

Abstract
Idiopathic pneumonia syndrome (IPS) is a non-infectious, inflammatory disorder of the lungs that occurs most often after fully myeloablative allogeneic hematopoietic stem cell transplantation (HSCT). IPS can be severe and is associated with high one year mortality rates despite existing therapies. The canonical NF-κB signaling pathway has previously been linked to several inflammatory disorders of the lung including asthma and lung allograft rejection. It has never been specifically targeted as a novel IPS treatment approach, however. Here we report that the IκB kinase 2 (IKK2) antagonist BAY 65-5811 or “Compound A”, a highly potent and specific inhibitor of the NF-κB pathway, was able to improve median survival times and recipient oxygenation in a well described mouse model of IPS. Compound A impaired the production of the pro-inflammatory chemokines CCL2 and CCL5 within the host lung after transplant. This resulted in significantly lower numbers of donor lung infiltrating CD4+ and CD8+ T cells, and reduced pulmonary inflammatory cytokine production after allograft. Compound A’s beneficial effects appeared to be specific for limiting pulmonary injury, as the drug was unable to improve outcomes in a B6 into B6D2 haplotype matched murine HSCT model in which recipient mice succumb to lethal acute graft-versus-host disease (GVHD) of the gastrointestinal tract. Collectively, our data suggest that the targeting of the canonical NF-κB pathway with a small molecule IKK2 antagonist may represent an effective and novel therapy for the specific management of acute lung injury that can occur after allogeneic HSCT.
Keywords: Idiopathic Pneumonia Syndrome, Graft vs. Host Disease, NF-kB pathway, Chemokines, Lymphocyte Trafficking
Introduction
Idiopathic pneumonia syndrome (IPS) refers to generalized, non-infectious, inflammatory lung injury occurring after hematopoietic stem cell transplantation (HSCT). The incidence of IPS has been estimated to range between 3 to 15%, is more common following myeloablative allogeneic transplants than reduced intensity or autologous HSCTs, and has been associated with the use of total body irradiation (TBI) and cyclophosphamide based conditioning regimens and concurrent acute GVHD1. IPS after allogeneic transplantation is often resistant to therapy with mortality rates of 60–80% being reported in some series, and new treatment approaches are needed.
The canonical NF-κB pathway is an important signaling cascade that is involved in multiple inflammatory pathways, and has been implicated in the pathogenesis of several pulmonary disorders including asthma and lung allograft rejection2,3. NF-κB itself is a dimer composed most commonly of a p50 and a RelA subunit. P50/RelA NF-κB is present in all cell types and is ordinarily sequestered in the cytoplasm in an inactive state by members of the IκB family of proteins4,5. Multiple proinflammatory signals including Tumor Necrosis Factor (TNF), CD40L, CD3/CD28, Lipopolysaccharide (LPS), and Interleukin-1 are able to induce the phosphorylation of IκB by activating IκB kinase (IKK), a heterotrimer composed of IKK1 and IKK2 catalytic subunits and a regulatory NEMO subunit. When this occurs, phosphorylated IκB is marked for ubiquination and ultimately degraded by the proteasome. This in turn liberates NF-κB and allows for its translocation to the nucleus where it induces the transcription of numerous inflammatory mediators6–9.
While specific inhibition of the canonical NF-κB signaling pathway has not been previously explored as a treatment for IPS, there has been considerable interest in antagonizing NF-κB as a therapy for graft-versus-host disease (GVHD) Bortezomib is a reversible antagonist of the proteasome with inhibitory properties against NF-κB and has been evaluated for the management of GVHD in both preclinical models and human clinical trials10–15. In mouse studies, bortezomib was found to be effective in preventing lethal acute GVHD. Its therapeutic index, however, was small. Repeated bortezomib administration during the early transplant period following an irradiation based conditioning regimen accelerated acute GVHD and exacerbated gastrointestinal pathology10,11. Nevertheless, several therapy trials in humans have suggested a benefit for bortezomib in the management of chronic GVHD12,14.
While active against NF-κB, bortezomib demonstrates numerous additional effects via its action on the proteasome. As a result, there has been interest in examining other compounds which more specifically block the canonical NF-κB pathway. In work by Vodanovic-Jankovic and colleagues, the authors evaluated the use of PS-1145, a direct inhibitor of IKK2, as a means for preventing acute GVHD in a mouse model system. There, PS-1145 appeared to be well tolerated and repeated doses given over the first week post-transplant did not reproduce the gastrointestinal pathology seen with bortezomib11.
Subsequent to this work a structurally distinct inhibitor of IKK2 termed BAY 65-5811 or “Compound A” became available which had previously demonstrated both anti-inflammatory and anti-tumor activity in vivo16,17. Compound A is considerably more potent than PS-1145 against IKK2 with an IC50 of only 4nM, and exhibits minimal off-target effects against a variety of intracellular protein kinases and phosphatases17. As a result, we set out to determine if this structure could improve upon the results previously obtained with bortezomib and PS-1145. Here, we report that Compound A was unable to prevent acute GVHD lethality in a B6 into B6D2 model where recipient death is primarily driven by gastrointestinal injury18. Subsequent in vivo trafficking studies, however, indicated that Compound A was very effective at attenuating donor T cell accumulation within pulmonary tissues. As a result, we examined the effects of compound A in a well described mouse model of IPS and found that the drug significantly improved median survival times and reduced hypoxemia. To our knowledge our data are the first to demonstrate that an IKK2 antagonist can improve IPS outcomes in a preclinical model, and suggest that the specific targeting of the canonical NFκB pathway might represent a new therapeutic approach for the management of acute lung injury after HSCT.
Methods
Mice
C57BL/6 (“B6”), B6xDBA/2 F1 (“B6D2”), and B10.BR mice were purchased from The Jackson Laboratory. Enhanced green fluorescent protein (eGFP) expressing B6 mice were generated as previously prescribed19. All experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at The University of North Carolina.
Compound A
Compound A (BAY 65-5811, Bayer Pharmaceuticals) was supplied by the laboratory of Dr. Albert Baldwin. For all in vitro and in vivo work, Compound A was suspended in 100% dimethylsulfoxide.
Mixed Lymphocyte Reaction (MLR)
MLRs using [3H] thymidine were performed as described previously20.
Transplantation Systems
Donor T cell depleted (TCD) bone marrow (BM) cells and whole CD25-depleted splenic conventional T cells (Tcons) were prepared as previously described21,22. Natural killer cell depleted splenocytes were prepared by subjecting freshly isolated murine splenocytes to a negative selection process using anti-NK1.1 antibodies coupled to ferromagnetic beads and a magnetic activated cell sorter (MACS) column (Miltenyi Biotec).
Stereomicroscopy
Recipient mice were imaged with a Zeiss Stereolumar V12 microscope with eGFP band-pass filter as room temperature. eGFP intensities were determined with Axiovision (Carl Zeiss) software. Specific exposure times and magnifications were as follows:
-
Day +7 eGFP+ Donor Tcon Imaging, Figure 3:
Colon, exp=5s, mag=12X; Liver, exp=6s, mag=40X; Lung, exp=10s, mag=17x
Organ eGFP quantification
Recipient organs were homogenized and absolute eGFP levels determined with an enzyme linked immunosorbent assay (ELISA) kit as has been described previously (Cell Biolabs)20,21.
Immunohistochemistry
Recipient lungs were perfused through with saline and then fixed in formalin. Paraffin imbedded tissue sections were then stained with a mouse monoclonal anti-phospho-IkBa-Ser32/36 antibody (Cell Signaling, Danvers MA) diluted 1:100 using a Mouse on Mouse Peroxidase Kit16.
Isolation of Leukocytes from Tissues
Mice were killed and perfused through the heart with saline. Organs were then digested with a collagenase solution and donor eGFP+ leukocytes isolated by centrifugation through a Percoll gradient as has been described previously22. Leukocytes were then stained and quantified using flow cytometry.
Organ Chemokine/Cytokine Analysis
Recipient mice were perfused through with PBS and their organs then removed and homogenized. Total cytokine levels were then determined by ELISA (eBioscience).
qRT-PCR
CD4+CD25− B6 Tcons were isolated using column purification. The cells were then activated in vitro with plate-bound anti-CD3 and anti-CD28 antibody in the presence of supplemental murine interleukin-2 (IL-2) at 100IU/ml. After 48 hours their total RNA was isolated for quantitative reverse transcription polymerase chain reaction (qRT-PCR) as previously described21.
Mouse Pulse Oximetry
Blood arterial oxygen saturation was tested in each mouse using a MouseOx Plus pulse oximeter (Starr Life Sciences; Oakmont, PA). Mice were anesthetized using isoflurane during the testing. A thigh sensor recorded data for a minimum of 22 seconds, and an average oxygen saturation level for that recorded time was calculated as a single data point in each mouse.
Antibodies for Cell Purifications and Flow Cytometry
The following antibodies were purchased from eBioscience: anti-CD4 (RM 4.5), CD8 (53-6-7), B220 (RA3-6B2), CD25 (PC61), NK1,1 (PK136), CD19 (1D3), CD11b (M1/70), Ly6C (HK1.4), Annexin V (VAA-33)
Statistical analysis
Survival curves were constructed using the method of Kaplan and Meier and median survival times compared using the log-rank test. Continuous variables were compared using the 2-tailed Student t test. P values less than 0.05 were considered significant. Unless otherwise indicated error bars represent SEM.
Results
Compound A inhibits T cell proliferation in a mixed lymphocyte reaction and NF-κB dependent TNF production in vivo
We began by examining the ability of Compound A to block T cell expansion in vitro. For this work we utilized a MLR with B6 Tcons as responder cells and irradiated haplotype matched B6D2 splenocytes as stimulator cells. As depicted in Figure 1A, Compound A was able to inhibit T cell activation and proliferation in a dose dependent fashion. In order to confirm that our IKK2 inhibitor was not causing direct T cell cytotoxicity, purified B6 Tcons were cultured for 48 hours in the presence of IL-2 with and without Compound A, and the cells were then evaluated for apoptotic markers. As shown in Figure 1B, the percentages of early apoptotic cells (Annexin V+ Propidium Iodide−) and late apoptotic cells (Annexin V+ Propidium Iodide+) cells were nearly identical in the two experimental groups. Finally, we evaluated the ability of Compound A to impair TNF production in mice in response to lipopolysaccharide (LPS) challenge. This is a well described method for evaluating the in vivo activity of IKK inhibitors because LPS induces TNF production through the NF-κB pathway17,23. As depicted in Figure 1C, serum TNF levels were greatly elevated in mice inoculated with LPS versus saline controls, and TNF production was blocked by Compound A in a dose dependent fashion. Collectively, these data confirmed the anti-inflammatory effects of Compound A that had been reported previously by other research groups17.
Figure 1. Compound A impairs Tcon proliferation in a mixed lymphocyte reaction and inhibits TNF production in vivo in response to LPS challenge.
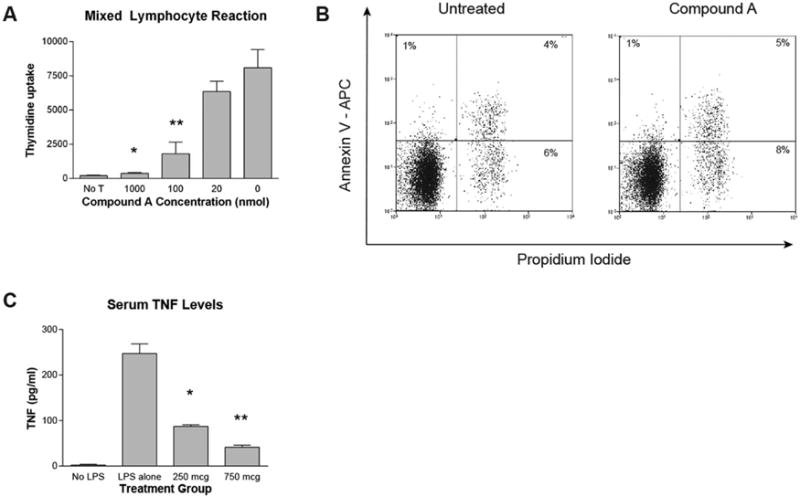
(A) 5×104 CD4+CD25− B6 responder cells were incubated with 5×104 irradiated TCD B6D2 stimulator splenocytes. During the last 16–20 hours of incubation 0.037 MBq (1 μCi) of [3H]thymidine was added to each well, and [3H]thymidine incorporation measured by scintillation counting. *P=0.0044 for comparison between 1000nM and 0 Compound A groups by Student’s t test. **P=0.0162 for comparison between 100nM and 0 Compound A groups. (B) 1×106 purified B6 CD4+CD25− Tcons were cultured for 48 hours in T cell media supplemented with murine IL-2 at 100 IUs/ml in the presence or absence of Compound A at 100nM. The cells were then collected and stained for flow cytometry with anti-Annexin V antibody and Propidium Iodide. Cell cultures were done in triplicate and representative flow cytometry plots for each culture condition are depicted. (C) Mice were administered saline or varying amounts of Compound A by IP injection. One hour later mice were given saline (No LPS group) or 200mcg of LPS by IP injection. Two hours later, the mice were killed and blood was collected by cardiac puncture. Serum TNF levels were then measured by ELISA. *P=0.0017 for comparison between 250mcg Compound A and LPS alone groups by Student’s t test. **P=0.0007 for comparison between 750mcg Compound A and LPS alone groups.
Compound A exacerbates GVHD lethality at high doses and is ineffective at lower doses
Next we examined the potential for Compound A to prevent GVHD lethality in vivo. For these experiments, we utilized a well described B6 into B6D2 acute GVHD model system where recipient lethality primarily results from gastrointestinal inflammation18. Notably, this is the same strain combination used in the MLR work described above. Based on the data depicted in Figure 1C, Compound A was dosed at 750mcg daily according to the administration schedules depicted in Figure 2A. As shown in Figure 2B, Compound A appeared to accelerate recipient mortality at this dose, with the greatest toxicity seen in the group receiving the highest dose intensity (treatment group 1). These data appeared to approximate those findings seen previously when bortezomib was administered over the first week following lethal irradiation11, and led us to hypothesize that Compound A was potentially exacerbating recipient gut injury by impairing post-irradiation epithelial repair.
Figure 2. Compound A accelerates recipient mortality at high doses and is ineffective in preventing acute GVHD at lower doses in a B6 into B6D2 model.
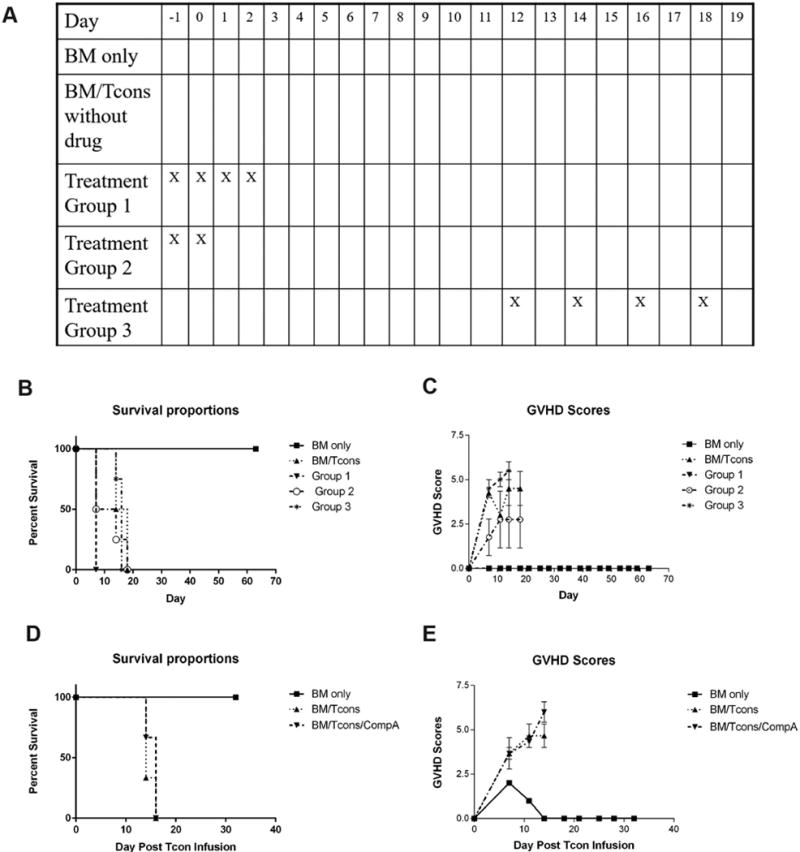
(A–C) B6D2 recipients were lethally irradiated to 950 rads on transplant day −1 and then administered 3×106 TCD BM cells +/− 4×106 CD25-depleted whole (CD4+ and CD8+) splenic Tcons from B6 donor mice on transplant day 0 by tail vein injection. Some recipient mice were also administered 750mcg of Compound A by IP injection according to the schema depicted (A). Recipient mice were followed for survival (B) and scored for GVHD twice weekly using a validated clinical scoring system (C). Animals were assigned a score from 0 to 2 for each of 5 GVHD parameters: weight loss, activity, fur ruffling, kyphosis, and skin lesions. Scores ranged from 0 (minimum) to 10 (maximum). Error bars depict standard error of the mean. n=4 mice per group. (D–E) B6D2 recipients were lethally irradiated to 950 rads on transplant day −1 and then administered 3×106 TCD B6 BM cells on day 0. Some mice were then administered 6×106 CD25-depleted whole splenic B6 Tcons as a DLI on transplant day +7 +/− Compound A dosed at 250mcg once daily by IP injection beginning on day +7. Mice were then followed for survival (D) and scored for GVHD twice weekly (E). n=3 mice per group.
In order to circumvent this issue, we modified our transplantation regimen so that the allo-reactive B6 donor Tcons were administered as a DLI on transplant day +7, one week removed from the irradiation conditioning regimen and bone marrow infusion. Daily Compound A dosing was similarly begun on transplant day +7. In addition we reduced our dose of Compound A by 66% in order to minimize drug toxicity. With this approach, we were able to induce a 100% mortality rate in our untreated controls. Compound A, however, had virtually no effect on GVHD scores or recipient survival when administered in this manner (Figure 2D, E). Thus, while Compound A did not exacerbate transplant outcomes at a daily dose of 250mcg, it also did not seem to offer any appreciable advantage. Following up on these data, we explored numerous other approaches to dosing the compound including oral gavage, subcutaneous administration, and prolonged low dose therapy via intraperitoneal injection. None of these strategies, however, consistently improved transplant outcomes in a B6 into B6D2 model system (data not shown).
Compound A inhibits donor T cell expansion within GVHD target organs and especially the lung
While the pro-inflammatory properties of the NF-κB pathway are well known, NF-κB signaling has also been implicated in the resolution of ongoing inflammatory responses in vivo24. As a result, we hypothesized that Compound A might be paradoxically promoting donor T cell expansion and/or survival after transplant. To explore this possibility, eGFP+ B6 donor T cells were transplanted into eGFP− B6D2 recipients, and donor T cell trafficking and expansion examined by stereofluorescence microscopy and quantified using an anti-eGFP ELISA approach. On transplant day +7, consistent with our in vitro MLR data depicted in Figure 1A, Compound A appeared to reduce donor T cell accumulation within GVHD target organs when administered at a dose of 250mcg daily. However, the differences between treated and untreated mice were not entirely consistent across organs. By both stereofluorescence microscopy and anti-eGFP ELISA, the greatest relative reduction in donor T cell accumulation was noted within pulmonary tissues (Figure 3A–E). Following up on these data, we repeated our in vivo trafficking experiment with a considerably lower dose of Compound A. When given at only 100mcg per day, the trafficking differences previously observed in the liver and colon were no longer apparent. Conversely, donor T cell accumulation within pulmonary tissues remained significantly lower than what was seen in untreated control animals (Figure 3F). Collectively, these data suggested that Compound A was producing two effects in vivo. First, at higher dosages it appeared to be generally impairing donor T cell expansion within all sites. However, given the efficacy differences observed between organs, it appeared that Compound A was also impairing donor T cell trafficking to individual GVHD target organs in a non-uniform way, with this effect being particularly pronounced in the lungs.
Figure 3. Compound A reduces donor Tcon accumulation within recipient organs after transplant and in particular the host lung.
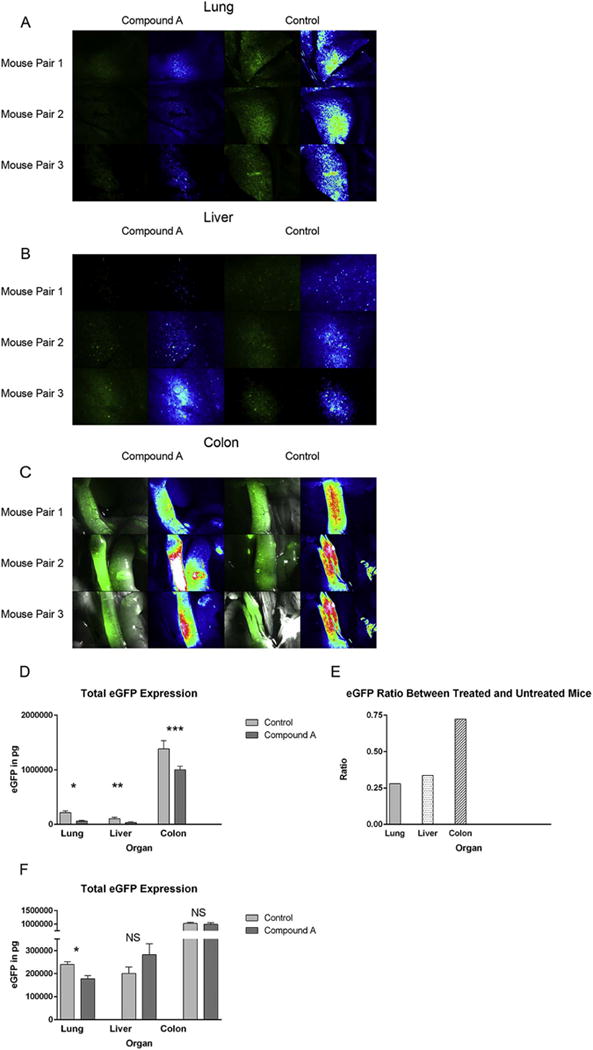
(A–E) B6D2 mice were lethally irradiated to 950 rads on transplant day −1 and then administered 3×106 WT B6 TCD BM cells plus 4×106 CD25-depleted whole Tcons obtained from eGFP+ B6 mice +/− Compound A dosed at 250mcg once daily by IP injection on transplant days 0–5. On transplant day +7, recipient mice were killed and their organs imaged by fluorescence stereomicroscopy (A–C). For each image, actual eGFP fluorescence is depicted on the left and a rainbow feature is shown on the right to indicate the strength of the eGFP signal (white>red>yellow>green>blue>black). Three untreated controls and three mice given Compound A were imaged. (D) Host organs were subsequently removed and homogenized, and total eGFP levels then measured in each organ by anti-eGFP ELISA. n=6 mice per treatment group. *P=0.0012 for total pulmonary eGFP comparison between control and Compound A groups by Student’s t test. **P=0.048. ***P=0.0355. (E) Mean organ eGFP level ratios between treated and untreated mice are also depicted. (F) B6D2 mice were lethally irradiated to 950 rads on day −1 and then administered 3×106 WT B6 TCD BM cells plus 4×106 CD25-depleted whole Tcons obtained from eGFP+ B6 mice +/− Compound A dosed at 100mcg once daily by IP injection on transplant days 0–7. On transplant day +7, recipient mice were killed and their organs removed and homogenized. Total eGFP levels were then measured for each organ by anti-eGFP ELISA. *P=0.0082.
Compound A inhibits NF-κB activity within pulmonary tissues in a model of IPS
Since Compound A demonstrated a particularly strong ability to impair donor T cell accumulation within pulmonary tissues, we reasoned that Compound A might be effective in improving transplant outcomes in a GVHD model system in which recipient mortality depends more on the development of acute lung injury. To examine this possibility, we made use of a B6 into B10.BR model using a combined TBI plus cyclophosphamide based conditioning regimen. This is a well described model of IPS in which recipient lung injury is dependent on the infusion of allo-reactive donor T cells and potentiated by pre-transplant cyclophosphamide25. Initial experiments began by examining NF-κB activity within the lungs of transplant recipients using immunohistochemistry. For this work, recipient lungs were stained with an antibody against phospho-IκB, the downstream target of IKK and therefore a marker for IKK-dependent NF-κB activity. In addition, consecutive slides were stained with CD3 to evaluate for T cell infiltration. As depicted in Figure 4, the lungs stained diffusely positive for phospho-IκB on transplant day +7 (Figure 4A) and to a lesser extent on day +21 (Figure 4B) in control mice not administered compound. Phospho-IκB staining was intense within epithelial cells lining the airways and did not entirely overlap with CD3 staining, indicating that a substantial proportion of overall pulmonary NF-κB activity was occurring within the host pulmonary parenchyma itself and not only within infiltrating donor T cells. Importantly, Compound A substantially reduced pulmonary phospho-IκB staining, confirming its ability to antagonize the NF-κB signaling pathway within the host lung after transplant.
Figure 4. In a murine model of IPS recipient lungs demonstrate diffuse NF-κB signaling for at least three weeks post-transplant and this activity is attenuated by Compound A administration.
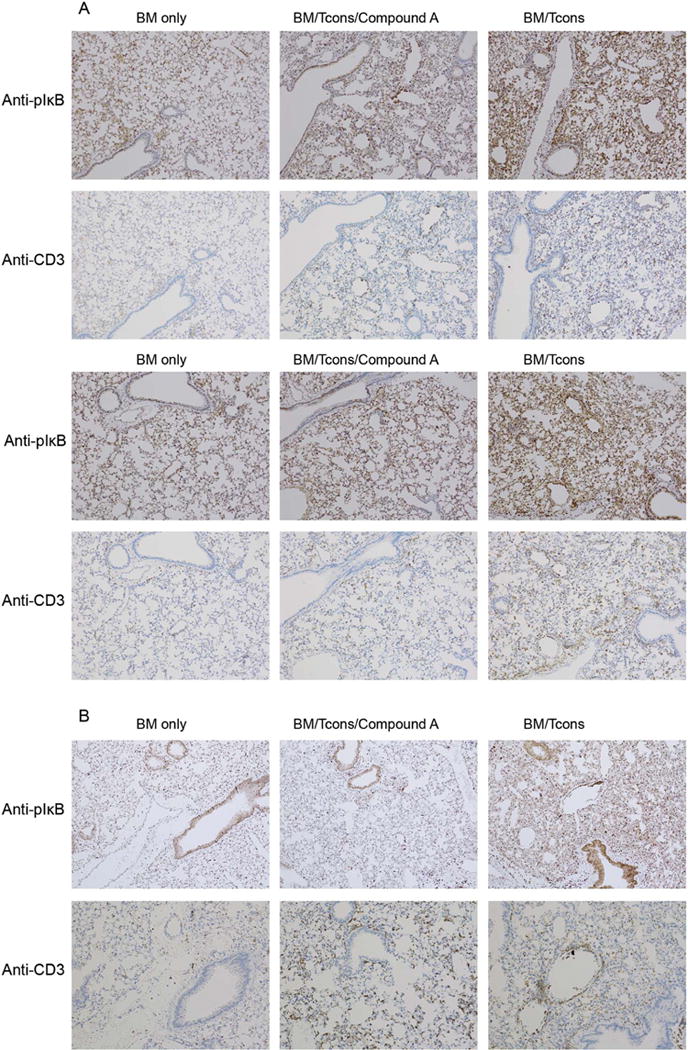
(A–B) B10.BR mice were administered cyclophosphamide at 120mg/kg by IP injection on transplant days −3 and −2. They were then irradiated to 700 rads on day −1 on a cesium irradiator. On day 0, recipient mice were administered 1×107 TCD BM cells from B6 mice +/− 7×106 NK1.1-depleted B6 splenocytes +/− Compound A dosed at 175mcg once daily by IP injection on days 0–6. (A) On day +7 the mice were killed and their hearts perfused through with saline. Their lungs were then removed and placed in formalin. The lungs were subsequently sectioned and stained for IHC using an anti-phospho-IκBα antibody. Consecutive slides were also stained with an anti-CD3 antibody. Two mice from each treatment group are depicted. (B) A third set of mice was transplanted similarly but administered daily Compound A on days 0–20. They were then killed on day +21 for IHC (B).
Compound A attenuates pulmonary chemokine production and impairs donor leukocyte accumulation within the host lung in an IPS model
Subsequent experiments focused on whether Compound A could substantially impair the accumulation of donor immune cells within pulmonary sites in a mouse IPS model. For this work, eGFP+ B6 mice served as bone marrow and splenocyte donors, and eGFP− B10.BR mice served as recipients. As seen in Figure 5A, total donor leukocyte accumulation within the host lungs as determined by eGFP ELISA was significantly impaired on transplant day +14 when mice were treated with daily doses of Compound A. Since in this model system whole splenocytes are used to generate an immune response versus purified Tcons, we were interested in determining which donor leukocyte subsets were impaired in their pulmonary trafficking and therefore contributing to the overall reduction in the observed lung eGFP signal. Using flow cytometry, we found that Compound A significantly reduced the number of CD4+ and CD8+ T cells accumulating within the host lung. Conversely, Compound A had a minimal effect on the pulmonary accumulation of donor CD11b+Ly6Cint neutrophils or CD11b+Ly6Chi inflammatory monocytes26, and did little to alter the very small number of pulmonary B cells that were observed at this time point (Figure 5B). Collectively, Compound A resulted in a reduction in the donor lymphoid to myeloid ratio within the lungs after transplant (Figure 5C).
Figure 5. Compound A reduces pulmonary chemokine levels and donor T cell accumulation within the host lung in a mouse IPS model.
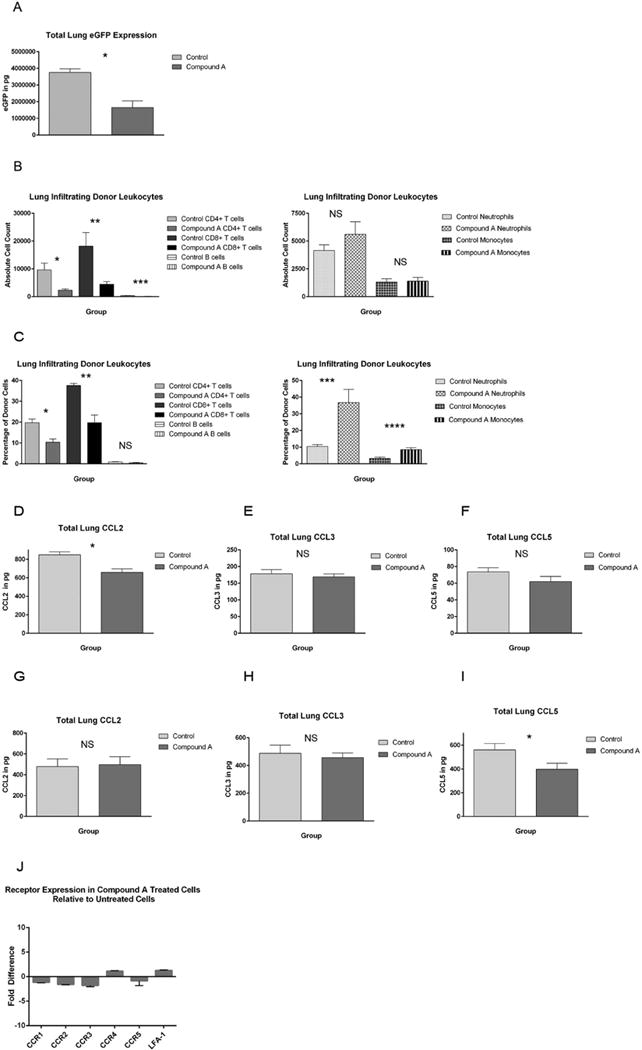
(A) B10.BR mice were administered cyclophosphamide at 120mg/kg by IP injection on transplant days −3 and −2. They were then irradiated to 700 rads on day −1. On day 0, recipient mice were administered 1×107 TCD BM cells from eGFP+ B6 mice + 7×106 NK1.1-depleted eGFP+ B6 splenocytes +/− Compound A dosed at 175mcg once daily by IP injection on days 0–13. The mice were killed on transplant day +14 and their left lung removed and homogenized. Total eGFP levels were then determined by ELISA. n=5 mice per group *P=0.0016 by Student’s t test. (B, C) B10.BR mice were transplanted as before and then killed on transplant day +14. Their right lung was then removed and digested using a collagenase/DNA-ase solution. Leukocytes were purified using a percoll gradient and infiltrating donor cell numbers then determined by flow cytometry. Donor cells were distinguished from residual host cells by virtue of their eGFP positivity. (B) Absolute numbers of donor CD4+ T cells, CD8+ T cells, CD19+ B cells, CD11b+Ly6cint neutrophils, and CD11b+Ly6chi inflammatory monocytes are depicted. *P=.0062 by Student’s t test. **P=0.0107. ***P=0.0012. n=8 mice per group. (C) The percentages of each cell type as a fraction of the total number of pulmonary infiltrating donor cells are also shown. *P=0.0015. **P=0.0007. ***P=0.0067. ****P=0.0033. (D–I) B10.BR mice were transplant as before and then killed on either day +7 (D–F) or day +14 (G–I). Their left lung was then homogenized and total chemokine levels determined by ELISA. n=9 control and 8 Compound A mice per group on day +7, and n=12 control and 11 Compound A mice per group on day +14. (D) *P=0.0014. (I) *P=0.0381. (J) Purified B6 CD4+CD25− Tcons were activated in vitro with plate-bound anti-CD3 and anti-CD28 antibody and murine IL-2 at 100 IUs/ml in the presence or absence of 100nM Compound A. The cells were expanded for 48 hours, and total RNA was then isolated. CCR1, CCR2, CCR3, CCR4, CCR5, and LFA-1 expression were then compared between Compound A treated and untreated cells using qRT-PCR. Fold changes for each receptor in treated cells relative to untreated cells are depicted. n=6 replicates per receptor.
Given the lower numbers of donor T cells seen in the host lung in those mice given Compound A, we hypothesized that Compound A might be blocking T cell trafficking into pulmonary sites by antagonizing the local production of lymphocyte recruiting chemokines. To explore this possibility we examined pulmonary protein levels of CCL2, CCL3, and CCL5, chemokines that have previously been implicated in T cell recruitment to the lung and are known to be regulated by the NF-κB pathway27–29. As seen in Figure 5D, pulmonary levels of CCL2 were significantly reduced on transplant day +7 in those mice given Compound A. With additional follow-up, overall CCL2 levels declined in both treated and control mice so that no differences were noted between the treatment groups by day +14 (Figure 5G). Conversely, CCL5 levels increased from transplant day +7 to day +14, and were significantly lower in Compound A mice at the latter time point (Figure 5F, I). Overall CCL3 levels were also noted to increase between transplant days +7 and +14, but were not significantly different between the two treatment groups at either time (Figure 5E, H).
Following up on these data, we also examined Compound A’s effects on the transcription by donor T cells of the corresponding receptors for the aforementioned chemokines using qRT-PCR. In addition, we evaluated the expression of LFA-1 in the presence and absence of compound since the LFA-1/ICAM-1 axis has previously been implicated in IPS pathogenesis30. As shown in Figure 5J, Compound A’s effects on the expression of these 6 receptors appeared to be relatively modest in vitro. Collectively these data would suggest that Compound A functions to alter donor T cell homing in vivo primarily via an effect on local chemokine generation rather than through a major alteration in cell surface trafficking receptor expression.
Compound A reduces inflammatory cytokine production within the lung after HSCT
Next we examined the effects of Compound A on inflammatory cytokine production after transplant. As depicted in Figure 6A, B, total pulmonary TNF and IFN-γ were both significantly reduced two weeks post-transplant in those mice treated with Compound A. In addition, we looked for changes in pulmonary IL-6 levels, a cytokine known to be critical for Th17 polarization and a skewing away from inducible Treg production. On transplant day +14, pulmonary IL-6 levels were also generally reduced, although this difference did not achieve statistical significance (Figure 6C). IL-17 levels were uniformly low in both treated and untreated mice at this time point and did not appear to be altered by Compound A administration (Figure 6D).
Figure 6. Compound A reduces inflammatory cytokine production within the host lung two weeks post-transplant.
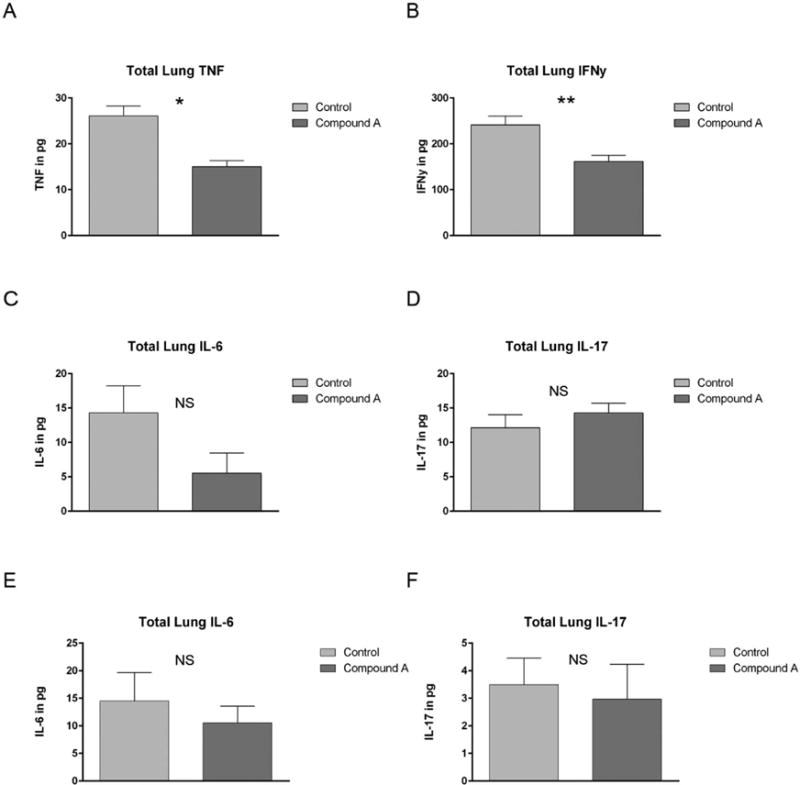
(A–D) B10.BR mice were administered cyclophosphamide at 120mg/kg by IP injection on transplant days −3 and −2. They were then irradiated to 700 rads on day −1. On day 0, recipient mice were administered 1×107 TCD BM cells from eGFP+ B6 mice + 7×106 NK1.1-depleted eGFP+ B6 splenocytes +/− Compound A dosed at 175mcg once daily by IP injection on days 0–13. The mice were killed on transplant day +14 and their left lung removed and homogenized. Total lung cytokine levels were then determined by ELISA. n=5 mice per group. (A) *P=0.0048 by Student’s t test for comparison of total lung TNF levels between untreated controls and those mice administered Compound A. (B)**P=0.0138 for IFN-γ comparison. (E–F) B10.BR mice were administered cyclophosphamide at 120mg/kg by IP injection on transplant days −3 and −2. They were then irradiated to 700 rads on day −1. On day 0, recipient mice were administered 1×107 TCD BM cells from eGFP+ B6 mice + 7×106 NK1.1-depleted eGFP+ B6 splenocytes +/− Compound A dosed at 175mcg once daily by IP injection on days 0–6. The mice were killed on transplant day +7 and their left lung removed and homogenized. Total lung cytokine levels were then determined by ELISA.
Since plasma IL-6 levels and Th17 numbers have previously been shown to peak within the first 1–2 weeks after transplant31, we also examined pulmonary levels of both cytokines on transplant day +7 (Figure 6 E, F). Again, a non-significant trend towards less IL-6 was noted following Compound A administration, with no change in low level IL-17 production. Collectively our data indicated that Compound A can effectively impair the generation of multiple pro-inflammatory cytokines within the lungs, but that it appears to have a marginal effect on the modest Th17 activity observed after transplant in this model system.
Compound A improves survival and arterial oxygenation in an IPS transplant model
Our previous data indicated that Compound A was able to impair the NF-κB signaling cascade within the host lung after transplant, and that this in turn resulted in a reduction in donor T cell accumulation and inflammatory cytokine production within pulmonary tissues. As a consequence, we set out to determine if Compound A could alter clinical outcomes after HSCT in an IPS model system. For this work, B10.BR mice were transplanted as before and then followed for survival and GVHD severity. Notably, we elected to administer a 21 day course of Compound A (versus indefinite dosing) in our treatment group due to drug cost and compound availability considerations. As shown in Figure 7A, B Compound A was able to significantly improve median survival times and to reduce overall GVHD scores during the period of compound administration. In addition, we evaluated the ability of Compound A to ameliorate a lung specific clinical parameter, arterial oxygenation, using a murine pulse-oximeter. As depicted in Figure 7C, Compound A was able to significantly reduce post-transplant hypoxia in treated mice versus untreated controls. Overall, these data indicated that Compound A could ameliorate lung injury and improve transplant outcomes in an aggressive murine IPS model.
Figure 7. Compound A prolongs median survival and improves arterial oxygenation in a murine IPS model.
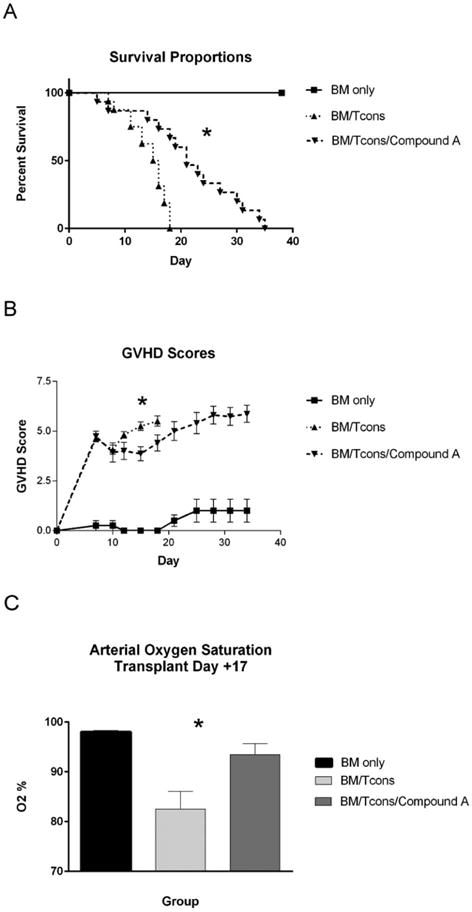
(A–C) B10.BR mice were administered cyclophosphamide at 120mg/kg by IP injection on transplant days −3 and −2. They were then irradiated to 700 rads on day −1. On day 0, recipient mice were administered 1×107 TCD BM cells from B6 mice +/− 5x106 NK1.1-depleted B6 splenocytes +/− Compound A dosed at 175mcg once daily by IP injection on days 0–21. The mice were followed for survival (A) and scored for GVHD twice weekly using a validated scoring system (B). On day +17, mice were anesthetized using isofluorane and arterial oxygen saturations determined using a mouse pulse oximeter placed on the hindleg (C). (A) *P<0.0001 for comparison of median survival times between the BM/Tcons and BM/Tcons/Compound A groups using the log rank test. (B) *P=0.0019 and *P=0.0292 for mean GVHD score comparison between BM/Tcon and BM/Tcon/Compound A groups on days +15 and +18 respectively by Student’s t test. (C) *P=0.017 for comparison of mean arterial oxygen saturations between the BM/Tcons and BM/Tcons/Compound A groups. n=4 BM only, 16 BM/Tcon, and 15 BM/Tcon/Compound A. Data are combined from two separate transplantation experiments.
Discussion
IPS refers to widespread non-infectious lung injury occurring after HSCT in the absence of cardiac or renal dysfunction. Mechanistically the pathogenesis of IPS is not entirely clear since there is no uniform pathognomonic correlate in human patients and there are numerous different mouse models of the disease process. Nevertheless, nearly all of the described preclinical model systems involve some degree of MCH incompatibility between the donor and recipient strains, implying that allo-immune reactivity is important. IPS is not typically seen in most murine acute GVHD models since mice usually succumb to gastrointestinal GVHD before lethal pulmonary pathology can develop. To circumvent this problem, investigators have modified their model systems to intensify pulmonary pathology. For example, in work by Hildebrandt et al. the authors used a B6 into B6D2 strain combination identical to that used for our initial Compound A experiments, but infused a lower T cell dose and a more intense pre-transplant irradiation regimen32. Our own research group has previously shown that the infusion of highly purified, ex-vivo polarized Th17 cells results in the development of GVHD manifestations that are particularly severe within pulmonary and cutaneous sites33. Consistent with these data, Varielias et al. have demonstrated IPS development in a B6 into B6D2 strain combination in the absence of functional IFN-γ, a Th1 polarizing cytokine and a well described inhibitor of Th17 generation. In this situation, Th17 numbers were enhanced within the lung, and this in turn resulted in acute pulmonary injury in a process that depended critically on IL-6 production18.
Here we utilized a well described mouse model of IPS in which IFN-γ signaling is preserved and pathology depends on the addition of cyclophosphamide to a standard TBI conditioning regimen. In our work, we found that a specific inhibitor of the canonical NF-κB pathway, Compound A, was able to block the influx of donor CD4+ and CD8+ T cells into pulmonary tissues. This resulted in a significant improvement in recipient oxygenation after transplant, and a prolongation of median survival times. Furthermore, in our standard B6 into B6D2 model donor T cell expansion was significantly reduced within pulmonary tissues.
The development of IPS has been shown to depend on the early production of proinflammatory mediators by the pulmonary parenchyma which leads to the recruitment of donor monocytes and T cells into the host lung. These cells in turn produce additional chemokines which augment the developing pulmonary inflammation in an autocrine fashion. CCL2, CCL3, CCL5 have been shown to promote leukocyte recruitment into pulmonary tissues and have been linked to IPS development in murine models34. Our own data demonstrate diffuse NF-κB activity within the host lung by transplant day +7. This is accompanied by an early peak in pulmonary CCL2 production which declines from day +7 to day +14, and the generation of CCL5 which conversely increases over the second transplant week. These data would be consistent with existing reports that describe early CCL2 production by the host pulmonary parenchyma, and the subsequent augmentation of the developing pulmonary inflammatory response by donor T cell derived CCL5. Here we show that Compound A significantly reduces pulmonary levels of both chemokines. Interestingly pulmonary levels of CCL3, a chemokine previously linked to enhanced IPS and inferior transplant outcomes35, were unaffected by Compound A administration.
Compound A, while effective in limiting pulmonary disease, was unable to prevent gastrointestinal dependent GVHD lethality in a standard B6 into B6D2 system. This is likely due to the fact that the NF-κB pathway is known to have protective effects on gastrointestinal epithelial cells, and that NF-κB inhibition can have variable effects on colonic inflammation36. For example, a peptide capable of binding to the regulatory NEMO subunit of IKK and inhibiting overall IKK function has been shown to ameliorate murine colitis37. However, the complete abrogation of the NF-κB pathway in the intestine through the simultaneous blockade of both of IKK’s catalytic subunits (IKK1 and IKK2) paradoxically results in severe intestinal inflammation in mice38. In the transplantation setting, beneficial anti-inflammatory effects in the gastrointestinal tract arising from NF-κB blockade could be partially offset by negative actions on the colonic epithelium.
Our results differed somewhat from data by Vodanovic-Jankovic et al11. There, the authors found that another IKK antagonist, PS-1145, reduced overall GVHD lethality in a B10.BR into B6 GVHD model with a low incidence of gastrointestinal pathology. Several factors could explain this discrepancy. First, their model system was different. When PS-1145 was administered over an extended 10 day course the donor/recipient strains were reversed and between 30–40% of the untreated control mice survived to the end of the transplantation period. Secondly, the two IKK inhibitors themselves are different. PS-1145 is less potent against IKK2 than Compound A (IC50=250nM vs. 4nM respectively) and also exhibits less activity against IKK1 (IC50>10,000nM vs. KiATP=135nM respectively)17,39,40. As a result, Compound A would be expected to be more active against IKK2 than PS-1145 within pulmonary tissues but perhaps less likely to ameliorate gut injury because of an overall greater net effect against the two catalytic subunits of IKK within colonic epithelial cells. Furthermore, the two antagonists exhibit different off target effects which could affect both their efficacy and toxicity17,39.
IPS is augmented by donor derived TNF in preclinical models32, and has been targeted therapeutically in human patients with the soluble TNF receptor etanercept. Efficacy data for etanercept, however, have been mixed. While the addition of etanercept to corticosteroids has been shown to improve short term disease response rates, one year survival remains poor with mortality figures approaching 80%41–43. Collectively these data would suggest that TNF blockade is suboptimal. Here we show that antagonizing NF-κB with Compound A not only reduces TNF levels within the host lung after transplant but also reduces pulmonary IFN-γ levels and lung infiltrating donor T cell numbers. Thus, Compound A targets multiple pathways of pulmonary inflammation beyond TNF alone and could offer a therapeutic advantage over existing therapies. Furthermore, agents specifically targeting the canonical NF-κB pathway might be combined with corticosteroids and/or TNF inhibitors to boost efficacy in a synergistic fashion.
In summary, we have found that the inhibition of the canonical NF-κB pathway with the IKK2 inhibitor Compound A can improve oxygenation and survival in a mouse model of IPS. While Compound A was ineffective at preventing lethality from acute gut GVHD, it did not appear to exacerbate gastrointestinal injury at doses below 10mg/kg/day and would appear to be a promising approach for the targeted management of IPS after allogeneic HSCT. In addition, our data when compared to existing reports10,11 highlight the variable and occasionally unpredictable treatment effects that can be observed with different inhibitors of the same inflammatory pathway after transplant, and underscore the overall complexity of the NF-κB signaling cascade.
Highlights.
A potent NF-κB inhibitor improved outcomes in a mouse model of idiopathic pneumonia syndrome
These effects were lung-specific and did not alter gastrointestinal graft-versus-host disease
The NF-κB antagonist blocked pro-inflammatory chemokine production in the host lung
Reduced donor T cell accumulation was observed within pulmonary tissues after transplant
The compound reduced pro-inflammatory cytokine production in the lungs
Acknowledgments
This work was supported by the following NIH grants: 5K12CA120780-05 and 5K08HL111205-04 to JMC; R01CA166794 and R01HL115761 to JSS
The authors would like to acknowledge The Flow Cytometry Core Facility and The Lineberger Comprehensive Cancer Center Animal Histology Core Facility at The University of North Carolina at Chapel Hill for their assistance with this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure Statement: The authors have nothing to disclose
References
- 1.Panoskaltsis-Mortari A, Griese M, Madtes DK, et al. An official American Thoracic Society research statement: Noninfectious lung injury after hematopoietic stem cell transplantation: Idiopathic pneumonia syndrome. Am J Respir Crit Care Med. 2011;183:1262–1279. doi: 10.1164/rccm.2007-413ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes PJ. Nuclear Factor-κB – A pivotal factor in chronic inflammatory diseases. NEJM. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 3.Ohmori K, Takeda S, Miyoshi S, et al. Attenuation of lung injury in allograft rejection using NF-κB decoy transfection-novel strategy for use in lung transplantation. European Journal of Cardio-thoracic Surgery. 2007;(27):23–27. doi: 10.1016/j.ejcts.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Siebenlist U, Franzoso G, Brown K. Structure, regulation, and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 5.Verma IM, Stevenson JK, Scharz EM, et al. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes and Development. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 6.O’Connell MA, Bennett BL, Mercurio F, et al. Role of IKK1 and IKK2 in Lipopolysaccharide signaling in human monocytic cells. Journal of Biological Chemistry. 1998;273:30410–30414. doi: 10.1074/jbc.273.46.30410. [DOI] [PubMed] [Google Scholar]
- 7.Kempiak SJ, Hiura TS, Nel AE. The Jun kinase cascade is responsible for activating CD28 response element of the IL-2 promoter: proof of cross-talk with the I kappa B kinase cascade. J Immunol. 1998;162(6):3176–3187. [PubMed] [Google Scholar]
- 8.Zandi E, Rothward DM, Delhase M, et al. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91(2):243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 9.Schwabe RF, Schnabl B, Kweon YO, et al. CD40 activates NF-kappa B and c-Jun N-terminal kinase and enhances chemokine secretion on activated human hepatic stellate cells. J Immunol. 2001;166:6812–6819. doi: 10.4049/jimmunol.166.11.6812. [DOI] [PubMed] [Google Scholar]
- 10.Sun K, Welniak LA, Panoskaltsis-Mortari A, et al. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. PNAS. 2004;101(34):8120–8125. doi: 10.1073/pnas.0401563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vodanovic-Jankovic S, Hari P, Jacobs P, et al. NF-κB as a target for the prevention of graft-versus-host disease: comparative efficacy of bortezomib and PS-1145. Blood. 2006;107(2):827–834. doi: 10.1182/blood-2005-05-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pai CS, Chen M, Mirsoian A, et al. Treatment of chronic graft-versus-host disease with bortezomib. Blood. 2014;14(10):1677–1688. doi: 10.1182/blood-2014-02-554279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caballero-Velazquez T, Sanchez-Abarca L, Guitieerez-Cosio S, et al. The novel combination of sirolimus and bortezomib prevents graft-versus-host disease but maintains the graft-versus-leukemia effect after allogeneic transplantation. Haematologica. 2012;97:1329–1337. doi: 10.3324/haematol.2011.058677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herrera A, Kim HT, Bindra B, et al. A phase II trial of bortezomib plus prednisone for initial therapy of chronic graft-versus-host disease. Biol Blood Marrow Transplantation. 2014;20(11):1737–1743. doi: 10.1016/j.bbmt.2014.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koreth J, Stevenson KE, Kim HT, et al. Bortezomib-based graft-versus-host disease prophylaxis in HLA-mismatched unrelated donor transplantation. J Clin Oncol. 2012;30(26):3202–3208. doi: 10.1200/JCO.2012.42.0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basseres DS, Ebbs A, Cogswell PC, et al. IKK is a therapeutic target in KRAS-inducted lung cancer with disrupted p53 activity. Genes and Cancer. 2014;5(1–2):41–55. doi: 10.18632/genesandcancer.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ziegelbauer K, Ganter F, Lukacs NW, et al. A selective novel low-molecular-weight inhibitor of IκB kinase-β (IKK-β) prevents pulmonary inflammation and shows broad antiinflammatory activity. British Journal of Pharmacology. 2005;145:178–192. doi: 10.1038/sj.bjp.0706176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varelias A, Gartlan KH, Kreijveld E, et al. Lung parenchyma-derived IL-6 promotes IL-17A-dependent acute lung injury after allogeneic stem cell transplantation. Blood. 2015;125(15):2435–2444. doi: 10.1182/blood-2014-07-590232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serody JS, Burkett SE, Panoskaltsis-Mortari A, et al. T-lymphocyte production of macrophage inflammatory protein-1 alpha is critical to the liver, lung, and spleen during graft-versus-host disease. Blood. 2000;96(9):2973–2980. [PubMed] [Google Scholar]
- 20.Coghill JM, Carlson MJ, Panoskaltsis Mortari A, et al. Separation of graft-versus-host disease from graft-versus-leukemia responses by targeting CC-chemokine receptor 7 on donor T cells. Blood. 2010;115(23):4914–4922. doi: 10.1182/blood-2009-08-239848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coghill JM, Fowler KA, West ML, et al. CC Chemokine receptor 8 potentiates donor Treg survival and is critical for the prevention of murine graft-versus-host disease. Blood. 2013;122(5):825–836. doi: 10.1182/blood-2012-06-435735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wysocki CA, Burkett SB, Panoskaltsis-Mortari A, et al. Differential roles for CCR5 expression on donor T cells during graft-versus-host disease base on pretransplant conditioning. J Immunol. 2004;173:845–854. doi: 10.4049/jimmunol.173.2.845. [DOI] [PubMed] [Google Scholar]
- 23.Murata T, Shimada M, Sakakibara S, et al. Discovery of novel and selective IKK-serine-threonine protein kinase inhibitors. Bioorg Med Chem Lett. 2003;13:913–918. doi: 10.1016/s0960-894x(02)01046-6. [DOI] [PubMed] [Google Scholar]
- 24.Lawrence T, Gilroy DW, Colville-Nash PR, et al. Possible new role for NF-κB in the resolution of inflammation. Nature Medicine. 2001;7(12):1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 25.Panoskaltsis-Mortari A, Taylor PA, Yaeger TM, et al. The critical early proinflammatory events associated with idiopathic pneumonia syndrome in irradiated murine allogeneic recipients are due to donor T cell infusion and potentiated by cyclophosphamide. J Clin Invest. 1997;100(5):1015–1027. doi: 10.1172/JCI119612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunay IR, Fuchs A, Sibley LD. Inflammatory monocytes but not neutrophils are necessary to control infection with toxoplasma gondii in mice. Infection and immunity. 2010;78(4):1564–1570. doi: 10.1128/IAI.00472-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wickremasinghe M, Thomas LH, O’Kane CM. Transcriptional mechanisms regulating alveolar epithelial cell-specific CCL5 secretion in pulmonary tuberculosis. J Biol Chem. 2004;279(26):27199–27210. doi: 10.1074/jbc.M403107200. [DOI] [PubMed] [Google Scholar]
- 28.Widmer U, Manogue KR, Cerami A, et al. Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1 alpha, and MIP-1 beta, members of the chemokine superfamily of proinflammatory cytokines. J Immunol. 1993;150(11):4966–5012. [PubMed] [Google Scholar]
- 29.Ueda A, Okuda A, Ohno S, et al. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. 1994;153(3):2052–2063. [PubMed] [Google Scholar]
- 30.Panoskaltsis-Mortari A, Hermanson JR, Haddad IY, et al. Intercellular Adhesion Molecule-1 (ICAM-1, CD54) deficiency segregates the unique pathophysiological requirements for generating idiopathic pneumonia syndrome (IPS) versus graft-versus-host disease following allogeneic murine bone marrow transplantation. Biology of Blood and Marrow Transplantation. 2001;7:368–377. doi: 10.1053/bbmt.2001.v7.pm11529486. [DOI] [PubMed] [Google Scholar]
- 31.Kappel LW, Goldberg GL, King CG, et al. IL-17 contributes to CD4-mediated graft-versus-host disease. Blood. 2009;113:945–952. doi: 10.1182/blood-2008-08-172155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hildebrandt GC, Olkiewicz KM, Corrion LA, et al. Donor derived TNF-α regulates pulmonary chemokine expression and the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood. 2004;104:586–593. doi: 10.1182/blood-2003-12-4259. [DOI] [PubMed] [Google Scholar]
- 33.Carlson MJ, West ML, Coghill JM, et al. In vitro differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood. 2009;113(6):1365–1374. doi: 10.1182/blood-2008-06-162420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panoskaltsis-Mortari A, Strieter RM, Hermanson JR, et al. Induction of monocyte-and T cell-attracting chemokines in the lung during the generation of idiopathic pneumonia syndrome following allogeneic murine bone marrow transplantation. Blood. 2000;96(3):834–839. [PubMed] [Google Scholar]
- 35.Panoskaltsis-Mortari A, Hermanson JR, Taras E, et al. Acceleration of idiopathic pneumonia syndrome (IPS) in the absence of donor MIP-1α (CCL3) after allogeneic BMT in mice. Blood. 2003;101:3714–3721. doi: 10.1182/blood-2002-08-2465. [DOI] [PubMed] [Google Scholar]
- 36.Wullaert A, Bonnet MC, Pasparakis M. NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Research. 2011;21:146–158. doi: 10.1038/cr.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibata W, Maeda S, Hikiba Y, et al. Curring edge: the I kappa B kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks inflammatory injury in murine colitis. J Immunol. 2007;179:2681–2685. doi: 10.4049/jimmunol.179.5.2681. [DOI] [PubMed] [Google Scholar]
- 38.Nenci A, Becker C, Wullaert A, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 39.Bain J, Plater L, Elliot M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karin M, Yamamoto Y, Wang QM. The Ikk NF-κB system: A treasure trove for drug development. Nature Reviews. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 41.Tizon R, Frey N, Heitjan DF, et al. High-dose corticosteroids with or without etanercept for the treatment of idiopathic pneumonia syndrome after allo-SCT. Bone Marrow Transplantation. 2012;47:1332–1337. doi: 10.1038/bmt.2011.260. [DOI] [PubMed] [Google Scholar]
- 42.Yanik GA, Ho VT, Levine JE, et al. The impact of soluble tumor necrosis factor receptor etanercept on the treatment of idiopathic pneumonia syndrome after allogeneic hematopoietic stem cell transplantation. Blood. 2008;112:3073–3081. doi: 10.1182/blood-2008-03-143412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frangoul H, Koyama T, Domm J. To the editor: Etanercept for treatment of idiopathic pneumonia syndrome after allogeneic hematopoietic stem cell transplantation. Blood. 2008;113:2868–2869. doi: 10.1182/blood-2008-09-177733. [DOI] [PubMed] [Google Scholar]