

Abstract
Intron length polymorphisms (ILPs), a type of gene-based functional marker, could themselves be related to the particular traits. Here, we developed a genome-wide cotton ILPs based on orthologs annotation from two sequenced diploid species, A-genome Gossypium arboreum and D-genome G. raimondii. We identified 10,180 putative ILP markers from 5,021 orthologous genes. Among these, 535 ILP markers from 9 gene families related to stress were selected for experimental verification. Polymorphic rates were 72.71% between G. arboreum and G. raimondii and 36.45% between G. hirsutum acc. TM-1 and G. barbadense cv. Hai7124. Furthermore, 14 polymorphic ILP markers were detected in 264 G. hirsutum accessions. Coupled with previous simple sequence repeats (SSRs) evaluations and salt tolerance assays from the same individuals, we found a total of 25 marker-trait associations involved in nine ILPs. The nine genes, temporally named as C1 to C9, showed the various expressions in different organs and tissues, and five genes (C3, C4, C5, C7 and C9) were significantly upregulated after salt treatment. We verified that the five genes play important roles in salt tolerance. Particularly, silencing of C4 (encodes WRKY DNA-binding protein) and C9 (encodes Mitogen-activated protein kinase) can significantly enhance cotton susceptibility to salt stress.
Introduction
In modern crop breeding, many traits of interest such as yield, quality, and resistance to biotic or abiotic stress need to be improved simultaneously to allow crops to survive in extreme environmental conditions and to safeguard the safety of crops. Since the majority of traits are complex and controlled by many genomic loci, each of which have small effects, molecular markers are the foundation for genomics-based crop improvement. Several types of molecular markers, such as restriction fragment length polymorphisms (RFLPs), sequence-characterized amplified regions (SCARs), simple sequence repeats (SSRs) and single nucleotide polymorphisms (SNPs), have been successfully used in molecular marker assisted-selection (MAS) in cotton1, 2, rice3, and wheat4. Functional markers (FMs) are a type of gene-based marker that was developed from sequence polymorphisms present in allelic variants of a functional gene at a given locus. FMs accurately discriminate between traits associated with alleles of a target gene, and are ideal molecular markers for MAS in breeding5. EST-SSRs (eSSRs), SSRs developed from expressed sequence tags (ESTs), and gene-based SNP markers, in which the discovery of SNPs is specific to candidate genes or transcript sequences, are the most widely used types of FMs in crop species6, 7.
As more genome information becomes available for crop species, intron-spanning markers have become an important type of FM and their development has greatly increased. Compared with exons, introns contain more variations due to a lower selection pressure during the evolutionary process. Intron length polymorphisms (ILPs) are the easiest identified molecular markers in introns. They can be conveniently detected by polymerase chain reaction (PCR), using primers designed for flanking exons. This technique is known as exon-primed intron-crossing PCR amplification (EPIC-PCR)8. The development of ILP markers is unique since they are gene-specific, co-dominant, hypervariable, neutral, convenient, and reliable. At first, ILP markers were used in a small number of crops, such as Medicago9, foxtail millet10, maize11, potato, and Solanum nigrum 12, and are restricted to a small number of genes. With more genome information released, Yang et al.13 conducted a more comprehensive study and extracted a total of 57,658 potential intron polymorphism (PIP) markers from 59 plant species, and created a web-based database of PIP markers (http://ibi.zju.edu.cn/pgl/pip/). The genome-wide development of ILP markers has also been reported in rice14 and foxtail millet15.
Cotton (Gossypium spp.) is the world’s most important natural textile fiber and is a significant oilseed crop. Four cultivated species have been independently domesticated: two tetraploids, G. hirsutum L. (AD)1 and G. barbadense L (AD)2, and two diploids, G. herbaceum L. (A1) and G. arboreum L. (A2)16. The ancestral A- and D-like genomes are thought to have diverged only 5–10 million years ago (MYA), and all allotetraploids were formed from interspecific hybridization events between an A-genome-like ancestral African species and a D-genome-like North American species 1–2 MYA17. Recently, the availability of data on the whole-genome of Gossypium in different cotton species, including G. raimondii (D5)18, 19, G. arboreum (A2)20, G. hirsutum acc. TM-1 (AD1)21, 22 and G. barbadense acc. 3–79 and Xinhai21 (AD2)23, 24, made it possible to develop cotton ILP markers at a genome-wide level. To date, no genome-wide exploitation of ILP markers has been reported in cotton. To achieve this, we screened for differences in the intron-lengths of orthologous A- and D-genome genes, which have a relatively high level of similarity, by comparing genome sequences and annotation information from G. raimondii and G. arboreum, and developed a large number of gene-based ILP markers. We selected a set of ILP markers representative of genes associated with abiotic stress response pathways to experimentally validate the levels of diversity between G. arboreum and G. raimondii or between two allotetraploid cultivated species (G. hirsutum acc. TM-1 and G. barbadense cv. Hai7124). Further, nine candidate ILP markers related to salt stress were confirmed by both multiple comparison and association mapping approaches using a set of natural Upland cotton accessions. We investigated the temporal and spatial expression profiles of the nine candidate genes associated with salt stress traits in different tissues and in response to salt stress treatment, and verified that the functional roles of five genes that are significantly induced by salt stress treatment by virus-induced gene silencing (VIGS) analysis. Our study not only provided genome-wide, gene-based ILPs marker resources in cotton, but also mined effectively the genes with salt-tolerance for developing abiotic-resistance cultivars in future cotton-breeding programs.
Results
Genome-wide comparison of orthologs between diploid and tetraploid cotton species
G. raimondii and G. arboreum genome annotation files were obtained from http://www.phytozome.net and http://cgp.genomics.org.cn, respectively, and were used to obtain the corresponding intron distributions. In the 37,505 protein-coding genes in the D-genome species, the number of introns was between 0 and 78, with 9,535 genes (25.42%) having no introns. In the 41,330 genes in the A-genome species, the number of introns was between 0 and 76, with 12,083 genes (29.23%) having no introns. The number and distribution of introns in the A- and D-genome cotton species is shown in Fig. S1.
The BLAST program was used to obtain A- and D-genome orthologous genes. In total, 9,598 genes from the D-genome were found to be highly homologous with 9,686 genes from the A-genome, according to the following rules: A-CDS/D-CDS ≥95%, A-mRNA/D-mRNA ≥80%. Using 9,598 genes from G. raimondii as probes, 2,528 gene sequences showed no differences between the A- and D-genome introns, and 44,966 introns from 7,070 genes showed at least one intron difference for each pair of orthologs, with the different intron-lengths ranging from 1 bp to 1,339 bp. Of these 44,966 introns, 13,683 (30.43%) had intron-length differences ranging from 10 bp to 1000 bp, and were used for further ILP marker development.
From an evolutionary point, one gene in the diploid G. raimondii correspond to one homologous gene in G. arboreum and two homeologs from the A and D subgenomes in tetraploid cotton species. So we investigated the distribution of the 7,070 genes in tetraploid cotton species using the whole genome sequence of G. hirsutum acc. TM-121 and G. barbadense acc. 3–7923. In total, 6989 and 7001 genes were found to exist homologous gene in the A and D subgenomes in G. hirsutum, respectively; and 6806 and 6821 genes were found to exist homologous genes in the A and D subgenomes in G. barbadense, respectively (Dataset S1), indicating more than 94% of 7070 genes can be used for ILP marker detection in tetraploid cotton species.
Based on the genome of the diploid cotton species G. raimondii 18, we investigated the physical location of the 7,070 genes with intron-length differences between the A- and D-genome orthologs. In total, 7,049 genes were mapped to 13 chromosomes, with an average gene density of 9.408 genes/Mb, and the remaining 21 genes were mapped to the 8 scaffolds. Chromosome distribution showed that the highest frequency of these genes were found on Chr. 9, which contained 935 genes (935/7049, 13.264%) and had 13.222 genes/Mb, followed by Chr. 7 (708 genes, 10.044%, 11.610 genes/Mb). The lowest average gene density was found on Chr. 10 (7.382 genes/Mb), and the lowest frequency of genes on Chr. 12 (331 genes, 4.696%) (Fig. 1; Table 1). These genes were unevenly distributed across each chromosome with an increasing density towards one end of the chromosome on Chr. 1, 6, 7, 8, and 9, and both ends on Chr. 2, 3, 4, 5, 10, and 13 (Fig. 1).
Figure 1.
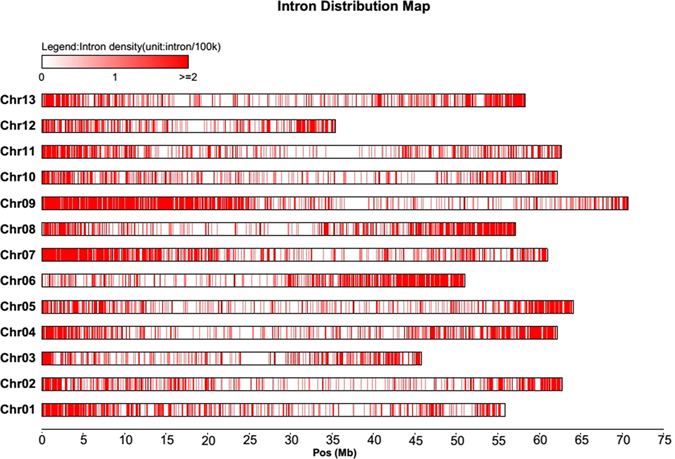
Chromosome distribution of 7,049 genes associated with ILP markers in G. raimondii. Each vertical short bar indicates the position of corresponding candidate gene.
Table 1.
Chromosome distribution of developed ILP markers in G. raimondii genome.
| Chr./Scaffold* | No. of genes | No. of genes developed markers | No. of primer pairs | Chromosome length (Mb) | Physical density (markers/Mb)% |
|---|---|---|---|---|---|
| Chr01 | 501 | 354 | 680 | 55.86823 | 8.967529 |
| Chr02 | 501 | 357 | 719 | 62.76943 | 7.981592 |
| Chr03 | 354 | 255 | 504 | 45.76565 | 7.735059 |
| Chr04 | 575 | 392 | 806 | 62.17826 | 9.247605 |
| Chr05 | 515 | 360 | 756 | 64.14041 | 8.029259 |
| Chr06 | 557 | 397 | 799 | 51.07452 | 10.90563 |
| Chr07 | 708 | 507 | 1056 | 60.98247 | 11.60989 |
| Chr08 | 606 | 449 | 917 | 57.12882 | 10.60761 |
| Chr09 | 935 | 693 | 1407 | 70.71302 | 13.22246 |
| Chr10 | 459 | 329 | 678 | 62.17517 | 7.382368 |
| Chr11 | 514 | 364 | 753 | 62.68101 | 8.200251 |
| Chr12 | 331 | 224 | 462 | 35.42995 | 9.342379 |
| Chr13 | 493 | 340 | 643 | 58.32116 | 8.453192 |
| Scaffold | 21 | / | / | / | / |
| Total | 7070 | 5021 | 10180 | 749.2281 | 9.40835 |
*The nomenclature of Chr./Scaffold in G. raimondii is from Paterson et al.18.
Bioinfomatic analysis of orthologs with intron difference
The 7,070 genes were further subjected to functional annotation. Of these, 5,695 (80.552%) were mapped to 24,683 GO terms using Gene ontology annotation and were categorized into the three main GO classes (biological processes, molecular functions, and cellular components) (Dataset S2). In detail, the majority of the GO terms were grouped into metabolic process and cellular process categories within the biological processes, binding and catalytic categories within the molecular functions, and cell and cell part categories within the cellular components.
Using Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, 2,948 sequences from 1,332 genes (18.84%) were assigned to 135 different metabolic pathways (Dataset S3, Table 2). Of them, 95.828% of sequences (2,825) were mapped to the metabolism GO class, and the remaining 4.172% were assigned to genetic information processing (26, 0.882%), environmental information processing (53, 1.798%) and organismal systems (44, 1.493%) classes. The sequences in the metabolism GO class were largely involved in carbohydrate metabolism (770 clusters, 27.257%), amino acid metabolism (460 clusters, 16.283%), and lipid metabolism (348 clusters, 12.319%). In the categories of genetic information processing, environmental information processing, and organismal systems, sequences were further assigned to translation, signal transduction, and immune system subcategories, respectively. As a comparison, we also mapped 9,670 sequences from 4,441 genes in G. raimondii to 143 different metabolic pathways (Dataset S3). We found that there was a strong linear correlation (R2 = 0.986) between the annotated metabolic pathways of the 2,948 sequences from 1,332 genes and the 9,670 sequences from 4,441 G. raimondii genes (Table 2, Dataset S3). In different metabolic pathways, the genes with the highest number of intron differences were related to signal transduction (53/147, 36.05%), followed by energy metabolism (224/647, 34.62%), amino acid metabolism (460/1356, 33.92%), carbohydrate metabolism (770/2310, 33.33%), and then immune system processes (44/212, 20.75%).
Table 2.
KEGG classification of 7,070 orthologs and their percentage in the whole genome.
| Classification* | Numbers in 7,070 genes | Numbers in G. raimondii genome | Percentage (%) |
|---|---|---|---|
| 1. Metabolism | 2825 | 9225 | 30.62 |
| 1.1 Carbohydrate metabolism | 770 | 2310 | 33.33 |
| 1.2 Energy metabolism | 224 | 647 | 34.62 |
| 1.3 Lipid metabolism | 348 | 1193 | 29.17 |
| 1.4 Nucleotide metabolism | 187 | 673 | 27.79 |
| 1.5 Amino acid metabolism | 460 | 1356 | 33.92 |
| 1.6 Metabolism of other amino acids | 110 | 355 | 30.99 |
| 1.7 Glycan biosynthesis and metabolism | 103 | 348 | 29.60 |
| 1.8 Metabolism of cofactors and vitamins | 167 | 608 | 27.47 |
| 1.9 Metabolism of terpenoids and polyketides | 88 | 296 | 29.73 |
| 1.10 Biosynthesis of other secondary metabolites | 203 | 747 | 27.18 |
| 1.11 Xenobiotics biodegradation and metabolism | 165 | 689 | 23.95 |
| 1.12 Chemical structure transformation maps | / | 3 | 0.00 |
| 2. Genetic Information Processing | 26 | 85 | 30.59 |
| 2.2 Translation | 26 | 85 | 30.59 |
| 3. Environmental Information Processing | 53 | 147 | 36.05 |
| 3.2 Signal transduction | 53 | 147 | 36.05 |
| 4. Organismal Systems | 44 | 212 | 20.75 |
| 4.1 Immune system | 44 | 212 | 20.75 |
*KEGG annotation and metabolism maps were performed with BLAST2GO.
The rate of ILP variation was then investigated in nine gene families relevant to stress responses: NAC and WRKY transcription factors, mitogen-activated protein kinase (MAPK), heat shock proteins (HSPs), cytochrome P450 (CYP450), WD40 repeat-containing proteins (WD40s), Zinc finger (ZnF), leucine-rich repeat (LRR), and aquaporin family proteins. Lower correlation coefficient (R2 = 0.1436) was detected when the number of genes with ILPs were compared to all genes in the G. raimondii genome. The most highly conserved gene family was the leucine-rich repeat family, where there were only 66 members showed ILPs for A- and D-genome orthologs with total 1514 leucine-rich repeats in G. raimondii genome. The NAC family was the next most highly conserved (27/313), followed by the MAPK (12/128), WRKY (22/220), P450 (49/477), WD40 (65/531), heat shock protein (47/309), aquaporin (12/59), and zinc finger (133/385) families. The relationship between the sequence variations and functional diversity among orthologs remains to be investigated.
Development and identification of ILP markers in cotton
The exon-primed intron-crossing PCR (EPIC-PCR) method8 was used to amplify intron polymorphisms. Based on bioinformatic analysis, we selected introns between 50 and 1000 bp in length with an intron length difference of 10–1000 bp, to develop ILP primers. Taking the D-genome sequence as reference and selecting polymorphic introns with at least 100 bp-length exons flanking the intronic region, a total of 10,180 ILP primers from 5,021 genes were developed (Dataset S4). Furthermore, we synthesized 535 ILP markers derived from nine gene families relevant to stress response, for experimental validation. Of them, 14 ILP markers were associated with the aquaporin protein family, 24 with the heat shock protein family, 25 with the NAC family, 27 with the WRKY family, 37 with the MAPK family, 59 with the P450 family, 74 with the leucine-rich repeat kinase family, 111 with the WD40 family, and 164 with the zinc finger transcription factor family (Dataset S5).
Of these 535 ILP markers, 411 primer pairs (76.82%) produced desirable and stable amplification products between G. herbaceum var. africanum and G. raimondii diploid species, with 54 (10.09%) failures in G. herbaceum var. africanum, 19 (3.55%) failures in G. raimondii, and 51 (9.53%) failures in both cotton species. The PCR products amplified by 389 primer pairs showed polymorphisms in the two diploid species, presenting a polymorphic rate of 72.71%. The highest polymorphic rate was in the WRKY transcription factor family (24/27; 88.89%); followed by the NAC family (21/25; 84%), the WD40 family (84/111; 75.68%) and the leucine-rich repeat family (56/74; 75.68%). P450 family members had the lowest polymorphic rate (37/59; 62.71%) (Table 3, Dataset S5).
Table 3.
ILP markers polymorphism between diploid A- and D-genome (A/D), or between tetraploid AtDt-genome (GhAtDt/GbAtDt) in nine tested gene family*.
| Gene family | No. of tested marker | A/D | GhAtDt/GbAtDt | ||
|---|---|---|---|---|---|
| No. of polymorphic marker | Polymorphic rate (%) | No. of polymorphic marker | Polymorphic rate (%) | ||
| Aquaporin | 14 | 10 | 71.43 | 9 | 64.29 |
| Heat shock proteins | 24 | 16 | 66.67 | 4 | 16.67 |
| NAC transcription factors | 25 | 21 | 84.00 | 6 | 24.00 |
| WRKY transcription factors | 27 | 24 | 88.89 | 15 | 55.56 |
| Mitogen-activated protein kinase | 37 | 26 | 70.27 | 12 | 32.43 |
| Cytochrome P450 | 59 | 37 | 62.71 | 14 | 23.73 |
| Leucine-rich repeat | 74 | 56 | 75.68 | 29 | 39.19 |
| WD40 repeat-containing proteins | 111 | 84 | 75.68 | 40 | 36.04 |
| Zinc finger | 164 | 115 | 70.12 | 66 | 40.24 |
| Total | 535 | 389 | 72.71 | 195 | 36.45 |
*A/D means two diploid cotton species, G. herbaceum var. africanum (A-genome) and G. raimondii (D-genome).
GhAtDt/GbAtDt means two allotetraploid cultivated species, G. barbadense cv. Hai7124 and G. hirsutum acc. TM-1.
We further used 535 ILP markers to screen allotetraploid interspecific polymorphisms of G. hirsutum acc. TM-1 and G. barbadense cv. Hai7124. Of these, 195 polymorphisms were detected, yielding a 36.45% polymorphic rate. We found that ILP markers associated with aquaporin, WRKY, zinc finger, leucine-rich repeat, and WD40 transcription factor families yielded the highest number of polymorphisms, with polymorphic rates of 64.29%, 55.56%, 40.24%, 39.19%, and 36.04%, respectively; while the heat shock protein family had the lowest polymorphic rate of 16.67%, with 4 of the tested markers found to be polymorphic (Table 3). As an example, an electropherogram of eight ILP markers (four WRKY and four leucine-rich repeat transcription factors) showed distinguishable A- and D-genome and/or At- and Dt-subgenome polymorphisms (Fig. 2A). The newly synthesized 535 ILP primer sequences, corresponding intron information, and data on polymorphisms between diploid cotton species G. herbaceum var. africanum and G. raimondii, and between tetraploid cultivated cotton species TM-1 and Hai7124, are presented in Dataset S5.
Figure 2.
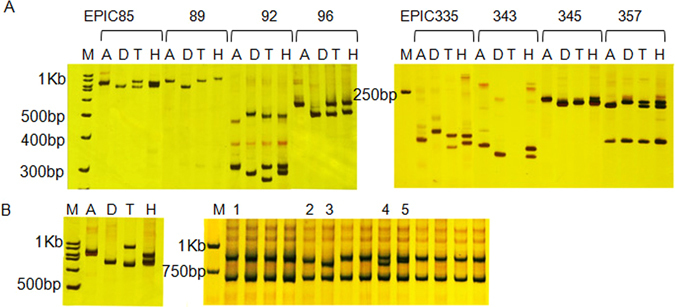
Electropherogram of detecting ILP markers polymorphism in cotton. (A) Electropherogram of ILP marker in four cotton species. M: marker; A: G. herbaceum var. africanum; D: G. raimondii; T: G. hirsutum acc. TM-1; H: G. barbadense cv. Hai7124. (B) Electropherogram of ILP marker EPIC211 in different G. hirsutum accessions. M: marker; A: G. herbaceum var. africanum; D: G. raimondii; T: G. hirsutum acc. TM-1; H: G. barbadense cv. Hai7124. Numbers 1–5 represents five alleles for EPIC211 detected in different G. hirsutum accessions.
Association analysis of salt stress traits in Upland cotton cultivars
Based on previous studies25, we investigated the ILPs from 264 G. hirsutum accessions. We firstly selected eight cotton varieties, derived from different germplasm pedigrees and three different ecological areas, to screen for polymorphic ILP markers. Polymorphisms were detected in 14 ILP markers and these were used to amplify alleles in the 264 accessions. In total, 41 alleles were detected, with 2.927 alleles per locus on average (ranging from two to five) and the highest number of alleles detected in EPIC211 (Fig. 2B). The average genetic diversity was 0.400 (ranging from 0.127 to 0.684) and the average polymorphism information content (PIC) was 0.345 (ranging from 0.122 to 0.623). Correlation between ILP markers and the ten salt stress tolerance traits, including relative germination rate (RGR) and germination percentage (RGP) at germinating stage, relative plant height (RPH), shoot dry matter (RSDM), root dry matter (RRDM), chlorophyll content (RCC), malondialdehyde (RMDA) content, and superoxide dismutase (RSOD), peroxidase (RPOD) and catalase (RCAT) enzyme activity at seedling stages, reported in Du et al.25, was assessed using multiple comparison and association analyses.
First, multiple comparisons were conducted to analyze the correlations between the ten salt stress traits and each polymorphic ILP marker using the LSR method in SPSS18.0. We found that there were 21 marker-trait correlations, involving 8 ILP markers and nine salt stress traits, showed significant differences between polymorphic loci and salt stress trait (P < 0.05). Of these, 13 marker-trait correlations reached statistical significance levels of P < 0.01 (Table S1).
Next, we analyzed the population structure of 264 G. hirsutum accessions using STRUCTURE V2.3.3 software based on 14 ILP markers and 145 SSR markers25. By comparing LnP (D) and ΔK, we selected K = 7 as the number of subpopulations, and corresponding Q-matrix data were used for the subsequent association mapping. The results showed that 18 marker-trait associations, involving 9 ILP markers and nine salt stress traits, were detected both from the MLM and GLM models (P < 0.05) (Table S2).
Integrated with the above two analysis, a total of 25 marker-trait associations involving 9 ILP markers for ten salt stress traits were detected (Table 4). Among these, two ILPs were from the WD40 family, two ILPs were from the leucine-rich repeat family, and the others were from MAPK, P450, WRKY, zinc finger, and aquaporin families, respectively. EPIC531, its gene encoding to plasma membrane intrinsic protein, was simultaneously associated with six salt stress traits: RCC, RPOD, RMDA, RPH, RRDM and RSDM. EPIC477, related to Zinc finger protein, was simultaneously associated with five salt stress traits: RCC, RPH, RSDM, RPOD, and RCAT. EPIC109 (Mitogen-activated protein kinase), EPIC309 (WD40 repeat-like protein) and EPIC356 (WRKY DNA-binding protein) were simultaneously associated with three salt stress traits. EPIC109 was associated with RCC, RPOD and RMDA; EPIC309 was associated with RCC, RPH and RPOD; and EPIC356 was associated with RSDM, RGR and RGP. EPIC50 (Leucine-rich repeat protein kinase family protein) was simultaneously associated with two salt stress traits (RRDM and RSDM). EPIC66 (Leucine-rich repeat protein kinase), EPIC211 (Cytochrome P450 protein) and EPIC274 (WD40 repeat-like protein) was associated with one salt stress trait each: RRDM, RPH and RMDA, respectively.
Table 4.
Association information on ILP marker-traits related to salt stress.
| Markers | Gene name | Gene ID | Sequence Description | Salt stress traits | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RCC | RPH | RRDM | RSDM | RSOD | RPOD | RCAT | RMDA | RGR | RGP | ||||
| EPIC50 | C1 | Gorai.005G026700 | Leucine-rich repeat protein kinase family protein | MC*, AM* | MC**, AM** | ||||||||
| EPIC66 | C2 | Gorai.007G047200 | Leucine-rich repeat protein kinase family protein | AM* | |||||||||
| EPIC274 | C3 | Gorai.006G261800 | Transducin/WD40 repeat-like superfamily protein | MC*, AM* | |||||||||
| EPIC356 | C4 | Gorai.012G051500 | WRKY DNA-binding protein 3 | MC* | MC**, AM** | MC**, AM** | |||||||
| EPIC531 | C5 | Gorai.009G418100 | Plasma membrane intrinsic protein 2 | MC*, AM* | MC** | MC* | AM* | MC**, AM* | MC**, AM** | ||||
| EPIC309 | C6 | Gorai.010G020600 | Transducin/WD40 repeat-like superfamily protein | MC**, AM* | AM* | MC* | |||||||
| EPIC211 | C7 | Gorai.011G060100 | Cytochrome P450 superfamily protein | MC*, AM* | |||||||||
| EPIC477 | C8 | Gorai.008G210800 | Zinc finger (C3HC4-type RING finger) family protein | MC**, AM* | MC**, AM* | MC** | MC** | AM* | |||||
| EPIC109 | C9 | Gorai.003G139900 | Mitogen-activated protein kinase 3 | MC* | MC**, AM* | MC**, AM* | |||||||
1MC and AM means marker- trait associations detected in multiple comparisons and association mapping analysis, respectively.
2* and **: significant difference P < 0.05 and P < 0.01, respectively.
3Abbreviation for 10 traits related to salt tolerance is relative chlorophyll content (RCC), relative plant height (RPH), relative root dry matter (RRDM), relative shoot dry matter (RSDM), relative SOD activity (RSOD), relative POD activity (RPOD), relative CAT activity (RCAT) and relative MDA content (RMDA), relative germination rate (RGR), relative germination percentage (RGP), respectively.
Potential function of candidate genes in cotton salt-tolerance
To confirm the relevance between the nine genes and salt tolerance, the salt induced expression patterns and functional characteristics based on VIGS analysis were investigated. We simplified to name the nine genes as C1-C9 with their information described in Table 4. The expression patterns of the nine genes showed diverse expression patterns in different tissues and organs of G. hirsutum acc. TM-1, including roots, stems, leaves, petals, anthers, ovules and fibers at three different developmental stages (0 days post-anthesis [dpa], 10 dpa and 21 dpa) (Fig. 3). C4 was a constitutively expressing gene in all tested tissues; C6 had low expression in different tissues and organs, especially in petal and anther; C9 was preferentially expressed in root and leaf, C1 and C8 were preferentially expressed in 10 DPA fibers; C2, C3, C5 and C7 was preferentially expressed in root, 0 DPA ovule, anther, stem, respectively. Then, we performed qRT-PCR to detect the differences in their expression abundance by 200 mM NaCl treatment in G. hirsutum cv. Jinmian 19. Five candidate genes (C3, C4, C5, C7 and C9) were significantly induced after salt treatment (Fig. 4), with the highest expression for C3 at 2 h, C4 at 0.5 h, C5 at 1 h, C7 at 24 h, and C9 at 12 h post treatment, respectively.
Figure 3.
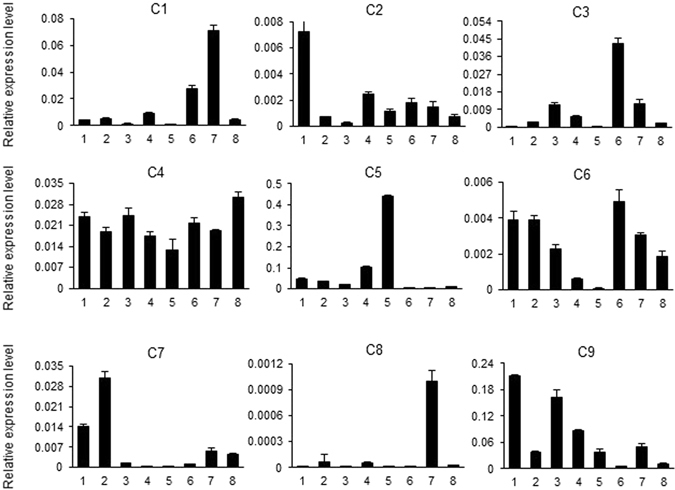
Expression patterns of nine candidate genes for salt-tolerance traits in different tissues and developmental stages. 1–8 indicate root, stem, leaf, petal, anther, 0 DPA ovule, 10 and 20 DPA fibers in TM-1, respectively. The error bars were calculated based on three biological replicates using standard deviation. The cotton histone3 (AF024716) gene was used as the reference gene.
Figure 4.
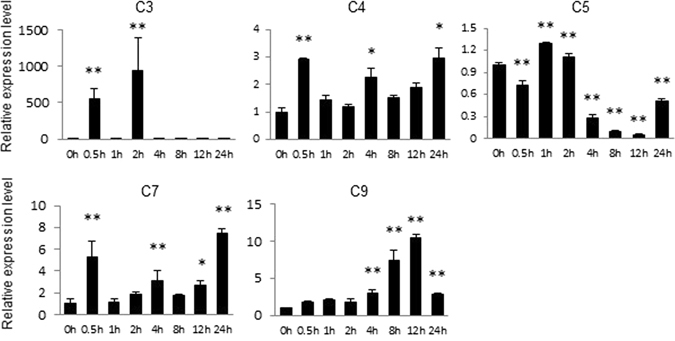
Expression analysis of five candidate genes induced after salt treatment. The error bars were calculated based on three biological replicates using standard deviation. The cotton histone3 (AF024716) gene was used as the reference gene. “*” at P < 0.05, “**” at P < 0.01.
To investigate the function of five candidate genes (C3, C4, C5, C7 and C9) in salt stress tolerance, we constructed TRV2:C3, TRV2:C4, TRV2:C5, TRV2:C7 and TRV2:C9 vectors to silence the endogenous genes in TM-1 seedlings, with TRV:00 as the mock treatment and TRV2:GhCLA1 (Cloroplastos alterados 1) as the technical control. More than 60 plants were infiltrated for each gene injection at 8 days post TM-1 seedlings. Two weeks later, the plants infiltrated with TRV2:GhCLA1 displayed a photobleached phenotype (Fig. 5A). Real-time PCR showed that the no-infiltration plants (CK) appeared high expression levels of the corresponding target gene, however, the transcripts of the target gene exhibited strong silencing in TRV2: target silenced gene infiltrated plants (Fig. 5B).
Figure 5.
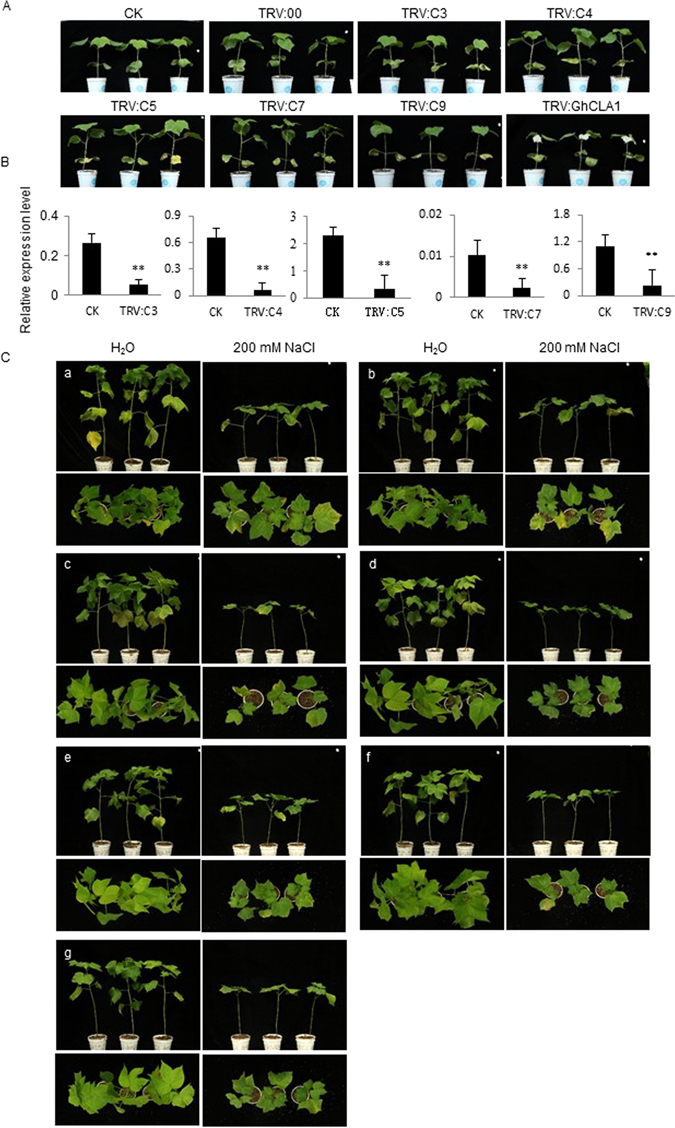
Plant phenotypes after target genes silencing with salt and mock treatments. (A) After two weeks infiltrated with TRV2: target silenced genes, the treated plants exhibited normal growth. TRV2:GhCLA1 plants exhibited a photobleaching phenotype as indicator. TRV: 00 as the mock treatment and no-infiltration plants as CK. (B) Gene expression of five candidate genes in silenced and control plant leaves by real-time PCR analysis. The error bars were calculated based on three biological replicates using standard deviation. The cotton beta-tubulin gene (GhTub1) was used as the reference gene. “*” at P < 0.05, “**” at P < 0.01. (C) Phenotypes of target gene-silenced and control plants after the salt stress treatment. Plants were treated with 200 mM NaCl for a month (H20 as control). a-g indicate CK (no-infiltration), TRV:00 (mock treatment), TRV:C3, TRV:C4, TRV:C5, TRV:C7 and TRV:C9, respectively. Upper: photographed from plants side; Below: photographed from plants top.
After the target gene was silenced, half of the plants per target gene-silenced plants were used for the salt stress treatment by irrigation 200 mM NaCl, and the others as water treatment control. A month later, there was no difference in growth between no-infiltration (CK), mock treated plants (TRV:00) and different target gene-silenced plant under tap water. However, cotton plants showed severe growth inhibition and lower true leaves defoliated under salt stress treatment. Especially, C3, C4, C7 and C9-silenced plants exhibited a serious true leaves defoliated than C5, no-infiltration (CK) and mock treated plants (TRV:00) (Fig. 5C). Next, we measured seven traits related to salt tolerance for salt stress treatment and water treatment control plants. Compared salt stress treatment with water control plants, the plant height, shoot dry matter weight, and root dry matter weight was significantly decreased, and SOD and POD activity, and Pro and H2O2 content was drastically increased. We calculated the difference between salt stress treatment and water control of target gene-silenced plants (TRV: target-silenced gene NaCl, and TRV: target-silenced gene H2O), and the difference between salt stress treatment and water control of no-infiltration plants (CK NaCl and CK H2O), and compared the statistics significance between them. As shown in Fig. 6, the difference between TRV: C4 NaCl and TRV: C4 H2O for C4-silenced plants were more distinct in the plant height, shoot dry matter weight, root dry matter weight, SOD activity and Pro content than that between CK NaCl and CK H2O. Similarly, the significant difference in the plant height, SOD and POD activity, and Pro and H2O2 content was detected in C9-silenced plants after salt treatment. SOD and POD activity in C7-silenced plants, the shoot dry matter weight in C3-silenced plants, and the H2O2 content in C5-silenced plants were also significantly changed, respectively. Integrated the phenotype with physiological data, silencing of C3, C4, C5, C7 and C9 can compromise cotton salt tolerance in different extent, especially C4 and C9, following C7, C3, and C5. The result indicated that increasing the gene expression for each have a potential utilization in cotton salt tolerance breeding.
Figure 6.
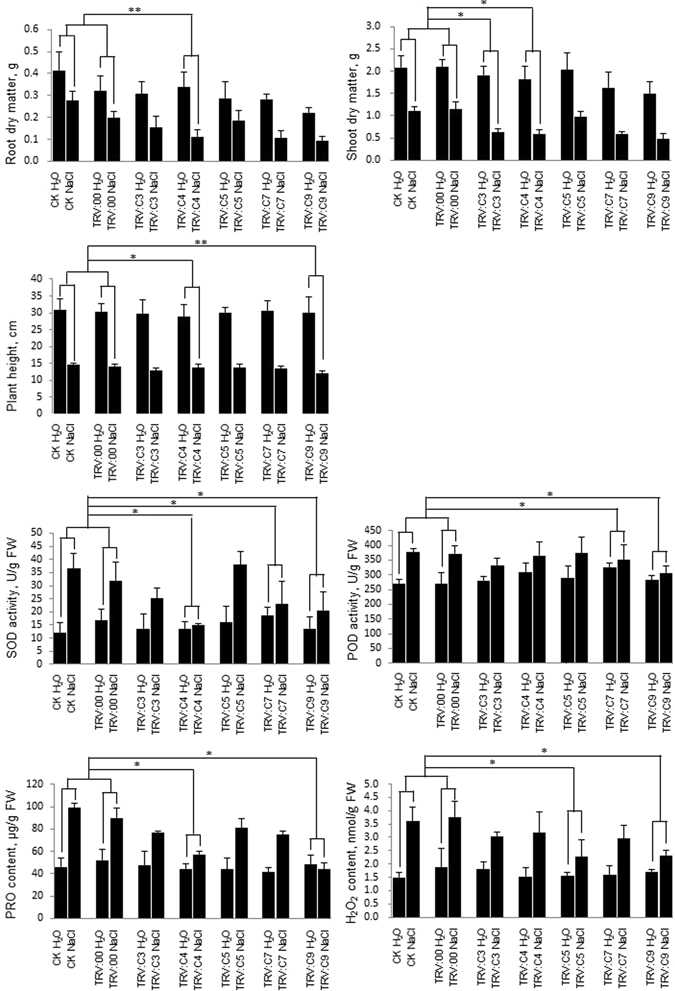
The difference of salt-tolerance parameters between gene-silenced and control plants in response to salt stress. Seven salt-tolerance parameters, including the plant height (cm), shoot dry matter weight (g), root dry matter weight (g), and SOD and POD (U/g FW) activity, and Pro (μg/g FW) and H2O2 (nmol/g FW) content were measured in leaves of gene-silenced and control plants with salt stress and water treatment. Plants were treated with 200 mM NaCl for a month (with water treatment as control). Significance level was compared between the difference between salt stress treatment and water control of target gene-silenced plants (TRV: target-silenced gene NaCl, and TRV: target-silenced gene H2O), and the difference between salt stress treatment and water control of no-infiltration plants (CK NaCl and CK H2O). “*”: at P < 0.05; “**”: at P < 0.01.
Discussion
Marker development in crop species is important in the facilitation of genomic-based crop improvement. In cotton, the most widely used type of DNA molecular markers is SSRs. Based on genome and EST sequence information from different Gossypium species, researchers have developed large quantities of SSR markers. In total, 17,448 publicly available SSRs have been deposited in the Cotton Database (http://www.cottondb.org/). These SSRs have been widely used in high-density genetic mapping26–28, target trait-related gene/QTL mapping29–37, association studies25, 38–49, and diversity analysis50, 51. As the large number of genomic sequence resources in Gossypium were released, the development of whole genome level markers, such as restriction-site associated DNA (RAD)52, 53 and insertion-deletion (InDel) and single nucleotide polymorphisms (SNPs)7, 54–57 has been initiated. FMs are a type of marker with the most potential to bridge the gap between structural polymorphisms and functional diversity, since these gene-derived markers are related to phenotypic variations. A pilot study for EST-based SSR, SNP, and InDel marker development and their utilization in tetraploid cotton genetic mapping has been carried out6, 7, however, the efficiency of marker development and the number of polymorphisms in designed primers were found to be relatively low using transcriptome data. High-throughput sequencing resources provide a better way to develop FMs in cotton. ILP markers are characterized as gene-specific, co-dominant, hypervariable, neutral, convenient, and reliable; however, to date, their genome-wide exploitation and application in cotton has not been reported. Here, we used whole-genome scaffold sequence information from the diploid cottons G. raimondii 18 and G. arboreum 20 to mine conserved orthologous sequences and develop functional ILP markers of orthologs between the A- and D-genomes. A total of 10,180 ILP markers from 5,021 orthologs were developed, and the polymorphism of 535 ILP markers associated with nine classes of gene family relevant to stress responses was validated experimentally. As a result, polymorphic rates of 72.71% and 36.45% between A/D-genome diploids and between AtDt tetraploid genome species were detected, respectively, implying the high efficiency of FM marker development using whole-genome scaffold sequence information.
The cotton lineage experienced an abrupt five- to sixfold ploidy increase approximately 60 MYA, shortly after its divergence from Theobroma cacao, and then an allopolyploidy event that reunited divergent Gossypium genomes approximately 1–2 MYA conferred a 30–36-fold duplication of ancestral angiosperm (flowering plant) genes in the cotton tetraploids G. hirsutum and G. barbadense 18. Whole genome duplication events brought about a substantial gene family expansion, and a larger number of paralogs (including tandem and segmental duplications) were generated. In the evolutionary process, these paralogs were with conserved domains but undergone various structural variations and functional diversity. In this study, we first verified orthologs between the A- and D-genomes for ILP markers development, and carried out in silico PCR analysis with strict no mismatch to confirm the primer specificity. As a result, of 10,180 ILP markers from 7,070 genes, 9,294 and 9,713 ILPs were mapped in At- and Dt-subgenome of TM-1 genome, respectively, and 94.35% (8,769) and 97.05% (9,426) were uniquely matched (Dataset S4). The result indicated that these ILP markers corresponded to a particular gene with few interference from paralogous genes, and could be used to verify the homologs polymorphism in tetraploid cotton species. This study also provides a reference for the development and application of whole-genome ILP FMs in other complex polyploid organisms.
Biotic and abiotic stresses, such as Verticillium dahliae, drought, heat, submergence, and high salinity, can severely affect plant growth and crop productivity, which leads to worldwide economic losses. To understand and improve stress responses and tolerances in crop species, researchers have focused on the signaling perception mechanisms, transcriptional regulation and expression of functional proteins in the stress response58. Previous studies found that NAC transcription factors are involved in plant development and biotic and abiotic stress regulation59; WRKY transcription factors are important components of many aspects in the plant defense systems and in plant abiotic stress responses60; HSPs, which were originally identified as heat-inducible gene products, are a family of highly conserved proteins that respond to a wide variety of stresses including oxidative and thermal stress61; CYP450 proteins function as growth and developmental signals and protect plants from various biotic and abiotic stresses62; WD40s play a key role in protein-protein and protein-DNA interactions by acting as scaffolding molecules, promoting protein activity and responsiveness to salt stress63; ZnF and LRR family proteins play regulatory roles in immune responses64, 65; aquaporins regulate the movement of water and other small molecules across plant vacuolar and plasma membranes, and are associated with plant tolerance of biotic and abiotic stresses66; MAPK cascade is one of the universal signaling pathways involved in responses to external stimuli, which play a crucial role in plant growth and development as well as biotic and abiotic stress responses67. In this study, we selected 535 ILP markers derived from these nine gene families that are related to stress responses for structural and functional identification.
In cotton, association analysis has been widely used in mining QTLs/genes related to the important agronomic traits, such as fiber qualities38–43, 46, yield and its components41, 44, 45, 48, salt stress25, 68, Verticillium wilt resistance47 and seed oil and protein contents50 etc. Nevertheless, further experiments need to be done to mine the genes and verify their function based on these marker-trait associations. In this study, we detected a total of 25 marker-trait associations involving 9 ILP markers for ten salt stress traits. Following these ILPs corresponding to genes, we found that five genes (C3, C4, C5, C7 and C9) played important roles in cotton salt tolerance through TRV-VIGS analysis. Taken together, the ILP markers associated with the traits with interest will accelerate the findings of functional genes and utilization in breeding program.
In this study, the functional role of C4 and C9 in salt stress were further confirmed in cotton, which were in line with previous evidence. C4 (EPIC356, gene ID: Gorai.012G051500 of G. raimondii), corresponding to WKY18 in G. raimondii 60, which encodes WRKY DNA-binding protein, was simultaneously associated with RGR and RGP in this study. Our previous study reported that under salt or drought treatment, WRKY18 expression levels were significantly increased, with a peak at 8 h of treatment with NaCl (200 mmol L−1), and at 8 h of treatment with PEG6000 (20%), indicating its role in abiotic stress tolerance60. C9 (EPIC109, gene ID: Gorai.003G139900 of G. raimondii), corresponding to MPK9 in G. raimondii 67, which encodes mitogen-activated protein kinase, was simultaneously associated with RPOD and RMDA. Zhang et al.67 found that MPK9 was constitutively expressed at high levels in both vegetative and reproductive organs. MPK9 can be induced by multi-stressors such as jasmonis acid, H2O2, salicylic acid, 4 °C, 37 °C, and wounding, and plays a role in plant defense responses and multiple stress-signaling pathways.
In summary, genome-wide ILP markers are powerful functional markers to bridge the gap between structural polymorphisms and functional diversity. The new gene-based FMs will be a useful resource for gene mining and breeding improvement for traits of interest via MAS in cotton. The methodology can also serve as a useful model for the development of FMs in other complex plant genomes. Further, five genes (C3, C4, C5, C7 and C9) were verified to be related to salt stress tolerance and have potential to improve salt tolerance in cotton abiotic-resistance breeding.
Materials and Methods
Development of putative ILP markers
The assembled genome sequences, annotated genes, transcripts and coding DNA sequences (CDSs) data were obtained from the genome annotation files for G. raimondii (http://www.phytozome.net) and G. arboreum (http://cgp.genomics.org.cn). The complete nuclear genome of 37,505 protein-coding genes in G. raimondii and 41,330 protein-coding genes in G. arboreum were extracted to identify conserved exons of single-copy genes. We extracted introns of a manageable size (<100 00 bp) as well as the corresponding flanking exons for further study. To detect orthologous genes in the two genomes, one-to-one orthologous relationship of genes between G. arboreum and G. raimondii were identified using reciprocal BLAST best hit with a high E-value (≥10−20). Further, transcripts and CDSs from the A genome were mapped to the D genome, and transcripts with an mRNA similarity ≥80% and a CDS similarity ≥95% were considered as orthologs. Orthologs with intron lengths between 50 and 1000 bp were investigated, and those with an intron length difference ranging from 10 bp to 1000 bp were used for the development of ILP orthologs primers. The scheme of ILP marker development is described in Fig. 7.
Figure 7.
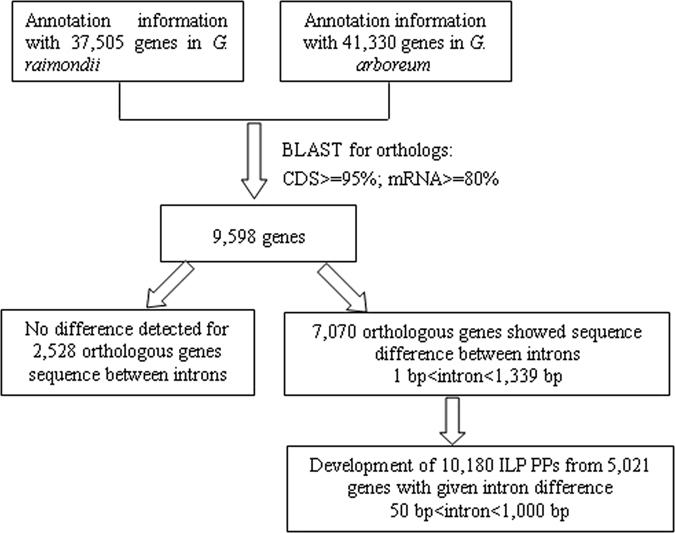
The development scheme of genome-wide ILP markers in this study.
Primer pairs were designed from the flanking exon sequences using Primer3 software (http://www-genome.wi.mit.edu/genome_sofware/other/primer3.html). Two perl scripts served as interface modules between MISA and Primer3; p3_in.pl for creating a primer3 input file which was submitted to Primer3, and p3_out.pl, which was used to calculate and merge the information. These two perl scripts were downloaded from MISA (http://pgrc.ipk-gatersleben.de/misa/). To verify the primer specificity, in silico PCR analysis (parameter:gap 0-mismach 0-product size 50–2000) was employed to examine the uniqueness of the primer in different cotton species. The primer pairs for experimental validation were synthesized by GenScript (Nanjing, China). All primer information, including the primer design parameters, is shown in Dataset S4.
After obtaining Gene Ontology (GO) annotations for orthologs, Web Gene Ontology Annotation Plot (WEGO) software was used to carry out GO functional classification and to characterize the distribution of gene functions at the macro level69.
Plant materials and treatments
A total of 267 cotton accessions for ILP marker analysis were collected for this study (Table S3). Firstly, two diploid cotton species, G. herbaceum var. africanum (A-genome) and G. raimondii (D-genome), were chosen for validating the efficiency of ILP markers. Secondly, two allotetraploid cultivated species (G. barbadense cv. Hai7124 and G. hirsutum acc. TM-1.), and 264 G. hirsutum germplasm accessions (including TM-1) were used for the analysis of ILP marker polymorphisms and their association with salt stress traits. Information on ten salt tolerance traits and 145 SSRs in 264 upland cotton cultivars was obtained from Du et al.25.
G. hirsutum L. acc TM-1, a genetic standard line of Upland cotton, was used for tissue/organ expression analysis. The plants were cultivated under normal field conditions. Petals and anthers were sampled on the day of flowering, and ovules and fibers were excised from developing flower buds or bolls on selected days post anthesis (dpa). Roots, stems and leaves were collected from two-week-old seedlings. The materials were quick-frozen in liquid nitrogen and stored at −70 °C before use.
G. hirsutum L. cv. Jinmian 19, which exhibits high tolerance to abiotic stress, was used for the salt stress treatments. Cotton seedlings (G. hirsutum L. cv. Jinmian 19) were grown in a growth chamber under greenhouse conditions at 28 °C under a 16 h light/8 h dark cycle for three weeks. Then the roots of cotton seedlings were irrigated with 200 mmol/L NaCl (ddH2O as a mock control). The leaves were harvested with three biological repeats at different time points (0, 0.5, 1, 2, 4, 8, 12 and 24 hours) after NaCl treatment, quick-frozen in liquid nitrogen and stored at −70 °C for RNA extraction.
PCR amplification, RNA isolation and real-time PCR analysis
Genomic DNA from the cotton accessions involved in the study was isolated as described by Paterson et al.70. PCR reactions were carried out with 0.5 units of Taq DNA polymerase (Tiangen), 50–100 ng of template DNA, 1 μL of each primer (5 μM/μL), 0.2 μL of each dNTPs (10 mM), 0.6 μL MgCl2 (25 mM), and 1 μL of 10 × PCR reaction buffer in a final volume of 10 μL. PCR amplifications were performed using a Peltier Thermal Cycler-225 (MJ Research), and the amplification conditions were as follows: an initial denaturation at 95 °C for 5 minutes, followed by 28 cycles of 30 s at 94 °C, 30 s at 58 °C, and 30 s at 72 °C, and a final extension of 10 min at 72 °C. PCR products were separated using 9% polyacrylamide gel electrophoresis (PAGE) as described by Zhang et al.71.
Total RNA was extracted using plant total RNA extraction kit (Bioer, Hangzhou, China). 200–500 ng RNA samples were reverse transcribed into cDNA by HiScript reverse transcriptase (Vazyme, Nanjing, China). Based on the candidate gene sequences, gene-specific real-time PCR primers were designed using Beacon Designer 7.0. The predicted amplified fragment lengths were between 75 and 200 bp, and the annealing temperatures were between 58 °C and 60 °C. The amplification reactions of real-time PCR were performed on an ABI Prism 7500 (Applied Biosystems, USA) with the light cycler fast start DNA Master SYBR Green I kit (Roche, Basel, Switzerland) and three replicates. The amplification reactions were as follows: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 58 °C for 15 s, and 72 °C for 30 s. The gene expression levels were calculated according to Livak and Schmittgen72. Two reference genes (GhHis3 73 with abundant transcripts and GhTub1 74 with moderate transcripts), were used simultaneously to confirm the real-time PCR results. The information for the real-time PCR primers were listed in Dataset S6.
Association analysis
Multiple comparison and association mapping methods were used to confirm marker-trait association analysis. The multiple comparisons were estimated using SPSS18.0 (http://www.spss.com.cn/), and were conducted using the least significant range (LSR) method for correlation analysis of ten salt stress traits and different marker alleles. STRUCTURE v2.3.3 software75 (http://pritch.bsd.uchicago.edu/software.html) was used to infer the population structure of 264 G. hirsutum accessions (K = 1 to 10, with five runs at each K) using a burn-in of 10,000 and a run length of 100,000. The most likely number of clusters (K) was selected by comparing LnP (D) and ΔK76. A mixed linear model (MLM) and a general linear model (GLM) were employed to construct marker-trait association tests using the TASSEL 2.0.1 software package77.
Virus induced gene silencing assays
G. hirsutum cv. TM-1 was used for VIGS analysis. The pTRV1 and pTRV2 vectors for VIGS analysis were generously provided by Dr. Libo Shan of Texas A & M University (College Station, TX, USA). GhCLA1 (Cloroplastos alterados 1), which encoded 1-deoxy-D-xylulose-5-phosphate synthase, was constructed pTRV2:GhCLA1 and acted as a control to verify the VIGS efficiency78. The primer pairs information used for the construction of VIGS vectors, the length of the segments of silence and corresponding base position are listed in Dataset S6. 201–430 bp PCR fragments for the corresponding target gene were amplified from TM-1 cDNA, double digested with EcoRI and XbaI, and inserted into enzyme sites for insertion into pTRV2 to make pTRV2 vectors with target silenced genes.
Agrobacterium were prepared and infiltrated as described by Cai et al.79. Eight-day-old TM-1 seedlings were infiltrated into two fully expanded cotyledons with a 1:1 mixture of Agrobacterium GV3101 carrying TRV1 and TRV2: target silenced gene. At the same time, cotton seedlings were infiltrated TRV1 and TRV2 or TRV1 and TRV2:GhCLA1 as the mock treatment and the technical control, respectively. All cotton seedlings (including untreated plants) were kept at 23/22 °C (day/night) in a growth chamber with a 16 h light/8 h dark cycle for two weeks before they were used for identification of target gene silencing and next salt stress treatment. VIGS experiments were repeated with more than 60 plants for each different injection events per gene.
Total RNA was extracted and reverse transcribed into cDNA from candidate gene-silenced and untreated plants’ fresh leaf tissue two weeks post-inoculation by plant total RNA extraction kit (Bioer, Hangzhou, China) and HiScript reverse transcriptase (Vazyme, Nanjing, China), respectively. Real-time PCR was used to analyze the relative levels of candidate genes expression.
After candidate gene silencing, half of the plants (~30 plants) per candidate gene-silenced were used for the salt stress treatment by irrigation 200 mM NaCl, and the other 30 plants were irrigated tap water as a mock control. For salt and water treatment, each ~10 plants were investigated as a replicate. A month later, each plant was measured for the plant height, shoot dry matter weight, and root dry matter weight. Further, the superoxide dismutase and peroxidase enzyme activities, proline and H2O2 content were estimated using corresponding assay kit (Jiancheng, Nanjing, China). All indexes were detected with three biological replicates and three experimental replicates.
Electronic supplementary material
Acknowledgements
This program was financially supported in part by Key R & D program in Jiangsu Province (BE2015360), the Fundamental Research Funds for the Central Universities (KYYJ201601), six talent peaks project in Jiangsu province (2015-NY-002), Qing Lan Project for Science and Technology Innovation Team in Jiangsu Province (No. 6) and JCIC-MCP (No. 10).
Author Contributions
Experiments were designed by W.Z.G. Experiments were performed by C.P.C., S.W., E.L.N. and C.Z.C., C.P.C. and W.Z.G. drafted the manuscript, W.Z.G. and C.P.C. revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Caiping Cai and Shuang Wu contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-00617-7
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Guo W, Zhang T, Sheng X, John Y, Kohel RJ. Development of SCAR marker linked to a major QTL for high fiber strength and its molecular marker assisted selection in Upland cotton. Crop Sci. 2003;6:2252–2256. doi: 10.2135/cropsci2003.2252. [DOI] [Google Scholar]
- 2.Khan MA, et al. Development of molecular markers linked to the ‘Fiesta’ linkage group 7 major QTL for fire blight resistance and their application for marker-assisted selection. Genome. 2007;50:568–577. doi: 10.1139/G07-033. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, et al. Application of identified QTL-marker associations in rice quality improvement through a design-breeding approach. Theor Appl Genet. 2007;115:87–100. doi: 10.1007/s00122-007-0545-x. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Chapman SC, Bonnett DG, Rebetzke GJ. Simultaneous selection of major and minor genes: use of QTL to increase selection efficiency of coleoptile length of wheat (Triticum aestivum L.) Theor Appl Genet. 2009;119:65–74. doi: 10.1007/s00122-009-1017-2. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, He Z, Appels R, Xia X. Functional markers in wheat: current status and future prospects. Theor Appl Genet. 2012;125:1–10. doi: 10.1007/s00122-012-1829-3. [DOI] [PubMed] [Google Scholar]
- 6.Guo W, et al. A microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium. Genetics. 2007;176:527–541. doi: 10.1534/genetics.107.070375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, et al. Development of EST-based SNP and InDel markers and their utilization in tetraploid cotton genetic mapping. BMC Genomics. 2014;15:1046. doi: 10.1186/1471-2164-15-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palumbi SR, Baker CS. Contrasting population structure from nuclear intron sequences and mtDNA of humpback whales. Mol Biol Evol. 1994;11:426–435. doi: 10.1093/oxfordjournals.molbev.a040115. [DOI] [PubMed] [Google Scholar]
- 9.Choi HK, et al. A sequence-based genetic map of Medicago truncatula and comparison of marker collinearity with M. sativa. Genetics. 2004;166:1463–1502. doi: 10.1534/genetics.166.3.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta S, et al. Development and utilization of novel intron length polymorphic markers in foxtail millet (Setaria italica (L.) P. Beauv.) Genome. 2011;54:586–602. doi: 10.1139/g11-020. [DOI] [PubMed] [Google Scholar]
- 11.Liu H, et al. Genome-scale identification of resistance gene analogs and the development of their intron length polymorphism markers in maize. Mol Breeding. 2012;29:437–447. doi: 10.1007/s11032-011-9560-3. [DOI] [Google Scholar]
- 12.Poczai P, et al. Development of intron targeting (IT) markers for potato and cross-species amplification in Solanum nigrum (Solanaceae) Am J Bot. 2010;97:e142–e145. doi: 10.3732/ajb.1000360. [DOI] [PubMed] [Google Scholar]
- 13.Yang L, et al. PIP: a database of potential intron polymorphism markers. Bioinformatics. 2007;23:2174–2177. doi: 10.1093/bioinformatics/btm296. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Zhao X, Zhu J, Wu W. Genome-wide investigation of intron length polymorphisms and their potential as molecular markers in rice (Oryza sativa L.) DNA Res. 2005;12:417–427. doi: 10.1093/dnares/dsi019. [DOI] [PubMed] [Google Scholar]
- 15.Muthamilarasan M, et al. Development of 5123 intron-length polymorphic markers for large-scale genotyping applications in foxtail millet. DNA Res. 2014;21:41–52. doi: 10.1093/dnares/dst039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Endrizzi JE, Turcotte EL, Kohel RJ. Genetics, cytology, and evolution of Gossypium. Adv Genet. 1985;23:271–375. [Google Scholar]
- 17.Wendel JF. New World tetraploid cottons contain Old World cytoplasm. Proc Natl Acad Sci USA. 1989;86:4132–4136. doi: 10.1073/pnas.86.11.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paterson AH, et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492:423–427. doi: 10.1038/nature11798. [DOI] [PubMed] [Google Scholar]
- 19.Wang K, et al. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;44:1098–1103. doi: 10.1038/ng.2371. [DOI] [PubMed] [Google Scholar]
- 20.Li F, et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet. 2014;46:567–572. doi: 10.1038/ng.2987. [DOI] [PubMed] [Google Scholar]
- 21.Zhang T, et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol. 2015;33:531–537. doi: 10.1038/nbt.3207. [DOI] [PubMed] [Google Scholar]
- 22.Li F, et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol. 2015;33:524–530. doi: 10.1038/nbt.3208. [DOI] [PubMed] [Google Scholar]
- 23.Yuan D, et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci Rep. 2015;5:17662. doi: 10.1038/srep17662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X, et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci Rep. 2015;5:14139. doi: 10.1038/srep14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du L, et al. Evaluation and exploration of favorable QTL alleles for salt stress related traits in cotton cultivars (G. hirsutum L.) Plos One. 2016;11:e0151076. doi: 10.1371/journal.pone.0151076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao L, et al. Toward allotetraploid cotton genome assembly: integration of a high-density molecular genetic linkage map with DNA sequence information. BMC Genomics. 2012;13:539. doi: 10.1186/1471-2164-13-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu JZ, et al. A high-density simple sequence repeat and single nucleotide polymorphism genetic map of the tetraploid cotton genome. G3/Genes Genom Genet. 2012;2:43–58. doi: 10.1534/g3.111.001552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Y, et al. Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genomics. 2011;12:15. doi: 10.1186/1471-2164-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang C, Ulloa M, Mullens TR, Yu JZ, Roberts PA. QTL analysis for transgressive resistance to root-knot nematode in interspecific cotton (Gossypium spp.) progeny derived from susceptible parents. PLoS One. 2012;7:e34874. doi: 10.1371/journal.pone.0034874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alfred Q, et al. Mapping of quantitative trait loci for oil content in cottonseed kernel. J Genet. 2012;91:289–295. doi: 10.1007/s12041-012-0184-0. [DOI] [PubMed] [Google Scholar]
- 31.Yu J, et al. Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Theor Appl Genet. 2013;126:275–287. doi: 10.1007/s00122-012-1980-x. [DOI] [PubMed] [Google Scholar]
- 32.Fang DD, et al. Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (Gossypium hirsutum L.) BMC Genomics. 2014;15:397. doi: 10.1186/1471-2164-15-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao Z, Wang P, Zhu X, Chen H, Zhang T. SSR marker-assisted improvement of fiber qualities in Gossypium hirsutum using G. barbadense introgression lines. Theor Appl Genet. 2014;127:587–594. doi: 10.1007/s00122-013-2241-3. [DOI] [PubMed] [Google Scholar]
- 34.Yu JZ, et al. Mapping genomic loci for cotton plant architecture, yield components, and fiber properties in an interspecific (Gossypium hirsutum L. × G. barbadense L.) RIL population. Mol Genet Genomics. 2014;289:1347–1367. doi: 10.1007/s00438-014-0930-5. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, et al. Genetic analysis of Verticillium wilt resistance in a backcross inbred line population and a meta-analysis of quantitative trait loci for disease resistance in cotton. BMC Genomics. 2015;16:577. doi: 10.1186/s12864-015-1682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu D, et al. Construction of a high-density genetic map and lint percentage and cottonseed nutrient trait QTL identification in upland cotton (Gossypium hirsutum L.) Mol Genet Genomics. 2015;290:1683–1700. doi: 10.1007/s00438-015-1027-5. [DOI] [PubMed] [Google Scholar]
- 37.Said JI, Lin Z, Zhang X, Song M, Zhang J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genomics. 2013;14:776. doi: 10.1186/1471-2164-14-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kantartzi SK, Stewart JM. Association analysis of fibre traits in Gossypium arboreum accessions. Plant Breeding. 2008;127:173–179. doi: 10.1111/j.1439-0523.2008.01490.x. [DOI] [Google Scholar]
- 39.Abdurakhmonov IY, et al. Molecular diversity and association mapping of fiber quality traits in exotic G. hirsutum L. germplasm. Genomics. 2008;92:478–487. doi: 10.1016/j.ygeno.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 40.Abdurakhmonov IY, et al. Linkage disequilibrium based association mapping of fiber quality traits in G. hirsutum L. variety germplasm. Genetics. 2009;136:401–417. doi: 10.1007/s10709-008-9337-8. [DOI] [PubMed] [Google Scholar]
- 41.Zeng L, Meredith WR, Jr., Gutiérrez OA, Boykin DL. Identification of associations between SSR markers and fiber traits in an exotic germplasm derived from multiple crosses among Gossypium tetraploid species. Theor Appl Genet. 2009;119:93–103. doi: 10.1007/s00122-009-1020-7. [DOI] [PubMed] [Google Scholar]
- 42.Kalivas A, Xanthopoulos F, Kehagia O, Tsaftaris AS. Agronomic characterization, genetic diversity and association analysis of cotton cultivars using simple sequence repeat molecular markers. Genet Mol Res. 2011;10:208–217. doi: 10.4238/vol10-1gmr998. [DOI] [PubMed] [Google Scholar]
- 43.Jena SN, et al. Analysis of genetic diversity, population structure and linkage disequilibrium in elite cotton (Gossypium L.) germplasm in India. Crop Pasture Sci. 2011;62:859–875. doi: 10.1071/CP11161. [DOI] [Google Scholar]
- 44.Zhang T, et al. Variations and transmission of QTL alleles for yield and fiber qualities in upland cotton cultivars developed in China. PLoS One. 2013;8:e57220. doi: 10.1371/journal.pone.0057220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mei H, Zhu X, Zhang T. Favorable QTL alleles for yield and its components identified by association mapping in Chinese Upland cotton cultivars. PLoS One. 2013;8:e82193. doi: 10.1371/journal.pone.0082193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai C, Ye W, Zhang T, Guo W. Association analysis of fiber quality traits and exploration of elite alleles in Upland cotton cultivars/accessions (Gossypium hirsutum L.) J Integr Plant Biol. 2014;56:51–62. doi: 10.1111/jipb.12124. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Y, Wang H, Chen W, Li Y. Genetic structure, linkage disequilibrium and association mapping of Verticillium wilt resistance in elite cotton (Gossypium hirsutum L.) germplasm population. PLoS One. 2014;9:e86308. doi: 10.1371/journal.pone.0086308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jia Y, et al. Association mapping for epistasis and environmental interaction of yield traits in 323 cotton cultivars under 9 different environments. PLoS One. 2014;9:e95882. doi: 10.1371/journal.pone.0095882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu G, et al. Association mapping of seed oil and protein contents in upland cotton. Euphytica. 2015;205:637–645. doi: 10.1007/s10681-015-1450-z. [DOI] [Google Scholar]
- 50.Guo WZ, Wang W, Zhou BL, Zhang TZ. Cross-species transferability of G. arboreum-derived EST-SSRs in diploid species of Gossypium. Theor Appl Genet. 2006;112:1573–1581. doi: 10.1007/s00122-006-0261-y. [DOI] [PubMed] [Google Scholar]
- 51.Zhu H, Zhang T, Yang L, Guo W. EST-SSR sequences revealed the relationship of D genome in diploid and tetraploid species in. Gossypium. Plant Sci. 2009;176:397–405. doi: 10.1016/j.plantsci.2008.12.007. [DOI] [Google Scholar]
- 52.Wang H, Jin X, Zhang B, Shen C, Lin Z. Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Res. 2015;22:147–160. doi: 10.1093/dnares/dsu047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, et al. Molecular mapping of restriction-site associated DNA markers in allotetraploid Upland cotton. PLoS One. 2015;10:e0124781. doi: 10.1371/journal.pone.0124781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lv Y, et al. Characterization of expressed sequence tags from developing fibers of Gossypium barbadense and evaluation of insertion-deletion variation in tetraploid cultivated cotton species. BMC Genomics. 2013;14:170. doi: 10.1186/1471-2164-14-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang S, et al. Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol. 2015;16:108. doi: 10.1186/s13059-015-0678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Logan-Young, C. J., Yu, J. Z., Verma, S. K., Perc, y. R. G. & Pepper, A. E. SNP discovery in complex allotetraploid genomes (Gossypium spp., Malvaceae) using genotyping by sequencing. Appl Plant Sci. 3, pii: apps.1400077 (2015). [DOI] [PMC free article] [PubMed]
- 57.Hulse-Kemp AM, et al. BAC-end sequence-based SNP mining in allotetraploid cotton (Gossypium) utilizing resequencing data, phylogenetic inferences, and perspectives for genetic mapping. G3/Genes Genom Genet. 2015;5:1095–1105. doi: 10.1534/g3.115.017749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakabayashi R, Saito K. Integrated metabolomics for abiotic stress responses in plants. Curr Opin Plant Biol. 2015;24C:10–16. doi: 10.1016/j.pbi.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 59.Tang Y, et al. Molecular characterization of novel TaNAC genes in wheat and overexpression of TaNAC2a confers drought tolerance in tobacco. Physiol Plant. 2012;144:210–224. doi: 10.1111/j.1399-3054.2011.01539.x. [DOI] [PubMed] [Google Scholar]
- 60.Cai C, et al. Genome-wide analysis of the WRKY transcription factor gene family in Gossypium raimondii and the expression of orthologs in cultivated tetraploid cotton. The Crop J. 2014;2:87–101. doi: 10.1016/j.cj.2014.03.001. [DOI] [Google Scholar]
- 61.Oksala NK, et al. Natural thermal adaptation increases heat shock protein levels and decreases oxidative stress. Redox Biol. 2014;3:25–28. doi: 10.1016/j.redox.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu J, Wang X, Guo W. The cytochrome P450 superfamily: Key players in plant development and defense. J Integr Agri. 2015;14:1673–1686. doi: 10.1016/S2095-3119(14)60980-1. [DOI] [Google Scholar]
- 63.Kong D, Li M, Dong Z, Ji H, Li X. Identification of TaWD40D, a wheat WD40 repeat-containing protein that is associated with plant tolerance to abiotic stresses. Plant Cell Rep. 2015;34:395–410. doi: 10.1007/s00299-014-1717-1. [DOI] [PubMed] [Google Scholar]
- 64.Ng A, Xavier RJ. Leucine-rich repeat (LRR) proteins: integrators of pattern recognition and signaling in immunity. Autophagy. 2011;7:1082–1084. doi: 10.4161/auto.7.9.16464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim GD, Cho YH, Yoo SD. Regulatory functions of evolutionarily conserved AN1/A20-like Zinc finger family proteins in Arabidopsis stress responses under high temperature. Biochem Biophys Res Commun. 2015;457:213–220. doi: 10.1016/j.bbrc.2014.12.090. [DOI] [PubMed] [Google Scholar]
- 66.Li J, et al. An aquaporin protein is associated with drought stress tolerance. Biochem Biophys Res Commun. 2015;459:208–213. doi: 10.1016/j.bbrc.2015.02.052. [DOI] [PubMed] [Google Scholar]
- 67.Zhang X, Wang L, Xu X, Cai C, Guo W. Genome-wide identification of mitogen-activated protein kinase gene family in Gossypium raimondii and the function of their corresponding orthologs in tetraploid cultivated cotton. BMC Plant Biol. 2014;14:345. doi: 10.1186/s12870-014-0345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saeed M, Guo W, Zhang T. Association mapping for salinity tolerance in cotton (Gossypium hirsutum l.) germplasm from us and diverse regions of china. Australian J Crop Sci. 2014;8:338–346. [Google Scholar]
- 69.Ye J, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paterson AH, Brubaker C, Wendel JF. A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep. 1993;11:122–127. doi: 10.1007/BF02670470. [DOI] [Google Scholar]
- 71.Zhang J, Guo WZ, Zhang TZ. Molecular linkage map of allotetraploid cotton (Gossypium hirsutum L. × Gossypium barbadense L.) with a haploid population. Theor Appl Genet. 2002;105:1166–1174. doi: 10.1007/s00122-002-1100-4. [DOI] [PubMed] [Google Scholar]
- 72.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 73.Xu Y, Wang J, Wang S, Wang J, Chen X. Characterization of GaWRKY1, a cotton transcription factor that regulates the sesquiterpene synthase gene (+)-delta-cadinene synthase-A. Plant Physiol. 2004;135:507–515. doi: 10.1104/pp.104.038612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mo H, et al. Cotton polyamine oxidase is required for spermine and camalexin signalling in the defence response to Verticillium dahlia. Plant J. 2015;83:962–975. doi: 10.1111/tpj.12941. [DOI] [PubMed] [Google Scholar]
- 75.Pritchard, J. K. & Wen, W. Documentation for STRUCTURE software. The University of Chicago Press, Chicago (2004).
- 76.Evanno G, Regaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 77.Bradbury PJ, et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 78.Wang XY, Lv K, Cai CP, Xu J, Guo WZ. Establishment and application of TRV-mediated virus-induced gene silencing in cotton. Acta Agronomica Sinica. 2014;40:1356–1363. doi: 10.3724/SP.J.1006.2014.01356. [DOI] [Google Scholar]
- 79.Cai C, et al. GhPSY, a phytoene synthase gene, is related to the red plant phenotype in upland cotton (Gossypium hirsutum L.) Mol Biol Rep. 2014;41:4941–4952. doi: 10.1007/s11033-014-3360-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.