

SUMMARY
This study reveals that high-copy satellite II (HSATII) sequences in the human genome can bind and impact distribution of chromatin regulatory proteins and that this goes awry in cancer. In many cancers, master regulatory proteins form two-types of cancer-specific nuclear bodies, caused by locus-specific deregulation of HSATII. DNA demethylation at the 1q12 mega-satellite, common in cancer, causes PRC1 aggregation into prominent Cancer-Associated Polycomb (CAP) bodies. These loci remain silent, whereas HSATII loci with reduced PRC1 become de-repressed, reflecting imbalanced distribution of UbH2A on these and other PcG-regulated loci. Large nuclear foci of HSATII RNA form and sequester copious MeCP2 into Cancer-Associated Satellite Transcript (CAST) bodies. Hence, HSATII DNA and RNA have an exceptional capacity to act as molecular sponges and sequester chromatin regulatory proteins into abnormal nuclear bodies in cancer. The compartmentalization of regulatory proteins within nuclear structure, triggered by demethylation of “junk” repeats, raises the possibility that this contributes to further compromise of the epigenome and neoplastic progression.
eTOC blurb
Satellite II is a prominent but poorly studied feature of human genomes. Hall et al. show HSATII DNA and RNA can sequester PRC1 and MeCP2. In cancer, PRC1-bodies form on the demethylated 1q12 mega-satellite while MeCP2-bodies form on HSATII RNA, potentially leading to further changes in the epigenome.
INTRODUCTION
Epigenetic changes are recognized as a major player in tumorigenesis (Feinberg, 2014; Sandoval and Esteller, 2012). Although attention has been focused on methylation and silencing of tumor suppressor genes, this often occurs concomitant with loss of methylation from other genomic regions, including pericentric satellites. High-copy tandem satellites repeat families (alpha, beta, SATI, II, III) constitute ~15% of the human genome. Alpha-satellite (α-SAT) is at the centromere of all chromosomes, and satellite III (HSATIII) on Chr 9 is linked to the heat-shock response and nuclear stress bodies (Biamonti and Vourc’h, 2010). However, many repeats are considered mere evolutionary relics and have been poorly studied. Perhaps the best example of this is human satellite II (HSATII), an exceptionally high-copy repeat with no raison d’etre. Small blocks of HSATII are found on the pericentromeres of 11 human chromosomes (Chr 2, 5, 7, 10, 13, 14, 15, 17 21, 22 & Y), but 1q12 on Chr1, and to a lesser extent 6q11 on Chr16, carry very large (~5–6 Mb) blocks of this ~26bp tandem repeat. There is frequent loss of methylation of HSATII in cancer (Ehrlich, 2009), but any significance of this is unknown. HSATII remains one of the most prominent but poorly studied features of the human genome.
Peri/centric heterochromatin is regulated by the polycomb group (PcG) complexes PRC1 and PRC2, which also control most early developmental pathways, and are commonly misregulated in cancer (Simon and Kingston, 2009). Peri/centric heterochromatin is generally silent, but can be expressed, in relation to cell cycle, heat-shock, and stress (Hall et al., 2012). When we found an unexpected association of the BRCA1 tumor-suppressor with satellite DNA during its replication (Pageau and Lawrence, 2006), we hypothesized that satellites might be expressed in cancer (Pageau et al., 2007). Studies subsequently reported peri/centric satellite expression in several cancers (Eymery et al., 2009; Ting et al., 2011; Zhu et al., 2011), but many questions remain as to the extent, specificity and significance of this. Does it reflect broad non-specific heterochromatic instability across all satellites, or might it show family or locus specificity? How does it arise, and, perhaps most importantly, does it have any impact on the cell? Study of satellites is limited using standard genomic approaches, as evidenced by the lack of the extraordinarily large 1q12 HSATII in human genome database. Here we use an approach to study satellite DNA and RNA which preserves molecular information within cell and nuclear/chromosome structure, and allows analysis not only of specific repeat families but specific loci of a given family within single cells.
Our results support a new paradigm for the significance of changes to satellite heterochromatin: the million or more copies of HSATII per cell have a pronounced capacity, at both the DNA and RNA levels, to absorb and impact the distribution of key epigenetic regulators across the nuclear genome. This goes awry in cancer, is linked to the common de-methylation of HSATII, and the “mega-satellite” at 1q12 may play a singular role.
RESULTS
Aberrant nuclear foci of satellite RNA are common in a wide-array of tumor types
We previously showed that a CoT-1 (highly repetitive) DNA probe (Britten and Kohne, 1968) detects an evenly distributed RNA signal across the euchromatic nucleoplasm by FISH (Fig 1A) (Hall et al., 2002; Hall et al., 2014). However, we noted some cell samples also contained very prominent CoT-1 RNA foci (Fig 1B–C, Fig S1A–B), which can be quite large (compare to XIST RNA; Fig 1D). In 22 cell lines surveyed, CoT-1 RNA foci were in 11 of 14 neoplastic lines, but none of 8 normals (Table S1).
Fig 1. Cancer cells contain bright nuclear HSATII RNA foci, not normal cells.

A) Primary fibroblasts with normal nucleoplasmic CoT-1 RNA signal. B–C) Cancer nuclei with large CoT-1 RNA foci. (A–B same scale. Bar 10μm). D) Cot-1 RNA foci versus the XIST RNA territory. E) HSATII RNA overlaps Cot-1 RNA foci. Scale bar 10μm. Red and green channel separated at right. F) H&E stain of breast tumor #2334T. G) HSATII RNA foci in cancerous cells of breast tumor #2334T. Inserts: DNA channel and close-up of region in (G). H) Mutant-P53 staining labels cancerous cells in #2334T tissue. DNA channel separated at right. I–K) Cancer cells (I–J) have aberrant HSATII RNA foci, normal cells (K) do not. Green channel for boxed regions separated at right. Exposure, magnification & scale bar (10um) same for all images. L) Single brightest pixel in 50 cells of two cell lines (IMR90/normal & U2OS/cancer). Threshold for each is 3X average minimum pixel intensity. Most normal cells fall below threshold, but few cancer cells. M) Linescans through three cancer (HCC-1937, MCF-7 & PC3), and two normal nuclei (Tig-1 & WS1) show the size (width) and intensity (height) of HSATII RNA foci. N) Total HSATII RNA signal above threshold for cancer (U2OS) and normal (IMR90) nuclei were quantified (integrated intensity) and the average plotted.
As CoT-1 is heterogeneous, probes to different repeat families (See Supplement) were used to identify specific repeats within the RNA foci. Although interspersed LINE1 or Alu (SINE) repeats were not present in CoT-1 RNA foci (Fig S1C–D), α-SAT (Fig S1E–F) and HSATII were (Fig 1E). These RNA foci are nuclear restricted, and several observations affirm these foci are single stranded RNA: detection without DNA denaturation (which will not detect even these highly abundant DNA sequences) (Lawrence 1989), loss of signal with RNase (Fig S1G–H & Fig S2D–E) or NaOH, specificity for certain cell lines, absence from mitotic chromosomes, and release of these HSATII RNA “bodies” into the cytoplasm of mitotic cells (Fig 6G & Fig S2F).
Fig 6. MeCP2 accumulates with HSATII RNA into CAST bodies.

A–B) MeCP2 bodies in breast cancer (A), and matched normal samples (B). C) MeCP2 bodies are coincident with HSATII RNA foci. Inserts: colors separated for box. D) Linescan of nucleus in image (C). E–F) MeCP2 and HSATII RNA in cultured cancer cells (E) and tumor tissue (F). Inserts: colors separated for image or outlined region. G–H) HSATII RNA foci released to cytoplasm in mitotic cells (G), are still associated with MeCP2 (H). Inserts: colors separated for CAST body (arrow). I) RNA-IP: HSATII RNA is pulled down with MeCP2 & SIN3A, but not MBNL1. J–K) HSATII probes made from MeCP2 (J) or SIN3A (K) RIP labels same HSATII RNA foci in cancer nuclei. L) Average number of L1 RNA foci per nucleus in cancer and normal cells. M) Average integrated intensity of HSATII DNA FISH signal in normal (Tig-1) and cancer (U2OS & PC3) cells using two different probes (“SatII Fitc” = Sat2-59nt probe & “SatII LNA” = Sat2-24nt LNA probe).
We examined α-Sat & HSATII expression by RNA FISH in 14 cancer cell lines and 8 non-cancer lines, 9 human effusion samples and 44 human solid tumors (with matched normal where available) for a wide array of tumor types (Table S1). Given that RNA preservation in pathology samples can be variable, we used poly-A RNA as a positive control. Satellite RNA foci were detectable in over half of the tissue samples (solid tumor and effusion) (Fig 1F–H), and 10 of 14 cancer lines, but in none of the normal tissue or lines (Table S1). Thus, aberrant nuclear satellite RNA foci are common in human cancers, in vivo and in vitro.
Satellite dysregulation is family specific, primarily HSATII and not tied to BRCA1 status
Does this misregulation reflect a generalized failure to maintain silencing on all satellites, or preferentially impact a specific repeat family or locus? This has implications for whether misregulated satellites are a consequence or contributor to neoplasia.
The conserved α-SAT repeat is easily distinguished on all chromosomes, however due to sequence similarity we tested a number of probes and labeling strategies to identify probes (and conditions) that discriminate HSATIII on Chr9 from HSATII on other chromosomes (Fig S1I–M). The HSATIII probe, and an antibody to Heat-shock Factor 1 (HSF1), confirmed that the RNA expressed in these unstressed cancer cells was HSATII (Fig S1I–O)(details in Supplement).
Numerous small dim nuclear foci of human α-SAT RNA were visible in all normal cell lines examined, including proliferative fibroblasts and post-mitotic myotube nuclei (Fig S2A–F & Table S1). Since our initial observations, centric satellite RNAs were reported at very low levels in normal cells (Bierhoff et al., 2014), supporting the accuracy and sensitivity of our in situ approach. However, these small foci were still distinct from the larger α-Sat foci that overlapped CoT-1 RNA foci in some cancer cells (Fig S2G–J).
In contrast to α-sat, HSATII RNA is essentially undetectable in normal cells using multiple HSATII probes (See Supplement)(Fig 1I–N & Table S1). Normal cells only occasionally had a tiny pinpoint of HSATII RNA fluorescence detectable with digital imaging. Conversely, HSATII RNA foci were quite large in cancer nuclei, and distinct enough to identify a single cell as cancerous. This was affirmed by quantitative digital imaging (Fig 1L–N), and showed U2OS cells with ~175 fold more HSATII RNA than normal cells (Fig 1N). The intensity and size (~0.4–1 micron) of these RNA conglomerations (Fig 1D, Table S3) indicates extremely high copies of the 26 bp HSATII sequence.
Examining the frequency of α-Sat and HSATII over-expression, we found α-SAT RNA foci were seen in only two of nine cancer lines (HT1080 and MDA-MB-436) (Fig S1E–F & Table S1), and 25% of tumor samples, whereas HSATII RNA was found, in 8 of 9 cancer lines (70–100% of cells), and about half of tumor and effusion samples (Table S1). Moreover, 50% of tumors with satellite RNA foci had HSATII exclusively (11/22 samples tested for both), and only 9% had α-SAT alone (and 41% with both). Zhu et al. (2011) reported α-SAT RNA expression in BRCA1 −/− breast cancers, and concluded loss of BRCA1 specifically causes this (due to loss of UbH2A deposition). However, BRCA1 (+) tumors were never examined, and our results show α-SAT RNA is neither specific to breast tumors nor to BRCA1 −/− tumors (Table S1), with two of three breast tumors testing normal for BRCA1 still exhibiting satellite RNA foci.
Thus, we find the HSATII family is preferentially expressed in cancer, and satellite expression (α-Sat or HSATII) does not closely correlate with BRCA1 status. Since the difference between normal and cancer cells is most marked for HSATII RNA, we focus on HSATII for the rest of this study.
Importantly, in many (~40%) HSATII positive tumors, HSATII RNA foci were consistently present in essentially all (Fig 1F–H), or most of the tumor cells, suggesting an early event. This, together with the specificity for HSATII, suggests satellite deregulation is not necessarily a sporadic consequence of neoplasia.
PRC1 redistributes into large cancer associated polycomb (CAP) bodies on 1q12, which remains repressed
We examined Polycomb Group (PcG) proteins, known to repress heterochromatin (including satellites), and are implicated in cancer pathogenesis (Sauvageau and Sauvageau, 2010).
Staining for BMI-1, RING1B and Phc1 (PRC1), and EED and EZH2 (PRC2) revealed PRC1 is aberrantly distributed in cancer cells (Fig 2 and Fig S2K), but generally not PRC2 (Fig S2L). BMI-1 labels a few prominent nuclear bodies in 88% of neoplastic lines and 44% of primary tumor samples, but not any normal tissues/lines (Fig 2A–G & Table S2). Since components of PRC1 can vary with cell type, these numbers (using BMI-1) may underestimate the frequency of PRC1 in these bodies. The large (0.4–1.5 microns) BMI-1 “bodies” were often 20 fold brighter than the surrounding nucleoplasm, whereas most normal cells had hundreds of tiny punctates (Fig 2H–L). The most prominent BMI-1 foci in non-cancer cells (e.g. immortalized RPE cells) had at most a “contrast ratio” of just 4:1 (Fig 2I & K). Importantly, the large cancer-associated PRC1 bodies consistently (~100%) and precisely co-localize with 1q12 HSATII DNA (and to a lesser extent HSATII on Chr 16q11) (Fig 3A).
Fig 2. BMI-1 aggregates in large aberrant foci, forming bodies.

A–B) BMI-1 protein accumulates into bodies in cancer cells. C–D) Normal fibroblasts only have faint punctate nucleoplasmic signal. Red channel enhanced at right. Exposures the same for A–C. E) Cancer cell lines contain BMI-1 bodies while non-cancer lines do not. F) Normal human tissue with uniform punctate distribution of BMI-1. G) The matched tumor sample contains BMI-1bodies. H–J) Low nucleoplasmic BMI-1 in fibroblasts (H), slightly higher levels in telomerase immortalized RPE cells (I), and large accumulations in cancer cells (J), with concomitant reduction in nucleoplasmic signal. (H–J same scale). K–L) BMI-1 linescans of the same cells in (H–J).
Fig 3. PRC1 aggregates on HSATII DNA at 1q12 maintaining repression while other loci become de-repressed.

A) The pUC1.77 probe labels HSATII on Chr1 and some on Chr16 (arrows), and correlates with BMI-1 “CAP” bodies. Channels from outlined region are separated at right. B–C) CAP bodies and HSATII RNA are mutually exclusive. Red and green channels of image B at right. D) CAP bodies and HSATII RNA foci in breast tumor tissue. E) HSATII RNA is expressed from small HSATII loci and not from the large 1q12 loci. Small loci are visible when DNA signal (of outlined region) is separated and enhanced at right. HSATII RNA accumulates adjacent to DNA loci (arrows). F–G) Sat2-7 probe labels only 1–2 RNA foci in U2OS cells (F) or in breast tumor #2334T (G). H) Chr7 specific centromere probe verifies Sat2-7 RNA is expressed from Chr7.
Our findings establish that these large PRC1aggregates on 1q12 are not normal nuclear structures, but a new epigenetic hallmark of cancer. Numerous small PcG puncta, widely dispersed in the nucleoplasm (termed “PcG bodies”), are found in all normal cells from Drosophila to humans, and are reported to help regulate gene expression (Bantignies and Cavalli, 2011; Geyer et al., 2011). PcG bodies have often been studied in cancer cells (widely assumed to model normal nuclear structures) (Dundr, 2012), where it was noted cancer and normal cells might differ in the number and size of their “PcG bodies” (Saurin et al., 1998). However, our analysis establishes that the few prominent bodies in cancer are a completely different structure, with different biological significance. To avoid confusion with the small, numerous and widely distributed PcG puncta in normal human cells, often referred to as “PcG bodies”, we refer to these large conglomerations of PRC1 proteins which specifically “cap” the HSATII locus at 1q12 in cancer cells as “CAP” bodies, for “cancer associated Polycomb” bodies.
We next examined the relationship between HSATII RNA foci and PRC1 accumulation into bodies, with two alternate possibilities: HSATII RNA might recruit PRC1 to 1q12, as do other ncRNAs such as XIST, or enrichment of PRC1 at 1q12 might maintain repression, while other loci, in the nucleoplasm with lower levels, may become de-repressed. Results show that the latter consistently occurs; HSATII RNA foci and CAP bodies are spatially separated (0% overlapping, 6% adjacent and 94% no association) (Fig 3B–D), and sequential hybridization to HSATII RNA and DNA (Byron et al., 2013) indicates that RNA foci emanate from small or medium HSATII DNA loci, but not the largest HSATII loci of 1q12 (Fig 3E). Importantly, when CAP bodies and HSATII RNA foci are both detected in a given tumor, they are typically in the same nuclei, suggesting a relationship. For example, in breast tumor #2334T, 80% CAP positive nuclei also contained RNA foci (e.g. Fig 3D), which is likely an underestimate since nuclei in the tissue are sliced.
Locus-specific HSATII sequence sub-types exist and may underlie differences in PRC1 accumulation or deregulation in cancer
The different biological behaviors of HSATII loci raise the question of whether there are variations in HSATII sequences at different loci. Since most HSATII sequence is not in the human genome build (hg19)(particularly 1q12), and it is difficult to establish locus specificity of small tandem repeats by sequence data, in situ hybridization allows us to investigate whether specific sequences are enriched at specific loci.
As detailed in the Supplement we examined a series of probes representing different permutations of divergent HSATII sequences, under varying hybridization stringencies. Because the 1q12 locus is so large, most sequences found on other chromosomes cross-hybridize to 1q12 and vice versa. However, from the larger pUC1.77 cloned sequence from 1q12 we identified a smaller 160bp PCR-generated sequence (Sat2-160) that showed specific hybridization to 1q12, with slight detection of 16q11. This probe did not detect HSATII RNA and correlated strongly with BMI-1 aggregates (Fig S2M–O), consistent with above results. While there is no known consensus sequence for mammalian PRC1 binding, this demonstrates that distinct subsets of HSATII sequences characterize different loci, which likely contributes to distinct biological behavior.
A separate question was whether we could identify specific HSATII sequences/loci that are expressed. As summarized in the Supplement, we identified an expressed sequence by RT-PCR (Fig S3A) that only labeled 1–2 RNA foci in U2OS nuclei (by RNA FISH), which have an unusually high number of RNA foci detected with the broader Sat2-24 LNA probe (Fig 3E–F & Table S3), suggesting locus specificity of the PCR-probe. This probe also labeled the more typical 1–2 RNA foci detected by the permissive LNA probe in tumor tissues (Fig 3G). We tentatively mapped the sequence to chromosome 7 (using hg19), however, since the probe labeled more than one chromosome by DNA FISH, specific centromere probes were used to confirm expression from chromosome 7(Fig 3H). Of five tumor samples that scored high for HSATII RNA, three expressed this sequence (now called Sat2-7)(3 positive breast tumors; #0853, #2004 & #2334, and 2 negative stomach tumors; #2539 & #2210). We also found HSATII sequences from Chr7 are also one of the more commonly expressed sequences in a pancreatic cancer deep-seq database (SRX056914) (Fig S3B), suggesting the Chr7 locus may be commonly misregulated in some cancers.
These findings demonstrate that there is a locus-specific distribution of HSATII sequence sub-types on different loci that may underlie differences in biological behavior. Further, the dysregulation of HSATII in cancer samples is not stochastic nor indiscriminate, but often shows locus and sequence specificity for a given tumor (e.g. Table S3). The HSATII locus at 1q12 consistently nucleates large CAP bodies and is marked by specific sub-types of HSATII not detected in RNA, whereas the Sat2-7 sequence we identified on Chr7p11 appears to be specifically expressed in several tumors studied here.
HSATII changes link to broader epigenetic instability in cancer from DNA demethylation to UbH2A distribution
We investigated whether changes in HSATII and CAP body formation relate to broader epigenetic instability in cancer, and found evidence they do, both in terms of their cause but also likely their consequences. DNA methylation is common in cancer and HSATII demethylation at 1q12 is a frequent and often early change (Ehrlich, 2009). We therefore tested the effects of inducing DNA demethylation on HSATII expression or potentially redistribution of PRC1.
We first examined cells of the rare and fatal ICF syndrome, where a DNMT3B mutation causes HSATII demethylation (Walton et al., 2014), and found PRC1 accumulated in large bodies that “cap” 1q12, in 61% of the ICF fibroblasts (Fig 4E, Fig S3C–D). We next treated normal human fibroblasts with 5-aza-2′-deoxycytidine, which we previously confirmed demethylates HSATII in these cells (Swanson et al., 2013). Remarkably, within 24 hours PRC1 accumulates into 1–2 large bodies over 1q12 (Fig 4A–C). Longer treatment increased the number of cells affected (Day 1=15%, Day 3=52%, Day 8=80%), and the number of bodies/cell (from 2 to 4, representing accumulation on Chr16q11). After several days a subset of cells (5–10%) began to express HSATII RNA from other loci (Fig 4D). Thus, loss of cytosine methylation on HSATII triggers massive recruitment of repressive PRC1 to 1q12, while other loci eventually become derepressed.
Fig 4. CAP bodies are induced by DNA de-methylation and PRC2 activity.

A–B) Fibroblasts treated with 5aza-d form CAP bodies. Red channel separated in (B). C) CAP bodies form over 1q12 in demethylated fibroblasts. Inserts: Colors separated for top body. D) HSATII is expressed following longer 5aza-d treatment. Insert: green channel of outlined region. E) CAP bodies are present in ICF cell nuclei. Inserts: Color channels separated for CAP body. F) EZH2 (PRC2) does not aggregate in CAP bodies following 5-aza. G) Low level H3K27me in controls, with enrichment on the inactive X-chromosome. H) Increased H3K27me following 5-aza treatment. I) H3K27me levels reduced with simultaneous treatment with EZH2-inhibitor and 5-aza. Equal exposures for G–I. J) Number of cells with CAP bodies on Day 3 and Day 5 following treatment with 5-aza (D3-A & D5-A) or 5-aza and EZH2-inhibitor (D3-A/Ei & D5-A/Ei).
Transient activity of PRC2 HMTase EZH2 is required to recruit PRC1 (Hernandez-Munoz et al., 2005). No consistent accumulation of EZH2 was seen on 1q12 in cancer cells (Fig S2L) nor following 5-aza2d treatment (Fig 4F), but an increase in nucleoplasmic H3K27me3 was seen (not limited to nor enriched on pericentromeres)(Fig 4G–H), suggesting PRC2 activation. Inhibition of EZH2 (3-deazaneplanocin) during 5-aza-2d treatment showed reduced H3K27me3 staining (confirming EZH2 inhibition)(Fig 4I), and slowed/reduced CAP body formation (Fig 4J), suggesting EZH2/PRC2 activity is likely involved in CAP body formation.
Corralling PRC1 into CAP bodies likely reduces its level elsewhere, which can be seen by IF in U2OS cells (Fig 2J), at low (10X) magnification in tumor sections (Fig 5A–B), and can be measured by line scan, when comparing neighboring nuclei with and without CAP bodies (Fig 5C–E; renal tumor). These findings point to a new biological significance for satellite demethylation in human cancers, due to the marked capacity of this extremely high-copy repeat at 1q12, to act as a molecular “sponge”, thereby impacting compartmentalization and accessibility of important nuclear factors.
Fig 5. CAP bodies sequester PRC1 affecting genomic distribution of UbH2A.

A) Low mag (10X) view of BMI-1 in tumor tissue. B) Magnification of area in box of Image A, show cells with CAP bodies (arrows) and reduced nucleoplasmic BMI-1. C–E) Tumor cells with CAP bodies have reduced nucleoplasmic BMI-1 compared to neighboring cells lacking CAP bodies. Green channel separated in (D), and linescan of cell 1 and 2 (E). F) ChIP-seq: Total HSATII reads with UbH2A in cancer versus normal cells. G) ChIP-seq: UbH2A levels on expressed (Sat2-7) versus repressed (1q12) HSAT II sequences in normal and cancer cells (total reads FigS3E). H–I) ChIP-seq: distribution of UbH2A across the genome in normal (H), and cancer (I) cells. J) ChIP-PCR: UbH2A enrichment on 1q12, HoxC5 and β-actin loci before and after 5aza treatment. K) RING1B/RNF2 on the EGLN3 gene (involved in hereditary breast and ovarian cancer) in cancer cells and normal foreskin and breast fibroblasts.
To assess potential downstream effects of PRC1 redistribution into CAP bodies we examined UbH2A distribution (laid down by PRC1), by ChiP, in cancer and normal cells. Reads mapping to HSATII increased sharply in cancer cells, from 8% in normal Tig-1 fibroblasts to 37% in U2OS osteosarcoma cells (Fig 5F). This is consistent with PRC1 accumulation at 1q12, which would dominate the HSATII sequence population. The distribution of UbH2A on 1q12 (Sat2-160) versus Chr7 (Sat2-7) satellites in normal cells is similar (since both loci are repressed), whereas in U2OS cancer cells the 1q12 sequences become disproportionately enriched for UbH2A over the expressed Chr7 sequence (Fig 5G & Fig S3E). Throughout the broader epigenome, UbH2A distributed more uniformally in Tigs (Fig 5H), whereas in U2OS more sequences deviate from average, particularly at the low ends (Fig 5I). Gene ontology analysis for genes with reduced UbH2A (4-fold or greater) in cancer cells suggested effects on genes involved in neural differentiation (Table S4A), consistent with the known role of BMI-1 (Corley and Kroll, 2015). Ting et al. also noted increased neural gene expression, a known poor prognostic indicator, was increased in cancers expressing HSATII (Ting et al., 2011). We also examined PRC1 distribution in published ChIP-seq datasets for RING1B/RNF2 (Pemberton et al., 2014; Rai et al., 2015) and found an increase in HSATII reads pulled down in cancer compared to normal, and proportionally more on 1q12 (Sat2-160) than Chr7 (Sat2-7) (Fig S3F). The genes that lost RING1b in cancer were also enriched for genes important to carcinogenesis (Table S4B & Fig 5K), illustrating how redistribution of PCR1 away from canonical genes could contribute to tumorigenesis (Wang et al., 2015).
We also used ChIP-PCR to examine UbH2A levels on three specific sequences in fibroblasts induced to form CAP bodies with 5-aza. UbH2A was again clearly increased on the 1q12 (Sat2-160), whereas HoxC5, an established PcG regulated gene, demonstrated a corresponding sharp loss of UbH2A, with no significant change in the beta-actin housekeeping gene (Fig 5J). These results further support that redistribution of PRC1 proteins into CAP bodies is associated with reduced access to repressive factors elsewhere, impacting UbH2A levels and making genes more vulnerable to de-repression over time (e.g. HSATII at Chr7).
MeCP2 accumulates with HSATII RNA transcripts into cancer-associated bodies
Since HSATII DNA repeats can “sponge-up” PRC1, it follows that the similarly large HSATII RNA foci, containing likely millions of copies of this small repeat, might likewise sequester specific RNA-binding or regulatory factors, potentially similar to “Toxic repeat RNAs” in other human diseases (see Discussion).
Therefore, we examined whether HSATII RNA foci might accumulate MBNL1 (sequestered in repeat RNA in myotonic dystrophy type I), which it did not, nor did HSATII RNA overlap the PNC domain that sequesters proteins in many cancers (Wen et al., 2013). Surprisingly, we discovered the methyl-DNA binding protein, MeCP2, is found in large nuclear bodies that precisely overlap the HSATII RNA foci. These large MeCP2 foci (Fig 6A) are distinct from the broadly distributed small puncta in normal cells (Fig 6B), and can be seen homogeneously distributed throughout many tumors at low magnification (Fig S3G–I). We initially examined MeCP2 due to its capacity to bind methylated DNA, and although MeCP2 foci were similar in size and number to both HSATII RNA foci and CAP bodies, detailed analysis clearly showed they were associated with the RNA, not DNA (Fig 6C–F) (using two different antibodies and several cancer samples). For example, MeCP2 exactly overlaps HSATII RNA foci (Fig 6D), and since HSATII RNA foci are immediately adjacent to the DNA locus (Fig 3E) MeCP2 is not over the DNA. In addition, MeCP2 continues to overlap HSATII RNA foci when the “body” enters the cytoplasm at mitosis (Fig 6G–H).
We further show that MeCP2 and HSATII RNA co-immunoprecipitate from cancer cells by RNA-IP (RIP). HSATII RNA was consistently enriched in MeCP2 pull downs over IgG controls (Fig 6I), whereas α-SAT RNA was not. RIP with MBNL1 did not pull down HSATII RNA (confirming in situ results), while Sin3A, a transcriptional regulator which can complex with MeCP2 (Jones et al., 1998) did pull it down in some experiments. Using the RNA pulled down by MeCP2 (or SIN3A) as probes, we detected HSATII RNA foci in cancer cells by RNA FISH (Fig 6J–K). Thus, the RNA foci of HSATII are not inert but also can bind and impact distribution of epigenetic factors. We term these large round aggregates of HSATII RNA with MeCP2 “cancer associated satellite transcript” (CAST) bodies, and identify them as another unique hallmark of cancer cells (in vitro and in vivo).
MeCP2 is known to repress expression of LINE1 elements (Erwin et al., 2014), which are inappropriately expressed in cancer (Rodic and Burns, 2013). We recently showed that L1 sequences are a major part of the repeat-rich “CoT-1 RNA” that associates with euchromatin, and may serve to maintain open chromatin (Hall et al., 2014). The peripheral heterochromatin compartment in most normal cells characteristically lacks this L1-rich CoT-1 RNA (Fig S3J–K), reflecting a stable heterochromatic state. However, in cancer cells, CoT-1/L1 RNA is detected throughout this peripheral compartment, indicating compromise of this heterochromatin (Fig S3L–M; (Pageau et al., 2007)). In addition, we find U2OS tumor cells have more L1 RNA foci in nuclei compared to normal cells (Tig-1) (Fig 6L), consistent with other reports of L1 activation in cancers. Hence the aberrant distribution of MeCP2 in cancer nuclear structure may be relevant to broader changes in cancer expression patterns.
Finally, we did not find evidence that HSATII RNA expression leads to massive tandem expansion of HSATII DNA loci, as a recent report suggests (Bersani et al., 2015). These authors postulate that HSATII cDNA (via reverse-transcription of RNA) is re-incorporated into and amplifies existing HSATII loci (e.g. up to 25 fold), since they saw no change in the number loci by DNA FISH. However, the relative size and intensity of DNA FISH signals is proportional to target size and much smaller differences (2–3 fold) are easily discernible (Lawrence et al., 1988). Using FISH and microfluorimetry to quantify the HSATII DNA signals in normal and cancer cells, we found no substantial differences in size, intensity, number of signals, or total HSATII signal (Fig 6M). This was quantified in U2OS osteosarcoma but similar results were observed in other cancers. We detect a slight increase in total HSATII DNA in U2OS, due to an extra copy of the large 1q12 satellite (not tandem amplification), as evident in Figure 3A.
The model and legend in Figure 6 summarizes and integrates several major findings in cancer nuclear structure and the epigenome which we show are connected to complex, and locus-specific biology of HSATII.
DISCUSSION
This work demonstrates multiple new properties of cancer cell nuclear structure, with implications for epigenetic changes in cancer, and at the same time points to the unanticipated biological importance of human HSATII DNA, a large, unexplored component of human genomes. We show that HSATII RNA accumulates in large nuclear foci in over half of all human cancers examined. This occurs both in vivo and in vitro, and is satellite family and locus specific. We also show an unexpected relationship between a specific HSATII DNA locus (1q12) and PRC1 distribution in cancer, directly related to DNA demethylation. Both HSATII RNA and demethylated HSATII DNA impact the nuclear distribution of two classes of important epigenetic regulators, corralling them into two types of aberrant nuclear bodies and reducing their accessibility to the rest of the nucleus. These findings support a major new concept for the biological importance of HSATII pericentric satellites, and their mis-regulation in cancer, whereby these exceptionally high-copy small tandem repeats, at both the DNA and RNA levels, can act as a molecular “sponge” which impacts the distribution of epigenetic regulators throughout the nuclear epigenome. This paves the way for new avenues of investigation into HSATII contribution to epigenomic regulation within nuclear structure, in cancer and possibly development.
While studying repetitive CoT-1 RNAs we independently discovered HSATII RNAs in cancers, and our results now affirm Ting et al. (2011) who reported abundant HSATII in RNAseq data from pancreatic cancer. Our study extends far beyond the initial observation of HSATII RNA expression to illuminate the fundamental biology in several major ways, giving rise to a new concept for the potential significance of HSATII changes via impact on the epigenome. We examined three satellite families (α-SAT, HSATII and HSATIII) in an array of tumor types and found preferential expression of HSATII. Zhu et al. (2011) reported α-SAT expression in BRCA1 dependent breast cancers and concluded this was due to BRCA1 loss. However, that study did not examine BRCA1(+) tumors for comparison. Our work clarifies that satellite mis-regulation is widespread in many tumors, irrespective of whether tumors maintain BRCA1, and HSATII RNA was more frequently expressed.
This study provides a new conceptual framework to investigate HSATII biology, but also demonstrates a powerful approach to study satellites, which can reveal biological relationships that would escape other approaches. Study of such highly-repetitive sequences is notoriously difficult by standard genomic approaches. The abundance of these simple repeats makes contamination a nearly intractable problem during extraction (reviewed in: (Vourc’h and Biamonti, 2011)), making it likely that HSATII DNA and RNA hybrids form in vitro rather than generated in vivo by a postulated reverse transcriptase (Bersani et al, 2015). Earlier evidence suggested that duplex molecules of highly repetitive sequences were impossible to avoid during RNA extractions (Fedoroff et al., 1977). Similarly, the abundance of HSATII RNA specifically in cancer may also complicate quantification of extracted satellite DNA. Using in situ hybridization we were able to study the 1q12 satellite (not in the genome build), and found no substantial HSATII amplification within existing loci.
Novel Biology and Unexplored Significance of Human Satellite II
The study of pericentric HSATII has lagged far behind the rest of the genome, but our results provide evidence for its biological significance, based on its capacity to amass and sequester cytological scale (i.e. huge) accumulations of epigenetic factors. Theoretically, if each 26 bp HSATII repeat in the two largest (~6 Mb) 1q12 loci could bind PRC1; just this locus alone could corral roughly 0.5 × 106 such factors. Our results also indicate that the reason PRC1 “piles up” on HSATII at 1q12 (disproportionately to size) is likely because it contains sequence subtypes not significantly detected on other chromosomes. Similarly, abundant HSATII repeat RNAs aggregated into large nuclear foci can also act as sponges for MeCP2 in cancer nuclei. The effects of HSATII on PRC1 and MeCP2 may be the “tip of the iceberg”, as these and other repeat sequences (half the genome) could potentially regulate distribution of chromatin regulatory factors.
This connection between HSATII DNA methylation, and PcG distribution has similarities to findings in normal mouse ESCs (Cooper et al., 2014) which showed that induced demethylation of mouse major-satellite DNA triggers recruitment of PRC1 and PRC2 to all mouse pericentromeres. PcG factors were also drawn away from canonical gene targets, bolstering the premise here that sequestration of factors on satellite repeats can impact other regions of the epigenome. However, there are interesting differences between our findings in human and those in mouse. In human, PRC1 preferentially localizes to the singular pericentric HSATII locus on 1q12 rather than all pericentromeres as in mice. Also, we generally did not find PRC2 accumulated with PRC1 on satellites as seen in mouse, although our results suggest PRC2 may be required for CAP body formation. Most importantly, findings here show that CAP body formation at 1q12 occurs naturally in vivo, is not a “normal” nuclear structure, and is a new hallmark of cancer seen in many tumors.
Given that this relationship to 1q12 is seen in vivo, albeit in a disease context, we postulate that it may have a normal function during development, such as global epigenomic programming in early development, or gametogenesis. Hypomethylation of satellites has been reported to be a hallmark of gametes, preimplantation embryonic cells, and extraembryonic tissues (e.g. (Kaneda et al., 2011; Zagradisnik and Kokalj-Vokac, 2000)). Similarly, HSATII RNA foci may have some normal role during development or in specific cell-types, potentially impacting distribution of MeCP2 or other regulators. Satellites have been reported to be transcribed in testis, brain, undifferentiated ESCs and pre-implantation embryos (Walton et al., 2014). Although MeCP2 is known to be a methyl-DNA “reader” mutated in Rhett’s syndrome, our findings bolster evidence that MeCP2 also binds RNA (Long et al., 2011; Young et al., 2005), and the specific affinity for HSATII RNA (contrasted to α-Sat) may be to regulate MeCP2 rather than visa versa.
HSATII DNA and Epigenetic Instability in Cancer
DNA methylation or PcG proteins are heavily studied in cancer, but the frequent hypomethylation of HSATII is not known to have a functional impact. The new HSATII biology shown here provides a nexus between changes in DNA methylation and polycomb repression (e.g. UbH2A). The 1q12 “mega-satellite” may play a particular role, as increased copies of 1q are especially frequent in cancer, particularly breast (Mertens et al., 1997). Our findings indicate that HSATII demethylation and 1q12 copy number may not only be caused by, but may contribute to epigenetic instability in cancer, possibly providing a survival advantage (e.g. a bigger “sponge” for PcG proteins).
HSATII demethylation and consequent CAP body formation is associated with aberrant distribution of ubH2AK119 more broadly. These observations shed light on the unexplained link between global DNA hypomethylation and derepression of specific polycomb target genes (Jin et al., 2008; Reddington et al., 2013), and may also explain why de-repression of PcG-targeted genes in ICF cells (or hypomethylated mouse cells) did not correlate with de-methylation of gene promoters (as expected), but with dramatic changes in their histone modifications (Jin et al., 2008). Hence, the collective evidence supports the possibility these changes in HSATII could further compromise heterochromatin on a broader scale. Based on knowledge of the inactive X-chromosome, loss of a single repressive histone mark would not be expected to result in immediate reactivation, since multiple chromatin modifications act synergistically to repress chromatin (Csankovszki et al., 2001). Instead, “leaky” maintenance and sporadic activation of different PcG regulated genes would occur in individual cells. We suggest this would generate a diversity of expression profiles with more opportunity for those that favor neoplastic progression (Carone and Lawrence, 2013; Timp and Feinberg, 2013), as changes in PcG regulated genes (Wang et al., 2015), and in UbH2A distribution can impact oncogenesis (Bhatnagar et al., 2014). Stochastic reactivation and clonal selection may also explain why not all HSATII loci express in the nucleoplasm low in PRC1, and why expression appears locus-specific for a given tumor.
In this context it is striking to consider that in a number of HSATII RNA positive tumors, cells throughout a given tumor show homogeneity of phenotype with respect to the number of HSATII RNA foci and particular loci expressed. Such results argue against this being a simple stochastic mismanagement of the cancer epigenome in different cells of a tumor, but rather indicate a clonal expansion of an early expression profile. Although it remains to be determined if some satellite expression can contribute to a selective advantage, we found the Chr7 locus expressed in multiple tumors in a small sample, and several studies have reported amplification of Chr7 pericentromere is common and may be a neoplastic “driver” in many types of solid tumors (including 80% of metaplastic breast cancers) (e.g. (Reis-Filho et al., 2006; Sanson et al., 2011)). We would also argue that the prevalence of PRC1 “capping” 1q12 in 50% of tumors is clearly non-random and thus early loss of DNA methylation on HSATII could promote sporadic reactivation of PcG regulated genes/loci in individual cells, anyone of which might favor inappropriate growth and neoplastic progression.
“Toxic repeat RNAs” in cancer can impact epigenetic regulators
This study provides evidence that the HSATII RNA expressed in cancer is not just an inert biomarker of epigenetic dysregulation, but can accumulate MeCP2 (and likely other regulatory factors) into aberrant nuclear bodies, possibly contributing to further epigenetic changes. HSATII RNA foci are highly reminiscent of nuclear “toxic repeat RNAs” in certain triplet repeat expansion disorders (myotonic dystrophy type I), in which sequestration of alternative splicing factors by repeat RNA is key to disease pathology (Sicot and Gomes-Pereira, 2013). Sequestration of MeCP2 is unlikely to be innocuous, as MeCP2 is a broad regulator, including miRNA processing (Woo and Kim, 2014) and LINE-1 repression (Yu et al., 2001), which is inappropriately expressed in many cancers (Carreira et al., 2014; Rodic and Burns, 2013). This would explain why we see increased L1 RNA in our cancer samples and why Ting et al (2011) noted L1 was over-expressed in cancers expressing HSATII.
Finally, these findings also have potential implications for cancer diagnosis and treatment. First, cancer-specific bodies discovered here may provide new biomarkers of epigenetic instability with potential prognostic utility. These bodies can be seen in a single cell of a pathology specimen, and our survey of tumors provides a preliminary indication that this is more common in higher grade tumors. It is also notable that the demethylating agents used here are in clinical development for cancer therapy (to reactivate tumor suppressor genes)(Kelly et al., 2010), however our results caution that demethylation of repeat sequences can impact master epigenetic regulators and amplify effects on the nuclear epigenome.
EXPERIMENTAL PROCEDURES
Cell lines, growth conditions & fixation
All cell lines (list in Supplement), were grown in conditions recommended by suppliers (ATCC, Cambrex, and Coriell). 5-aza-2′deoxycytidine (0.2ug/ml) and 3-Deazaneplanocin A (EZH2 inhibitor)(5μM) was added fresh daily to asynchronously growing cultures. Our standard fixation protocols have been detailed previously (Byron et al., 2013; Johnson et al., 1991; Tam et al., 2002)(details in Supplement).
FISH and IF
Our standard hybridization conditions for RNA, DNA, simultaneous DNA/RNA, and simultaneous DNA or RNA and IF detection was performed as previously described (e.g. (Byron et al., 2013))(details in Supplement). Sat2-24nt LNA oligo was used for most images unless otherwise indicated. List of probes are in the Supplement. Oligos were usually hybridized at 15% formamide, unless higher stringency hybridizations were needed (40% and 50% formamide).
RNA-IP
RIPs were carried out essentially as described (Jones et al., 1998; Long et al., 2011) (details in Supplement).
ChIP-Seq & ChIP-PCR
ChIP and Illumina paired end deep sequencing was performed as previously described (Yildirim et al., 2012) with some modification (details in Supplement). Deep sequencing data was mapped to human genome build hg19 using Bowtie. Data normalization and peak calling was performed over a 10kb sliding window using SeqMonk.
Statistics
For the ChIP qPCR data, 95% confidence intervals were calculated (p<.05) when comparing between groups (treated and control). Data including error bars of 95% confidence for B-actin gene are shown. ChIP-seq peaks was computed assuming a Poisson distribution and a cutoff of p < 10-5. Peak overlap defined to be within 50 bp of each summit. P-values for Disease Ontology enrichment were computed using a binomial distribution, corrected for multiple hypothesis testing by the Benjamini and Hochberg procedure.
Microscopy and Digital Imaging
An Axiovert 200 or an Axiophot Zeiss microscope was used equipped with a 100X PlanApo objective (NA 1.4) and Chroma 83000 multi-bandpass dichroic and emission filter sets (Brattleboro, VT), set up in a wheel to prevent optical shift. Images were captured with the Zeiss AxioVision software, and an Orca-ER camera (Hamamatsu, NJ).
Data and Software Availability
UbH2A Chip-seq accession #XXX
Supplementary Material
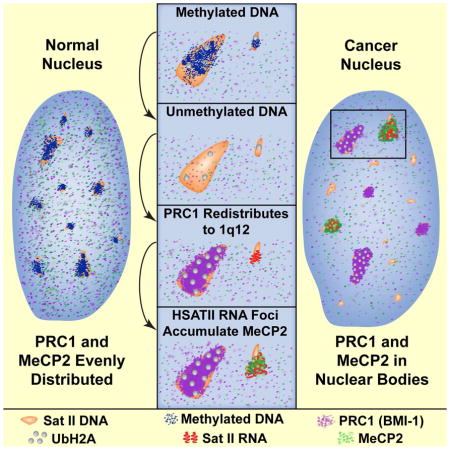
Fig 7. Demethylated HSATII DNA and aberrant HSATII RNA foci can sequester copious epigenetic factors and impact the genomic balance of heterochromatin marks.

HSATII DNA loci in normal cells are methylated, ubiquitinated and repressed. When HSATII DNA becomes de-methylated, PRC1 proteins aggregate specifically at HSATII mega-satellite on 1q12 (and sometimes 16q11), where it increases UbH2A levels and maintains repression. Meanwhile, other HSATII loci in the nucleoplasm lower in PRC1 become de-repressed and begin to express HSATII RNA, which in turn binds large amounts of MeCP2. Thus, high copy HSATII DNA and RNA can accumulate large amounts of epigenetic factors into cancer-associated PcG (CAP) bodies and cancer-associated satellite transcript (CAST) bodies, and impact their accessibility in the cancer epigenome.
HIGHLIGHTS.
HSATII misregulation is common in cancers and is locus and tumor specific
Demethylation of HSATII DNA at the 1q12 mega-satellite sequesters PRC1 into CAP bodies
Large RNA foci from other specific HSATII loci accumulate MeCP2 into CAST bodies
Sequestration of master regulators by HSATII may further compromise the epigenome
Acknowledgments
This work was supported by NIH grants #GM107604 and #GM053234 supplemented by #IRG 93-033 from the American Cancer Society. We thank Dr. Cherie Taglienti from the UMass Tumor Tissue Bank, Heather Kolpa and Alvin Gomez for early FISH experiments, and Takako Jones for help with HSATII RIP.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, J.B.L. and L.L.H.; Investigation, M.B., D.M.C. and L.L.H., early experiments: G.P.P., RNF2 ChIP-seq analysis: T.W.; RNA-IP: P.J., Pathologist: A.F.; Writing, L.L.H., J.B.L. and D.M.C.; Methodology, J.B.L. and L.L.H.; Visualization, M.B., L.L.H., and D.M.C.; Funding Acquisition, J.B.L.; Supervision, J.B.L.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bantignies F, Cavalli G. Polycomb group proteins: repression in 3D. Trends Genet. 2011;27:454–464. doi: 10.1016/j.tig.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Bersani F, Lee E, Kharchenko PV, Xu AW, Liu M, Xega K, MacKenzie OC, Brannigan BW, Wittner BS, Jung H, et al. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc Natl Acad Sci U S A. 2015;112:15148–15153. doi: 10.1073/pnas.1518008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, Gazin C, Chamberlain L, Ou J, Zhu X, Tushir JS, Virbasius CM, Lin L, Zhu LJ, Wajapeyee N, et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature. 2014;516:116–120. doi: 10.1038/nature13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biamonti G, Vourc’h C. Nuclear stress bodies. Cold Spring Harb Perspect Biol. 2010;2:a000695. doi: 10.1101/cshperspect.a000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhoff H, Postepska-Igielska A, Grummt I. Noisy silence: non-coding RNA and heterochromatin formation at repetitive elements. Epigenetics. 2014;9:53–61. doi: 10.4161/epi.26485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ, Kohne DE. Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science. 1968;161:529–540. doi: 10.1126/science.161.3841.529. [DOI] [PubMed] [Google Scholar]
- Byron M, Hall LL, Lawrence JB. A multifaceted FISH approach to study endogenous RNAs and DNAs in native nuclear and cell structures. Curr Protoc Hum Genet. 2013;Chapter 4(Unit 4):15. doi: 10.1002/0471142905.hg0415s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone DM, Lawrence JB. Heterochromatin instability in cancer: from the Barr body to satellites and the nuclear periphery. Semin Cancer Biol. 2013;23:99–108. doi: 10.1016/j.semcancer.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira PE, Richardson SR, Faulkner GJ. L1 retrotransposons, cancer stem cells and oncogenesis. FEBS J. 2014;281:63–73. doi: 10.1111/febs.12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, De Marco V, Elderkin S, Koseki H, Klose R, et al. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 2014;7:1456–1470. doi: 10.1016/j.celrep.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley M, Kroll KL. The roles and regulation of Polycomb complexes in neural development. Cell Tissue Res. 2015;359:65–85. doi: 10.1007/s00441-014-2011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csankovszki G, Nagy A, Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol. 2001;153:773–784. doi: 10.1083/jcb.153.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr M. Nuclear bodies: multifunctional companions of the genome. Curr Opin Cell Biol. 2012;24:415–422. doi: 10.1016/j.ceb.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–259. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin JA, Marchetto MC, Gage FH. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci. 2014;15:497–506. doi: 10.1038/nrn3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eymery A, Horard B, El Atifi-Borel M, Fourel G, Berger F, Vitte AL, Van den Broeck A, Brambilla E, Fournier A, Callanan M, et al. A transcriptomic analysis of human centromeric and pericentric sequences in normal and tumor cells. Nucleic Acids Res. 2009;37:6340–6354. doi: 10.1093/nar/gkp639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff N, Wellauer PK, Wall R. Intermolecular duplexes in heterogeneous nuclear RNA from HeLa cells. Cell. 1977;10:597–610. doi: 10.1016/0092-8674(77)90092-7. [DOI] [PubMed] [Google Scholar]
- Feinberg A. DNA methylation in cancer: three decades of discovery. Genome Med. 2014;6:36. doi: 10.1186/gm553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer PK, Vitalini MW, Wallrath LL. Nuclear organization: taking a position on gene expression. Curr Opin Cell Biol. 2011;23:354–359. doi: 10.1016/j.ceb.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall LE, Mitchell SE, O’Neill RJ. Pericentric and centromeric transcription: a perfect balance required. Chromosome Res. 2012;20:535–546. doi: 10.1007/s10577-012-9297-9. [DOI] [PubMed] [Google Scholar]
- Hall LL, Byron M, Sakai K, Carrel L, Willard HF, Lawrence JB. An ectopic human XIST gene can induce chromosome inactivation in post differentiation human HT-1080 cells. Proc Natl Acad Sci U S A. 2002;99:8677–8682. doi: 10.1073/pnas.132468999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall LL, Carone DM, Gomez AV, Kolpa HJ, Byron M, Mehta N, Fackelmayer FO, Lawrence JB. Stable C0T-1 repeat RNA is abundant and is associated with euchromatic interphase chromosomes. Cell. 2014;156:907–919. doi: 10.1016/j.cell.2014.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Munoz I, Taghavi P, Kuijl C, Neefjes J, van Lohuizen M. Association of BMI1 with polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol Cell Biol. 2005;25:11047–11058. doi: 10.1128/MCB.25.24.11047-11058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, Fields CR, Delmas AL, Liu X, Qiu J, et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet. 2008;17:690–709. doi: 10.1093/hmg/ddm341. [DOI] [PubMed] [Google Scholar]
- Johnson CV, Singer RH, Lawrence JB. Fluorescent detection of nuclear RNA and DNA: Implication for genome organization. Methods Cell Biol. 1991;35:73–99. [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Kaneda M, Akagi S, Watanabe S, Nagai T. Comparison of DNA methylation levels of repetitive loci during bovine development. BMC Proc. 2011;5(Suppl 4):S3. doi: 10.1186/1753-6561-5-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JB, Villnave CA, Singer RH. Sensitive, high-resolution chromatin and chromosome mapping in situ: presence and orientation of two closely integrated copies of EBV in a lymphoma line. Cell. 1988;52:51–61. doi: 10.1016/0092-8674(88)90530-2. [DOI] [PubMed] [Google Scholar]
- Long SW, Ooi JY, Yau PM, Jones PL. A brain-derived MeCP2 complex supports a role for MeCP2 in RNA processing. Biosci Rep. 2011;31:333–343. doi: 10.1042/BSR20100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens F, Johansson B, Hoglund M, Mitelman F. Chromosomal imbalance maps of malignant solid tumors: a cytogenetic survey of 3185 neoplasms. Cancer Res. 1997;57:2765–2780. [PubMed] [Google Scholar]
- Pageau GJ, Hall LL, Ganesan S, Livingston DM, Lawrence JB. The disappearing Barr body in breast and ovarian cancers. Nat Rev Cancer. 2007;7:628–633. doi: 10.1038/nrc2172. [DOI] [PubMed] [Google Scholar]
- Pageau GJ, Lawrence JB. BRCA1 foci in normal S-phase nuclei are linked to interphase centromeres and replication of pericentric heterochromatin. J Cell Biol. 2006;175:693–701. doi: 10.1083/jcb.200602055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton H, Anderton E, Patel H, Brookes S, Chandler H, Palermo R, Stock J, Rodriguez-Niedenfuhr M, Racek T, de Breed L, et al. Genome-wide co-localization of Polycomb orthologs and their effects on gene expression in human fibroblasts. Genome Biol. 2014;15:R23. doi: 10.1186/gb-2014-15-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K, Akdemir KC, Kwong LN, Fiziev P, Wu CJ, Keung EZ, Sharma S, Samant NS, Williams M, Axelrad JB, et al. Dual Roles of RNF2 in Melanoma Progression. Cancer Discov. 2015;5:1314–1327. doi: 10.1158/2159-8290.CD-15-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddington JP, Perricone SM, Nestor CE, Reichmann J, Youngson NA, Suzuki M, Reinhardt D, Dunican DS, Prendergast JG, Mjoseng H, et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. 2013;14:R25. doi: 10.1186/gb-2013-14-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis-Filho JS, Pinheiro C, Lambros MB, Milanezi F, Carvalho S, Savage K, Simpson PT, Jones C, Swift S, Mackay A, et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J Pathol. 2006;209:445–453. doi: 10.1002/path.2004. [DOI] [PubMed] [Google Scholar]
- Rodic N, Burns KH. Long interspersed element-1 (LINE-1): passenger or driver in human neoplasms? PLoS Genet. 2013;9:e1003402. doi: 10.1371/journal.pgen.1003402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22:50–55. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Sanson M, Hosking FJ, Shete S, Zelenika D, Dobbins SE, Ma Y, Enciso-Mora V, Idbaih A, Delattre JY, Hoang-Xuan K, et al. Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum Mol Genet. 2011;20:2897–2904. doi: 10.1093/hmg/ddr192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurin AJ, Shiels C, Williamson J, Satijn DP, Otte AP, Sheer D, Freemont PS. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J Cell Biol. 1998;142:887–898. doi: 10.1083/jcb.142.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicot G, Gomes-Pereira M. RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim Biophys Acta. 2013;1832:1390–1409. doi: 10.1016/j.bbadis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- Swanson EC, Manning B, Zhang H, Lawrence JB. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol. 2013;203:929–942. doi: 10.1083/jcb.201306073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam R, Shopland LS, Johnson CV, McNeil J, Lawrence JB. Applications of RNA FISH for visualizing gene expression and nuclear architecture. Vol. 260. New York: Oxford University Press; 2002. [Google Scholar]
- Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13:497–510. doi: 10.1038/nrc3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331:593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourc’h C, Biamonti G. Transcription of Satellite DNAs in Mammals. Prog Mol Subcell Biol. 2011;51:95–118. doi: 10.1007/978-3-642-16502-3_5. [DOI] [PubMed] [Google Scholar]
- Walton EL, Francastel C, Velasco G. Dnmt3b Prefers Germ Line Genes and Centromeric Regions: Lessons from the ICF Syndrome and Cancer and Implications for Diseases. Biology (Basel) 2014;3:578–605. doi: 10.3390/biology3030578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Qin JJ, Voruganti S, Nag S, Zhou J, Zhang R. Polycomb Group (PcG) Proteins and Human Cancers: Multifaceted Functions and Therapeutic Implications. Med Res Rev. 2015;35:1220–1267. doi: 10.1002/med.21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Wang C, Huang S. The perinucleolar compartment associates with malignancy. Front Biol (Beijing) 2013;8 doi: 10.1007/s11515-013-1265-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo JS, Kim VN. MeCP2 caught moonlighting as a suppressor of MicroRNA processing. Dev Cell. 2014;28:477–478. doi: 10.1016/j.devcel.2014.02.015. [DOI] [PubMed] [Google Scholar]
- Yildirim E, Sadreyev RI, Pinter SF, Lee JT. X-chromosome hyperactivation in mammals via nonlinear relationships between chromatin states and transcription. Nat Struct Mol Biol. 2012;19:56–61. doi: 10.1038/nsmb.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci U S A. 2005;102:17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Zingler N, Schumann G, Stratling WH. Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res. 2001;29:4493–4501. doi: 10.1093/nar/29.21.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagradisnik B, Kokalj-Vokac N. Hypomethylation of alphoid DNA and classical satellite DNA on chromosome 1, 9, 16 and Y in extraembryonic tissue. Pflugers Arch. 2000;440:R190–192. [PubMed] [Google Scholar]
- Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477:179–184. doi: 10.1038/nature10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.