

Abstract
Atherosclerosis is considered the major cause of heart attack, stroke and gangrene of the extremities, which is responsible for 50% of all mortality in Western countries. The pathogenesis and causes of atherosclerosis remain elusive. Recent studies highlight inflammation as a contributing factor for atherosclerosis in all stages of the disease process. In this study, we demonstrate that the treatment of human umbilical vein endothelial cells (HUVECs) with ghrelin inhibits the oxidized low-density lipoprotein (oxLDL)-induced inflammatory response, In addition, treatment with ghrelin led to the accumulation of uncoupling protein 2 (UCP2) in the cells, thus decreasing reactive oxygen species (ROS) generation. Moreover, the siRNA-mediated knockdown of UCP2 expression suggested that the inhibitory effects of ghrelin on the inflammatory response relied on its ability to induce the accumulation of cellular UCP2 levels. Further analysis indicated that the accumulation of UCP2 in the ghrelin-treated cells was due to the ability of ghrelin to inhibit the ubiquitination of UCP2 and prevent UCP2 degradation, resulting in the extended protein half-life of UCP2. On the whole, our data indicate that ghrelin inhibits the oxLDL-induced inflammatory response in HUVECs, and may thus have potential for use as an anti-atherosclerotic agent. Our data may also provide valuable insight into the pathogenesis of atherosclerosis.
Keywords: atherosclerosis, uncoupling protein 2, ghrelin, ubiquitination, inflammatory response, low-density lipoprotein
Introduction
Atherosclerosis, which is also known as arteriosclerotic vascular disease (ASVD), refers to a specific form of arteriosclerosis in which an artery wall thickens as a result of the invasion and accumulation of white blood cells (WBCs) (foam cells) and the proliferation of intimal smooth-muscle cells, creating a fibrofatty plaque (1). Atherosclerosis is considered the major cause of heart attack, stroke and gangrene of the extremities, and is responsible for 50% of all mortality across Western countries (1).
The pathogenesis and causes of atherosclerosis are highly complex and remain exclusive to date. For a long period of time, atherosclerosis was considered a metabolic disease and its development was traditionally based on thecholesterol hypothesis due to the accumulation of atherogenic lipoproteins in the blood vessel wall (2,3). Atherosclerosis is associated with other metabolic diseases, such as diabetes and dyslipoproteinemia (4,5). However, in recent years, it was discovered that inflammation may be a contributing factor for atherosclerosis and this may thus provide new insight into the mechanisms responsible for the disease (2,3).
In a previous review, it was suggested that constituents of oxidatively modified (oxidized) low-density lipoprotein (oxLDL) induce a local inflammatory response (6). Pro-inflammatory stimuli in endothelial cells (ECs) trigger the expression of adhesion molecules, such as P-selectin and vascular cell adhesion molecule-1 (VCAM-1), which results in the attachment of circulating monocytes or lymphocytes (7–9). In macrophages, the expression of scavenger receptors in response to inflammatory cytokines subsequently increases, transforming them into lipid-laden foam cells following the endocytosis of modified lipoprotein particles; macrophage-derived foam cells drive lesion progression via the continuation of the secretion of pro-inflammatory cytokines (3). There are data to suggest a central role for inflammation in both early atherogenesis and in the progression of lesions (10). Therefore, the circulating markers of inflammation are considered as an indicator of atherosclerosis (10). On the other hand, the role of inflammation also implies a potential therapeutic target for atherosclerosis.
Ghrelin is a peptide hormone produced by ghrelinergic cells in the gastrointestinal tract and acts as a neuropeptide in the central nervous system (11). However, a recent study suggested that ghrelin may be a potent anti-inflammatory mediator and a promising therapeutic agent in the treatment of inflammatory diseases or injury (12). It has been shown that low ghrelin serum levels are significantly associated with advanced carotid atherosclerosis in patients with type 2 diabetes (13). Another study also demonstrated that the administration of ghrelin attenuated inflammation, oxidative stress, and apoptosis during and after the development of non-alcoholic fatty liver disease (14). Therefore, it is interesting to note that ghrelin also plays a role in the prognosis of atherosclerosis.
In this study, we demonstrate that the treatment of human umbilical vein endothelial cells (HUVECs) with ghrelin inhibits the oxLDL-induced inflammatory response via the upregulation of uncoupling protein (UCP)2. Treatment of the HUVECs with ghrelin inhibited the ubiquitin-mediated degradation of UCP2, while its mRNA level was unaffected by ghrelin. Our data highlight the potential use of ghrelin as an anti-atherosclerotic agent, as it inhibited the oxLDL-induced inflammatory response in HUVECs. Our data may also provide further insight into the pathogenesis of atherosclerosis.
Materials and methods
Cells and chemicals
Human umbilical vein endothelial cells (HUVECs, ATCC® CRL-1730™) were purchased from ATCC (Manassas, VA, USA) and maintained in Kaighn's Modification of Ham's F-12 Medium (ATCC® 30–2004™) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA). oxLDL was purchased from Cleveland HeartLab (Cleveland, OH, USA) and used to treat the cells at a concentration of 50 µg/ml. Ghrelin was purchased from Cayman Chemical (Ann Arbor, Michigan, USA). UCP2 siRNA (h) (sc-42682) and scramble siRNA (control siRNA; sc-37007) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The transfection of the cells with siRNA was carried out using Lipofectiamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Cell treatment
For the cell treatment with different drugs, ghrelin was used to treating cells at doses of 1, 5, 10, 20, 40 and 50 nM 24 h prior oxLDL stimulation. The oxLDL was purchased from Cleveland HeartLab and used to treat the cells at a concentration of 50 µg/ml for 36 h. Mock cells were the cells without any treatment.
Reverse transcription-quantitative PCR (RT-qPCR)
The RNA isolation from the HUVECs was conducted using TRIzol reagent (Invitrogen) according to manufacturer's instructions. The synthesis of cDNA was conducted using AMV reverse transcriptase (Promega, Madison, WI, USA) according to instructions provided by the manufacturer. Quantitative PCR (qPCR) detection for the transcripts of target genes with SYBR-Green mix (Life Technologies, Carlsbad, CA, USA) was carried out as previously described (15,16). The conditions for qPCR cycle were as follows: 98°C for 5 min for denaturation, 98°C for 30 sec, 72°C for 1 min, then repeated for 40 cycles. Transcripts of ribosomal protein L32 (RPL32) were also amplified from the same sample to serve as an internal control for normalization purposes. Relative gene expression was quantified by the 2−ΔΔCT method as previously described (17). The primers for qPCR detection are listed in Table I.
Table I.
Primers used in this study.
| Primer name | Primer sequence (5′→3′) |
|---|---|
| IL-6 | F: GACAACTTTGGCATTGTGG |
| R: ATGCAGGGATGATGTTCTG | |
| CCL-2 | F: CCCCAGTCACCTGCTGTTAT |
| R AGATCTCCTTGGCCACAATG | |
| ICAM-1 | F: GGCCTCAGTCAGTGTGA |
| R AACCCCATTCAGCGTCA | |
| VCAM-1 | F: TACTCCCGTCATTGAGGATATTGG |
| R CTCCTTCACACACATAGACTCC | |
| RPL32 | F: CAACTGGCCATCAGAGTCAC |
| R GTGCACATGAGCTGCCTACT |
IL-6, interleukin-6; CCL-2, C-C motif chemokine ligand 2; VCAM-1, vascular cell adhesion molecule-1; RPL32, ribosomal protein L32; F, forward; R, reverse.
Determination of reactive oxygen species (ROS) generation
The dihydroethidium (DHE) fluorescence-based assay was used to determine ROS generation and was conducted as previously described (18). Briefly, cells with the indicated treatments were first washed twice with PBS, 0.5 µM DHE was then added to the cells and the cells were incubated under 37°C for 20 min. After the incubation, cells were washed again twice with PBS to remove the free DHE probe. DHE-derived fluorescence at 570 nm which was generated by each group was recorded by the VICTOR™ X5 Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA).
Western blot analysis
The cells subjected to the indicated treatments were lysed by the Laemmli sample buffer as previously described (16,19). The protein of lysate was then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis was performed as previously described (19). Briefly, following SDS-PAGE, the separated proteins were transferred onto PVDF membranes and probed with rabbit anti-UCP2 antibody and mouse anti-ubiquitin (sc-166553) (both from Santa Cruz Biotechnology, Inc.). Specific reactions were detected using goat anti-rabbit IgG or goat anti-mouse IgG conjugated with horseradish peroxidase (Sigma, St. Louis, MO, USA) and revealed by a chemiluminescence substrate (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were also blotted with tubulin antibody (sc-33749; Santa Cruz Biotechnology, Inc.) to normalize protein loading. The chemiluminescence signal was recorded using the ChemiDoc MP imaging system (Bio-Rad Laboratories). The luminescence signal was captured and analyzed using the Image Lab Program (version 6.1).
Protein half-life assay
Cycloheximide is a translation inhibition drug which is generally used to determine protein half-life by monitoring the protein degradation speed. Briefly, 100 µg cycloheximide (Sigma) was added to cells seeding in 12-well plates for the indicated times (12, 24 and 36 h). The cells were then harvested for SDS-PAGE and western blot analysis was performed to determine the level of UCP2.
Protein ubiquitination assay
To examine the ubiquitination status of UPC2, the immunoprecipitation (IP) for UCP2 was conducted first as previously described with modifications (20). Briefly, the HUVECs subjected to the indicated treatment were lysed with lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) followed by the addition of ubiquitin aldehyde (Boston Biochem, Inc., Cambridge, MA, USA), a specific inhibitor of ubiquitin C-terminal hydrolases, at a final concentration of 2.53 µM. The lysate was clarified by centrifugation at 14,000 × g for 5 min at 4°C. Following centrifugation, the cell lysate was incubated with UCP2 antibody (Santa Cruz Biotechnology, Inc.) followed by incubation with protein G-agarose (KPL, Inc., Gaithersburg, MD, USA). To detect the ubiquitination UCP2, The IP samples containing UCP2 were subjected to western blot analysis with ubiquitin antibody (Santa Cruz Biotechnology, Inc.).
Statistical analysis
The experimental results were plotted and analyzed for statistical significance using the Excel program (Microsoft, Seattle, WA, USA). Data are represented as the means ± SD. Differences in indicators between treatment samples, such as the cellular RNA level between the groups in the presence or absence of oxLDL or ghrelin, were assessed using the Student's t-test. A two-tailed P-value of <0.05 was considered to indicate a statistically significant difference.
Results
Ghrelin inhibits the oxLDL-induced inflammation response in HUVECs
Recent studies have suggested that ghrelin may be a potent anti-inflammatory mediator and that the administration of ghrelin attenuates inflammation, oxidative stress and apoptosis during and after the development of non-alcoholic fatty liver disease (12,14). Combined with previous observations that the ghrelin serum levels are significantly associated with advanced carotid atherosclerosis in patients with type 2 diabetes (13), we wished to determine whether ghrelin inhibits the oxLDL-induced inflammatory response in HUVECs. In order to examine our hypothesis, in the present study, the HUVECs were treated with 50 nM ghrelin for 24 h, and were then stimulated by oxLDL to induce the inflammatory response. Pro-inflammatory cytokines, chemokines and related molecule expression such as interleukin-6 (IL-6), C-C motif chemokine ligand 2 (CCL-2), intercellular adhesion molecule-1 (ICAM-1) and VCAM-1 were examined by RT-qPCR. Based on our results, stimulation with oxLDL alone led to the significant upregulation in the expression levels of these molecules (Fig. 1A). However, in the cells pre-treated with ghrelin, the expression of these cytokines and related molecules was decreased (Fig. 1A), which suggested an inhibitory effect of ghrelin on the oxLDL-induced inflammatory response. Moreover, to confirm our data, treatment of the HUVECs with a gradient dose of ghrelin was also conducted. As shown in Fig. 1B, the increase in the expression levels of inflammatory cytokines and related molecules induced by oxLDL was suppressed as the concentration of ghrelin increased, which was consistent with our above-mentioned observation.
Figure 1.

Ghrelin inhibits the oxidized low-density lipoprotein (oxLDL)-induced inflammatory response in human umbilical vein endothelial cells (HUVECs). (A) HUVECs were either pre-treated with ghrelin or left untreated, followed by oxLDL stimulation. RT-qPCR was conducted to determine the mRNA levels of IL-6, CCL-2, ICAM-1 and VCAM-1. (B) HUVECs were pre-treated with gradiatint concentrations of ghrelin followed by oxLDL stimulation. RT-qPCR was conducted to monitor the mRNA levels of IL-6, CCL-2, ICAM-1 and VCAM-1. Quantitative experiments were repeated at least 3 times. *P<0.05, significant differences between different groups.
Ghrelin upregulates UCP2 to suppress ROS generation
Studies have demonstrated that the oxLDL-induced inflammatory response always correlates with the generation of ROS (21,22). Therefore, in this study, we examined whether the oxLDL-induced generation of ROS can be inhibited by ghrelin. By utilizing DHE fluorescence-based ROS assay, our data demonstrated that in the HUVECs pre-treated with ghrelin, oxLDL-induced ROS generation was inhibited (Fig. 2A), which fulfilled our expectations. As a natural byproduct of the normal oxygen metabolism in physiological conditions, ROS are involved in many cell signaling processes, such as cell proliferation, inflammation, apoptosis and phagocytosis (23). However, the cellular ROS level can be markedly increased during cell responses to stress and increases in ROS levels cause oxidative stress, which results in damage to the cells (24).
Figure 2.

Ghrelin upregulates uncoupling protein 2 (UCP2) to decrease reactive oxygen species (ROS) generation. (A) Human umbilical vein endothelial cells (HUVECs) were either pre-treated with ghrelin or left untreated, followed by oxidized low-density lipoprotein (oxLDL) stimulation. The cellular ROS level was measured in indicated groups. (B) Western blot analysis for the UCP2 protein level in HUVECs treated with ghrelin. Untreated cells was included as a control. (C) Gradient treatment of HUVECs with ghrelin for the observed the dose-depended upregulation of UCP2 at the protein level. Quantitative experiments were repeated at least 3 times. *P<0.05, significant differences between different groups.
On the other hand, the UCP2 family is involved in the downregulation of the oxidative phosphorylation by increasing membrane proton conductance (25). Therefore, the function of UCPs is considered a defense mechanism with which to attenuate ROS-induced damage. Among the members of the UCP family, UCP2 can be identified in a variety of tissues (26), while UCP1 and UCP3 have a more tissue-specific expression in brown adipose tissue and skeletal muscle, respectively (26–28). Thus, we then examined whether UCP2 expression is upregulated in ghrelin-treated cells, leading to a reduced of oxLDL induced ROS level in HUVECs. Our results revealed that the UCP2 protein level was significantly upregulated in the ghrelin-treated cells (Fig. 2B). Moreover, a concentration-dependent upregulation of UCP2 expression by ghrelin treatment was also observed, which suggested that the inhibitory effects of ghrelin on ROS generation were due to the upregulation of UCP2.
siRNA-mediated knockdown of UCP2 expression antagonizes the inhibitory effects of ghrelin on ROS generation
To further understand the role that UCP2 plays during the ghrelin-mediated inhibition of ROS generation and the inflammatory response, siRNA transfection was employed for the knockdown of UCP2 expression in HUVECs. The knockdown efficiencty of the UCP2 siRNA was examined by western blot analysis (Fig. 3A). With the knockdown of UCP2 expression, the inhibitory effects of ghrelin on ROS generation were markedly suppressed, and the ROS level induced by oxLDL in the cells treated with ghrelin and transfected with UCP siRNA was similar to the level in the cells treated with oxLDL alone (Fig. 3B). Moreover, using ICAM-1 as an indicator of the oxLDL-induced inflammatory response, the knockdown of UCP2 also antagonized the ghrelin-mediated inhibition of ICAM-1 expression. Taken together, these data suggest that ghrelin upregulates UCP2, thus inhibiting the oxLDL-induced inflammatory response.
Figure 3.

siRNA-mediated knockdown of uncoupling protein 2 (UCP2) expression antagonizing ghrelin caused reactive oxygen species (ROS) reduction. (A) Western blot analysis for UCP2 protein level in human umbilical vein endothelial cells (HUVECs) subjected to the indicated treatments. Cells were transfected with siRNA first, and then ghrelin was added to the siRNA-transfected cells. Cells from indicated groups were harvested 24 h later and examined to determine the UCP2 protein level. (B) Quantitative analysis of the ROS level in the indicated groups. (C) The ICAM-1 mRNA level in cells from indicated groups. Experiments were repeated 3 times. *P<0.05, significant differences between different groups.
Ghrelin extends the protein half-life of UCP2 without affecting its mRNA level
Since our data suggested that UCP2 was be upregulated by ghrelin treatment, we then examined whether the accumulation of UCP2 was the result of the upregulation of its mRNA expression. However, to our surprise, with 24 h of treatment of the HUVECs with ghrelin, the UCP2 mRNA level between the treatment groups and the control cells was similar (Fig. 4A), which suggested that ghrelin treatment did not cause the upregulation of UCP2 gene expression and implied that UCP2 functions at the post-translational level of UCP2 mRNA. To further confirm our findings, we examined whether the protein half-life of UCP2 was extended by ghrelin. A protein translation inhibitor, cycloheximide was added to the HUVECs to inhibit mRNA translation as previously described (20). The cells were then harvested at different time points to examine the protein level of UCP2. As shown in Fig. 4B, without ghrelin treatment, the UCP2 protein level began to decrease at 24 h after cycloheximide treatment. However, in the ghrelin-treated cells, the protein level of UCP2 was not significantly altered until 36 h after cycloheximide treatment. Taken together, these data demonstrated that ghrelin treatment extended the protein half-life of UCP2 rather than promoting the transcription of UCP2 mRNA.
Figure 4.

Ghrelin extends the protein half-life of uncoupling protein 2 (UCP2) without affecting the UCP2 mRNA level. (A) Treatment with ghrelin did not alter the mRNA level in human umbilical vein endothelial cells (HUVECs). (B) UCP2 half-life assay. Cells were treated with ghrelin for 24 h first, then cycloheximide was added to the cells to block mRNA translation. Both the ghrelin-treated or untreated cells were harvested at different time points to monitor the changes in the UCP2 level for the determination of the UCP2 half-life. Quantitative experiments were repeated 3 times. *P<0.05, significant differences between different groups.
Ghrelin prevents UCP2 degradation by inhibiting UCP2 ubiquitination
Protein ubiquitination is considered as the major pathway for cellular protein degradation (29). Although UCP2 is located in the inner membrane of the mitochondria, it has been demonstrated that mitochondrial proteins, including UCP2 can be degraded by the ubiquitin-proteasome system as well (30). As our data demonstrated that ghrelin extended the protein half-life of UCP2, we further examined the ubiquitination status of UCP2 to explain the reason leading to the accumulation of UCP2 in ghrelin-treated cells. As shown in Fig. 5A, treatment of the HUVECs with ghrelin significantly inhibited the ubiquitination level of UCP2 when compared with the controls. Moreover, the ubiquitination level of the whole cell lysate demonstrated that ghrelin did not alter the ubiquitination status of the whole cell, which suggested that the decreased ubiquitination level of UCP2 appeared to be specific (Fig. 5B). These observations suggested that ghrelin specifically extended the UCP2 half-life by inhibiting UCP2 ubiquitination, which leads to UCP2 degradation. On the whole, our data demonstrated that ghrelin prevented UCP2 degradation, thus reducing the cellular ROS level induced by oxLDL, and inhibiting the oxLDL-induced inflammatory response in HUVECs.
Figure 5.
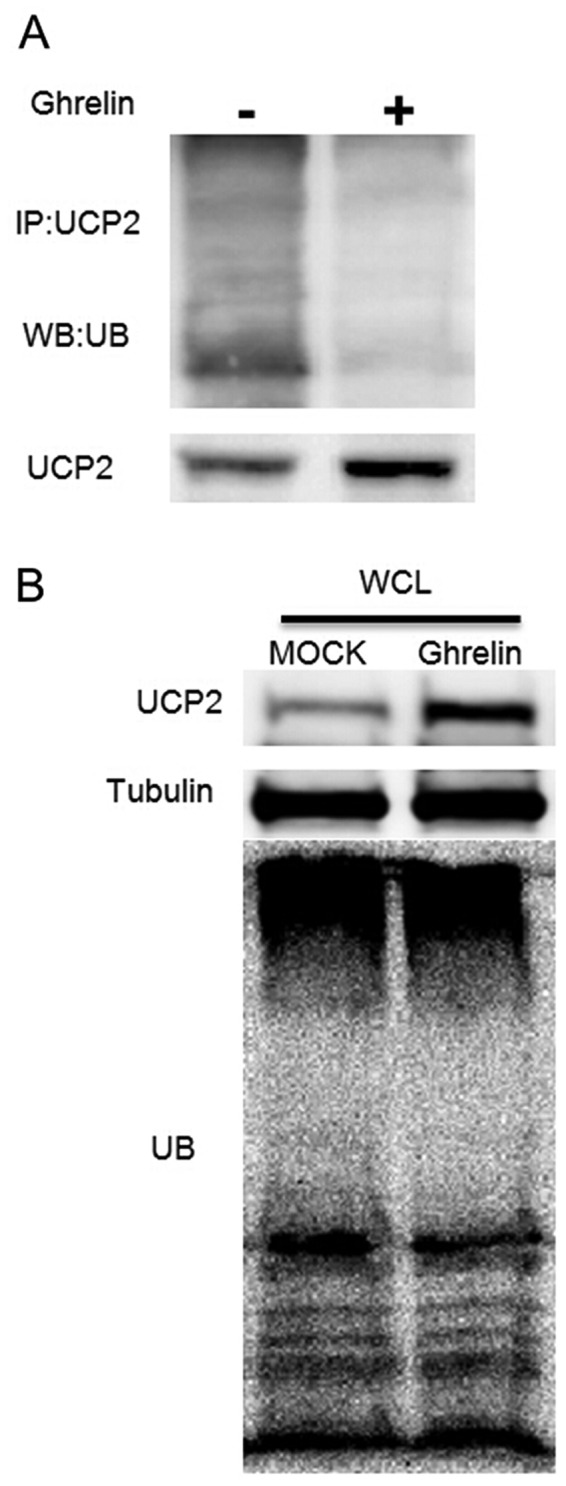
Ghrelin prevents uncoupling protein 2 (UCP2) degradation by inhibiting UCP2 ubiquitination. (A) Ubiquitination assay of UCP2: human umbilical vein endothelial cells (HUVECs) treated with ghrelin for 24 h, and cells were then lysed for immunoprecipitation (IP) by anti-UCP2 antibody. Anti-ubiquitin antibody was used for probing the ubiquitination of UCP2. (B) Western blot analysis of the total ubiqutination level in HUVECs; ghrelin untreated cells were used as controls.
Discussion
In the present study, we demonstrated that stimulation of HUVECs with oxLDL induced the expression of pro-inflammatory cytokines and adhesion molecules, and increased ROS generation. However, pro-treatment of the HUVECs with ghrelin prevented the oxLDL-induced inflammatory response via the upregulation of UCP2. Further analysis demonstrated that the ubiquitination of UCP2 was inhibited by ghrelin, therefore preventing the degradation of UCP2, while the mRNA level of UCP2 was not altered.
The UCP family is a sub-category of the mitochondrial anion-carrier proteins superfamily, which can be both found in animal and plant species (27). The genome of the mammalian species encodes 5 UCP homologues (UCP1 to 5), with UCP1 to 3 demonstrating highest sequence similarity (27,31,32). Generally, UCPs locate in the inner membrane of the mitochondria to regulate the thermogenic proton leak (27). Compared with UCP1 and UCP3, which have a more tissue-specific expression in brown adipose tissue and skeletal muscle (26–28), the expression of UCP2 is more universal (26). The biochemical function of UCPs is to downregulate oxidative phosphorylation by increasing membrane proton conductance (25). The uncoupling function increases respiration and leads in the decreased generation of superoxide (25,33).
To date, while UCP1 is still a key regulator of adaptive thermogenesis, UCP2 and UCP3 also have important functions, decreasing ROS which are produced by electron transport (34,35). With the function of regulating ROS, accumulating evidence suggests that UCP2 is involved in many diseases, such as neurodegenerative diseases, cardiovascular diseases, type 2 diabetes and cancer (36). A previous study demonstrated that the rapid degradation of UCP2 was mediated by the ubiquitin-proteasome system (30). However, as a protein located in the inner membrane of the mitochondria and no mitochondrial protein export machinery has been identified (30), the mechanisms through which UCP2 can be transported to the cytoplasm for ubiquitin-mediated degradation are still unclear, since the ubiquitin-proteasome system is a cytosolic degradation machinery. Moreover, the E3-ligease of ubiquitin mediated degradation for UCP2 is still unclear. Therefore, the mechanisms through which ghrelin prevents UCP2 from ligating with poly-ubiquitin chains warrant further investigation. There may be other cellular proteins involved in this process as well.
In conclusion, our data suggest a novel role of ghrelin in preventing the oxLDL-induced inflammatory response in HUVECs. Our findings may aid in the understanding of the pathogenesis of atherosclerosis.
References
- 1.Ross R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Ruiz F, Becker MA. Inflammation: A possible mechanism for a causative role of hyperuricemia/gout in cardiovascular disease. Curr Med Res Opin. 2015;31(Suppl 2):9–14. doi: 10.1185/03007995.2015.1087980. [DOI] [PubMed] [Google Scholar]
- 3.Packard RR, Libby P. Inflammation in atherosclerosis: From vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008;54:24–38. doi: 10.1373/clinchem.2007.097360. [DOI] [PubMed] [Google Scholar]
- 4.Funk SD, Yurdagul A, Jr, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: Lessons from type 1 diabetes. Int J Vasc Med. 2012;2012:569654. doi: 10.1155/2012/569654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schanberg LE, Sandborg C. Dyslipoproteinemia and premature atherosclerosis in pediatric systemic lupus erythe-matosus. Curr Rheumatol Rep. 2004;6:425–433. doi: 10.1007/s11926-004-0021-4. [DOI] [PubMed] [Google Scholar]
- 6.Miller YI, Chang MK, Binder CJ, Shaw PX, Witztum JL. Oxidized low density lipoprotein and innate immune receptors. Curr Opin Lipidol. 2003;14:437–445. doi: 10.1097/00041433-200310000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Cybulsky MI, Gimbrone MA., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb. 1993;13:197–204. doi: 10.1161/01.ATV.13.2.197. [DOI] [PubMed] [Google Scholar]
- 9.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farmer JA, Torre-Amione G. Atherosclerosis and inflammation. Curr Atheroscler Re. 2002;4:92–98. doi: 10.1007/s11883-002-0031-5. [DOI] [PubMed] [Google Scholar]
- 11.Dickson SL, Egecioglu E, Landgren S, Skibicka KP, Engel JA, Jerlhag E. The role of the central ghrelin system in reward from food and chemical drugs. Mol Cell Endocrinol. 2011;340:80–87. doi: 10.1016/j.mce.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 12.Baatar D, Patel K, Taub DD. The effects of ghrelin on inflammation and the immune system. Mol Cell Endocrinol. 2011;340:44–58. doi: 10.1016/j.mce.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 13.Kadoglou NP, Sailer N, Moumtzouoglou A, Kapelouzou A, Tsanikidis H, Vitta I, Karkos C, Karayannacos PE, Gerasimidis T, Liapis CD. Visfatin (nampt) and ghrelin as novel markers of carotid atherosclerosis in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2010;118:75–80. doi: 10.1055/s-0029-1237360. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Hai J, Li L, Chen X, Peng H, Cao M, Zhang Q. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine. 2013;43:376–386. doi: 10.1007/s12020-012-9761-5. [DOI] [PubMed] [Google Scholar]
- 15.Patel D, Opriessnig T, Stein DA, Halbur PG, Meng XJ, Iversen PL, Zhang YJ. Peptide-conjugated morpholino oligomers inhibit porcine reproductive and respiratory syndrome virus replication. Antiviral Res. 2008;77:95–107. doi: 10.1016/j.antiviral.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol. 2010;84:11045–11055. doi: 10.1128/JVI.00655-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Peshavariya HM, Dusting GJ, Selemidis S. Analysis of dihydroethidium fluorescence for the detection of intracellular and extracellular superoxide produced by NADPH oxidase. Free Radic Res. 2007;41:699–712. doi: 10.1080/10715760701297354. [DOI] [PubMed] [Google Scholar]
- 19.Nan Y, Wang R, Shen M, Faaberg KS, Samal SK, Zhang YJ. Induction of type I interferons by a novel porcine reproductive and respiratory syndrome virus isolate. Virology. 2012;432:261–270. doi: 10.1016/j.virol.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nan Y, Ma Z, Wang R, Yu Y, Kannan H, Fredericksen B, Zhang YJ. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J Virol. 2014;88:8696–8705. doi: 10.1128/JVI.01228-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Yin Y, Zhou Z, He M, Dai Y. OxLDL-induced IL-1 beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflam-masome activation. Inflamm Res. 2014;63:33–43. doi: 10.1007/s00011-013-0667-3. [DOI] [PubMed] [Google Scholar]
- 22.Magwenzi S, Woodward C, Wraith KS, Aburima A, Raslan Z, Jones H, McNeil C, Wheatcroft S, Yuldasheva N, Febbriao M, et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood. 2015;125:2693–2703. doi: 10.1182/blood-2014-05-574491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salganik RI. The benefits and hazards of antioxidants: controlling apoptosis and other protective mechanisms in cancer patients and the human population. J Am Coll Nutr. 2001;20(Suppl 5):464S–475S. doi: 10.1080/07315724.2001.10719185. [DOI] [PubMed] [Google Scholar]
- 24.Lobo V, Patil A, Phatak A, Chandra N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn Rev. 2010;4:118–126. doi: 10.4103/0973-7847.70902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Echtay KS. Mitochondrial uncoupling proteins - what is their physiological role? Free Radic Biol Med. 2007;43:1351–1371. doi: 10.1016/j.freeradbiomed.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Donadelli M, Dando I, Fiorini C, Palmieri M. UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol Life Sci. 2014;71:1171–1190. doi: 10.1007/s00018-013-1407-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krauss S, Zhang CY, Lowell BB. The mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol. 2005;6:248–261. doi: 10.1038/nrm1592. [DOI] [PubMed] [Google Scholar]
- 28.Lin CS, Klingenberg M. Characteristics of the isolated purine nucleotide binding protein from brown fat mitochondria. Biochemistry. 1982;21:2950–2956. doi: 10.1021/bi00541a023. [DOI] [PubMed] [Google Scholar]
- 29.Xu L, Qu Z. Roles of protein ubiquitination and degradation kinetics in biological oscillations. PLoS One. 2012;7:e34616. doi: 10.1371/journal.pone.0034616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azzu V, Brand MD. Degradation of an intramitochondrial protein by the cytosolic proteasome. J Cell Sci. 2010;123:578–585. doi: 10.1242/jcs.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, Bouillaud F, Seldin MF, Surwit RS, Ricquier D, Warden CH. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat Genet. 1997;15:269–272. doi: 10.1038/ng0397-269. [DOI] [PubMed] [Google Scholar]
- 32.Boss O, Samec S, Paoloni-Giacobino A, Rossier C, Dulloo A, Seydoux J, Muzzin P, Giacobino JP. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997;408:39–42. doi: 10.1016/S0014-5793(97)00384-0. [DOI] [PubMed] [Google Scholar]
- 33.Azzu V, Brand MD. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci. 2010;35:298–307. doi: 10.1016/j.tibs.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richard D, Picard F. Brown fat biology and thermogenesis. Front Biosci (Landmark Ed) 2011;16:1233–1260. doi: 10.2741/3786. [DOI] [PubMed] [Google Scholar]
- 35.Garlid KD, Jabůrek M, Jezek P, Varecha M. How do uncoupling proteins uncouple? Biochim Biophys Acta. 2000;1459:383–389. doi: 10.1016/S0005-2728(00)00175-4. [DOI] [PubMed] [Google Scholar]
- 36.Mattiasson G, Sullivan PG. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid Redox Signal. 2006;8:1–38. doi: 10.1089/ars.2006.8.1. [DOI] [PubMed] [Google Scholar]