

Abstract
Inflammation with macrophage infiltration is a key feature of atherosclerosis. Although the mechanisms had been unclear, emerging evidence unveiled that NLRP3 inflammasomes, which regulate caspase-1 activation and subsequent processing of pro-IL-1β, trigger vascular wall inflammatory responses and lead to progression of atherosclerosis. NLRP3 inflammasomes are activated by various danger signals, such as cholesterol crystals, calcium phosphate crystals, and oxidized low-density lipoprotein in macrophages, to initiate inflammatory responses in the atherosclerotic lesion. Recent studies have further clarified the regulatory mechanisms and the potential therapeutic agents that target NLRP3 inflammasomes. In this study, we reviewed the present state of knowledge on the role of NLRP3 inflammasomes in the pathogenesis of atherosclerosis and discussed the therapeutic approaches that target NLRP3 inflammasomes.
Keywords: Cholesterol, Cytokines, Inflammation, Interleukin-1, Leukocytes
Introduction
Atherosclerosis is a chronic inflammatory disease characterized by lipid deposition, leukocyte infiltration, and proliferation of vascular smooth muscle cells (VSMCs)1, 2). The inflammatory nature of atherosclerosis has been proven by several findings as follows: 1) the presence of inflammatory cells, mainly macrophages, which infiltrate into the atherosclerotic plaques and the correlation between the number of these cells and disease severity; 2) the presence of inflammatory cytokines and chemokines in all stages of atherosclerosis3); and 3) the widely accepted association between cardiovascular events and serum inflammatory markers, such as C-reactive protein and interleukin (IL)-63–6). Until recently, however, the molecular basis of these inflammatory responses in atherosclerotic lesions has not been elucidated.
Inflammation is defined as the process by which the body responds to injury or infection and is mediated by components of the innate immune system, such as neutrophils and macrophages. The innate immune system has been recognized as first-line defense against invading pathogens and plays a primary role in acute host defense; this concept was referred to as the “self–non-self theory” or the “stranger theory”7). In the 1990s, Polly Matzinger suggested a new concept called the “danger theory”8), which claimed that immune responses can be activated by danger signals, released from injured tissues or cells, as well as by the presence of pathogens. These danger signals can be recognized by pattern recognition receptors (PRRs) and stimulate immune responses similar to those triggered by pathogens. This danger theory has recently received considerable attention because it may explain activation of the innate immune system in the absence of pathogens. PRRs are evolutionary conserved and expressed in the innate immune system cells, such as macrophages, neutrophils, and dendritic cells, and can identify two types of molecular patterns: pathogen-associated molecular patterns (PAMPs) and damage/danger-associated molecular patterns (DAMPs). PRRs are classified into at least four groups: Toll-like receptors (TLR), nucleotide-binding oligomerization domain-like receptors (NLRs), retinoic acid-inducible gene (RIG)-I-like receptors, (RLRs), and C-type lectin receptors (CLRs)9). Theses clusters of PRRs exhibit distinct intracellular localization and recognize different PAMPs and DAMPs. In particular, NLR pyrin domain containing 3 (NLRP3), a member of the NLR family, recognizes various DAMPs to form an NLRP3 inflammasome molecular complex in the cytosol and leads to inflammatory responses. Indeed, recent studies demonstrated that NLRP3 inflammasome-driven inflammatory responses contributed to the progression of atherosclerosis10–13). This study highlighted the present state of knowledge on the role of NLRP3 inflammasomes in the pathogenesis of atherosclerosis.
Inflammasomes
An inflammasome is a cytoplasmic complex containing multiple proteins, which is formed in response to DAMPs or PAMPs, and serves as a molecular platform for activation of the cysteine protease caspase-114). It typically contains one NLR protein, an apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and the cysteine protease caspase-1. Since caspase-1 was originally identified as an IL-1 converting enzyme (ICE)15), inflammasome-mediated activation of caspase-1 promotes processing of the IL-1β precursor, a potent inflammatory cytokine, and induces mature IL-1β release to cause tissue inflammatory responses. Similar to IL-1β, caspase-1 can process pro-IL-18 into its mature form, which is another IL-1 family cytokine16). Several types of inflammasome complexes have been reported; among the NLRs, NLRP1, NLRP3, NLRC4, NLRP6, and NLRP12 can participate in the inflammasomes17). Similar to NLRs, the pyrin and HIN domain-containing protein (PYHIN) family, including absence in melanoma 2 (AIM2) and IFN-γ-inducible protein 16 (IFI16), is also a component of inflammasomes. In general, inflammasomes are named according to the specific NLR that it contains; therefore, NLRP3 inflammasomes are composed of NLRP3, ASC, and caspase-1 (Fig. 1).
Fig. 1.
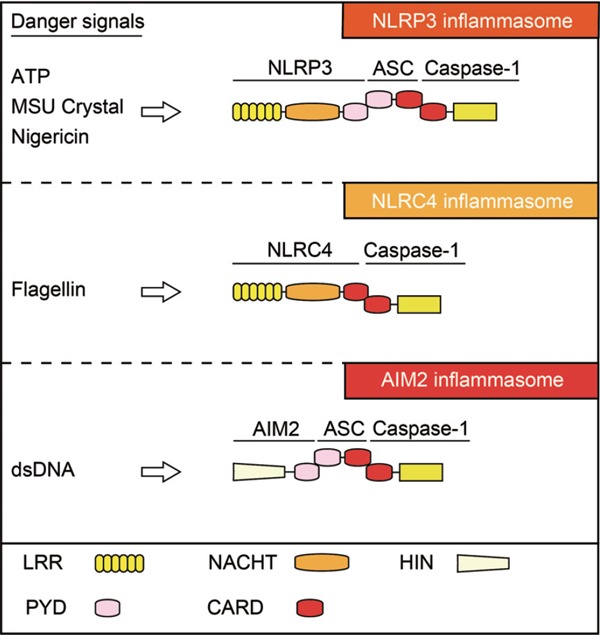
Representative inflammasomes and their components
Several PRRs that recognize distinct DAMPs form the inflammasome complex, which serves as the molecular platform to activate caspase-1. NLRP3 inflammasomes are composed of NLRP3, ASC, and caspase-1. NLRP3 binds ASC via PYD-PYD interaction. Subsequently, ASC binds caspase-1 via CARD–CARD interaction. NLRC4 inflammasomes are composed of NLRC4 and caspase-1, whereas AIM2 inflammasomes are composed of AIM2, ASC, and caspase-1. ATP, adenosine triphosphate; CARD, caspase recruitment domain; DAMPs, damage/danger-associated molecular patterns; HIN, hematopoietic interferon-inducible protein with a 200-amino-acid repeat; LRR, leucine-rich repeats; MSU, monosodium urate; NACHT, found in NAIP, CITA, HET-E ,and TP1; NLR, nucleotide-binding oligomerization domain-like receptor; PRRs, pattern recognition receptors; PYD, pyrin domain.
NLRP3 Inflammasomes
Unlike other inflammasomes, NLRP3 inflammasomes are activated by endogenous or exogenous DAMPs and are involved in the process of sterile inflammation17). As described above, NLRP3 inflammasomes are composed of NLRP3, ASC, and caspase-1, which have conserved domains for homophilic interaction (Fig. 1). NLRP3 contains three domains: C-terminal leucine-rich repeats (LRRs), a central nucleotide domain termed as the NACHT domain, and an N-terminal effector domain (pyrin domain [PYD]). ASC contains an N-terminal PYD and a C-terminal caspase recruitment domain (CARD). Caspase-1 contains a CARD and catalytic domains (p10 and p20). When cells are stimulated by DAMPs, NLRP3 assembles by the NACHT domain to provide a scaffold for ASC oligomerization by their interaction between PYDs. In turn, because NLRP3 lacks CARD and cannot recruit caspase-1 except in the presence of ASC, the oligomerized ASC interacts with caspase-1 via CARD homophilic interaction and induces caspase-1 auto-activation to exert pro-inflammatory effects by its proteolytic activity. As mentioned above, caspase-1 also processes pro-IL-18 to its bioactive mature form. In addition, recent studies showed that caspase-1 cleaves gasdermin D (GSDMD) to induce pyroptosis, which is an inflammatory programmed cell death accompanied by increased plasma membrane permeability18, 19). Although IL-1β has no signal sequence for exocytosis and the mechanism of its release is still unknown, this pyroptosis-mediated membrane permeabilization is critical for IL-1β release20).
Although various endogenous and exogenous danger signals, such as extracellular adenosine triphosphate (ATP), monosodium urate (MSU) crystal, and silica, are known to activate NLRP3 inflammasomes, the precise mechanism of NLRP3 recognition of DAMPs remains unclear21, 22). Nevertheless, several common upstream pathways, including potassium (K+) efflux, generation of mitochondrial reactive oxygen species (ROS), and lysosomal destabilization, have been reported to be important for activation of NLRP3 inflammasomes23). Of these, it has been well accepted that lysosomal destabilization and a subsequent cathepsin release is responsible for NLRP3 inflammasome activation by particulate matters. In addition, K+ efflux inhibition prevents NLRP3 inflammasome activation in response to most or all NLRP3 stimuli, including particulate matters, suggesting K+ efflux as a common trigger of its activation24, 25). The production and release of IL-1β is regulated in two-steps: Transcriptional synthesis of pro-IL-1β and proteolytic processing into a mature form by inflammasomes (Fig. 2). The transcriptional regulation of IL-1β mRNA is mediated by PRRs or cytokine receptors, including TLRs and IL-1 receptor (signal 1), and is known as priming. Apart from NF-kB-mediated mRNA induction of IL-1β and NLRP3, activation of these receptors has also been shown to prime the NLRP3 by posttranscriptional regulation, such as ubiquitination and deubiquitination26, 27). Thereafter, the accumulated pro-IL-1β in the cytosol is rapidly processed by activated caspase-1, which is activated by NLRP3 inflammasomes (signal 2). Thus, this two-step system is considered to be necessary for tight regulation of the potent inflammatory cytokine IL-1β to maintain inflammatory homeostasis.
Fig. 2.
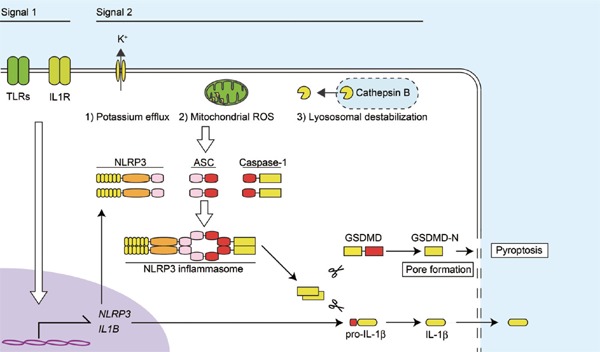
Mechanisms of NLRP3 inflammasome-driven IL-1β release
IL-1β release is regulated in two-steps: Transcriptional synthesis of pro-IL-1β and proteolytic processing into its mature form by inflammasomes. The transcriptional regulation of IL-1β mRNA is mediated by TLRs and IL-1 receptor (signal 1), which also provides NLRP3 mRNA. Then, the NLRP3 inflammasome-activated caspase-1 processes accumulated pro-IL-1β and induces the release of IL-1β (signal 2). The common upstream pathways of NLRP3 inflammasomes include potassium efflux, generation of mitochondrial ROS, and lysosomal destabilization and leakage of cathepsin B. Activated caspase-1 also cleaves GSDMD, whose processed N-terminal fragment (GSDMD-N) increases plasma membrane permeability, resulting in pyroptosis. ROS, reactive oxygen species; TLRs, Toll-like receptors.
At first, NLRP3, which is also known as CIAS1 gene that encodes the cryopyrin protein, was identified by a gain-of-function mutation in a gene that was associated with autoinflammatory syndrome, cryopyrin-associated periodic syndrome (CAPS). CAPS includes three different conditions, including familial cold autoinflammatory syndrome, Muckle–Wells syndrome, and chronic infantile neurological cutaneous and articular syndromes28). Since CAPS is an extremely rare genetic disease, only a few studies on NLRP3 inflammasomes had been conducted. In 2006, however, uric acid crystals in gout were discovered to activate NLRP3 inflammasomes22). This discovery motivated further studies on the association between NLRP3 inflammasomes and sterile inflammatory diseases. Indeed, other research workers and we have revealed a pivotal role of NLRP3 inflammasome in the pathophysiology of cardiovascular and renal diseases, including atherosclerosis, vascular injury, aortic aneurysm, myocardial infarction, chronic kidney disease, and acute kidney injury10, 11, 29–33). Furthermore, in the last decade, this premise was further supported by studies that described NLRP3 inflammasomes as a key mediator of other diseases associated with sterile inflammation, including type 2 diabetes and metabolic syndrome34, 35).
NLRP3 Inflammasome in Atherosclerosis
Role of NLRP3 Inflammasomes in Atherosclerosis
Many clinical and experimental studies have reported that IL-1β plays a crucial role in the progression of atherosclerosis and identified IL-1β as a proatherogenic cytokine36–38). These findings suggested that NLRP3 inflammasomes may contribute to the progression of atherosclerosis. In 2010, Duewell et al.10) were the first to show the important role of NLRP3 inflammasomes in the progression of atherosclerosis. Using atherosclerosis-prone, low-density lipoprotein receptor-deficient (LDLR-/-) mice, they produced chimeric mice whose bone marrows were transplanted with NLRP3-/-, ASC-/-, or IL-1α/β-/- bone marrow cells and showed that the lack of these inflammasome-related molecules in the bone marrow-derived inflammatory cells decreased atherosclerotic lesion development. Because cholesterol crystal formation is present in the early stages and can be detected in all stages of atherosclerosis, Duewell et al.10) focused on cholesterol crystals as the candidate of DAMPs and found that these crystals strongly activated NLRP3 inflammasomes in macrophages. Interestingly, they further showed that the presence of oxidized low-density lipoprotein (LDL), which exhibits highly atherogenic properties, can lead to cholesterol crystallization and activation of priming signals to induce NLRP3 and pro-IL-1β expressions; these indicated that oxidized LDL could sufficiently provide signal 1 and signal 2 to induce IL-1β release. According to a later study by Sheedy et al.39), incorporation of oxidized LDL via a scavenger receptor CD36 provoked intracellular cholesterol crystallization. Together with the findings that the lack of IL-1β decreased the severity of atherosclerosis in ApoE-/- mice36), which are frequently used as another atherosclerosis-prone model, these observations indicated that NLRP3 inflammasome-driven IL-1β release contributed to the progression of atherosclerosis. However, Menu et al.40) reported that neither NLRP3 and ASC nor caspase-1 deficiency could prevent macrophage infiltration and atherosclerosis in ApoE-/- mice. Using a similar experimental model, we observed that caspase-1 deficiency clearly reduced macrophage infiltration and development of atherosclerotic lesions in ApoE-/- mice11). The reason for this discrepancy is not entirely clear, but the more atherogenic diet used in that study, in comparison with our study, may have influenced the immune status and inflammatory responses. In addition to the studies mentioned above, several studies have described the role of NLRP3 inflammasomes in the progression of atherosclerosis (Table 1). It should be noted that the conventional caspase-1-/- mouse strain lacks both caspase-1 and caspase-11 because these genes were quite close in the genome to have been segregated.
Table 1. Effects of NLRP3 inflammasome deficiency on progression of atherosclerosis in mice.
| Reference | Model | Diet | Deficient gene | Lesion size | ||
|---|---|---|---|---|---|---|
| Duewell et al. | 2010 | Ref. 8 | Ldlr-/- | WTD (0.15% Cho) | Nlrp3, Asc (BMT) | decrease |
| Menu et al. | 2011 | Ref. 38 | Apoe-/- | HFD (1.25% Cho) | Nlrp3, Asc, Casp1/11 | no change |
| Usui et al. | 2012 | Ref. 9 | Apoe-/- | WTD (0.15% Cho) | Casp1/11 | decrease |
| Gage et al. | 2012 | Ref. 10 | Apoe-/- | HFD (1.5% Cho) | Casp1/11 | decrease |
| Hendrikx et al. | 2015 | Ref. 11 | Ldlr-/- | HFD (0.2% Cho) | Casp1/11 (BMT) | decrease |
Abbreviation: BMT, bone marrow transplant; Cho, cholesterol; HFD, high fat diet; WTD, western diet
Mechanisms of NLRP3 Inflammasome Activation in Atherosclerosis
The mechanism of cholesterol crystal-induced NLRP3 inflammasome activation has been described. Since vascular wall macrophage infiltration is the hallmark of atherosclerosis4, 41), studies have mainly focused on the role of NLRP3 inflammasomes in macrophages. Indeed, the components of NLRP3 inflammasomes are expressed by macrophages and are highly upregulated by priming signals (e.g., TLR and IL-1R). Duewell et al.8) found that cholesterol crystals were phagocytosed by macrophages, but induced phagolysosomal destabilization and rupture because phagocytosis cannot progress appropriately. This process has been known as frustrated phagocytosis wherein the phagocytosed crystals cannot be engulfed or digested. In such cases, lysosomal rupture induces leakage of the lysosomal enzyme cathepsin B, resulting in NLRP3 inflammasome activation (Fig. 3). Similar results were reported by Rajamaki et al.42) on human macrophages. Furthermore, a recent study suggested that the complement system was involved in NLRP3 inflammasome activation by cholesterol crystals43). Calcium phosphate is also a particulate matter, which can accumulate in atherosclerotic lesions and is associated with vascular calcification. In this regard, other investigators and we showed that calcium phosphate crystals, including hydroxyl apatite and tricalcium phosphate, were also phagocytosed and activated NLRP3 inflammasomes through lysosomal rupture and cathepsin B11). At present, however, the mechanism of NLRP3 inflammasome in sensing cathepsin B release by crystal-induced lysosomal destabilization remains undefined.
Fig. 3.
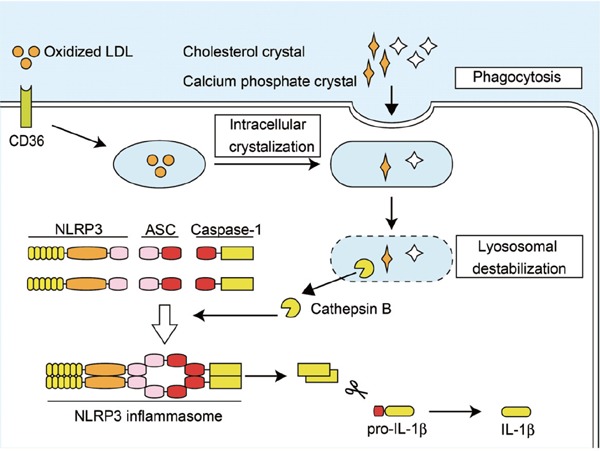
Proposed mechanism of NLRP3 inflammasome activation in atherosclerosis
Cholesterol crystals are incorporated into the lysosome by phagocytosis or formed intracellularly when oxidized LDL is incorporated with CD36. Overloaded crystals cause lysosomal destabilization and rupture, which induce leakage of the lysosomal enzyme cathepsin B, resulting in the activation of NLRP3 inflammasomes. Calcium phosphate crystals also activate NLRP3 inflammasomes through a similar mechanism. LDL, low-density lipoprotein.
Recent studies uncovered an association between NLRP3 inflammasomes and metabolic disease-related lipid species, such as saturated fatty acids (SFAs), ceramide, and 7-ketocholesterol34, 35, 40). Palmitic acid and stearic acid, the major species of SFAs, activate NLRP3 inflammasomes via mitochondrial ROS production and lysosomal destabilization35, 44). An imbalance between SFAs and unsaturated fatty acids (USFAs) was assumed to play a causative role in this response because USFAs could inhibit SFA-induced activation of NLRP3 inflammasomes45, 46).
Regulation of NLRP3 Inflammasomes in Atherosclerosis
The negative regulation of NLRP3 inflammasomes is a potential therapeutic target for atherosclerosis. Recent studies have shown that autophagy, an intracellular degradation system to maintain cellular homeostasis, negatively regulated NLRP3 inflammasome activation through several mechanisms47, 48). Because mitochondrial ROS is important for NLRP3 inflammasome activation in response to many stimuli, clearance of damaged mitochondria by autophagy (mitophagy) can inhibit its activation47). Another study showed that autophagy was able to capture and degrade the assembled complex of NLRP3 inflammasomes via its ubiquitination and modulate its activity48). Indeed, autophagy-defective Atg 5-/- mice developed accelerated atherosclerosis, accompanied by enhanced activation of NLRP3 inflammasomes49). Furthermore, because lysosome is an essential organelle for autolysosome formation and degradation of a particulate matter, activation of lysosome biogenesis by overexpression of transcription factor EB (TFEB) in macrophages has been shown to inhibit cholesterol crystal-induced NLRP3 inflammasome activation and attenuate the progression of atherosclerosis50).
Phosphorylation of protein kinase A (PKA) was reported to be a negative regulator of NLRP inflammasomes51). Transmembrane G protein-coupled receptor 5 (TGR5) bile acid receptor-induced PKA activation leads to the ubiquitination of NLRP3, which is associated with the PKA-induced NLRP3 phosphorylation (Ser 291). Although TGR5 agonists prevent the progression of atherosclerosis by increased cholesterol efflux and alleviated inflammatory responses, antiinflammatory mechanisms were not fully elucidated52). Therefore, further studies are necessary to clarify the precise mechanisms of NLRP3 inflammasome regulation in atherosclerosis.
Concluding Remarks
Increasing evidence indicated the importance of NLRP3 inflammasomes in the pathophysiology of sterile inflammatory diseases. In the progression of atherosclerosis, cholesterol and/or calcium phosphate crystals activate NLRP3 inflammasomes and induce subsequent IL-1β release, leading to vascular inflammation. In a similar manner, using a wire-mediated vascular injury mice model, we previously demonstrated that ASC was upregulated at the site of vascular injury and that its deficiency significantly attenuated IL-1β expression and neointimal formation after vascular injury29). In addition, inflammasome activation in bone marrow-derived inflammatory cells was demonstrated to be essential for neointimal formation in bone marrow chimeric mice. More recently, we found that NLRP3 inflammasome activation by mitochondrial oxidative stress on adventitial macrophages led to the development of abdominal aortic aneurysm in ApoE-/- mice infused with angiotensin II30). The importance of inflammasomes in adventitial macrophages was further supported by the finding that ASC was expressed in the cells of patients with abdominal aortic aneurysm. Therefore, these findings strongly suggested that NLRP3 inflammasomes were a key mediator of inflammation-related vascular diseases. A better understanding of the molecular mechanisms underlying NLRP3 inflammasome activation and development of specific inhibitors will offer new therapeutic or preventive options and break new grounds for studying the role of inflammation in atherosclerosis as well as in other vascular diseases.
Conflict of Interest
None.
References
- 1). Tall AR, Yvan-Charvet L: Cholesterol, inflammation and innate immunity. Nat Rev Immunol, 2015; 15: 104-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Bennett MR, Sinha S, Owens GK: Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res, 2016; 118: 692-702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3). Weber C, Noels H: Atherosclerosis: current pathogenesis and therapeutic options. Nat Med, 2011; 17: 1410-1422 [DOI] [PubMed] [Google Scholar]
- 4). Libby P, Lichtman AH, Hansson GK: Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity, 2013; 38: 1092-1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Nagasawa SY, Ohkubo T, Masaki K, Barinas-Mitchell E, Miura K, Seto T, El-Saed A, Kadowaki T, Willcox BJ, Edmundowicz D, Kadota A, Evans RW, Kadowaki S, Fujiyoshi A, Hisamatsu T, Bertolet MH, Okamura T, Nakamura Y, Kuller LH, Ueshima H, Sekikawa A, Group E-JS : Associations between inflammatory markers and subclinical atherosclerosis in middle-aged white, Japanese-American and Japanese men: The ERA-JUMP Study. J Atheroscler Thromb, 2015; 22: 590-598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Kumakura H, Fujita K, Kanai H, Araki Y, Hojo Y, Kasama S, Iwasaki T, Ichikawa S, Nakashima K, Minami K: High-sensitivity C-reactive protein, lipoprotein(a) and homocysteine are risk factors for coronary artery disease in Japanese patients with peripheral arterial disease. J Atheroscler Thromb, 2015; 22: 344-354 [DOI] [PubMed] [Google Scholar]
- 7). Pradeu T, Cooper EL: The danger theory: 20 years later. Front Immunol, 2012; 3: 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Matzinger P: Tolerance, danger, and the extended family. Annu Rev Immunol, 1994; 12: 991-1045 [DOI] [PubMed] [Google Scholar]
- 9). Takeuchi O, Akira S: Pattern recognition receptors and inflammation. Cell, 2010; 140: 805-820 [DOI] [PubMed] [Google Scholar]
- 10). Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E: NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature, 2010; 464: 1357-1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11). Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Hida S, Sagara J, Taniguchi S, Takahashi M: Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western dietfed apolipoprotein E-deficient mice. Biochem Biophys Res Commun, 2012; 425: 162-168 [DOI] [PubMed] [Google Scholar]
- 12). Gage J, Hasu M, Thabet M, Whitman SC: Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can J Cardiol, 2012; 28: 222-229 [DOI] [PubMed] [Google Scholar]
- 13). Hendrikx T, Jeurissen ML, van Gorp PJ, Gijbels MJ, Walenbergh SM, Houben T, van Gorp R, Pottgens CC, Stienstra R, Netea MG, Hofker MH, Donners MM, Shiri-Sverdlov R: Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr-/- mice. FEBS J, 2015; 282: 2327-2338 [DOI] [PubMed] [Google Scholar]
- 14). Martinon F, Burns K, Tschopp J: The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell, 2002; 10: 417-426 [DOI] [PubMed] [Google Scholar]
- 15). Black RA, Kronheim SR, Merriam JE, March CJ, Hopp TP: A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1β. J Biol Chem, 1989; 264: 5323-5326 [PubMed] [Google Scholar]
- 16). Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K, Kurimoto M, Tanimoto T, Flavell RA, Sato V, Harding MW, Livingston DJ, Su MS: Activation of interferongamma inducing factor mediated by interleukin-1β converting enzyme. Science, 1997; 275: 206-209 [DOI] [PubMed] [Google Scholar]
- 17). Lamkanfi M, Dixit VM: Mechanisms and functions of inflammasomes. Cell, 2014; 157: 1013-1022 [DOI] [PubMed] [Google Scholar]
- 18). Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM: Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature, 2015; 526: 666-671 [DOI] [PubMed] [Google Scholar]
- 19). Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F: Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature, 2015; 526: 660-665 [DOI] [PubMed] [Google Scholar]
- 20). Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, Spiller D, White M, Daniels MJ, Mortellaro A, Penalver M, Paszek P, Steringer JP, Nickel W, Brough D, Pelegrin P: Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ, 2016; 23: 1219-1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM: Cryopyrin activates the inflammasome in response to toxins and ATP. Nature, 2006; 440: 228-232 [DOI] [PubMed] [Google Scholar]
- 22). Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature, 2006; 440: 237-241 [DOI] [PubMed] [Google Scholar]
- 23). Guo H, Callaway JB, Ting JP: Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med, 2015; 21: 677-687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G: K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity, 2013; 38: 1142-1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). He Y, Hara H, Nunez G: Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci, 2016; 41: 1012-1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26). Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES: Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem, 2012; 287: 36617-36622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J: Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell, 2013; 49: 331-338 [DOI] [PubMed] [Google Scholar]
- 28). Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD: Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet, 2001; 29: 301-305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29). Yajima N, Takahashi M, Morimoto H, Shiba Y, Takahashi Y, Masumoto J, Ise H, Sagara J, Nakayama J, Taniguchi S, Ikeda U: Critical role of bone marrow apoptosis-associated speck-like protein, an inflammasome adaptor molecule, in neointimal formation after vascular injury in mice. Circulation, 2008; 117: 3079-3087 [DOI] [PubMed] [Google Scholar]
- 30). Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Yoshimura K, Aoki H, Tsutsui H, Noda T, Sagara J, Taniguchi S, Takahashi M: Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol, 2015; 35: 127-136 [DOI] [PubMed] [Google Scholar]
- 31). Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U: Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation, 2011; 123: 594-604 [DOI] [PubMed] [Google Scholar]
- 32). Komada T, Usui F, Shirasuna K, Kawashima A, Kimura H, Karasawa T, Nishimura S, Sagara J, Noda T, Taniguchi S, Muto S, Nagata D, Kusano E, Takahashi M: ASC in Renal Collecting Duct Epithelial Cells Contributes to Inflammation and Injury after Unilateral Ureteral Obstruction. Am J Pathol, 2014; 184: 1287-1298 [DOI] [PubMed] [Google Scholar]
- 33). Komada T, Usui F, Kawashima A, Kimura H, Karasawa T, Inoue Y, Kobayashi M, Mizushina Y, Kasahara T, Taniguchi S, Muto S, Nagata D, Takahashi M: Role of NLRP3 Inflammasomes for Rhabdomyolysis-induced Acute Kidney Injury. Sci Rep, 2015; 5: 10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34). Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD: The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med, 2011; 17: 179-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP: Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol, 2011; 12: 408-415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M: Lack of interleukin-1β decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol, 2003; 23: 656-660 [DOI] [PubMed] [Google Scholar]
- 37). Chi H, Messas E, Levine RA, Graves DT, Amar S: Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model: pharmacotherapeutic implications. Circulation, 2004; 110: 1678-1685 [DOI] [PubMed] [Google Scholar]
- 38). Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL, Owens GK: Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest, 2012; 122: 70-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ: CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol, 2013; 14: 812-820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40). Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J: Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis, 2011; 2: e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41). Komohara Y, Fujiwara Y, Ohnishi K, Shiraishi D, Takeya M: Contribution of macrophage polarization to metabolic diseases. J Atheroscler Thromb, 2016; 23: 10-17 [DOI] [PubMed] [Google Scholar]
- 42). Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund KK: Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One, 2010; 5: e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, Lappegard KT, Brekke OL, Lambris JD, Damas JK, Latz E, Mollnes TE, Espevik T: Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol, 2014; 192: 2837-2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Weber K, Schilling JD: Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem, 2014; 289: 9158-9171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45). L'Homme L, Esser N, Riva L, Scheen A, Paquot N, Piette J, Legrand-Poels S: Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J Lipid Res, 2013; 54: 2998-3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46). Finucane OM, Lyons CL, Murphy AM, Reynolds CM, Klinger R, Healy NP, Cooke AA, Coll RC, McAllan L, Nilaweera KN, O'Reilly ME, Tierney AC, Morine MJ, Alcala-Diaz JF, Lopez-Miranda J, O'Connor DP, O'Neill LA, McGillicuddy FC, Roche HM: Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1β secretion and insulin resistance despite obesity. Diabetes, 2015; 64: 2116-2128 [DOI] [PubMed] [Google Scholar]
- 47). Zhou R, Yazdi AS, Menu P, Tschopp J: A role for mitochondria in NLRP3 inflammasome activation. Nature, 2011; 469: 221-225 [DOI] [PubMed] [Google Scholar]
- 48). Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH: Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol, 2012; 13: 255-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49). Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF: Autophagy links inflammasomes to atherosclerotic progression. Cell Metab, 2012; 15: 534-544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50). Emanuel R, Sergin I, Bhattacharya S, Turner JN, Epelman S, Settembre C, Diwan A, Ballabio A, Razani B: Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol, 2014; 34: 1942-1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51). Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, Zheng M, Zhang X, Xia D, Ke Y, Lu L, Wang D: Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity, 2016; 45: 802-816 [DOI] [PubMed] [Google Scholar]
- 52). Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, Rizzo G, Gioiello A, Adorini L, Pellicciari R, Auwerx J, Schoonjans K: TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab, 2011; 14: 747-757 [DOI] [PMC free article] [PubMed] [Google Scholar]