

A three-dimensional culture system enables maintenance of pluripotent stem cells (PSCs) and formation of PSC-derived organoids.
Keywords: pluripotent stem cell, three-dimensional culture, organoid, bone, osteoblast
Abstract
The development of in vitro models for the maintenance and differentiation of pluripotent stem cells (PSCs) is an active area of stem cell research. The strategies used so far are based mainly on two-dimensional (2D) cultures, in which cellular phenotypes are regulated by soluble factors. We show that a 3D culture system with atelocollagen porous scaffolds can significantly improve the outcome of the current platforms intended for the maintenance and lineage specification of mouse PSCs (mPSCs). Unlike 2D conditions, the 3D conditions maintained the undifferentiated state of mouse embryonic stem cells (mESCs) without exogenous stimulation and also supported endoderm, mesoderm, and ectoderm differentiation of mESCs under serum-free conditions. Moreover, 3D mPSC–derived mesodermal cells showed accelerated osteogenic differentiation, giving rise to functional osteoblast-osteocyte populations within calcified structures. The present strategy offers a 3D platform suitable for the formation of organoids that mimic in vivo organs containing various cell types, and it may be adaptable to the generation of ectoderm-, mesoderm-, and endoderm-derived tissues when combined with appropriate differentiation treatments.
INTRODUCTION
Offering greater physiological relevance than conventional two-dimensional (2D) cultures, 3D systems have become a promising alternative for bridging the gap between in vitro cultures and living tissues. Cell-cell and cell–extracellular matrix (ECM) interactions have been recognized to be fundamentally different between 2D and 3D cultures; as a consequence of this difference, the molecular pathways that regulate cell behaviors are also altered, leading to distinct biological outcomes, such as cell phenotypes and functions (1). A classic and clinically relevant example of a dimensionality-mediated cell response was shown in murine mammary tumor sublines that are highly resistant to cyclophosphamide, an anticancer drug. The drug-resistant phenotypes were not observed in cells growing as monolayers (2). However, cells cultured under 3D conditions recapitulated the drug resistance properties that previously had been seen only in vivo. Thus, 3D cultures may offer an attractive tool for stem cell research, in which the control of cell phenotypes is often required (3–5).
Mouse pluripotent stem cells (mPSCs) are invaluable resources for drug screening, disease studies, and developmental investigations. Initially, the self-renewal of mPSCs, including mouse embryonic stem cells (mESCs) and mouse induced PSCs (miPSCs), was achieved in monolayer cultures with mitotically inactivated mouse embryonic fibroblast. Leukemia inhibitor factor (LIF) was then identified as the factor that could maintain the pluripotency of mPSCs (6–8). Nowadays, the ground state of mPSCs is achieved using the combination of LIF and two inhibitors, CHIR99021 (CHIR) and PD032591 (PD) (2iLIF), under defined conditions (9).
The use of small molecules to control stem cell fates is also increasingly common, exploring areas such as reprogramming, cell proliferation, differentiation, dedifferentiation, and transdifferentiation in various stem cell schemes (10–13). We recently reported an effective stepwise differentiation protocol for osteoblast differentiation from PSCs that uses four small-molecule inducers under serum-free and feeder-free conditions (14). However, this method was established under the conventional 2D culture.
We hypothesized that the combination of the abovementioned protocol with appropriate 3D culture conditions could improve the maintenance and subsequent osteoblast differentiation of mPSCs. Although several studies have successfully differentiated PSCs into osteoblasts (15–22), none of the strategies utilized in these studies combine the advantages of both 3D culture and defined conditions using small-molecule approaches. Here, we characterized mPSCs cultured with atelocollagen scaffolds and compared them with those cultured in conventional gelatin-coated plates in terms of pluripotency maintenance and differentiation under defined conditions.
RESULTS
Maintenance of mPSCs in the 3D culture
To investigate whether 3D cultures can maintain mPSCs under serum-free, defined conditions, we seeded mESCs into an atelocollagen sponge scaffold and cultured them for 5 days in the 2iLIF-supplemented medium (2iLIF+). Rapidly growing colonies were observed throughout the scaffold from days 1 to 5, as shown by the cross-sectional view in Fig. 1A. We further evaluated cell proliferation by performing a metabolic activity assay. The mESCs cultured under 3D conditions (3D mESCs) displayed a higher proliferation rate than those cultured under conventional 2D conditions (2D mESCs), that is, using gelatin-coated plates (Fig. 1B). These results suggest that the 3D culture can maintain the growth of mESCs under the serum-free, defined conditions applied in this study.
Fig. 1. Characterization of mESCs cultured under 2D (plates) or 3D (scaffolds) conditions.
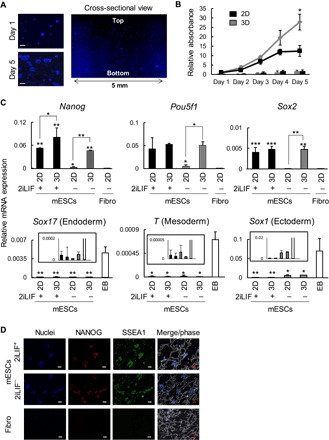
(A) Nuclei 4′,6-diamidino-2-phenylindole (DAPI) staining on sections of scaffolds seeded with mESCs. The staining was performed on frozen sections. Scale bars, 100 μm. (B) Cell proliferation of mESCs cultured under the 2iLIF condition. Calorimetric assay was performed on the indicated days. The absorbance (bars) and the absorbance relative to day 1 (lines) are means ± SD from three independent experiments. *P < 0.05 in 3D-cultured mESCs versus 2D-cultured mESCs. (C) mRNA expression determined by reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis in mESCs cultured under 2D or 3D conditions in the presence (+) or absence (−) of 2iLIF. The Sox17, T, and Sox1 expression data are shown in the rectangular box with the smaller scale of the y axis. *P < 0.05, **P < 0.01, and ***P < 0.001 as indicated or versus NIH3T3 (Fibro) or EB. Data are means ± SD from four independent experiments. (D) Protein expressions of pluripotency markers (NANOG and SSEA1) in mESCs cultured under the 3D condition in scaffolds in the presence (+) or absence (−) of 2iLIF. Immunostaining was performed on frozen sections. Nuclei were stained with DAPI. Scale bars, 100 μm.
We next evaluated the maintenance of mESC pluripotency under 3D conditions with the 2iLIF+ medium by analyzing mRNA expression. 3D mESCs strongly expressed pluripotency markers; they expressed Pou5f1 and Sox2 at levels comparable to those expressed by 2D mESCs, and the Nanog expression was significantly higher in 3D mESCs than in 2D mESCs (Fig. 1C). Note that the 3D mESCs maintained the expressions of all tested pluripotency markers in the absence of 2iLIF (2iLIF−), whereas the 2D mESCs showed down-regulation of those genes in the 2iLIF− medium (Fig. 1C, top panel).
The expressions of T and Sox17, which are mesoderm and endoderm marker genes, respectively, were not altered in any of the culture conditions (Fig. 1C, bottom panel). The expression of Sox1 (an ectoderm marker) was slightly up-regulated in the 2iLIF− medium compared with the 2iLIF+ medium in both 2D and 3D cultures (Fig. 1C, bottom panel), which is consistent with previous reports suggesting that 2i-cultured mESCs preferentially differentiate into ectoderm lineages rather than other lineages (6). However, Sox1 expression levels were still significantly lower in those cultures than in embryoid bodies (EBs) (Fig. 1C, bottom panel). In addition, the protein expressions of NANOG, OCT4, and SOX2 as well as SSEA1, a well-known carbohydrate antigenic epitope of undifferentiated cells, were observed in 3D mESCs under both 2iLIF+ and 2iLIF− conditions (Fig. 1D and fig. S1A).
To determine whether the maintenance of pluripotency in the 2iLIF− medium was due to the nature of the scaffold material (atelocollagen) or the 3D condition itself, we compared gene expression of mESCs cultured on atelocollagen-coated plates with that under 3D conditions using the scaffold without 2iLIF (fig. S1B). Analogous to the conventional 2D system with gelatin-coated plates, the mESCs cultured on atelocollagen-coated plates showed down-regulation of all tested pluripotency markers in the absence of 2iLIF, whereas those cultured under 3D conditions maintain their expressions even in the absence of 2iLIF (fig. S1B).
Thus, 3D culture, not the scaffold material, is likely to contribute to the maintenance of mESC pluripotency in the 2iLIF− medium. These results indicate that 3D culture can maintain mESCs in an undifferentiated state with the self-renewal capacity, which is comparable to 2D culture on plates, even in the absence of 2iLIF.
Lineage specification of mPSCs in the 3D culture
We next examined whether 3D culture could allow mESCs to differentiate into three lineages in vivo and in vitro. The in vivo differentiation was assessed by a teratoma assay. mESCs maintained in 2D or 3D in 2iLIF+ or 2iLIF− medium were subcutaneously implanted in nude mice (Fig. 2A). Teratoma formation and histological features were evaluated 8 weeks after the implantation. The mESCs from both the 2D and 3D cultures in the 2iLIF+ medium formed teratomas containing tissues from three germ layers with similar incidences and timing of appearance (Fig. 2B).
Fig. 2. Differentiation of mESCs cultured under 2D (plates) or 3D (scaffolds) conditions.
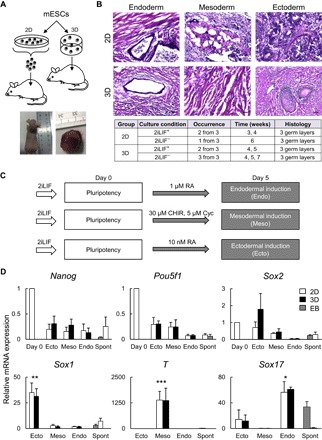
(A) Schematic representation of the in vivo implantation procedure and gross appearance of a teratoma in a nude mouse. (B) Representative pictures of teratomas derived from mESCs cultured under 2D and 3D conditions (upper) and the table showing the occurrence of teratomas (lower). Sections were stained with hematoxylin and eosin (H&E). Scale bars, 100 μm. (C) A schematic of the protocols used for in vitro early differentiation of mESCs. RA, retinoic acid; Cyc, cyclopamine. (D) mRNA expression determined by RT-qPCR analysis in the 2D or 3D differentiation cultures of mESCs; EBs and spontaneously differentiated mESCs (Spont) in 2D cultures were used for controls. Data are means ± SD from four independent experiments. *P < 0.05 in Endo-induced mESCs versus Meso- and Ecto-induced mESCs, Spont, and EB; **P < 0.01 in Ecto versus Endo, Meso, Spont, and EB; and ***P < 0.001 in Meso versus Endo, Ecto, Spont, and EB.
The teratomas derived from mESCs cultured in the 2iLIF− medium also contained tissues from three germ layers. However, the teratoma incidence was lower in the mice injected with mESCs from the 2D culture compared to the mice injected with those from the 3D culture (Fig. 2B). These data suggest that pluripotency of mESCs from the 3D culture is comparable to that from the 2D culture and that the 3D culture is likely to depend less on 2iLIF than the 2D culture does to maintain mESC pluripotency.
The in vitro differentiation of mESCs was successfully achieved in the 3D culture. The 3D culture differentiated mESCs into ectodermal, mesodermal, and endodermal cells, as the 2D culture did (Fig. 2, C and D). The protocols for the differentiation into each lineage are shown in Fig. 2C. After 5 days of differentiation, Sox1, T, and Sox 17, which are specific markers for each lineage, were selectively up-regulated in the 3D and 2D cultures by the corresponding differentiation treatments, whereas pluripotency markers (Nanog, Sox2, and Pou5f1) were down-regulated (Fig. 2D).
The mRNA expression of the lineage markers was higher in the 3D cultures than in the spontaneously differentiated 2D mESCs and the EBs (Fig. 2D). Thus, the 3D culture successfully maintained the pluripotency of mESCs, which is evidenced by the expression of pluripotency markers, the in vivo formation of teratomas containing three germ layers, and the in vitro specification of the cells into the three lineages.
Osteogenic differentiation of mPSCs in the 3D culture
We then examined whether the 3D culture could be applied to not only lineage specification but also the generation of terminally differentiated cells. We took advantage of the serum-free and feeder-free strategy for osteoblast induction that we reported in a 2D culture (14). Here, the mESCs maintained in the 3D culture were directly subjected to this strategy (Fig. 3A). Figure 3B shows the gene expression pattern in the osteogenic 3D culture compared with those in the osteogenic 2D culture as a positive control, using the abovementioned strategy. The pluripotency markers Nanog and Pou5f1 were progressively down-regulated from days 0 to 26 in both the osteogenic 2D and 3D cultures without any significant difference between the two cultures. The expression patterns of the mesoderm and osteoblast differentiation markers showed similar trends in the osteogenic 2D and 3D cultures.
Fig. 3. Osteogenic differentiation of mESCs cultured under 2D (plates) or 3D (scaffolds) conditions.
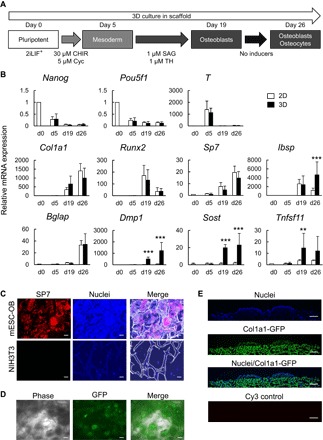
(A) A schematic of the strategy for inducing osteogenic differentiation of mPSCs. SAG, smoothened agonist. (B) mRNA expression determined by RT-qPCR analysis in the osteogenic 2D or 3D cultures of mESCs. Data are means ± SD from six independent experiments. **P < 0.01 and ***P < 0.001 in 3D-cultured mESCs versus 2D-cultured mESCs. (C) SP7 protein expression in the osteogenic 3D culture of mESCs (mESC-OB, day 26) and NIH3T3 cultured on scaffolds (negative control). Immunostaining was performed for SP7 on frozen sections. Nuclei were stained with DAPI. Scale bars, 50 μm. (D) Green fluorescent protein (GFP) expression in the osteogenic 3D culture of 2.3-kb Col1a1-GFP mESCs (day 26). GFP signal was assessed in the whole scaffold by fluorescence microscopy. Scale bars, 100 μm. (E) Cross-sectional view of the surface of the scaffold in the osteogenic 3D culture of 2.3-kb Col1a1-GFP mESCs (day 26). Immunostaining was performed for GFP on frozen sections. Nuclei were stained with DAPI. Scale bars, 250 μm.
After a transient up-regulation of T at the mesoderm induction phase (day 5; no significant difference between the 2D and 3D cultures), Col1a1, Sp7, and Ibsp were gradually up-regulated throughout the osteoblast induction phase. Runx2 was up-regulated during osteoblast differentiation (day 19) and then down-regulated after maturation (day 26), whereas Bglap, a marker for mature osteoblasts, was up-regulated on day 26. In contrast, we found significant differences in osteocyte markers on days 19 and 26 between the osteogenic 2D and 3D cultures: Compared to day 0, Dmp1 and Sost were highly expressed in the osteogenic 3D culture but not in the 2D culture.
Osteocytes and osteoblasts are reported as a main source of the receptor activator of nuclear factor κB ligand {RANKL; official symbol, Tnfsf11 [tumor necrosis factor (ligand) superfamily, member 11]}, a key stimulator of osteoclastogenesis (23–25). Here, the Tnfsf11 expression was up-regulated in the 3D culture compared to the 2D culture, a finding that also confirms the presence of osteocytes and osteoblasts in the osteogenic 3D culture.
We performed immunohistochemistry to detect SP7 protein expression in sectioned scaffolds and used 2.3-kb Col1a1-GFP mESCs (14, 26) to monitor osteoblast differentiation in situ. Because the 2.3-kb rat Col1a1 promoter fragment is activated specifically in osteoblasts (27, 28), GFP expression driven by the fragment can be used as an indicator of osteoblast differentiation. The nuclear localization of SP7 was detected in most of the cells within the scaffold (Fig. 3C), and 2.3-kb Col1a1-GFP was gradually increased throughout the culture and largely observed on day 26 (Fig. 3D). In addition, cross-sectional views revealed superficial cell clusters strongly expressing GFP similar to those in the 2D culture (Fig. 3, D and E).
To further examine whether bone ECM is synthesized by the osteoblastic population generated from mESCs in the 3D culture, we analyzed the cell-scaffold complex using H&E staining, von Kossa staining, and scanning electron microscopy (SEM) on day 26 of the culture (Fig. 4A). We found a positive relationship between mineralization of the scaffold and cell density; higher bone ECM deposition was observed at the surface and peripheries of the scaffolds than at the center, where less viable cells were visualized (compare left and right panels in fig. S2).
Fig. 4. Characterization of the mESC-derived cell-scaffold complex after osteogenic differentiation (day 26) and the coculture with BM cells (day 26 + 7).
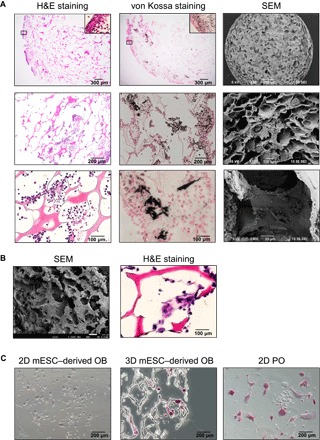
(A) Representative pictures of H&E staining, von Kossa staining with nuclear fast red, and SEM in the osteogenic 3D culture of mESCs. Insets show fivefold magnification views of regions marked by rectangular boxes. (B) Representative pictures of H&E staining and SEM in 3D cocultures of the mESC-derived osteoblast/osteocyte population and BM cells. (C) TRAP staining in cocultures of the 3D mESC– or 2D mESC–derived osteoblasts (OB) and BM cells. Coculture of PO and BM cells in 2D is shown as a positive control.
The von Kossa staining with nuclear fast red staining revealed cells embedded in the calcified ECM within the scaffold (Fig. 4A). SEM examination revealed highly confluent, flattened cells with a large amount of produced ECM; they lined the pore walls and filled the spaces between them (Fig. 4A). A higher magnification highlights numerous nodule-like structures (fig. S3A). The morphology of these structures is similar to that of calcium-rich agglomerates commonly found in previous studies on 3D osteogenic cultures (29–31).
The induction of osteocyte markers and Tnfsf11 in the osteogenic 3D cultures (Fig. 3B) led us to speculate that the mESC-derived osteoblast/osteocyte population in the culture may contribute to the osteoclast differentiation from precursors. To address this question, we seeded murine bone marrow (BM) cells in the osteogenic 3D and 2D cultures on day 26 and cocultured them with the mESC-derived osteoblast/osteocyte population for an additional 7 days. Coculture with primary osteoblasts (POs) was used as a positive control. The attachment of the osteoclast precursors to the scaffolds was confirmed by SEM (compare Fig. 4B with Fig. 4A and see also fig. S3B). Tartrate-resistant acid phosphatase (TRAP)–positive multinucleated cells, a key feature of osteoclasts, were detected in the 3D and PO cocultures but not in the 2D coculture (Fig. 4, B and C). Thus, mESCs successfully underwent small molecule–mediated, terminal differentiation into mature osteoblasts in the 3D cultures. Unlike the 2D cultures, the osteogenic 3D cultures further induced maturation of osteoblasts into osteocytes, which could support osteoclastogenesis from precursors.
Last, we confirmed that the present 3D culture platform can also be useful to differentiate miPSCs into osteoblasts. When we cultured miPSCs under the 3D conditions, following the same procedure as the one used for mESCs (fig. S4), the miPSC culture showed phenotypes that were similar to those of the mESC culture in terms of expression profiles of pluripotency (Nanog and Pou5f1), mesoderm (T), osteoblast (Col1a1, Runx2, Ibsp, and Bglap), and osteocyte markers (Dmp1, Sost, and Tnfsf11) (fig. S4A); SP7 protein expression (fig. S4B); and calcification of the cell-scaffold complex. Collectively, our findings demonstrate that the present 3D culture platform maintains mPSCs, achieves lineage specifications, and differentiates the specified population into terminally differentiated cells.
DISCUSSION
We developed a fully defined strategy in which mPSCs are maintained and differentiated into specific lineages in vitro in a 3D manner. Our results suggest that the 3D microenvironment plays a positive role in pluripotency maintenance and the differentiation of mPSCs. In particular, our 3D culture system showed superiority to conventional 2D culture in maintaining the pluripotency of mESCs, and it produced a unique milieu for enhanced osteoblast differentiation and the maturation of mPSCs. We also suspect that 3D culture allows the cells to self-organize and interact with fewer restrictions than the conventional 2D culture, resulting in a cell-cell signaling landscape more compatible with the physiological conditions.
Our 3D culture system appeared to substitute for exogenous signaling manipulation using 2i and LIF in mESCs for pluripotency maintenance. As shown here, several types of 3D cultures do not require extrinsic growth factors to sustain mESCs in the undifferentiated state. Analogous to our results, mESCs encapsulated in 3D in poly(ethylene glycol) (PEG)–based hydrogels maintained the undifferentiated state following the removal of LIF, whereas 2D mESCs rapidly differentiated under the same conditions (32). Moreover, the conjugation of PEG-based hydrogels with specific adhesion peptides allowed long-term maintenance of mESC pluripotency (as much as 4 weeks) in the absence of LIF (33). These reports suggest that the dimensionality difference affects the critical signals for pluripotency maintenance.
The abovementioned differences between 2D and 3D cultures in our experiments are likely to depend more on spatial conformation changes rather than on ECM composition; the similar gene expression patterns observed here in the mESCs maintained in gelatin-coated plates and those cultured on atelocollagen-coated plates exclude the possibility that the material itself induced significant changes in gene expression. Consistent with our present findings, Wei et al. (34) reported that mESCs cultured with different biomaterials or the ECM showed a high degree of similarity in gene expression, whereas a larger variation in gene expression was found between 2D and 3D cultures regardless of the materials that they used. In addition, examination of a set of pluripotency-related genes revealed that the characteristics of 3D mESCs were closer to those of 2D mESCs on feeder cells than 2D mESCs cultured with 2i-LIF medium (34). More recently, Nava et al. (35) described that a 3D nanoengineered substrate was able to maintain pluripotency of mESCs in the absence of soluble factors by providing biophysical constraint. They hypothesized that the confinement effect, due to the 3D microarchitecture of the substrate, induced genetic reprogramming of cells by controlling their cytoskeletal tension. Thus, several studies, including ours, suggest that 3D culture is suitable for maintenance of mESC pluripotency and, more importantly, that the biological effects of the spatial conformation change can be dominant over those of exogenous signaling factors in this context. Future studies should further examine the use of 3D platforms for the routine maintenance of mESCs as an attractive alternative to the current growth factor–based systems. Long-term cultures of mESCs under 3D conditions, through several passages and mouse generation from these cells, are required to fully validate this model.
Under early differentiation treatments, 3D mESCs achieved lineage specification at comparable induction efficiency with those in 2D culture. Although we did not find any significant difference in mRNA expression between mESCs differentiated in 2D and 3D cultures, 2D-established protocols successfully allowed the generation of endodermal, mesodermal, and ectodermal cells in 3D culture without additional optimization. Thus, our strategy may allow us to develop 3D functional tissues and, ultimately, organs, when simply combined with induction protocols that were used in 2D. We were able to generate 3D bone-like tissue using our 2D-established osteogenic protocol (14).
Unlike 2D culture, our combined 3D small molecule–based system induced the maturation of osteoblasts into functional osteocytes within calcified structures, and the mESC-derived osteoblast/osteocyte population was able to induce osteoclast differentiation from progenitors. These findings are relevant to studies involving late osteoblast functions (36, 37). Additional investigations will aim to fully reproduce the bone cell microenvironment by combining our 3D culture system with osteoclast precursors also derived from mPSCs. This approach may lead to a better understanding of the bone remodeling process, for example, in comparison analyses of healthy and disease conditions.
A 3D coculture system derived from human iPSCs cultured with a hydroxyapatite, poly(lactide-co-glycolide), and poly(l-lactic acid) composite scaffold was recently reported (20). Equivalent to our results, this previous study showed the benefit of a 3D culture over a 2D culture in terms of osteoblast functions and maturation in vitro and the advantage of the presence of both osteoblasts and osteoclasts in in vivo bone formation (20). Because our strategy is based on a serum-free 3D platform, it may have significant advantages in the screening of drugs for osteoporosis and in basic research regarding skeletal development by increasing the accuracy of experimental outcomes.
Our findings also highlight several differences between 2D and 3D cultures that may be exploited to control stem cell phenotypes. Less or no use of growth factors for mPSC maintenance and shorter time to obtain functionally mature cells from mPSCs are two examples of benefits that our 3D culture system can offer to stem cell research. However, 3D cultures have a common limitation: the heterogeneity of the cell population. Cells located on the surface of the scaffolds, which are highly exposed to the medium, are typically viable and proliferate, whereas cells within the construct receiving less oxygen and nutrients from the medium are less active or necrotic (38, 39). Because the outcome of our 3D system is highly dependent on the cellular intake of the small molecules from the medium, core and outer cells may experience different stages of differentiation, resulting in mixed populations. The use of a bioreactor to perfuse the culture medium through the porous constructs may give us homogeneous populations representing more precise osteoblast differentiation by reducing this gradient and enhancing the delivery of the chemical inducers as well as oxygen transport (39–45).
MATERIALS AND METHODS
Maintenance of mPSCs
We established and maintained mPSCs, including mESCs and miPSCs, as described earlier (14). The mPSCs were maintained on gelatin-coated culture dishes in a humidified atmosphere with 5% CO2 at 37°C in the 2iLIF-supplemented medium (9). The basal medium (2iLIF−) consisted of Dulbecco’s modified Eagle’s medium (DMEM) high glucose/F-12 (11330-032, Gibco), neurobasal medium (21103-049, Gibco), N2 supplement (17502-048, Gibco), B27 supplement (17504044, Gibco), bovine serum albumin (BSA) fraction V (A3059, Sigma-Aldrich), and penicillin-streptomycin solution (P4458, Sigma-Aldrich). To create the 2iLIF+ culture medium, we added PD032591 (1 μM; 163-24001, Wako) and CHIR99021 (3 μM; 039-20831, Wako) to the basal medium with LIF (1000 units/ml; ESG1107, Millipore). Accutase (AT104, Innovative Cell Technologies Inc.) was used to dissociate the cells.
The 3D culture of mPSCs in atelocollagen sponges
Commercially available atelocollagen scaffolds (Mighty, KKN-CSM-50, Koken Co.) were immersed in 2iLIF− medium for 24 hours before use. A 10-μl droplet containing 105 mESCs or miPSCs was carefully formed on top of each scaffold. The samples were then placed into low-adhesive 24-well plates (Z721077, Sigma) (two or three samples per well) in the incubator for 3 hours to allow cell attachment. Subsequently, 2 ml of the 2iLIF+ or 2iLIF− medium was added to each well; cells in the scaffolds were cultured for another 24 hours, until any subsequent treatments were begun. For the comparison of the pluripotency between mESCs cultured under 2D conditions and those cultured under 3D conditions, the cells were incubated for 5 days in the 2iLIF+ or 2iLIF− medium. The culture medium was changed every day. In 2D cultures, 2 × 105 mESCs were seeded on 10-cm dishes coated with gelatin or atelocollagen solution (KOU-IPC-30, Koken Co.). Cells cultured under 2D and 3D conditions were used for the RT-qPCR, metabolic activity assay, teratoma formation assay, and immunostaining.
In vitro lineage specification of mESCs
For 3D cultures, mESCs were seeded into atelocollagen sponges, as described above. For 2D cultures, mESCs were seeded on gelatinized plates at 100,000 cells/cm2. The 2iLIF− medium was used as a basal medium for the small molecule–mediated differentiation of mESCs into three lineages. Retinoic acid (R2625-5MG, Sigma) was added to the medium for ectoderm (10 nM) and endoderm (1 μM) differentiation (46, 47). Mesoderm induction was achieved by culturing cells with 30 μM CHIR and 5 μM cyclopamine (BML-GR334, Enzo Life Sciences) (14). The spontaneous differentiation of 2D mESCs was induced by culturing cells without any inducer. All differentiation procedures described above lasted 5 days. EBs were formed by the hanging drop culture method, as described (48), and cultured for 2 weeks. Total RNA extraction for RT-qPCR was performed in all groups.
Differentiation of mESCs into osteoblasts via mesoderm
mPSCs were differentiated into osteoblasts as described (15). Briefly, 2D-cultured mESCs were seeded at 100,000 cells/cm2 and miPSCs at 300,000 cells/cm2 onto gelatin-coated six-well plates. For 3D cultures, mPSCs were seeded into atelocollagen sponges, as described above. The mPSCs under all of the conditions were cultured for 24 hours in the 2iLIF+ medium before differentiation. The 2iLIF− medium was used during mesoderm and osteoblast differentiation. Mesoderm induction (from days 0 to 5) was achieved with CHIR and cyclopamine, as described above. For the induction of osteoblast differentiation, mPSCs were cultured for 14 days (from days 5 to 19) in the 2iLIF− culture medium supplemented with ascorbic acid phosphate (50 μg/ml) (AsAP; A4034, Sigma-Aldrich), 10 mM β-glycerophosphate (β-GP; G9422, Sigma-Aldrich), 0.1 μM dexamethasone (Dex; 41-18861, Wako), 1 μM SAG (566660, Calbiochem), and 1 μM TH (synthesized by Takeda Chemical Industries and distributed upon request). For osteoblast maturation, the cells were cultured for an additional 7 days (from days 19 to 26) in the 2iLIF− medium supplemented with AsAP, β-GP, and Dex without any inducers. The culture medium was changed every day. Total RNA extraction for RT-qPCR was performed in cells cultured under 2D and 3D conditions at days 0, 5, 19, and 26. Scaffolds with mPSC-derived osteoblasts were cryosectioned for immunostaining and von Kossa staining. Scaffolds with mESC-derived osteoblasts were subjected to the coculture with BM cells and analyzed with SEM and H&E staining.
PO isolation
POs were isolated as described (15). Briefly, calvarias from P1-P3 Crl:CD1 (ICR) mice were digested with 0.1% collagenase/0.2% dispase in DMEM at 37°C for 50 min. Cells suspended in the solution were collected every 10 min by centrifugation. Cells in the fractions 4 and 5 were used as POs.
Coculture system
Murine BM cells were flushed out from the tibias and femurs of 8-week-old ICR male mice (Charles River Laboratories), centrifuged, and immediately seeded in the osteogenic 3D and 2D cultures (day 26). A coculture with POs on plates (2D) was also prepared as a positive control. BM cells were cocultured in 10% fetal bovine serum/minimum essential medium containing 10 nM 1α,25-dihydroxyvitamin D3 and 100 nM prostaglandin E2 for 7 days. Fixed cells in scaffold or plates were stained for TRAP, as described (49).
RNA extraction and RT-qPCR
Total RNA was prepared as described (50) and was reverse-transcribed into single-strand complementary DNA using a ReverTra Ace qPCR RT Master Mix kit (FSQ-301, Toyobo) according to the manufacturer’s instructions. qPCR (40 cycles of amplification) was performed using a FastStart Universal SYBR Green Master kit (4913850, Roche) on a 7500 Fast Real-Time PCR System (Applied Biosystems). The expression of target genes was normalized to that of the reference gene (Actb), and the normalized expressions are shown relative to those of calibrator samples (day 0); the data of the calibrator samples represent the 1× expression of target genes normalized to Actb. PCRs were run in duplicate. The primer sequences are listed in table S1.
Metabolic activity assay
Quantification of the cell proliferation was performed using a Cell Counting Kit (CK04-01, Dojindo Molecular Technologies) according to the manufacturer’s instructions with minor modifications. Briefly, 5000 cells were seeded onto a gelatin-coated 96-well plate or an atelocollagen scaffold and cultured in a humidified incubator (37°C, 5% CO2). The colorimetric solution (CCK-8) was added to each well, followed by 2-hour incubation. Absorbance at 450 nm was measured using a microplate reader.
Teratoma formation and histological analysis
Cells that dissociated from plates or cell-scaffold complexes were subcutaneously implanted into the back of 5-week-old BALB/c nude female mice (Charles River Laboratories). Eight weeks after the implantation, the tumors were dissected and fixed with fresh 4% paraformaldehyde (PFA)/phosphate-buffered saline (PBS) overnight at 4°C. Paraffin-embedded teratomas were sectioned and stained with H&E. All experimental and surgical procedures involving animals were performed in accord with the protocol approved by the Animal Care and Use Committee of the University of Tokyo (#KA14-3).
Immunofluorescence
Cells were fixed with scaffolds with fresh 4% PFA/PBS at room temperature for 1 hour and then incubated overnight with 30% sucrose/PBS solution. The scaffolds were then embedded in optimal cutting temperature compound to prepare cryosections (10 to 12 μm). After blocking with PBS containing 0.1% Triton X-100, 3% BSA (A7906, Sigma), and 1% heat-inactivated sheep serum (S2263, Sigma) at room temperature for 1 hour, the sections were incubated at 4°C overnight with the following primary antibodies: rabbit anti-OCT3/4 (1:200; sc-9081, Santa Cruz Biotechnology), rabbit anti-NANOG (1:200; RCAB0001P, ReproCELL), rabbit anti-SOX2 (1:500; AB5603, Millipore), mouse anti-SSEA1 (1:50; sc-21702, Santa Cruz Biotechnology), and rabbit anti-SP7 (1:300; ab22552, Abcam). The sections were then incubated for 1 hour at room temperature with the following secondary antibodies: anti-rabbit [immunoglobulin G (IgG)] Alexa Fluor 546 (1:500; A-11035, Invitrogen) and anti-mouse IgG (H+L) Alexa Fluor 488 (1:400; A-11001A, Invitrogen). Nuclei were stained with VECTASHIELD Mounting Medium containing DAPI (H-1200, Vector Laboratories). Images were collected using a BIOZERO-9000 (Keyence) fluorescence microscope.
Detection of calcification
Von Kossa staining was performed as described in cryosectioned scaffolds (49).
Scanning electron microscopy
Cell-scaffold complex were fixed with 2.5% glutaraldehyde at 4°C for 4 hours. Dehydration was performed by a 20-min sample immersion into an ethanol series in distilled water (50, 60, 70, 80, 90, and 100%) at room temperature. Samples were then transferred into a fresh solution of 1:2 100% ethanol/hexamethyldisilazane (HDMS; 440191, Sigma) for 20 min. Samples were air-dried overnight while immersed in 100% HDMS. Gold-sputtered samples were observed and photographed at 15-kV accelerating voltage using a scanning electron microscope (6300F or 7500F, JEOL).
Supplementary Material
Acknowledgments
We thank A. Yamaguchi, S. Onodera, T. Sakai, K. Kushiro, S. Ohtsuka, T. Saito, and F. Yano for their helpful inputs and K. Morii, R. Yonemoto, and N. Nagumo for providing technical assistance. Funding: This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (26713054, 26221311, 15K15732, and 16H06312), the Graduate Program for Leaders in Life Innovation, Core-to-Core Program A (Advanced Research Networks), the Center for NanoBio Integration, the Center of Innovation Program, the Takeda Science Foundation Research Grant, Uehara Memorial Foundation Research Grant, and Mochida Memorial Foundation for Medical and Pharmaceutical Research. Author contributions: D.Z., K.K., and S.O. conceived the project. D.Z. and K.K. performed the experiments. D.Z., K.K., A.C.L., H.H., U.C., and S.O. analyzed and interpreted the data. D.Z., H.H., U.C., and S.O. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602875/DC1
fig. S1. Pluripotency maintenance of mESCs cultured under 3D (scaffolds) conditions in the presence or absence of 2iLIF, related to Fig. 1.
fig. S2. Representative pictures of H&E staining in the osteogenic 3D culture of mESCs on day 26.
fig. S3. Representative SEM pictures in the osteogenic 3D culture of mESCs at day 26.
fig. S4. Osteogenic differentiation of miPSCs cultured under 2D (plates) or 3D (scaffolds) conditions.
table S1. Primer sequences (forward and reverse, 5′-3′) used for RT-qPCR.
REFERENCES AND NOTES
- 1.S. A. Lelièvre, M. J. Bissell, Reviews in Cell Biology and Molecular Medicine (Wiley-VCH Verlag GmbH & Co. KGaA, 2006). [Google Scholar]
- 2.Teicher B. A., Herman T. S., Holden S. A., Wang Y. Y., Pfeffer M. R., Crawford J. W., Frei E. III, Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science 247, 1457–1461 (1990). [DOI] [PubMed] [Google Scholar]
- 3.Beckstead B. L., Santosa D. M., Giachelli C. M., Mimicking cell-cell interactions at the biomaterial-cell interface for control of stem cell differentiation. J. Biomed. Mater. Res. A 79, 94–103 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Meng X., Leslie P., Zhang Y., Dong J., Stem cells in a three-dimensional scaffold environment. Springerplus 3, 80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kraehenbuehl T. P., Langer R., Ferreira L. S., Three-dimensional biomaterials for the study of human pluripotent stem cells. Nat. Methods 8, 731–736 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Marks H., Kalkan T., Menafra R., Denissov S., Jones K., Hofemeister H., Nichols J., Kranz A., Francis Stewart A., Smith A., Stunnenberg H. G., The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149, 590–604 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith A. G., Heath J. K., Donaldson D. D., Wong G. G., Moreau J., Stahl M., Rogers D., Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 336, 688–690 (1988). [DOI] [PubMed] [Google Scholar]
- 8.Williams R. L., Hilton D. J., Pease S., Willson T. A., Stewart C. L., Gearing D. P., Wagner E. F., Metcalf D., Nicola N. A., Gough N. M., Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 336, 684–687 (1988). [DOI] [PubMed] [Google Scholar]
- 9.Ying Q.-L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A., The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding S., Schultz P. G., A role for chemistry in stem cell biology. Nat. Biotechnol. 22, 833–840 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Li W., Ding S., Small molecules that modulate embryonic stem cell fate and somatic cell reprogramming. Trends Pharmacol. Sci. 31, 36–45 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Schugar R. C., Robbins P. D., Deasy B. M., Small molecules in stem cell self-renewal and differentiation. Gene Ther. 15, 126–135 (2008). [DOI] [PubMed] [Google Scholar]
- 13.KalantarMotamedi Y., Peymani M., Baharvand H., Nasr-Esfahani M. H., Bender A., Systematic selection of small molecules to promote differentiation of embryonic stem cells and experimental validation for generating cardiomyocytes. Cell Death Discov. 2, 16007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanke K., Masaki H., Saito T., Komiyama Y., Hojo H., Nakauchi H., Lichtler A. C., Takato T., Chung U.-i., Ohba S., Stepwise differentiation of pluripotent stem cells into osteoblasts using four small molecules under serum-free and feeder-free conditions. Stem Cell Reports 2, 751–760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian X.-F., Heng B.-C., Ge Z., Lu K., Rufaihah A. J., Fan V. T.-W., Yeo J.-F., Cao T., Comparison of osteogenesis of human embryonic stem cells within 2D and 3D culture systems. Scand. J. Clin. Lab. Invest. 68, 58–67 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Hesse E., Hefferan T. E., Tarara J. E., Haasper C., Meller R., Krettek C., Lu L., Yaszemski M. J., Collagen type I hydrogel allows migration, proliferation, and osteogenic differentiation of rat bone marrow stromal cells. J. Biomed. Mater. Res. A 94, 442–449 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takitoh T., Kato Y., Nakasu A., Tadokoro M., Bessho M., Hirose M., Ohgushi H., Mori H., Hara M., In vitro osteogenic differentiation of HOS cells on two types of collagen gels. J. Biosci. Bioeng. 110, 471–478 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Bilousova G., Jun D. H., King K. B., De Langhe S., Chick W. S., Torchia E. C., Chow K. S., Klemm D. J., Roop D. R., Majka S. M., Osteoblasts derived from induced pluripotent stem cells form calcified structures in scaffolds both in vitro and in vivo. Stem Cells 29, 206–216 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buttery L., Bielby R., Howard D., Shakesheff K., Osteogenic differentiation of embryonic stem cells in 2D and 3D culture. Methods Mol. Biol. 695, 281–308 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Jeon O. H., Panicker L. M., Lu Q., Chae J. J., Feldman R. A., Elisseeff J. H., Human iPSC-derived osteoblasts and osteoclasts together promote bone regeneration in 3D biomaterials. Sci. Rep. 6, 26761 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews B. G., Naot D., Callon K. E., Musson D. S., Locklin R., Hulley P. A., Grey A., Cornish J., Enhanced osteoblastogenesis in three-dimensional collagen gels. Bonekey Rep. 3, 560 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang H., Shih Y.-R. V., Nakasaki M., Kabra H., Varghese S., Small molecule–driven direct conversion of human pluripotent stem cells into functional osteoblasts. Sci. Adv. 2, e1600691 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F., Penninger J. M., Takayanagi H., Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Takahashi N., Akatsu T., Udagawa N., Sasaki T., Yamaguchi A., Moseley J. M., John Martin T., Suda T., Osteoblastic cells are involved in osteoclast formation. Endocrinology 123, 2600–2602 (1988). [DOI] [PubMed] [Google Scholar]
- 25.Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O’Brien C. A., Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohba S., Ikeda T., Kugimiya F., Yano F., Lichtler A. C., Nakamura K., Takato T., Kawaguchi H., Chung U. I., Identification of a potent combination of osteogenic genes for bone regeneration using embryonic stem (ES) cell-based sensor. FASEB J. 21, 1777–1787 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Kalajzic I., Kalajzic Z., Kaliterna M., Gronowicz G., Clark S. H., Lichtler A. C., Rowe D., Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J. Bone Miner. Res. 17, 15–25 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Bogdanovic Z., Bedalov A., Krebsbach P. H., Pavlin D., Woody C. O., Clark S. H., Thomas H. F., Rowe D. W., Kream B. E., Lichtler A. C., Upstream regulatory elements necessary for expression of the rat COL1A1 promoter in transgenic mice. J. Bone Miner. Res. 9, 285–292 (1994). [DOI] [PubMed] [Google Scholar]
- 29.Depan D., Pesacreta T. C., Misra R. D. K., The synergistic effect of a hybrid graphene oxide–chitosan system and biomimetic mineralization on osteoblast functions. Biomater. Sci. 2, 264–274 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Yan L., Jiang D.-m., Study of bone-like hydroxyapatite/polyamino acid composite materials for their biological properties and effects on the reconstruction of long bone defects. Drug Des. Devel. Ther. 9, 6497–6508 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Z. A. Bakar, B. F. Hussein, N. M. Mustapha, Cockle shell-based biocomposite scaffold for bone tissue engineering, in Regenerative Medicine and Tissue Engineering—Cells and Biomaterials, D. Eberli, Ed. (InTech, 2011), pp. 365–390. [Google Scholar]
- 32.Caiazzo M., Okawa Y., Ranga A., Piersigilli A., Tabata Y., Lutolf M. P., Defined three-dimensional microenvironments boost induction of pluripotency. Nat. Mater. 15, 344–352 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Lee S. T., Im Yun J., van der Vlies A. J., Kontos S., Jang M., Pyo Gong S., Yong Kim D., Lim J. M., Hubbell J. A., Long-term maintenance of mouse embryonic stem cell pluripotency by manipulating integrin signaling within 3D scaffolds without active Stat3. Biomaterials 33, 8934–8942 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Wei J., Han J., Zhao Y., Cui Y., Wang B., Xiao Z., Chen B., Dai J., The importance of three-dimensional scaffold structure on stemness maintenance of mouse embryonic stem cells. Biomaterials 35, 7724–7733 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Nava M. M., Piuma A., Figliuzzi M., Cattaneo I., Bonandrini B., Zandrini T., Cerullo G., Osellame R., Remuzzi A., Raimondi M. T., Two-photon polymerized “nichoid” substrates maintain function of pluripotent stem cells when expanded under feeder-free conditions. Stem Cell Res. Ther. 7, 132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moon Y. J., Yun C.-Y., Choi H., Ka S.-O., Ryul Kim J., Park B.-H., Cho E.-S., Smad4 controls bone homeostasis through regulation of osteoblast/osteocyte viability. Exp. Mol. Med. 48, e256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim H.-Y., Yoon J.-Y., Yun J.-H., Cho K.-W., Lee S.-H., Rhee Y.-M., Jung H.-S., Lim H. J., Lee H., Choi J., Heo J.-N., Lee W., No K. T., Min D., Choi K.-Y., CXXC5 is a negative-feedback regulator of the Wnt/β-catenin pathway involved in osteoblast differentiation. Cell Death Differ. 22, 912–920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holy C. E., Shoichet M. S., Davies J. E., Engineering three-dimensional bone tissue in vitro using biodegradable scaffolds: Investigating initial cell-seeding density and culture period. J. Biomed. Mater. Res. 51, 376–382 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Cartmell S. H., Porter B. D., García A. J., Guldberg R. E., Effects of medium perfusion rate on cell-seeded three-dimensional bone constructs in vitro. Tissue Eng. 9, 1197–1203 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Bancroft G. N., Sikavitsas V. I., van den Dolder J., Sheffield T. L., Ambrose C. G., Jansen J. A., Mikos A. G., Fluid flow increases mineralized matrix deposition in 3D perfusion culture of marrow stromal osteoblasts in a dose-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 99, 12600–12605 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vance J., Galley S., Liu D. F., Donahue S. W., Mechanical stimulation of MC3T3 osteoblastic cells in a bone tissue-engineering bioreactor enhances prostaglandin E2 release. Tissue Eng. 11, 1832–1839 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Holtorf H. L., Jansen J. A., Mikos A. G., Modulation of cell differentiation in bone tissue engineering constructs cultured in a bioreactor. Adv. Exp. Med. Biol. 585, 225–241 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Volkmer E., Drosse I., Otto S., Stangelmayer A., Stengele M., Kallukalam B. C., Mutschler W., Schieker M., Hypoxia in static and dynamic 3D culture systems for tissue engineering of bone. Tissue Eng. Part A 14, 1331–1340 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Alvarez-Barreto J. F., Landy B., VanGordon S., Place L., DeAngelis P. L., Sikavitsas V. I., Enhanced osteoblastic differentiation of mesenchymal stem cells seeded in RGD-functionalized PLLA scaffolds and cultured in a flow perfusion bioreactor. J. Tissue Eng. Regen. Med. 5, 464–475 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Iannetti L., D’Urso G., Conoscenti G., Cutrì E., Tuan R. S., Raimondi M. T., Gottardi R., Zunino P., Distributed and lumped parameter models for the characterization of high throughput bioreactors. PLOS ONE 11, e0162774 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Micallef S. J., Janes M. E., Knezevic K., Davis R. P., Elefanty A. G., Stanley E. G., Retinoic acid induces Pdx1-positive endoderm in differentiating mouse embryonic stem cells. Diabetes 54, 301–305 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Engberg N., Kahn M., Petersen D. R., Hansson M., Serup P., Retinoic acid synthesis promotes development of neural progenitors from mouse embryonic stem cells by suppressing endogenous, Wnt-dependent nodal signaling. Stem Cells 28, 1498–1509 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Höpfl G., Gassmann M., Desbaillets I., Differentiating embryonic stem cells into embryoid bodies. Methods Mol. Biol. 254, 79–98 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Ohba S., Kawaguchi H., Kugimiya F., Ogasawara T., Kawamura N., Saito T., Ikeda T., Fujii K., Miyajima T., Kuramochi A., Miyashita T., Oda H., Nakamura K., Takato T., Chung U.-i., Patched1 haploinsufficiency increases adult bone mass and modulates Gli3 repressor activity. Dev. Cell 14, 689–699 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Hojo H., Ohba S., Yano F., Saito T., Ikeda T., Nakajima K., Komiyama Y., Nakagata N., Suzuki K., Takato T., Kawaguchi H., Chung U.-i., Gli1 protein participates in Hedgehog-mediated specification of osteoblast lineage during endochondral ossification. J. Biol. Chem. 287, 17860–17869 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602875/DC1
fig. S1. Pluripotency maintenance of mESCs cultured under 3D (scaffolds) conditions in the presence or absence of 2iLIF, related to Fig. 1.
fig. S2. Representative pictures of H&E staining in the osteogenic 3D culture of mESCs on day 26.
fig. S3. Representative SEM pictures in the osteogenic 3D culture of mESCs at day 26.
fig. S4. Osteogenic differentiation of miPSCs cultured under 2D (plates) or 3D (scaffolds) conditions.
table S1. Primer sequences (forward and reverse, 5′-3′) used for RT-qPCR.