

Abstract
Rationale:
Kennedy disease (KD) is also known as spinal bulbar muscular dystrophy. As KD has similar symptoms with most neuromuscular diseases, so it is difficult to make a rapid diagnosis clinically.
Patient concerns:
We report a case of a 43-year-old male with progressive limb proximal weakness without family history. Physical examination showed gynecomastia, erectile dysfunction, bilateral tendon reflex and quadriceps weakness, and tongue muscle atrophy.
Diagnoses:
Laboratory examination found increased creatine kinase, impaired glucose tolerance, and abnormal lactic acid values. There was no mutation or copy number variant in SMN1 gene and related mitochondrion genes tested, even with the use of multiplex ligation probe- dependent amplification technique. Diagnosis was confirmed with genetic analysis which displayed trinucleotide CAG (glutamine)- repeat expansion in the androgen-receptor gene.
Interventions and Outcomes:
The patient achieved good prognosis with symptomatic treatment after diagnosis.
Lessons:
To diagnose KD, clinicians should pay more attention to differentiate KD and myasthenia gravis, mitochondrial myopathy, and amyotrophic lateral sclerosis. Gene analysis was the key in detecting this rare confusing disease in the patient.
Keywords: case report, genetic analysis, Kennedy disease
1. Introduction
Kennedy disease (KD) is also known as spinal bulbar muscular dystrophy; it is a late-onset X-linked recessive hereditary nervous system degenerative disease. The incidence rate of it is about 1 per 100,000, and the high incidence of KD is shown in Wazard region in Finland.[1,2] The disease is mainly manifested with lower motor neuron damage clinically, and may be associated with incomplete androgen insensitivity syndrome, sensory nerve, and endocrine system involvement. As KD has similar symptoms with most neuromuscular diseases, so it is difficult to make a rapid diagnosis clinically. Patient in this case once went through the similar experience.
2. Case report
The patient in this case is a male 43 years old, who has been suffering from limb weakness for 6 years. The patient began to have the proximal limb muscle weakness and muscle bundle twitch 6 years ago, especially after activities. The weakness symptom at night was usually worse than early in the morning; the lower limbs were more obvious. Symptoms had got worse significantly in recent 2 years, exercise endurance had decreased, and walking distance also shortened year by year. The patient could only walk about 1000 m currently. He had no similar family history. Magnetic resonance imaging (MRI) excluded the lumbar or spinal cord pathological changes, but found that hip muscles were atrophic. After admission, physical examination showed mild bilateral breast development, weakened bilateral tendon reflex symmetry, and normal bilateral shades. Quadriceps strength was 4-level, and bilateral tongue muscle atrophy, fibrillation, and bilateral pathological signs were negative. Blood routine, tumor markers, rheumatoid immunity, and autoimmune indicators were normal. Blood biochemistry showed glucose: 6.7 mmol/L; oral glucose tolerance test suggested impaired glucose tolerance (the blood glucose level of forearm venous blood 2 hours after the meal was 8.9 mmol); triglyceride: 1.82 mmol/L; lactic acid: 3.0 mmol/L; lactate dehydrogenase (LDH): 151 IU/L; creatine kinase (CK): 658 IU/L; creatine kinase-MB isoenzyme (CK-MB): 32.1 IU/L. Chest computed tomography (CT) was conducted to exclude “myasthenia gravis,” and no found of thymic tumors. Electromyography (EMG) showed neurogenic damage, and repeated electrical nerve stimulation found no characteristic changes. Bromide pyridine experimental treatment was invalid. Muscle pathological biopsy was carried out at the right quadriceps muscle, showing muscle fiber atrophy and degeneration (Fig. 1A–C). Multiplex ligation probe-dependent amplification (MLPA) technique of spinal muscular atrophy indicated that the following: no detection of heterozygous deletion or homozygous deletion mutations of exon 7 and 8 of SMN1 gene. Mitochondrial DNA deletion mutation (MLPA) technique showed the following: no deletion mutations of mitochondrial DNA were detected. In addition, the following mitochondrial DNA point mutations were not detected: m3243A, m3250T, m3252A, and m3271T. The minimum exercise test of lactic acid and pyruvic acid showed the following: the lactic acid values were 3.0, 3.1, and 2.4 mmol/L at 1, 2, and 3 hours, respectively. The results were compared with that of the control group (P < .05), whereas the lactic acid value of the control group was 0.5–2.2 mmol/L. Myelopathy, myasthenia gravis, spinal muscular atrophy, and mitochondrial myopathy were basically ruled out, combined with comprehensive medical history, clinical manifestations, and the results of various examinations. Although patient had no family history of genetic disease, he indeed had mild symptoms of myasthenia, impaired glucose tolerance, mild breast development, and erectile dysfunction, which met many points of the KD, so the genetic testing was performed. KD gene test results showed that the number of trinucleotide repeat (CAG) sequence in exon 1 of androgen receptor was 42 (Fig. 1D). According to the European Federation of Neurological Sciences guidelines, it is noted that the CAG repeat sequence ≥35 is the standard for the diagnosis of KD.[3] Thus, this patient was diagnosed as suffering from KD.
Figure 1.
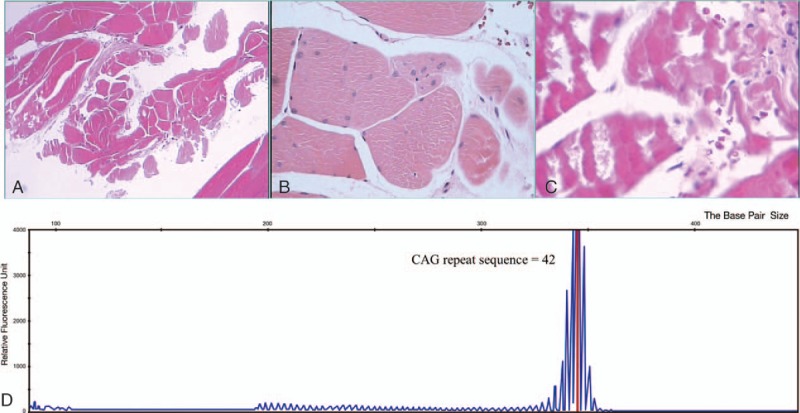
Muscle pathological biopsy of the right quadriceps muscle showing muscle fiber atrophy and degeneration (A), increase of internalized nuclei (B), and few lymphocytic infiltrates in muscle interstitial tissue (C). Genetic analysis found trinucleotide CAG (glutamine)-repeat expansion in the androgen-receptor gene (D).
Currently, there is no standard treatment for KD, but the prognosis is good. Patients are expected to achieve good prognosis with symptomatic treatment after diagnosis.
This work was approved by the Medical Ethical Committee of Zhujiang Hospital of Southern Medical University, and the informed consent was obtained from the patient.
3. Discussion
Kennedy disease is a rare neurological disease; statistics showed that it takes an average of 5.5 years from the first onset of myasthenia symptoms to the clear diagnosis,[4] which can also be easily misdiagnosed. For example, patients with lumbar spondylosis can also have symptoms of lower limbs weakness, and the majority of pathological changes can be effectively shown through MRI. On the contrary, the KD MRI showed no spinal lesions, and muscle atrophy can be found by scanning weak limbs.
Symptoms of myasthenia gravis usually occurred or exacerbated after exertion, with the characteristics of better in morning, but worse at night. Tendon reflex is generally not affected, repeated nerve electrical stimulation is positive, and thymus CT can find thymic hyperplasia and hypertrophy. Treatment with cholinesterase inhibitors is effective.[5] However, myasthenia symptoms of KD do not show the fluctuated phenomenon of better in morning, but worse at night, tendon reflex is diminished or disappeared, repeated nerve electrical stimulation has no characteristic changes, thymus is normal, and cholinesterase inhibitor treatment is also ineffective.
In addition to the performance of muscle weakness and decreased endurance, the levels of lactic acid, CK, and LDH in patients who suffered from mitochondrial myopathy were also significantly increased, which can be seen from the blood biochemistry. EMG showed myogenic damage, the muscle biopsy rarely showed muscle atrophy, and the gomoritrichrome staining showed the ragged-red fiber. Maternal genetic characteristics can be found in family history.[6,7] LDH level of KD was generally normal, EMG showed neurogenic damage, and muscle biopsy showed muscle atrophy. X chromosome recessive genetic characteristics may be revealed by analyzing family history.
The most difficult one to be diagnosed is motor neuron disease. As both the amyotrophic lateral sclerosis (ALS) and KD are middle-aged onset, and with similar clinical manifestations, EMG and muscle biopsy results. The difference lies in the fact that ALS shows typical and simultaneous upper and lower neurons injury, the pathological signs are positive, and feelings are normal; besides, both CK and CK-MB levels are normal, but with rapid progression, patients usually die of respiratory muscle paralysis or pulmonary infection within 3 to 5 years.[8] On the contrary, KD shows spinal cord submucosal motor neuron damage,[9] the pathological signs are negative, and sensory disturbances may also be associated with[10]; besides, the endocrine system is abnormal, CK level is significantly increased, and the course of disease is progressively developed, so good prognosis can be achieved. The diagnosis can be confirmed with gene detection.
As shown in Table 1, KD lacks characteristic clinical manifestation, for which it could be misdiagnosed as other neurogenic muscular disease. It could indicate KD, if patent had the proximal limb muscle atrophy, weakness, and muscle bundle twitch, especially with lingualis atrophy and weakness, the significant increase of CK and CK-MB, which is obviously higher than other muscular diseases, or EMG showed neurogenic damage and the lower amplitude of SNAP, muscle biopsy showed neurogenic atrophy. Especially, it means a higher possibility to be KD on the condition that the feature above occurs with the incomplete androgen insensitivity syndrome or the disorder of metabolic function. When some or all of the clinical features mentioned above are present, a gene test is necessary for the exclusion or diagnosis of KD.
Table 1.
The differential diagnosis among KD, myasthenia gravis, and ALS.

Kennedy disease has been prevalent for more than 40 years, but due to its atypical clinical manifestations, misdiagnosis and missed diagnosis are quite common clinically, so inappropriate treatment can easily be caused. On one hand, it will increase the economic burden of patients, but more importantly, it can also increase the psychological burden due to alleviated symptoms. Therefore, if lower motor neuron damage of spinal cord medullary is found clinically, it is necessary to perform the electrophysiology, muscle pathology, and genetic testing to diagnose KD rigorously and treat it as early as possible.
Footnotes
Abbreviations: ALS = amyotrophic lateral sclerosis, CK = creatine kinase, CK-MB = creatine kinase-MB isoenzyme, CT = computed tomography, DNA = deoxyribonucleic acid, EMG = electromyography, KD = Kennedy disease, LDH = lactate dehydrogenase, MLPA = multiplex ligation probe-dependent amplification, MRI = magnetic resonance imaging, SMN1 = survival of motor neuron 1 gene.
Y.C., P.L., and Z.L. made equal contribution to the study.
The authors report no conflicts of interest.
References
- [1].Fan HC, Ho LI, Chi CS, et al. Polyglutamine(PolyQ) diseases: genetics to treatments. Cell Transplant 2014;23:441–58. [DOI] [PubMed] [Google Scholar]
- [2].Weydt P, Sagnelli A, Rosenbohm A, et al. Clinical trials in spinal and bulbar muscular atrophy: past, present, and future. J Mol Neurosci 2016;58:379–87. [DOI] [PubMed] [Google Scholar]
- [3].Burgunder JM, schols L, Baets J, et al. EFNS guidelines for the molecular diagnosis of neurogenetic disorders: motoneuron, peripheral nerve and muscle disorders. Eur J Neurol 2011;18:207–17. [DOI] [PubMed] [Google Scholar]
- [4].Rhodes LE, Freeman BK, Auh S, et al. Clinical features of spinal and bulbar muscular atrophy. Brain 2009;132:3242–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sussman J, Farrugia ME, Maddison P. Myasthenia gravis: association of British Neurologists’ management guidelines. Pract Neurol 2015;15:199–206. [DOI] [PubMed] [Google Scholar]
- [6].Pfeffer G, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med 2013;45:4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Petty TKH, Harding AE, Motgan-Hughes JA. The clinical features of mitochondrial myopathy. Brain 1986;109:915. [DOI] [PubMed] [Google Scholar]
- [8].Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic Lateral Scler 2009;1:293–9. [DOI] [PubMed] [Google Scholar]
- [9].Fischbech KH, Lieberman A, et al. Androgen receptor mutation in Kennedy's disease. Philos Trans Roy Soc 1999;354:1075–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Grunseich C, Rinaldi C, Fischbeck KH. Spinal and bulbar muscular atrophy: pathogenesis and clinical management. Oral Dis 2013;20:6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Traynor BJ, Codd MB, Corr B, et al. Amyotrophic lateral sclerosis mimic syndromes: a population-based study. Arch Neurol 2000;57:109–13. [DOI] [PubMed] [Google Scholar]
- [12].Grunseich C, Rinaldi C, Fischbeck KH. Spinal and bulbar muscular atrophy: pathogenesis and clinical management. Oral Dis 2014;20:6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Finsterer J. Perspectives of Kennedy's disease. J Neurol Sci 2010;298:1–0. [DOI] [PubMed] [Google Scholar]