hsp40 to disaggregate protein assemblies in vitro 43, 44. Specifically, Hsp110 prevented mutant SOD1 from activating p38 MAPK in axoplasm20 and overexpression of Hsp110 extended the survival of mutant SOD1 transgenic mice45. Co-perfusion of substoichiometric amounts of Hsp110 to FUS R521G (1:4) significantly attenuated the inhibitory effect of mutant-FUS on both anterograde and retrograde FAT rates, which were similar to those observed in FUS WT-perfused axoplasms (Fig. 2A,B,F). These data suggest that a misfolded conformation of mutant-FUS may underlie its inhibitory effect on FAT20.
Next, we attempted to identify the putative misfolded mutant FUS species. Recombinant FUS protein stocks used for squid axoplasm studies were analyzed using electron microscopy (EM). There was no visible protein precipitate, nor were any proteins in a hydrogel-like state. Instead, EM images revealed a mixture of oligomeric and higher-ordered protein assemblies for both FUS WT (Fig. 3A) and mutant FUS proteins (Fig. 3B–D). This morphological heterogeneity precluded the identification of a specific mutant FUS species that would trigger the defects in FAT.
Figure 3.

The morphology of FUS species assessed by electron microscopy. Representative electron microscopy (EM) images of negatively stained FUS WT (A), G230C (B), R521G (C) and R495X (D) proteins. Low magnification images are shown in the top row (scale bar represents 0.5 μm). High magnification images are shown for insets (dashed squares) in the bottom row (scale bar represents 200 nm). FUS exhibits heterogeneous morphologies across all samples.
Inhibition of p38 MAPK rescues impaired axon outgrowth in mammalian motor neurons expressing mutant FUS
Axonal defects have been reported in cortical neurons ectopically expressing mutant FUS46, and endogenous expression of mutant FUS in motor neurons derived from patient iPSCs recapitulated ALS-relevant phenotypes such as hyperexcitability47. Here, we sought to determine whether mutant FUS-induced toxicity within mammalian motor neurons involves p38 MAPK activity as demonstrated for squid axoplasm (Figs 1 and 2). To this end, murine motor neurons were transiently transfected at day in vitro (DIV) 2 with FLAG/HA-FUS WT, R521G and P525L in the presence of either MW069 (20 μM), an inhibitor with higher selectivity for p38 MAPK than SB203580, or a structurally related, inactive analogue of MW069 (MW069_inactive, 20 μM) as a negative control48. Motor neurons were co-transfected with the green fluorescent protein (GFP) to facilitate visualization of axonal outgrowth using live cell imaging (Fig. 4A and Supplemental Video S1). In the presence of MW069_inactive, axon outgrowth was significantly impaired in motor neurons expressing FUS P525L compared to those expressing FUS WT (Fig. 4B,C). Strikingly, axonal outgrowth rates in FUS P525L expressing motor neurons were restored to WT FUS levels in the presence of the active MW069 inhibitor (Fig. 4B,C). Axon outgrowth speed was also reduced in FUS R521G expressing motor neurons treated with MW069_inactive, although this reduction was not statistically significant compared to FUS WT with MW069_inactive treatment (p = 0.32). Moreover, the active MW069 inhibitor had no effect on axon outgrowth speed in FUS R521G expressing motor neurons (Fig. 4C). These latter results are suggestive of a toxic function of FUS R521G within the nucleus, where this protein is predominately expressed within intact motor neurons, that is unrelated to p38 MAPK activation (Fig. 4A). Supporting this idea, FUS P525L is severely mislocalized to the cytoplasm33 and expressed at detectable levels within the axon (Fig. 4A). Analogous to the direct perfusion of mutant FUS proteins into squid axoplasm (Figs 1 and 2), the presence of mutant FUS within axons of intact motor neurons was sufficient to induce toxicity through a mechanism involving p38 MAPK activity. Importantly, the MW069 inhibitor also rescues the inhibition of FAT by mutant FUS in squid axoplasm (Fig. 4D).
Figure 4.

Mutant FUS impairs axon outgrowth in motor neurons through a mechanism involving p38 MAPK activity. Murine motor neurons were transiently co-transfected with FLAGHA-FUS WT, R521G or P525L and green fluorescent protein (GFP) at 2 days in vitro (DIV). Motor neurons were cultured in 20 μM MW069, a potent and selective p38 MAPK inhibitor, or an inactive analogue, MW069_inactive. (A) FUS WT and R521G are predominately expressed within the nucleus of motor neurons, where as FUS P525L is expressed in the nucleus and within axons (white arrowheads). FUS P525L localization is similar whether motor neurons are treated with MW069_inactive or MW069 (+MW069). (B) Montages corresponding to live cell imaging of axon outgrowth (white arrow) over a 60 min time course for the indicated condition. Note that the growth cone of a FUS P525L expressing motor neuron is stalled in the presence of MW069_inactive (middle panel) relative to the active form of MW069 (bottom panel). See Supplemental Video S1 for live cell imaging of all conditions. (C) Quantification of axon outgrowth speed compiled from n = 3 biological experiments normalized to the WT + MW069_inactive condition. Statistical significance (**p < 0.01) of pertinent comparisons are indicated. Additional significant comparisons include FUS WT + MW069 versus FUS R521G + MW069_inactive (p < 0.05) and FUS WT + MW069 versus FUS P525L + MW069_inactive (p < 0.0001). (D) The inhibition of FAT by FUS R521G is blocked by MW069 in squid axoplasm. The motility plot for FUS R521G in the absence of MW069 is shown in Fig. 1G.
Increased activation of p38 MAPK is detected in human post-mortem ALS tissue
To evaluate the disease relevance of findings in the squid axoplasm preparation, we investigated the relationship between mutant FUS expression and p38 MAPK activation in nerve tissue samples obtained from ALS-FUS cases. Specifically, we examined post-mortem CNS tissues derived from ALS individuals harboring different FUS mutations (Supplemental Table S2), including R521G, H517Q, a splice site mutation (ex14del) that causes skipping of exon14, P525L and a base pair deletion (bp1408del) (A1-A5, respectively, in Fig. 5). Both ex14del and bp1408del are predicted to cause a frame shift, resulting in FUS proteins comprised of 477 and 526 amino acids, respectively, but without the correct FUS sequence at the C-terminus. All cases exhibited both upper and lower motor neuron pathology at autopsy (Supplemental Table S2), consistent with FUS pathology reported in both motor cortex and spinal cord (SpC) tissues42.
Figure 5.

Increased levels of phosphorylated p38 MAPK in human post-mortem CNS tissues derived from individuals with ALS-FUS. Immunoblot analysis of frozen post-mortem brain (A) and spinal cord (B) tissues derived from non-disease control (C1-C11) and ALS individuals harboring FUS mutations (A1-R521G; A2-H517Q; A3- ex14del; A4-P525L and A5- bp1408del) with the indicated antibodies (see Supplemental Table S2 for detailed patient information). (C) Quantification of phosphorylated (active) p38 (p-p38) by immunoblot analysis revealed higher levels of p-p38 MAPK in ALS cases (cases with levels above the control mean are indicated by red squares), compared to control cases. P-p38 values were normalized to total p38 levels. (D) Quantification of FUS levels in brain normalized to Gapdh. ALS cases with FUS levels above the control mean are indicated by red squares. (C,D) Although, there appears to be a trend in the data, differences between ALS cases and controls did not reach statistical significance due to the small sample size and inherent variability of human postmortem samples.
Next, we evaluated p38 MAPK activation in post-mortem brain and SpC tissues by immunoblotting using an antibody that selectively recognizes catalytically active forms of p38 (p-p38) phosphorylated by upstream MKKs. A phosphorylation-independent antibody against p38 MAPK (total p38) served as internal control for expression and loading. An anti-GAPDH antibody served as an additional loading control for each analysis. Notably, when compared to control tissues there were elevated levels of p-p38 relative to total p38 MAPK in both ALS motor cortex (Fig. 5A) and SpC (Fig. 5B) tissues, with a more pronounced effect in brain samples than in SpC. Three out of five ALS cases (A2-4) expressed elevated levels of p-p38 in tissue derived from the motor cortex (Fig. 5A), with p-p38 levels above the mean of the control set (Fig. 5C). Additional immunoblot analyses were performed on the brain samples. For example, a qualitative direct correlation between p-p38 and p-Hsp27, a target of p-p3849, provided independent evidence of increased p38 activation in these samples (Fig. 5A). Interestingly, three of the five ALS cases (A1, A3 and A4), all expressing mutant forms of FUS that are expected to exhibit cytoplasmic mislocalization1, 30, 33, express relatively high levels of FUS (Fig. 5A,D). This trend is consistent with the notion that mislocalized FUS fails to properly autoregulate its expression50. Two FUS bands were detected in the immunoblot for A3, the ALS case with the highest FUS levels; the faster migrating band is attributed to the mutant (477 amino acids) form, whereas the slower migrating protein (526 amino acids) is produced from the wild-type allele (Fig. 5A). The differences between control and ALS did not reach statistical significance for p-p38 (Fig. 5C; p = 0.16) or FUS (Fig. 5D; p = 0.053) biochemical quantification, likely due to the relatively small number of human cases that were available for this study and the inherent variability of human postmortem tissue, but the trend was consistent with increased p-p38 levels.
Paraffin-fixed motor cortex tissue was available for a subset of our ALS (A2 and A3) and control (C3 and C12) cases that could be used for immunostaining. Consistent with results from the immunoblot analysis, immunostaining of motor cortex tissue revealed higher p-p38 immunoreactivity from ALS cases, compared to control cases (Fig. 6). Tissues were co-stained with SMI31, an antibody reactive for phosphorylated neurofilament H that can be used to assess neuroaxonal integrity51. As expected from the marked degeneration of neurites that characterizes ALS pathology52, SMI31 reactivity was markedly reduced in both ALS cases compared to controls (Fig. 6 and Supplemental Figure S3). Quantification of the ratio of p-p38 to SMI31 revealed a statistically significant difference between controls (0.044 +/− 0.010) and ALS (0.243 +/− 0.017, p < 0.0001; Supplemental Figure S4). Elevated levels of phosphorylated p38 MAPK in affected ALS-FUS tissues shown here are consistent with reports of elevated p38 MAPK in SOD112, 22, 53 and sporadic16 forms of ALS.
Figure 6.

Active p38 MAPK in neurons within post-mortem motor cortex tissues from individuals diagnosed with ALS-FUS. Brain sections from paraffin-embedded tissue samples obtained from two control (C3 and C12) and two ALS-FUS (A2 and A3) cases were probed with antibodies against phosphorylated, catalytically active p38 (P-p38; red), phosphorylated neurofilament H (SMI31; green) as a marker of neuroaxonal integrity, and the nuclear stain DAPI (blue). Scale bar represents 10 μm. Low magnification images are shown in Supplemental Figure S3 . The P-p38 signal is higher, and SMI31 signal lower, in both ALS cases compared to controls; see Supplemental Figure S4 for quantification.
Discussion
The identification of mutant FUS-specific phenotypes have been elusive, in part because FUS WT overexpression elicits a toxic phenotype in many model organisms34, 54–56. Important insights into the mechanism of FUS-mediated pathogenesis were recently revealed by two independent studies, which compared FUS knockout mice to a series of transgenic mouse lines expressing the human FUS transgene at or below endogenous FUS levels4, 5. Both studies demonstrated that a loss of FUS expression does not cause motor neuron degeneration, whereas cytoplasmic mislocalization of FUS due to deletions or point mutations within the nuclear localization signal induced age-dependent, motor neuron degeneration4, 5. Collectively, these in vivo studies strongly implicated a toxic gain of function mechanism in FUS-mediated ALS pathogenesis. However, specific toxic effects of mutant FUS in the cytoplasm remained unknown. The present study using isolated squid axoplasm sheds new light on this question, as we demonstrate that mutant, but not WT forms of FUS, impair FAT through a mechanism dependent on activation of p38 MAPK (Fig. 7).
Figure 7.
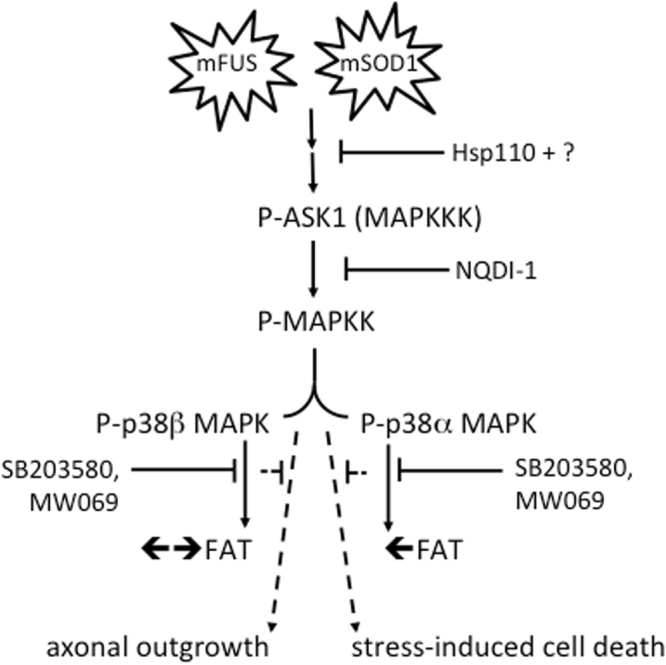
Model for aberrant activation of p38 MAP kinase(s) by mutant and misfolded ALS-associated proteins. Available data (from this study and others10, 12, 20) support a model whereby mutant and misfolded forms of FUS (mFUS; left) and SOD1 (mSOD1; right) induce the aberrant activation of the p38 MAPK pathway. Hsp110 likely synergizes with other chaperones to ameliorate the effects of mSOD1 and mFUS, possibly upstream of ASK1 and additional unidentified factors. While mSOD1 and mFUS converge on p38 MAPK activation, perfusion of these proteins into squid axoplasm have differential effects on FAT; mFUS inhibits both anterograde (←) and retrograde (→) FAT (Figs 1 and 2) whereas mSOD1 only inhibits anterograde FAT10, 12, 20, consistent with inhibition of the beta and alpha isoforms of P38 MAPK, respectively12. In addition to FAT inhibition in squid (solid lines), mFUS- and mSOD1-induced activation p38 MAPK can manifest different phenotypes in mammalian systems (dashed lines), including but not limited to inhibition of axon outgrowth (Fig. 4) and enhanced susceptibility to cell stress12. This figure is adapted from Song et al. 20.
Axonal transport defects are a common pathological feature of neurodegenerative disorders24, and represent an early, pre-symptomatic event in motor neuron diseases including familial and sporadic forms of ALS24, 57. Evidence in humans and ALS transgenic model organisms support a ‘dying back’ pattern for motor neuron death that is initiated by pathogenic events within the distal axon, including the breakdown of neuromuscular junctions (NMJs)11, 57. Consistent with a common disease phenotype associated with different genetic causes of ALS, dysfunctional NMJs preceded the loss of neuronal cell bodies in mutant FUS mice5. Transient expression of the cytoplasmically mislocalized FUS P525L variant resulted in reduced axon outgrowth in primary motor neurons (Fig. 4), consistent with observations of abnormal axonal morphologies in primary cortical neurons expressing mutant FUS46. In addition, recent work using Drosophila larval motor neurons reported a reduction in anterograde transport of MBOs induced by both WT and the cytoplasmically mislocalized P525L mutant FUS variant58. While physiologically relevant concentrations of FUS WT did not affect FAT in isolated axoplasm (Figs 1 and 2), the Drosophila study involved overexpression of FUS, which in some cases can cause toxicity34, 54–56. The effects of FUS WT on FAT in Drosophila may also represent an epiphenomenon of FUS-modulated changes in gene expression or other nuclear activities3, 59. Supporting this view, a trend toward reduced axon outgrowth speed was observed in motor neurons expressing the predominately nuclear FUS R521G variant (Fig. 4), but this effect was not mitigated by p38 MAPK inhibition as observed for the predominantly cytoplasmic FUS P525L mutant variant (Fig. 4). Additional work is needed to evaluate the extent to which nuclear dysfunction-specific mechanisms of FUS contribute to FAT alterations and impaired axon outgrowth within intact neurons.
Unlike other models, isolated squid axoplasm affords a unique, ex vivo experimental system to directly compare the effects of FUS WT and ALS-linked variants because proteins are perfused directly into axoplasm at the same concentrations. Regardless of whether these variants are predominately expressed in the nucleus (G203C), cytoplasm (R495X), or somewhere in between (R521G) within intact cells, they all inhibited FAT to a similar extent, implicating a common toxic feature among FUS mutants. The protective effect of Hsp110 (Fig. 2) further raised the possibility that a misfolded conformation underlies the toxic effect of mutant FUS on FAT. Hsp110 is nucleotide exchange factor for Hsp(c)70, and together with Hsp40 these chaperones comprise a mammalian disaggregase system that dissolves amorphous protein aggregates in vitro 43, 44. FUS variants are known to aggregate in vitro and in vivo 34, although FUS aggregation is not required to induce motor neuron degeneration in vivo 4, 5. Interestingly, FUS has been shown to undergo liquid-liquid phase separation and assemble into a hydrogel35–38. Here, all FUS samples were soluble (i.e., not visibly precipitated or in a hydrogel-like state) before they were perfused into squid axoplasm or subjected to EM. Nonetheless, EM analysis revealed the presence of soluble, self-associated FUS species in both FUS WT and mutant FUS samples (Fig. 3), precluding our ability to assign the ‘toxic’ FUS species to a particular size or morphology of FUS aggregate, as reported for other neuropathogenic proteins60. Our studies do not exclude the possibility that mutant FUS proteins acquire a toxic conformation within the squid axoplasm, but this possibility is difficult to investigate with current methods. Further, recombinant FUS is largely disordered, precluding structural analyses of the isolated full-length protein3. Thus, conformational properties of mutant FUS that confer toxicity await further study.
Inhibition of FAT by mutant FUS occurs through a mechanism that involves activation of the p38 MAPK pathway (Fig. 2). Active p38 MAPK was previously shown to phosphorylate and inhibit kinesin-1 subunits of conventional kinesin, providing a mechanistic basis by which p38 MAPK activation impairs FAT12. FUS overexpression was recently correlated with glycogen synthase kinase 3β (GSK-3β) kinase activation, which also affects anterograde FAT61, although this effect was not mutant-specific62. Nonetheless, these observations support the notion that disease-causing neuropathogenic proteins may exert toxicity through inappropriate activation of kinase-signaling cascades. Although our studies evaluate axon-autonomous effects, mutant forms of FUS and SOD1 would likely affect other p38 MAPK-related processes within intact mammalian neurons in vivo, including but not limited to transcription and activation of apoptotic pathways (Fig. 7). Intriguingly, ALS-linked forms of SOD1, a functionally and structurally dissimilar protein compared to FUS, similarly impaired FAT in squid axoplasm through activation of a p38 MAPK pathway10, 12, 20. In fact, multiple groups12, 21–23 have reported increased levels of active p38 MAPK in the SOD1-G93A transgenic ALS mouse model, as well as in human sporadic and familial ALS forms16, 18. Here, we detected elevated levels of phosphorylated p38 MAPK in the motor cortex of human ALS-FUS cases (Figs 5 and 6). Increased levels of active p38 MAPK in disease models of different ALS variants suggests that specific molecular components within the p38 MAPK axis may represent viable therapeutic targets to treat ALS19. Indeed, inhibition of p38 MAPK activity using poorly penetrant inhibitors correlates with improved motor neuron survival in ALS-SOD1 transgenic mice22, 53. The development of highly specific, brain-penetrant MAPK inhibitors should facilitate a better evaluation of p38 MAPK contributions to ALS-related phenotyopes in vivo 19. Collectively, findings in this work provide a novel conceptual framework for the development of therapeutic strategies aimed to prevent the adverse consequences of p38 MAPK activation in ALS and other neurodegenerative disorders.
Materials and Methods
Recombinant FUS expression and purification
FUS WT, R521G, R495X or G230C constructs were cloned into pGEX-6P1 vector (GE Life Sciences) and expressed in Rosetta DE3 cells (Novagen) as follows. Single bacterial colonies were inoculated into 5 ml growth medium (luria broth with 100 µg/ml ampicillin and 34 µg/ml chloramphenicol) and incubated at 37 °C while shaking for 8 h, after which these cultures were used to inoculate 150 ml growth medium (intermediate culture) that was then incubated at 30 °C for ~24 h. The intermediate cultures were used to inoculate 2L growth medium to an OD600 of 0.1–0.2. Cultures were grown at 20 °C until an OD600 ~0.8, at which time IPTG (Isopropyl β-D-1-thiogalactopyranoside) and ZnCl2 were added to a final concentration of 150 µM and 50 µM, respectively, and cells were grown for an additional 22 h at 20 °C. Cells were harvested by centrifugation and cell pellets were resuspended in lysis buffer (50 mM Tris pH 8.0 with 1 mM Dithiothreitol (DTT), 0.1 mM Ethylenediaminetetraacetic acid (EDTA) and protease inhibitor cocktail (Roche)). Cells were lysed by sonication on ice in the presence of 10 µg/ml RNase A. GST-fusion proteins were purified from cleared lysates with Glutathione-Agarose (Sigma, #G4510) resin according to the manufacturer’s instructions and stored at −80 °C.
Squid axoplasm vesicle motility assays
Vesicle motility assays were performed as described previously12, 20, 27. GST-FUS proteins alone or in combination with either pharmacological inhibitors or Hsp110 protein were employed at the following final concentrations: 2.5 µM GST-FUS proteins, 5 µM of SB203580 (EMD Millipore; 55-938-91MG), 0.5 µM of SP600125 (EMD Millipore; 42-011-95M), 10 µM of MW-069 (a gift from Dr. M. Watterson, Northwestern University), 20 µM NQDI1 (R&D Systems; 4429) and 0.6 µM HSP110 (a gift from Dr. A. Horwich, Yale University). FAT rates shown in Figs 1 and 2 were plotted using Prism (GraphPad software). The slope of the linear best-fit line for each individual axoplasm was calculated and the mean of slopes for each condition was plotted as bar graphs using Prism.
Transmission Electron Microscopy
Samples (10 µl) containing purified 12.5 µM GST-FUS protein was adsorbed onto a carbon stabilized, Formvar support film applied to a 200 mesh copper grid for 30 seconds. The grids were then washed with water and negatively stained with 1% aqueous uranyl acetate in water as described63. The stained grids were analyzed using a Philips CM10 transmission electron microscope and images were captured at both low (20,500X) and high (87,000X) magnification using a Gatan Erlangshen 785 CCD digital camera system.
Axon outgrowth measurements in primary motor neurons
Motor neurons were isolated from embryonic (E)12.5 mouse embryos and cultured as described64. FLAGHA-FUS expression cassette vectors were generated from pFRT-TO-DEST-FLAGHA-FUS-WT, which was obtained from Addgene (#26373). ALS-linked mutations were introduced with the QuikChange II Mutagenesis kit (Stratagene; 200523) according to the manufacturer’s instructions. Cells were transfected at 2 days in vitro (DIV)65 with green fluorescent protein (GFP) and FLAGHA-tagged FUS constructs in a 1:2 ratio with NeuroMag (OZ Biosciences) as previously described65. FLAGHA-FUS plasmids were generated from pFRT-TO-DEST-FLAGHA-FUS-WT obtained from Addgene (#26373). The QuikChange II Mutagenesis kit (Stratagene; 200523) was used to introduce the R521G and P525L mutations according to the manufacture’s instructions. Twenty µM MW069 or its inactive form (MW069_inactive) were added to the culture medium after transfection to inhibit p38 activity48 and maintained throughout the experiment for a total of ~18–24 hours. Cells were imaged at 3 days in vitro (DIV) using a Nikon TiE widefield microscope equipped with temperature- and CO2-controlled environmental chamber. Neurobasal medium was replaced with Hybernate E Low Fluorescence (BrainBits) 1 hour prior to imaging to reduce autofluorescence and light toxicity. GFP was used to identify transfected cells. Movies were acquired with a 20x lens at a rate of 1 frame every 10 minutes for 1 hour. The speed of axon outgrowth was measured using the ImageJ plugin MTrackJ66, 67. Data from three independent, biological replicates were normalized to the condition with FUS WT in the presence of MW069_inactive. Statistical analysis (2-way ANOVA followed by Tukey’s multiple comparisons test) was performed using Graph Pad Prism 6 to establish statistical differences (p < 0.05) between conditions. To assess FLAGHA-FUS expression, cells were post-fixed in 4% paraformaldehyde for 15 minutes and stained with an anti-HA antibody (Cell Signaling - C29F4; 1:500) overnight at 4 °C. Secondary antibody (donkey anti-Rabbit conjugated with Alexa594; Jackson ImmunoResearch) was incubated at room temperature for 1 hour.
Processing of post-mortem tissues
Frozen post-mortem tissues were a generous gift from Dr. Robert H. Brown, Jr. (University of Massachusetts Medical School). Frozen tissue samples (~100 mg) were transferred to 2 mL borosilicate glass tube (Wheaton, #358028) containing lysis buffer (50 mM Tris–HCl (pH 7.5) supplemented with 0.5 M NaCl, 1% NP-40, 1% deoxycholic acid, 0.1% SDS, 2 mM EDTA, protease and phosphatase inhibitors (Roche)). Tissues were homogenized at 4 °C using a motorized tissue grinder with motor (Wheaton,) and the resulting lysates cleared by centrifugation prior to total protein determination using a bicinchonic acid (BCA) based colorimetric assay (ThermoScientific). Thirty micrograms of total protein was loaded onto gels for SDS-PAGE and immunoblot analyses. The following antibodies were employed: anti-phospho-p38 MAPK (Cell Signaling, #9215), anti-total-p38 MAPK (Sigma, #M0800) and anti-GAPDH (Sigma, #G8795). Densitometry was performed using the Odyssey infrared imaging systems software (LI-COR).
Paraffin embedded tissues were acquired through the Alzheimer’s Disease Research Center at the Massachusetts General Hospital (Charlestown, MA). Paraffin was removed using progressive dilutions of alcohol and Xylene as described68. Tissue sections were permeabilized with 0.2% Triton-X-100 in TBS and blocked with TBS plus 2.5% goat serum (Sigma-Aldrich, G9023). Primary antibodies used were anti-phospho p38 MAPK (P-p38) (Cell Signaling CS#4511; 1:200) and SMI-31 (Covance SMI-31R, 1:1000). Secondary antibodies were goat anti-rabbit Alexa 647 serum (Invitrogen, A21244) and goat anti-mouse Alexa 488 serum (Invitrogen, A21235), both used at a 1:1000 dilution. Slides were dried and mounted using Dapi Fluoromont-G (SoutherBiotech) hard mounting media. Images were acquired using an LSM-710 confocal microscope (Zeiss). For quantitative immunofluorescence analysis, images corresponding to five regions of interest (ROIs) per case were acquired from motor cortex using the same settings. Ten images (2 cases × 5 ROIs) gathered from each channel (647 nm for P-p38 and 488 nm for SMI-31 antibodies, respectively) were processed and average pixel counts determined using standard auto-threshold algorithms embedded in ImageJ software (http://imagej.nih.gov/ij/). Statistical analysis (unpaired student t -test) with Graph Pad Prism 6 (La Jolla, CA) was used to establish statistical differences (p < 0.05) between control versus ALS-FUS cases. All procedures were repeated twice with similar results.
Electronic supplementary material
Acknowledgements
We would like to thank Drs. Arthur L. Horwich and Wayne Fenton (Yale University) for providing recombinant Hsp110 protein; Dr. Robert H. Brown Jr. (University of Massachusetts Medical School; UMMS), Diane McKenna-Yasek (UMMS), Peter Sapp (UMMS) and Isabel Constantino (Massachusetts General Hospital) for their procurement of human tissue samples; Dr. Catherine Douthwright (UMMS) for help with protein preparations; D. Martin Watterson (Northwestern University) for the active and inactive forms of MW-069, Athena Diagnostics (Marlborough, MA) for sequencing analysis; Ms. Katherine Gall (from Bosco lab, UMMS) for help with immunoblot analyses; Dr. Gregory Hendricks (Electron Microscopy Core facility, UMMS) for help with electron microscopy; Ms. Laura Kaushansky and Ms. Maeve Tischbein (from Bosco lab, UMMS) for generation of FLAG-HA FUS constructs; Dr. Louise Maranda (UMMS) for help with data analysis; and the Proteomics and Mass Spectrometry facility (UMMS) for their assistance. We are grateful for funding from NIH/NINDS (R01 NS078145, R01 NS090352, and R21 NS091860 to D.A.B., R01 NS066942A and R21 NS096642 to G.M., R01NS023868 and R01NS041170 to S.T.B.), the ALS Therapy Alliance/CVS Pharmacy (to D.A.B. and G.M.) and the ALS Association (to C.F. and J.M.).
Author Contributions
R.R.K.S., C.F., Y.S., M.R., G.M., S.T.B. and D.A.B. designed the experiments; R.R.K.S., C.F., R.G., J.M., Y.S., M.R., S.P., I.A., G.M., S.T.B. and D.A.B. performed the experiments; R.R.K.S., C.F., R.G., J.M., Y.S., M.R., G.M., S.T.B. and D.A.B. analyzed the data; J.E.L. contributed resources; R.R.K.S., G.M., S.T.B. and D.A.B. wrote the manuscript. All authors approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Reddy Ranjith K. Sama, Claudia Fallini and Rodolfo Gatto contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-00091-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gerardo Morfini, Email: gmorfini@uic.edu.
Scott T. Brady, Email: stbrady@uic.edu
Daryl A. Bosco, Email: Daryl.Bosco@umassmed.edu
References
- 1.Kwiatkowski TJ, Jr., et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 2.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sama RR, Ward CL, Bosco DA. Functions of FUS/TLS From DNA Repair to Stress Response: Implications for ALS. ASN Neuro. 2014;6:1–18. doi: 10.1177/1759091414544472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scekic-Zahirovic J, et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016;35:1077–97. doi: 10.15252/embj.201592559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma A, et al. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun. 2016;7:10465. doi: 10.1038/ncomms10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shiihashi, G. et al. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain (2016). [DOI] [PubMed]
- 7.Yasuda, K. & Mili, S. Dysregulated axonal RNA translation in amyotrophic lateral sclerosis. Wiley Interdiscip Rev RNA (2016). [DOI] [PMC free article] [PubMed]
- 8.Bosco DA, LaVoie MJ, Petsko GA, Ringe D. Proteostasis and movement disorders: Parkinson’s disease and amyotrophic lateral sclerosis. Cold Spring Harbor perspectives in biology. 2011;3:a007500. doi: 10.1101/cshperspect.a007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bilsland LG, et al. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci USA. 2010;107:20523–8. doi: 10.1073/pnas.1006869107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosco DA, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer LR, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–40. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Morfini GA, et al. Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase. PLoS One. 2013;8:e65235. doi: 10.1371/journal.pone.0065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999;819:120–31. doi: 10.1016/S0006-8993(98)01351-1. [DOI] [PubMed] [Google Scholar]
- 14.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–6. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 15.Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol. 1997;139:1307–15. doi: 10.1083/jcb.139.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendotti C, et al. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J Neuropathol Exp Neurol. 2004;63:113–9. doi: 10.1093/jnen/63.2.113. [DOI] [PubMed] [Google Scholar]
- 17.Correa SA, Eales KL. The Role of p38 MAPK and Its Substrates in Neuronal Plasticity and Neurodegenerative Disease. J Signal Transduct. 2012;2012:649079. doi: 10.1155/2012/649079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picher-Martel V, Dutta K, Phaneuf D, Sobue G, Julien JP. Ubiquilin-2 drives NF-kappaB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol Brain. 2015;8:71. doi: 10.1186/s13041-015-0162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy SM, et al. Targeting human central nervous system protein kinases: An isoform selective p38alphaMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem Neurosci. 2015;6:666–80. doi: 10.1021/acschemneuro.5b00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song Y, et al. Molecular chaperone Hsp110 rescues a vesicle transport defect produced by an ALS-associated mutant SOD1 protein in squid axoplasm. Proc Natl Acad Sci USA. 2013;110:5428–33. doi: 10.1073/pnas.1303279110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holasek SS, et al. Activation of the stress-activated MAP kinase, p38, but not JNK in cortical motor neurons during early presymptomatic stages of amyotrophic lateral sclerosis in transgenic mice. Brain Res. 2005;1045:185–98. doi: 10.1016/j.brainres.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 22.Lee JK, et al. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc Natl Acad Sci USA. 2013;110:12066–71. doi: 10.1073/pnas.1300894110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tortarolo M, et al. Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol Cell Neurosci. 2003;23:180–92. doi: 10.1016/S1044-7431(03)00022-8. [DOI] [PubMed] [Google Scholar]
- 24.Morfini GA, et al. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–86. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morfini G, et al. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nature neuroscience. 2006;9:907–16. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- 26.Morfini GA, et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nature neuroscience. 2009;12:864–71. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song Y, Kang M, Morfini G, Brady ST. Fast axonal transport in isolated axoplasm from the squid giant axon. Methods Cell Biol. 2016;131:331–48. doi: 10.1016/bs.mcb.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Chu Y, et al. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain. 2012;135:2058–73. doi: 10.1093/brain/aws133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gatto RG, et al. Analysis of YFP(J16)-R6/2 reporter mice and postmortem brains reveals early pathology and increased vulnerability of callosal axons in Huntington’s disease. Hum Mol Genet. 2015;24:5285–98. doi: 10.1093/hmg/ddv248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bosco DA, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Human molecular genetics. 2010;19:4160–75. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–26. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- 32.Waibel S, et al. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: a clinico-genetic study in Germany. Eur J Neurol. 2012;20:540–6. doi: 10.1111/ene.12031. [DOI] [PubMed] [Google Scholar]
- 33.Dormann D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO journal. 2010;29:2841–57. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun Z, et al. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011;9:e1000614. doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burke KA, Janke AM, Rhine CL, Fawzi NL. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol Cell. 2015;60:231–41. doi: 10.1016/j.molcel.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato M, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–67. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murakami T, et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron. 2015;88:678–90. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel A, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–77. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 39.Dini Modigliani S, Morlando M, Errichelli L, Sabatelli M, Bozzoni I. An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA-FUS regulatory circuitry. Nat Commun. 2014;5:4335. doi: 10.1038/ncomms5335. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–30. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brady ST, Lasek RJ, Allen RD. Fast axonal transport in extruded axoplasm from squid giant axon. Science. 1982;218:1129–31. doi: 10.1126/science.6183745. [DOI] [PubMed] [Google Scholar]
- 42.Mackenzie IR, et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011;122:87–98. doi: 10.1007/s00401-011-0838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One. 2011;6:e26319. doi: 10.1371/journal.pone.0026319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torrente, M. P. & Shorter, J. The metazoan protein disaggregase and amyloid depolymerase system: Hsp110, Hsp70, Hsp40, and small heat shock proteins. Prion7 (2014). [DOI] [PMC free article] [PubMed]
- 45.Nagy M, Fenton WA, Li D, Furtak K, Horwich AL. Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc Natl Acad Sci USA. 2016;113:5424–8. doi: 10.1073/pnas.1604885113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Groen EJ, et al. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet. 2013;22:3690–704. doi: 10.1093/hmg/ddt222. [DOI] [PubMed] [Google Scholar]
- 47.Wainger BJ, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munoz L, et al. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guay J, et al. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. 1997;110:357–68. doi: 10.1242/jcs.110.3.357. [DOI] [PubMed] [Google Scholar]
- 50.Zhou Y, Liu S, Liu G, Ozturk A, Hicks GG. ALS-Associated FUS Mutations Result in Compromised FUS Alternative Splicing and Autoregulation. PLoS Genet. 2013;9:e1003895. doi: 10.1371/journal.pgen.1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schirmer L, Antel JP, Bruck W, Stadelmann C. Axonal loss and neurofilament phosphorylation changes accompany lesion development and clinical progression in multiple sclerosis. Brain Pathol. 2011;21:428–40. doi: 10.1111/j.1750-3639.2010.00466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geevasinga N, Menon P, Ozdinler PH, Kiernan MC, Vucic S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat Rev Neurol. 2016;12:651–661. doi: 10.1038/nrneurol.2016.140. [DOI] [PubMed] [Google Scholar]
- 53.Dewil M, dela Cruz VF, Van Den Bosch L, Robberecht W. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1(G93A)-induced motor neuron death. Neurobiol Dis. 2007;26:332–41. doi: 10.1016/j.nbd.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 54.Casci I, Pandey UB. A fruitful endeavor: modeling ALS in the fruit fly. Brain Res. 2015;1607:47–74. doi: 10.1016/j.brainres.2014.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ju S, et al. A Yeast Model of FUS/TLS-Dependent Cytotoxicity. PLoS Biol. 2011;9:e1001052. doi: 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mitchell JC, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013;125:273–88. doi: 10.1007/s00401-012-1043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murray LM, Talbot K, Gillingwater TH. Review: neuromuscular synaptic vulnerability in motor neurone disease: amyotrophic lateral sclerosis and spinal muscular atrophy. Neuropathol Appl Neurobiol. 2010;36:133–56. doi: 10.1111/j.1365-2990.2010.01061.x. [DOI] [PubMed] [Google Scholar]
- 58.Baldwin, K. R., Godena, V. K., Hewitt, V. L. & Whitworth, A. J. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum Mol Genet (2016). [DOI] [PMC free article] [PubMed]
- 59.Schwartz JC, Cech TR, Parker RR. Biochemical Properties and Biological Functions of FET Proteins. Annu Rev Biochem. 2015;84:355–79. doi: 10.1146/annurev-biochem-060614-034325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanaan NM, et al. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci. 2011;31:9858–68. doi: 10.1523/JNEUROSCI.0560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–93. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoica, R. et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep (2016). [DOI] [PMC free article] [PubMed]
- 63.Harris, J. R. & De Carlo, S. Negative staining and Cryo-negative staining: Applications in Biology and Medicine (2014). [DOI] [PubMed]
- 64.Oprea GE, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320:524–7. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fallini C, Bassell GJ, Rossoll W. High-efficiency transfection of cultured primary motor neurons to study protein localization, trafficking, and function. Mol Neurodegener. 2010;5:17. doi: 10.1186/1750-1326-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- 67.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cunningham, R. E. Deparaffinization and Processing of Pathologic Material (2010). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.