

Abstract
Background/Aims
Pneumonia is a significant risk factor for the development of venous thrombosis (VT). Cell-adhesion molecules (CAMs) are linked to the pathogenesis of both pneumonia and VT. We hypothesized that remote infection would confer a prothrombogenic milieu via systemic elevation of CAMs.
Methods
Lung injury was induced in wild-type (C57BL/6) mice by lung contusion or intratracheal inoculation with Klebsiella pneumoniae or saline controls. K. pneumoniae-treated mice and controls additionally underwent inferior vena cava (IVC) ligation to generate VT.
Results
Lung-contusion mice demonstrated no increase in E-selectin or P-selectin whereas mice infected with K. pneumoniae demonstrated increased circulating P-selectin, ICAM-1, VCAM-1 and thrombin-antithrombin (TAT) complexes. Mice with pneumonia formed VT 3 times larger than controls, demonstrated significantly more upregulation of vein-wall and systemic CAMs, and formed erythrocyte-rich thrombi.
Conclusion
Elevated CAM expression was identified in mice with pneumonia, but not lung contusion, indicating that the type of inflammatory stimulus and the presence of infection drive the vein-wall response. Elevation of CAMs was associated with amplified VT and may represent an alternate mechanism by which to target the prevention of VT.
Keywords: Cell-adhesion molecules, Selectins, Venous thrombosis, Venous thromboembolism, Pneumonia, Thrombogenesis, P-selectin, E-selectin, ICAM-1
Introduction
Pneumonia represents a leading cause of death in the USA [1], and venous thromboembolism (VTE) represents the leading cause of preventable hospital death [2]. Several large-population studies have established a link between pneumonia and subsequent VTE [3, 4]. Among medical and surgical patients, pneumonia confers the highest risk of all infections for the development of subsequent inhospital VTE [3, 4]. However, the pathophysiology underlying this relationship remains poorly defined.
Similar molecular mechanisms underlie both diseases: cell-adhesion molecules (CAMs) have been shown to play a central role in venous thrombogenesis [5] and normal pulmonary defense. Venous thrombus initiation is dependent upon leukocyte rolling and firm adhesion via P-selectin (P-sel), E-sel and the recently described intracellular-adhesion molecule-1 (ICAM-1) [6–8]. ICAM-1 has been shown to play a critical role in innate pulmonary defense, mediating leukocyte transmigration across endothelial and alveolar epithelial cells, and the host response to Klebsiella infection [9, 10].
Early polymorphonuclear neutrophil (PMN) cell activation is a hallmark of both diseases. Activated PMNs have both procoagulant and antimicrobial functions via the release of elastase and cathepsin G, which degrade tissue factor pathway inhibitor (TFPI) [11], and also of nuclear material to form neutrophil extracellular traps (NETs) [12]. NETs have been implicated as an important mammalian defense mechanism against bacteria [13], and are elevated in experimental Klebsiella pneumonia [14] and venous thrombosis (VT) [15, 16]. A critical process involved with NETosis is the citrullination of histone H3 [17]. Mice lacking NETs have smaller nonstasis thrombi [16].
Gram-negative pneumonias make up a majority of hospital-acquired pneumonia. Klebsiella is an important pathogen in this group and is particularly associated with high mortality. In a report published in 2008, out of 5,960 VAP infections, Klebsiella pneumoniae was among the top 5 most common pathogens (446 cases) [18]. Given the clinical precedent, we hypothesized that Gram-negative pneumonia would confer an increase in the early venous thrombogenic response. Here, we characterize the circulating and vein-wall response to pneumonia and to lung injury without infection. For the first time, we combine well-defined animal models of pneumonia and VT to demonstrate amplified VT in the setting of pneumonia.
Methods
Animals
Male mice [C57BL/6, 20–25 g, wild-type (WT), Charles River Breeding Laboratories, Wilmington, Mass., USA] were used for all studies. All animals were housed within the Unit for Laboratory Animal Medicine, which is an AAALAC internationally accredited facility at the University of Michigan. All animal protocols were approved by the University of Michigan Committee for the Use and Care of Animals.
Lung Contusion Mouse Model
Lung contusion (LC) was induced in anesthetized WT mice as described previously [19]. Briefly, after induction of anesthesia, the mouse was placed in a left lateral position and, using a cortical contusion impactor, the right chest was struck along the posterior axillary line 1.3 cm above the costal margin using a velocity of 5.8 m/s adjusted to a depth of 10 mm. Mice were then allowed to recover spontaneously. Mice were euthanized at 24 and 48 h after LC, plasma and IVC were snap-frozen in liquid nitrogen for analysis. Control mice did not undergo LC.
Klebsiella Pneumonia Mouse Model
WT C57BL/6 mice were subjected to intratracheal (i.t.) inoculation with K. pneumoniae (LD50: 1 × 103 CFU; strain 43816, serotype 2, ATTC) or equal volume of sterile saline (0.9% normal or PBS, as indicated) [20–22]. Mice were euthanized at various time points with identical specimen-processing to LC mice.
Klebsiella Pneumonia Mouse Model of VT
Following the measurement of circulating CAM levels, K. pneumoniae (1 × 103 CFU) or vehicle (sterile saline at equal volume i.t.) was administered 24 h prior to VT to correspond with peak circulating CAM levels (fig. 1–3). VT was formed by generating stasis blood flow by IVC ligation [23–25]. Mice were anesthetized via 2% inhaled isoflurane and midline laparotomy was performed. Venous side and dorsal branches were interrupted, and the infrarenal IVC was ligated with 7–0 prolene sutures (Ethicon Inc., Somerville, N.J., USA) to generate stasis thrombosis. Fascia and skin were secured with 5–0 vicryl sutures and mice recovered under a warming lamp. Mice were euthanized 2 h after IVC ligation to coincide with initial solid VT formation, and corresponding to the thrombogenesis rather than resolution process [15]. Prior to processing, the IVC and thrombus were measured (centimeters) and weighed (grams) en bloc as well as separately, and then snap-frozen in liquid nitrogen. Plasma, as appropriate for the assay, was collected in EDTA and snap-frozen for later analysis.
Fig. 1.
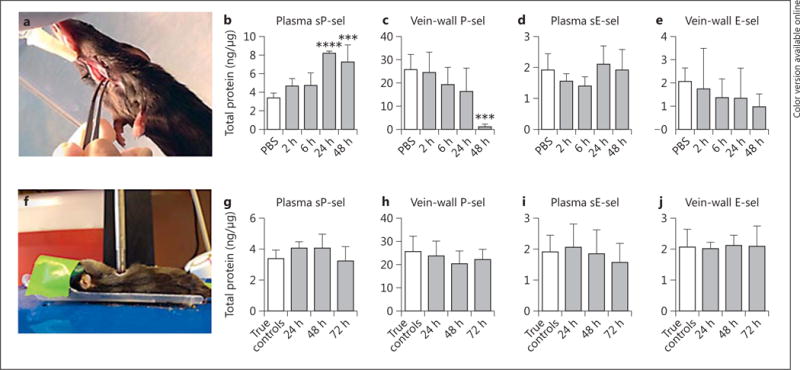
Circulating levels of P-sel and E-sel were captured at various time points after both K. pneumoniae inoculation (a–e) and LC (f–j). After K. pneumoniae (a) or sterile saline inoculation via i.t. injection, circulating sP-sel (b, n = 5–6, PBS vs. 48 h p < 0.001, PBS vs. 24 h p < 0.0001, 2 h vs. 24 h p < 0.001, 6 h vs. 24 h p < 0.001, 2 h vs. 48 h p < 0.01, 6 h vs. 48 h p < 0.01) and sE-sel (c, n = 3–6, all p = n.s.) were measured. Concurrently, vein-wall P-sel (d, n = 6, PBS vs. 48 h p < 0.001) and E-sel (e, n = 4–6, all p = n.s.) were quantified via ELISA. Compared to pulmonary injury from pneumonia, sterile lung injury (f, induction of LC) did not induce changes in circulating (g, h, n = 5–6, all p = n.s.) or vein-wall selectins (i, j, n = 5–6, p = n.s.). *** p ≤ 0.001; **** p ≤ 0.0001.
Fig. 3.
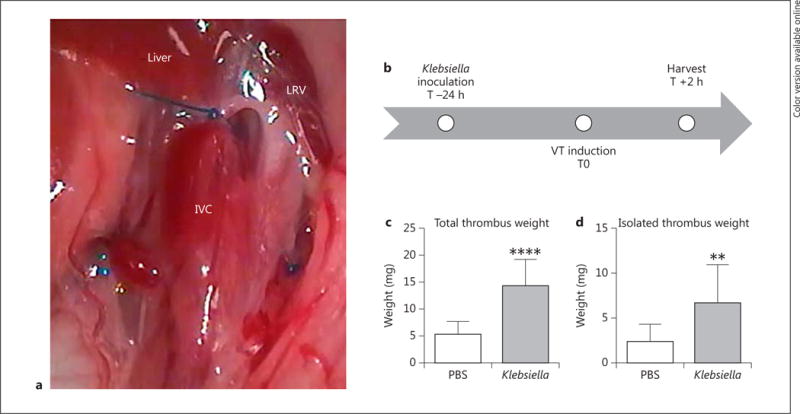
Mice underwent VT via stasis of the infrarenal IVC (a, note prolene suture caudal to LRV) at 24 h after inoculation of PBS or K. pneumoniae (b). Total thrombus weight was measured postthrombosis (including the vein wall, c, n = 9–11, p < 0.0001) as well as isolated thrombus weight (d, thrombus extracted from the vein wall, n = 10–11, p = 0.0085). LRV = Left renal vein. ** p ≤ 0.01; **** p ≤ 0.0001.
Inhibitor Studies
ICAM-1 interactions with the activated lymphocyte associated antigen-1 (LFA-1) receptor were inhibited using the cIBR peptide, a cyclic peptide inhibitor derived from residues 1–21 of the domain I N-terminus of ICAM-1 [26]. WT C57/BL6 mice were subjected to i.p. cIBR 12 h before surgery and redosed at the completion of surgery (150 μg/150 μl, Siahaan lab).
Selectins were similarly inhibited with GMI-1070 (10 mg/kg administered 12 h prior, at the time of surgery completion, Glycomimetics, Rockville, Md., USA). GMI-1070 is a pan-selectin inhibitor designed to mimic the bioactive conformation of the sialyl Lex carbohydrate-binding domain of E-sel and the sulfate interactions of P-sel and L-sel. GMI-1070 has primary activity against E-sel, with second and tertiary activity against P-sel and L-sel [27].
Enzyme-Linked Immunosorbance Assay
Thrombi and vein wall were homogenized, sonicated, and then centrifuged at 13,200 g for 30 min. The supernatant was collected for ELISA analysis. Vein wall and thrombus P-sel, E-sel, ICAM-1 and VCAM-1 (DuoSet ELISAs, R&D Systems, Minneapolis, Minn., USA), circulating soluble (s)P-sel, sE-sel, sICAM-1 and sVCAM-1 (Quantikine ELISA, R&D Systems) and thrombin-antithrombin (TAT) complex (My BioSource, San Diego, Calif., USA) were quantified using commercially available kits according to the manufacturer’s instructions and measured on a BioTek Synergy 2 plate reader (Winooski, Vt., USA) at 450 nm.
Western Blots
Protein was isolated from IVC segments or thrombus using RIPA buffer (ThermoScientific, Rockford, Ill., USA) with dissolved cOmplete ULTRA mini tablets (Roche, Mannheim, Germany). Proteins were electrophoretically separated on NuPAGE 4–12% Bis Tris gels (Invitrogen, Carlsbad, Calif., USA) and blotted onto PVDF membranes (Millipore, Billerica, Mass., USA). Nonspecific binding was blocked with starting block (TBS) blocking buffer (ThermoScientific). Antibodies used included: anti-H3-Cit (1/500 dil., Abcam, Cambridge, Mass., USA), anti-cathepsin G (1/20,000 dil., Novus, Littleton, Colo., USA), anti-TFPI (2 μg/ml dil., Novus), anti-GAPDH (1/1,000 dil., Santa Cruz, Dallas, Tex., USA), anti-fibrin (clone 59D8, 1/1,000 dil., a gift from Dr. Charles Esmon, Oklahoma Medical Research Foundation, Oklahoma City) [25]. Immunoreactive bands were visualized with SuperSignal West Pico.
Chemiluminescent Substrate (ThermoScientific) and densitometry was performed using Image J software. Optical densities were summed and normalized to GAPDH.
Thrombus Lysate Red Blood Cell Content
Thrombus lysate was diluted with 20 m M sodium phosphate at pH 7.4 and 150 m M NaCl (PBS) by 10-fold. Absorbance was measured at 575 nm as previously described to reflect red blood cell (RBC) mass [25].
Statistical Analysis
All data are represented as mean ± standard deviation (SD). Paired Student’s 2-tailed t test or one-way ANOVA with Dunnett’s multiple comparisons, as appropriate, were used to evaluate differences amongst experimental groups and controls (GraphPad Prism v6.01, San Diego, Calif., USA). p < 0.05 was considered significant. Independent statistical review, numbers needed for significance and protocols were used to determine animal numbers (n). A power of 80% and α = 0.05 requires n = 5 for immunologic tests.
Results
Klebsiella Pneumonia, but Not Sterile Pulmonary Injury, Increases Circulating Cell Adhesion Molecules
Circulating levels of CAMs were measured at various time points after K. pneumoniae inoculation or LC. Controls were treated with i.t. saline (PBS, Klebsiella group) or were left uninjured (LC). Time points were chosen per typical time course of insult [21–25, 28]. K. pneumoniae infection, administered via i.t. injection (fig. 1a), increased circulating levels of sP-sel by >2-fold (p < 0.001), but not levels of sE-sel, (fig. 1b, c) compared with saline controls at 24 and 48 h following inoculation. Vein-wall P-sel decreased by 21-fold over time, reaching a nadir at 48 h (p < 0.001), corresponding to the time of maximal circulating P-sel (fig. 1d). Similar to plasma E-sel, vein-wall E-sel levels were not significantly altered in the mice with pneumonia (fig. 1e). Mice undergoing LC served as a comparison group with pulmonary inflammation but without infection; they had similar plasma and vein-wall levels of P-sel and E-sel when compared to noninjured controls (fig. 1g, h).
Given that ICAM-1 has recently been shown to contribute to microvascular VT model [8], we chose to further examine ICAM-1 and VCAM-1 in the Klebsiella model (fig. 2). Experimentally, the contribution of VCAM-1 to the development of VT is unclear, although limited human data suggests that it is elevated during in vivo thrombosis [29, 30]. Similar to P-sel, plasma sICAM-1 elevation of 2.1-fold (p = 0.0028; fig. 2 a) occurred concomitantly with a 6.6-fold decrease of vein-wall protein (p = 0.0320; fig. 2c). Moderate elevations were observed in circulating sVCAM-1 late (fig. 2b) and in the vein wall at the mid-time point (fig. 2d).
Fig. 2.
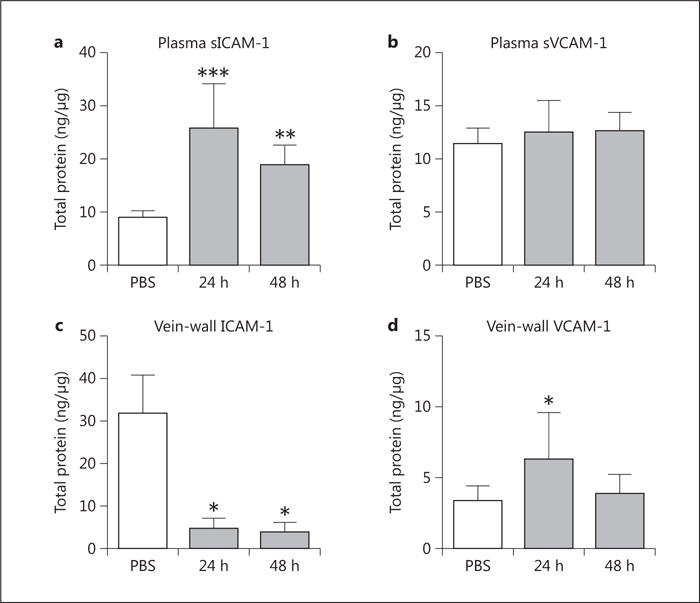
ICAM-1 and VCAM-1 were measured in the plasma and vein wall at various time points following induction of K. pneumonia. Circulating plasma sICAM-1 (a, n = 6–8, PBS vs. 24 h p = 0.0006, PBS vs. 48 h p = 0.0028) and sVCAM-1 (b, n = 6–8, p = n.s.) were quantified. Vein-wall measurements of ICAM-1 (c, n = 3–7, PBS vs. 24 h p = 0.0320, PBS vs. 48 h p = 0.0294) and VCAM-1 (d, n = 4–8, PBS vs. 24 h p = 0.0469) were similarly measured via ELISA. * p ≤ 0.05; * * p ≤ 0.01; * * * p ≤ 0.001.
VT and Postthrombotic Circulating CAMs Amplified during Active Pneumonia
Thrombus size was measured to quantify the effects of remote pulmonary infection on venous thrombogenesis. Total thrombus weight (thrombus + IVC wall) and isolated thrombus weight (thrombus only) were measured. In mice infected with Klebsiella, levels of circulating sP-sel (fig. 1b) and sICAM-1 (fig. 2a) were highest at 24 h after inoculation, therefore we chose to induce VT via IVC ligation (fig. 3a) at this time point. A schematic of the experimental design is illustrated in figure 3b. An early time point of 2 h postthrombosis was chosen to examine the effect of pneumonia on early thrombus formation, as this has previously been shown to approximate the time of early stable thrombus formation in murine VT [15]. Klebsiella infection resulted in thrombi 3-fold larger than in saline controls (fig. 3c, d).
To measure the early systemic immune response to VT and pneumonia, we quantified CAM expression at 2 h following thrombosis. Circulating levels of measured CAMs were 1.6- to 3.6-fold elevated in Klebsiella pneumonia mice compared to saline controls, with significantly higher levels of plasma sP-sel, sE-sel, sICAM-1 and sVCAM-1 following VT (fig. 4a–d). The vein wall demonstrated no change in P-sel (fig. 4e) in response to VT, but there were similar elevations in E-sel, ICAM-1 and VCAM-1 (fig. 4f–h).
Fig. 4.
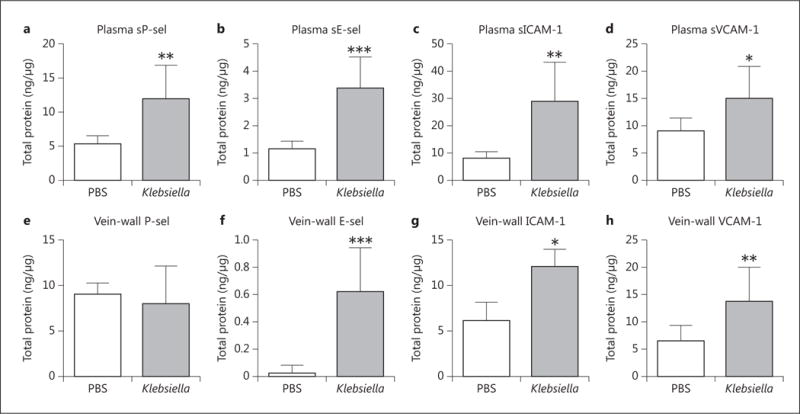
Circulating CAM response was measured by ELISA 2 h postthrombosis, with elevations found in plasma sP-sel (a, n = 10, p = 0.0023), sE-sel (b, n = 10, p = 0.0002), sICAM-1 (c, n = 10, p = 0.0010) and sVCAM-1 (d, n = 10, p = 0.0114). The vein wall was isolated from the adherent thrombus and similarly analyzed for CAM content. e P-sel was not significantly changed (n = 9–10, p = n.s.). Elevations were seen in vein-wall E-sel (f, n = 8–10, p = 0.0002), ICAM-1 (g, n = 10, p = 0.0138) and VCAM-1 (h, n = 10, p = 0.0051). *p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001.
Venous Thrombi Formed under Conditions of Pneumonia Have Altered Composition
As a measure of systemic thrombotic response, we found that circulating TAT complex levels in Klebsiella-treated mice were increased compared to in controls (fig. 5a) prethrombosis. Postthrombosis, RBC content makes up a significant mass of early VT (<48 h) in the stasis VT model [25]. RBC content was elevated by 30% in thrombus lysate of Klebsiella-treated mice compared to controls (p < 0.01, fig. 5b). Markers of PMN-mediated thrombosis were quantified by Western blotting for cathepsin G, citrullinated (cit)-H3, and TFPI [15, 31]. Klebsiella-infected mice demonstrated 1.5-fold elevation in thrombus cit- H3 compared to saline controls (p = 0.045, fig. 5c). Levels of TFPI and cathepsin G in thrombus lysates were not significantly different in Klebsiella-infected mice and saline controls (data not shown). Thrombus fibrin was nonsignificantly elevated by 33% in the setting of pneumonia compared to controls (fig. 5d).
Fig. 5.
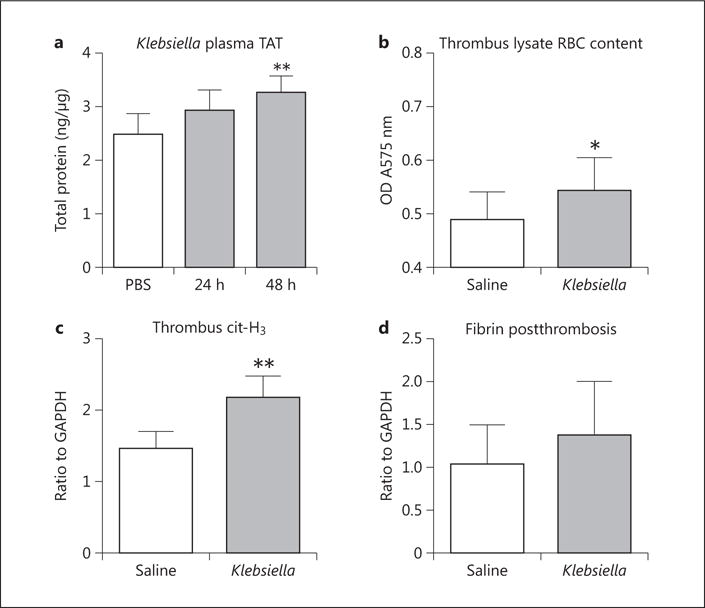
As a measure of a prothrombotic state, plasma TAT complexes were measured in saline and K. pneumoniae-inoculated mice at various intervals. a TAT was increased 48 h after Klebsiella inoculation (n = 6–8, p < 0.01). Thrombus profile was quantified 2 h postthrombosis. b Thrombus RBC content was elevated (n = 4–5, p = 0.0481). c Extracellular cit-H3, a marker of NETs, was increased (n = 5, p = 0.0045) postthrombosis. d Thrombus fibrin content was nonsignificantly elevated Compared to saline controls (n = 4–5, p = 0.374). *p ≤ 0.05; **p ≤ 0.01.
Inhibition of CAMs Attenuates Thrombotic Response to Pneumonia-Associated VT
To determine whether the elevations in CAMs were merely associated with the acute infectious process of pneumonia or played a causal role in the amplified thrombogenesis seen in these animals, the mice were treated with inhibitors to CAMs (fig. 6). Both inhibitors were administered prior to and at the time of induction of thrombosis. Combined P/E-sel inhibition resulted in a modest, nonsignificant decrease in thrombus size. However, ICAM-1 inhibition resulted in a significant decrease of 37% in average thrombus weight (p < 0.05).
Fig. 6.
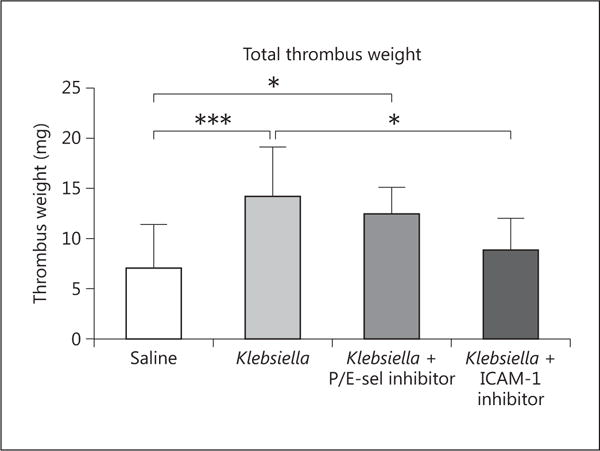
Mice were treated with saline or K. pneumoniae and harvested at 2 h postthrombosis. Additional K. pneumoniae-inoculated mice were treated with inhibitors to P/E-sel or ICAM-1 i.t. Klebsiella-infected mice formed larger thrombi compared to saline-treated mice (n = 11, p < 0.001). A nonsignificant decrease of 12% in average thrombus weight was seen in the P/E-sel inhibited Klebsiella-inoculated mice (p < 0.05 compared to saline, n = 10–11). Disruption of ICAM-1/LFA interactions resulted in a significantly smaller thrombus weight (n = 7–11, p < 0.05). *p ≤ 0.05; ***p ≤ 0.001.
Discussion
Pneumonia is amongst the most frequently diagnosed conditions worldwide and a major cause of death [32]. Pneumonia confers a major risk for VTE development with associated morbidity and mortality [33]. Current VTE prophylaxis regimens may be ineffective at preventing VTE formed under these conditions and thus an improved understanding of the underpinning molecular mechanisms is necessary [34]. With a series of experiments, examining for the first time large-vein-wall response to Gram-negative pneumonia and VT formed under conditions of pneumonia, we demonstrate that upregulation in deep-vein-wall CAMs is elevated with lung inflammation secondary to Gram-negative infection but not under conditions of sterile lung inflammation from mechanical lung injury. Secondly, Gram-negative pneumonia amplified VT and circulating CAMs compared to saline controls. Thirdly, thrombi formed during Gram-negative pneumonia had an altered composition, including a greater proportion of RBCs and extracellular cit-H3.
VTE remains a difficult problem to study experimentally as, despite being a common source of morbidity and mortality in humans, no other animal spontaneously develops pathological VT. While a strong human clinical correlation exists between pneumonia and VT, to date, there is no relevant murine data to suggest if mice develop remote spontaneous venous thrombi with models of pneumonia or LC. In our experience, using these models for over a decade, it is clear that these animals do not have IVC thrombi, as we typically transect the IVC at the time of sacrifice and have not observed any large clots. Locally, increased fibrin deposition occurs within the lungs, consistent with enhanced pulmonary coagulation [35]. This particular combination of well-established mouse models, Klebsiella pneumonia and stasis VT represents a unique utilization of team science to approach a frequent and morbid clinical scenario.
The CAMs P-sel, E-sel and ICAM-1 are well-established mediators of VT as demonstrated by the protection of mice lacking P-sel or E-sel or those treated with ICAM-1 antibody from VT [5, 8, 15]. Multiple human studies have validated P-sel as a reliable marker for the development of VT [36]. Similarly, elevated sICAM-1 levels in patients with VT have been associated with recurrent VT and postthrombotic syndrome [37, 38]. Currently, a dual E-sel and P-sel inhibitor is in clinical trials for the treatment of human deep-vein thrombosis (DVT) (NCT02271113). Besides VT, these CAMs have been found to be elevated in humans with endotoxemia [39] and in murine models of infections such as cecal ligation and puncture [40–42]. Similarly, inhibition of leukocyte interactions via disruption of CAM interactions has been shown to decrease cytokine storm and lethality in a mouse model of cecal ligation and puncture [43].
We show that plasma CAMs sP-sel and sICAM-1 are elevated in the setting of pneumonia, peaking at 24 h after insult. Interestingly, the vein-wall content of both CAMs was depressed at this time point. Under conditions of thrombosis, exposure of endothelial cells to thrombin results in endothelial activation, with exocytosis of P-sel containing Weibel-Palade bodies and increased surface expression of CAMS [44]. It is feasible that the immediate translocation of Weibel-Palade bodies results in depleted vein-wall stores of P-sel as seen in our pneumonia model. Inflammatory stimuli such as thrombin, leukotrienes and histamine induce the rapid translocation of Weibel-Palade bodies to the surface of endothelial cells, translocating P-sel to the surface. In addition, thrombin-activated platelets release P-sel to the surface. We also discovered that, in the absence of thrombosis, pneumonia-treated mice had elevated circulating TAT complex, perhaps from microvascular thrombosis in the pulmonary circulation [45]. This raises the possibility that distant microvascular thrombosis may promote a systemic prothrombotic environment, which may underlie what is observed in humans with pneumonia-related VTE. We have previously shown in Delta cytoplasmic tail (^CT) mice, which exhibit high levels of circulating sP-sel with no difference in vein-wall P-sel compared to WT, that VT is amplified [46]. This suggests that circulating CAMs, likely via microparticle-mediated interactions, may induce or amplify thrombosis independent of vein-wall CAMs.
Following induction of VT, mice treated with K. pneumoniae had an exaggerated thrombotic response, characterized by global elevation of nearly all circulating and vein-wall CAMs, and erythrocyte-rich thrombi nearly 3 times the size of those in the saline-treated mice. Activation of the Factor XIII-fibrinogen axis is a factor in VT formation, entrapping erythrocytes and forming larger thrombi [25]. Previous experiments have demonstrated that selectin deficiency results in decreased fibrin deposition in murine VT [47]. Conversely, an amplified selectin response such as that demonstrated in the pneumonia model-thrombosed mice would be expected to have augmented the deposition of fibrin and the concurrent trapping of erythrocytes.
Systemic inflammation from a remote infection has been well-described, and multiple studies have shown that remote organ damage is associated with the elevation of CAMs and leukocyte activation [48, 49]. Neutrophils are central to infection responses and VT as well as mediating remote organ injury. Activated PMNs mediate thrombogenic activities through NETs and via elastase and cathepsin G breakdown of TFPI [11, 15, 16]. How dominant a role these mechanisms play is likely dependent on the type of inflammatory insult and the VT model. We found that the NET marker of cit-H3 was elevated in mice with pneumonia compared to controls, but there was no difference in TFPI. While not directly proving causality for the exaggerated VT response in Klebsiella-infected mice, these experiments suggest that PMNs activated by a remote infection may play a role in modulating the stasis VT response. Recent data demonstrate that PMNs activated by remote infection can re-enter the circulation [50], and this could explain the increased thrombotic propensity observed in this study.
Several models of DVT exist for study in mice. We chose to use the stasis model as this most reliably produces VT for analysis and is less susceptible to operator technique [23]. Moreover, stasis VT is dependent on CAMs [51], which were significantly elevated with the pneumonia insult. Disruption of NET-mediated early thrombogenesis does not occur in stasis VT [52], as is possible in other models such as partial IVC stenosis [16], making it difficult to ascertain the exact role of NETs in pneumonia-related VT. Though trauma is an independent predictor of subsequent development of VTE, the model of a survivable, spontaneously breathing animal with precise, small-volume LC may not by itself be a robust stimulator of systemic inflammatory response that is required for the development of VTE. We also chose only the early time point due to morbidity and mortality of untreated pneumonia and the timing of peak circulating CAMs. We also study community-acquired MRSA and this may be the subject of further investigation, but the Klebsiella model was used here to reflect high degree of severity of lung disease and our familiarity with the various virulence factors involved in its pathogenicity. It is important to note that the pathogenicity and virulence factors associated with Gram-negative infections are fundamentally different from Gram-positive infections, and so the results of our study should not be generalized to all types of pneumonia.
Translationally, these data suggest the possibility of condition-specific pathways of DVT formation; infection/inflammation may result in differing CAM-leukocyte interactions than in VT formed under conditions such as May-Thurner (stasis), malignancy, device-related VT or VT associated with coagulation abnormalities. Specifically, CAM inhibitor experiments as performed in this paper preliminarily suggest a possible major role for ICAM-1 in the amplified thrombotic response seen with acute Gram-negative pneumonia. While pharmacomechanical VTE prophylaxis is effective, critically ill patients often develop VTE with appropriate thromboprophylaxis [34]. Our experimental data suggest that activation of the vein wall, and possibly PMN activation, plays a role in the mouse model of Gram-negative pneumonia-associated VT and lays the foundation for future studies on differential molecular pathway targets in Gram-negative pneumonia-associated DVT.
Acknowledgments
We appreciate the technical expertise and gift of P/E selectin inhibitor from Dr. Daniel Myers DVM. This study was supported by grants HL092129 (P.K.H.), NIH 5 T32 HL076123-09 (A.O.), NIH 2 T32HL007853 (Y.K.) and HL089407 (T.W.) and the Eliza-beth Anne Baiardi Research Fund.
Footnotes
Presented in part at the Academic Surgical Congress, 4 February 2014 in San Diego, Calif., USA.
Disclosure Statement
None of the authors report any conflicts of interest.
References
- 1.Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke. 2011;42:2351–2355. doi: 10.1161/STROKEAHA.111.621904. [DOI] [PubMed] [Google Scholar]
- 2.Shojania KGDB, McDonald KM, Wachter RM, Markowitz AJ. Making health care safer: a critical analysis of patient safety practices. Evid Rep Technol Assess. 2001:1–668. [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt M, Horvath-Puho E, Thomsen RW, Smeeth L, Sorensen HT. Acute infections and venous thromboembolism. J Intern Med. 2012;271:608–618. doi: 10.1111/j.1365-2796.2011.02473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gangireddy C, Rectenwald JR, Upchurch GR, Wakefield TW, Khuri S, Henderson WG, Henke PK. Risk factors and clinical impact of postoperative symptomatic venous thromboembolism. J Vasc Surg. 2007;45:335–341. doi: 10.1016/j.jvs.2006.10.034. discussion 341–342. [DOI] [PubMed] [Google Scholar]
- 5.Wakefield TW, Myers DD, Henke PK. Mechanisms of venous thrombosis and resolution. Arterioscler Thromb Vasc Biol. 2008;28:387–391. doi: 10.1161/ATVBAHA.108.162289. [DOI] [PubMed] [Google Scholar]
- 6.Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118:6743–6751. doi: 10.1182/blood-2011-07-343566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myers D, Jr, Farris D, Hawley A, Wrobleski S, Chapman A, Stoolman L, Knibbs R, Strieter R, Wakefield T. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J Surg Res. 2002;108:212–221. doi: 10.1006/jsre.2002.6552. [DOI] [PubMed] [Google Scholar]
- 8.Darbousset R, Thomas GM, Mezouar S, Frere C, Bonier R, Mackman N, Renne T, Dignat-George F, Dubois C, Panicot-Dubois L. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012;120:2133–2143. doi: 10.1182/blood-2012-06-437772. [DOI] [PubMed] [Google Scholar]
- 9.Basit A, Reutershan J, Morris MA, Solga M, Rose CE, Jr, Ley K. ICAM-1 and lFA-1 play critical roles in LPS-induced neutrophil recruitment into the alveolar space. Am J Physiol Lung Cell Mol Physiol. 2006;291:L200–L207. doi: 10.1152/ajplung.00346.2005. [DOI] [PubMed] [Google Scholar]
- 10.Qin L, Quinlan WM, Doyle NA, Graham L, Sligh JE, Takei F, Beaudet AL, Doerschuk CM. The roles of CD11/CD18 and ICAM-1 in acute Pseudomonas aeruginosa-induced pneumonia in mice. J Immunol. 1996;157:5016–5021. [PubMed] [Google Scholar]
- 11.Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887–896. doi: 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 12.Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury Chevance A, Zhang K, Conly J, Kubes P. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 14.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brill A, Fuchs TA, Savchenko A, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136–144. doi: 10.1111/j.1538-7836.2011.04544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. 2013;110:8674–8679. doi: 10.1073/pnas.1301059110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK, National Healthcare Safety Network Team; Participating National Healthcare Safety Network Facilities NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol. 2008;29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 19.Suresh MV, Yu B, Lakshminrusimha S, Machado-Aranda D, Talarico N, Zeng L, Davidson BA, Pennathur S, Raghavendran K. The protective role of MNTBAP in oxidant-mediated injury and inflammation in a rat model of lung contusion. Surgery. 2013;154:980–990. doi: 10.1016/j.surg.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolgachev VA, Yu B, Reinke JM, Raghavendran K, Hemmila MR. Host susceptibility to Gram-negative pneumonia after lung contusion. J Trauma Acute Care Surg. 2012;72:614–622. doi: 10.1097/TA.0b013e318243d9b1. discussion 622–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hemmila MR, Fan MH, Kim J, Sun JM, Steinstraesser L, Gong KQ, Arbabi S, Minter RM, Remick DG, Su GL, Wang SC. Improved survival in mice given systemic gene therapy in a Gram-negative pneumonia model. J Trauma. 2005;58:1110–1118. doi: 10.1097/01.ta.0000170855.37686.91. discussion 1118. [DOI] [PubMed] [Google Scholar]
- 22.Ipaktchi K, Mattar A, Niederbichler AD, Kim J, Hoesel LM, Hemmila MR, Su GL, Remick DG, Wang SC, Arbabi S. Attenuating burn wound inflammation improves pulmonary function and survival in a burn-pneumonia model. Crit Care Med. 2007;35:2139–2144. doi: 10.1097/01.ccm.0000280568.61217.26. [DOI] [PubMed] [Google Scholar]
- 23.Diaz JA, Obi AT, Myers DD, Jr, Wrobleski SK, Henke PK, Mackman N, Wakefield TW. Critical review of mouse models of venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:556–562. doi: 10.1161/ATVBAHA.111.244608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obi AT, Diaz JA, Ballard-Lipka NL, Roelofs KJ, Farris DM, Lawrence DA, Wakefield TW, Henke PK. Plasminogen activator-1 overexpression decreases experimental postthrombotic vein wall fibrosis by a non-vitronectin-dependent mechanism. J Thromb Haemost. 2014;12:1353–1363. doi: 10.1111/jth.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aleman MM, Byrnes JR, Wang JG, Tran R, Lam WA, Di Paola J, Mackman N, Degen JL, Flick MJ, Wolberg AS. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124:3590–3600. doi: 10.1172/JCI75386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson ME, Yakovleva T, Hu Y, Siahaan TJ. Inhibition of ICAM-1/LFA-1-mediated heterotypic T-cell adhesion to epithelial cells: design of ICAM-1 cyclic peptides. Bioorg Med Chem Lett. 2004;14:1399–1402. doi: 10.1016/j.bmcl.2003.09.100. [DOI] [PubMed] [Google Scholar]
- 27.Magnani JL. The discovery, biology, and drug development of sialyl Lea and sialyl Lex. Arch Biochem Biophys. 2004;426:122–131. doi: 10.1016/j.abb.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Suresh MV, Yu B, Machado-Aranda D, Bender MD, Ochoa-Frongia L, Helinski JD, Davidson BA, Knight PR, Hogaboam CM, Moore BB, Raghavendran K. Role of macrophage chemoattractant protein-1 in acute inflammation after lung contusion. Am J Respir Cell Mol Biol. 2012;46:797–806. doi: 10.1165/rcmb.2011-0358OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quarmby J, Smith A, Collins M, Cederholm-Williams S, Burnand K. A model of in vivo human venous thrombosis that confirms changes in the release of specific soluble cell adhesion molecules in experimental venous thrombogenesis. J Vasc Surg. 1999;30:139–147. doi: 10.1016/s0741-5214(99)70186-2. [DOI] [PubMed] [Google Scholar]
- 30.Smith A, Quarmby JW, Collins M, Lockhart SM, Burnand KG. Changes in the levels of soluble adhesion molecules and coagulation factors in patients with deep vein thrombosis. Thromb Haemost. 1999;82:1593–1599. [PubMed] [Google Scholar]
- 31.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartlett JG, Mundy LM. Community-acquired pneumonia. N Engl J Med. 1995;333:1618–1624. doi: 10.1056/NEJM199512143332408. [DOI] [PubMed] [Google Scholar]
- 33.Obi AT, Pannucci CJ, Nackashi A, Abdullah N, Alvarez R, Bahl V, Wakefield TW, Henke PK. Validation of the Caprini venous thromboembolism risk assessment model in critically ill surgical patients. JAMA Surg. 2015;150:941–948. doi: 10.1001/jamasurg.2015.1841. [DOI] [PubMed] [Google Scholar]
- 34.Pannucci CJ, Obi A, Alvarez R, Abdullah N, Nackashi A, Hu HM, Bahl V, Henke PK. Inadequate venous thromboembolism risk stratification predicts venous thromboembolic events in surgical intensive care unit patients. J Am Coll Surg. 2014;218:898–904. doi: 10.1016/j.jamcollsurg.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 35.de Stoppelaar SF, van ’t Veer C, Claushuis TA, Albersen BJ, Roelofs JJ, van der Poll T. Thrombocytopenia impairs host defense in Gram-negative pneumonia-derived sepsis in mice. Blood. 2014;124:3781–3790. doi: 10.1182/blood-2014-05-573915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antonopoulos CN, Sfyroeras GS, Kakisis JD, Moulakakis KG, Liapis CD. The role of soluble P selectin in the diagnosis of venous thromboembolism. Thromb Res. 2014;133:17–24. doi: 10.1016/j.thromres.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Cushman M, Callas PW, Allison MA, Criqui MH. Inflammation and peripheral venous disease. The San Diego Population Study. Thromb Haemost. 2014;112:566–572. doi: 10.1160/TH13-10-0860. [DOI] [PubMed] [Google Scholar]
- 38.Shbaklo H, Holcroft CA, Kahn SR. Levels of inflammatory markers and the development of the post-thrombotic syndrome. Thromb Haemost. 2009;101:505–512. [PubMed] [Google Scholar]
- 39.Zonneveld R, Martinelli R, Shapiro NI, Kuijpers TW, Plotz FB, Carman CV. Soluble adhesion molecules as markers for sepsis and the potential pathophysiological discrepancy in neonates, children and adults. Crit Care. 2014;18:204. doi: 10.1186/cc13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin L, Batra S, Douda DN, Palaniyar N, Jeyaseelan S. CXCL1 contributes to host defense in polymicrobial sepsis via modulating T cell and neutrophil functions. J Immunol. 2014;193:3549–3558. doi: 10.4049/jimmunol.1401138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roller J, Wang Y, Rahman M, Schramm R, Laschke MW, Menger MD, Jeppsson B, Thorlacius H. Direct in vivo observations of P-selectin glycoprotein ligand-1-mediated leukocyte-endothelial cell interactions in the pulmonary microvasculature in abdominal sepsis in mice. Inflamm Res. 2013;62:275–282. doi: 10.1007/s00011-012-0575-y. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Jin S, Wang C, Jiang R, Wan J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World J Surg. 2010;34:1676–1683. doi: 10.1007/s00268-010-0493-5. [DOI] [PubMed] [Google Scholar]
- 43.Wong KF, Wo J, Ho D, Poon RT, Casasnovas JM, Luk JM. Prophylactic uses of integrin CD18-betaA peptide in a murine polymicrobial peritonitis model. World J Gastroenterol. 2010;16:2648–2656. doi: 10.3748/wjg.v16.i21.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pearson JD. Normal endothelial cell function. Lupus. 2000;9:183–188. doi: 10.1191/096120300678828299. [DOI] [PubMed] [Google Scholar]
- 45.Ware LB, Camerer E, Welty-Wolf K, Schultz MJ, Matthay MA. Bench to bedside: targeting coagulation and fibrinolysis in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L307–L311. doi: 10.1152/ajplung.00157.2006. [DOI] [PubMed] [Google Scholar]
- 46.Myers DD, Jr, Rectenwald JE, Bedard PW, Kaila N, Shaw GD, Schaub RG, Farris DM, Hawley AE, Wrobleski SK, Henke PK, Wakefield TW. Decreased venous thrombosis with an oral inhibitor of P selectin. J Vasc Surg. 2005;42:329–336. doi: 10.1016/j.jvs.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 47.Sullivan VV, Hawley AE, Farris DM, Knipp BS, Varga AJ, Wrobleski SK, Thanapron P, Eagleton MJ, Myers DD, Fowlkes JB, Wakefield TW. Decrease in fibrin content of venous thrombi in selectin-deficient mice. J Surg Res. 2003;109:1–7. doi: 10.1016/s0022-4804(02)00041-0. [DOI] [PubMed] [Google Scholar]
- 48.Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83:536–545. doi: 10.1189/jlb.0607373. [DOI] [PubMed] [Google Scholar]
- 49.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–3777. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 50.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12:761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Myers DD, Jr, Henke PK, Bedard PW, Wrobleski SK, Kaila N, Shaw G, Meier TR, Hawley AE, Schaub RG, Wakefield TW. Treatment with an oral small molecule inhibitor of P selectin (PSI-697) decreases vein wall injury in a rat stenosis model of venous thrombosis. J Vasc Surg. 2006;44:625–632. doi: 10.1016/j.jvs.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 52.El Sayed H, Kerensky R, Stecher M, Mohanty P, Davies M. A randomized phase II study of xilonix, a targeted therapy against interleukin 1alpha, for the prevention of superficial femoral artery restenosis after percutaneous re-vascularization. J Vasc Surg. 2016;63:133–141.e1. doi: 10.1016/j.jvs.2015.08.069. [DOI] [PubMed] [Google Scholar]