

Abstract
We have learned that low-density lipoprotein (LDL) cholesterol is the cause of atherosclerosis from various aspects, including a single case with familial hypercholesterolemia, other cases with different types of Mendelian dyslipidemias, large-scale randomized controlled trials using LDL cholesterol lowering therapies, and Mendelian randomization studies using common as well as rare variants associated with LDL cholesterol levels. There is no doubt that determinations of genotypes in lipid-associated genes have contributed not only to the genetic diagnosis for Mendelian dyslipidemias but also to the discoveries of novel therapeutic targets. Furthermore, recent studies have shown that such genetic information could provide useful clues for the risk prediction as well as risk stratification in general and in particular population. We provide the current understanding of genetic analyses relating to plasma lipids and coronary artery disease.
Keywords: Dyslipidemia, Familial hypercholesterolemia, PCSK9, Coronary artery disease
Introduction
Familial hypercholesterolemia (FH; OMIM #143890) is characterized by the triad of (1) primary hyper-LDL-cholesterolemia, (2) tendon xanthomas, and (3) premature coronary artery disease (CAD)1, 2). Since the identification of LDL receptor in 1970s, as the cause of FH, three different genes have been shown to cause this clinical phenotype. Genetic analyses for the patients with FH have provided not only definitive diagnosis but also the chances of cascade screening for their offsprings3). Moreover, investigations for the genetic background for FH led to the development of novel therapeutic targets, such as proprotein convertase subtilisin/kexin type 9 (PCSK9) and apolipoprotein B (ApoB). Moreover, observations on other Mendelian dyslipidemias exhibiting LDL cholesterol elevation, such as sitosterolemia, and autosomal recessive hypercholesterolemia (ARH) convince us the causal role of LDL cholesterol on atherosclerosis4–6). Recent advances of comprehensive genetic analyses made us realize the genetic architecture comprising rare variants with large effect sizes causing Mendelian dyslipidemia and common variants with small effect sizes causing polygenic dyslipidemias7). Due to such advances, we are now understanding that determination of rare as well as common genetic variants associated with lipids and CAD are quite useful not only in research settings but also in clinical settings.
Lessons from Patients with FH and Other Mendelian Dyslipidemias
(1). LDL Cholesterol as a Causal Factor for the Development of CAD
FH is one of the most common monogenic Mendelian inherited disorders characterized by excess deposition of cholesterol in tissues leading to tendon xanthomas and premature CAD1, 2). It has been well known that FH is caused by genetic mutations of LDL receptor or its related proteins, including ApoB, or PCSK9, although a fraction of patients with clinical FH were unexplained by mutations in those genes1, 2). It is of note that even a single encounter with a patient with FH harboring an LDL receptor mutation could tell us that LDL cholesterol is a causal factor of CAD. Moreover, the fact that individuals with different mutation(s) in different gene(s) that significantly elevated LDL cholesterol level exhibit premature CAD strongly convince us that LDL cholesterol is the causal factor of CAD, including ARH caused by LDL receptor adaptor protein 1 (LDLRAP1) gene mutations and sitosterolemia caused by ATP-binding cassette subfamily G member 5/8 (ABCG5/ABCG8) gene mutations (Fig. 1). Investigations on such extreme cases exhibiting Mendelian form of inheritance caused by (a) striking mutation(s) are one of the best ways to understand the roles of particular molecules and the causal relationship between a biomarker and an outcome.
Fig. 1.

Mendelian dyslipidemias exhibiting hyper LDL cholesterolemia. A. Autosomal recessive hypercholesterolemia caused by LDL receptor adaptor protein 1 gene mutations. B. Sitosterolemia caused by ATP-binding cassette subfamily G member 5 gene mutations.
(2). Clinical Impact of Genetic Analyses for FH-Associated Gene
Currently, genetic analyses for FH-associated genes are mostly conducted in research settings in most of the country, including Japan, since its cost cannot be covered by health insurance. Moreover, such analyses have been suggested as a pure diagnostic tool when the clinical diagnosis of FH is unclear by most of the guidelines8–10). Based on such backgrounds, few data had existed regarding the clinical impact of determination of genetic status of FH in the patients with elevated LDL cholesterol level, which could aid us for risk stratification in the patients suspected as FH. In this regard, we have assessed the prevalence of an FH mutation among those with severe hypercholesterolemia and investigated whether CAD risk varies according to mutation status beyond the observed LDL cholesterol level and found that FH mutation status had substantially increased risk for CAD (Fig. 2)11). Given that genetic analyses for FH-associated genes needed to be assessed only once in a lifetime could help identifying the individuals with the highest risk far before the development of other traditional acquired risk factors, such as hypertension and diabetes among the patients with elevated LDL cholesterol level, it may be reasonable that such costs be covered by insurance.
Fig. 2.

Impact of clinical signs and genetic diagnosis of familial hypercholesterolemia on risk of coronary artery disease. Odds ratios were calculated using logistic regression after adjustment for age, sex, hypertension, diabetes, smoking, and LDL cholesterol levels.
(3). Clinical Application of whole Exome Sequencing for Mendelian Dyslipidemias
Usefulness of whole exome sequencing (WES) has been shown in many Mendelian inherited diseases, especially, in cases with recessive form of inheritance12, 13). We have demonstrated that WES is feasible even where DNA is available in only one individual if the recessive form of inheritance is firmly established in a family with Tangier disease caused by ATP-binding cassette transporter A1 gene mutations12). Moreover, WES could identify causative mutations in different genes simultaneously, where patients suffered from different Mendelian inherited diseases, such as sitosterolemia and familial Mediterranean fever5). Furthermore, we also identified a rare case with heterozygous FH caused by de novo LDL receptor mutation exhibiting recessive form of inheritance using WES14).
(4). Challenges of Identifying of Novel FH Genes
We could not determine causative mutations within established three FH genes (LDL receptor, ApoB, and PCSK9) in up to 40% of the cases with clinically diagnosed as FH. Such a fact has motivated researchers to identify novel genes causing this situation among the patients with genetically undetermined FH. We conducted WES on 213 selected family members from 41 kindreds with suspected Mendelian inheritance of extreme levels of LDL cholesterol or high-density lipoprotein (HDL) cholesterol (mainly LDL cholesterol); however, we failed to find any novel genes related with this disease (Fig. 3)15). Several potential reasons could be considered. First, it is still hard to determine structural variations by WES. Accordingly, further improvements in the application of approaches to discover structural variations using WES are needed. Secondary, large numbers of shared rare alleles within families obfuscated causal variant identification. We still need a large pedigree to identify a causal gene in a dominant mode of inheritance. Third, we found that ∼15% of the families suspected as Mendelian dyslipidemia could be polygenic carrying a significant burden of common lipid-related alleles, rather than one or two critical mutations. It is still hard to define polygenic Mendelian dyslipidemias, since not all variations contributed to this phenotype have not been discovered.
Fig. 3.

Yield of whole exome sequencing for Mendelian dyslipidemias. The graph shows the percentage of final status using whole exome sequencing of the 41 families suspected as Mendelian dyslipidemias.
(5). Polygenic FH
In 2013, Humphries and colleagues have demonstrated that the phenotype as FH could be caused by the excess of LDL cholesterol-raising common variants in a substantial proportion of patients with FH without a known mutation, in contrast to a situation where a single rare variant causes significant elevation in LDL cholesterol level (Fig. 4). Similar results were obtained in our analyses mentioned earlier, and we estimated up to 15% of the families suspected as Mendelian dyslipidemia could be polygenic cause16).
Fig. 4.

Monogenic familial hypercholesterolemia (FH) and polygenic FH. X-axis represents LDL cholesterol value. Y-axis represents frequency. A single rare variant with large effect size or multiple single-nucleotide polymorphisms with modest effect sizes could elevate LDL cholesterol level substantially.
Heritability from Common and Rare Variants
Plasma lipids and lipoproteins are heritable risk factors for CAD, with heritability estimates ranging from 40% to 60%. Genome-wide association studies (GWASs) for plasma lipids and CAD have successfully identified 157 gene regions for plasma lipids17) and 58 for CAD18). Despite the success of GWAS, single-nucleotide polymorphisms (SNPs) at these loci explain only a modest proportion of the heritability—20%–25% of the heritability for plasma lipids and < 10% of the heritability of CAD. These observations have led to the general question of how to account for the unexplained heritability. To estimate such contributions, several rare variant association studies investigating on rare variants and lipids using whole genome sequencing and imputation-based approach have been performed. Those investigations have found that rare and low-frequency variants account for 3%–4% of the variance explained in lipids19–21). In contrast, we have demonstrated that fair portion of the missing heritability for lipid traits can be explained by multiple common variants at each known locus22).
Genetic Risk Score for Risk Prediction of CAD
It is well known that CAD is highly heritable motivating the inclusion of family history of CAD as an option for risk assessment in patients with borderline risk. Moreover, researchers have tried to identify genetic clues associated with CAD and found as many as 58 independent loci associated with CAD. However, any guideline so far does not recommend the use of any genetic marker for the risk assessment of CAD due to lack of evidence23). In this regard, Kathiresan and colleagues initially organized an interesting study investigating if aggregating information of individual SNPs (at that time 13 SNPs) could increase power of risk prediction in 201024). After that, updated genetic risk scores (GRSs) using increasing number of SNPs showed substantial clinical usefulness beyond traditional risk factors, including family history (Table 1). It is of note that such GRS could improve risk prediction far beyond family history information25). Moreover, we can assess GRS quantitatively far before the development of traditional risk factors26). Interestingly, a recent study showed that a favorable lifestyle was associated with a nearly 50% lower relative risk of CAD than was an unfavorable lifestyle among individuals at high GRS27). Another study showed that individuals with the highest GRS derived the largest relative and absolute clinical benefit from statin therapy28). Those facts collectively suggested that genetic predisposition to CAD is not deterministic, or rather, genetic risk might be attenuated by a favorable lifestyle and/or statin therapy. Accordingly, it may be a prime time for us to consider GRS as a risk assessment tool in clinical settings.
Table 1. Comparison of power between traditional risk factors and GRS.
| Baseline characteristics | HR (95% CI) | P value |
|---|---|---|
| Age (per 10-year increment) | 1.86 (1.74 – 1.98) | 2.8 × 10−83 |
| Men | 2.50 (2.28 – 2.74) | 1.4 × 10−84 |
| Body mass index (top vs. bottom quintile) | 1.07 (0.92 – 1.24) | 0.38 |
| Smoking | 1.81 (1.66 – 1.98) | 1.1 × 10−38 |
| Hypertension | 1.69 (1.53 – 1.86) | 1.6 × 10−25 |
| Prevalent diabetes mellitus | 2.43 (2.12 – 2.78) | 4.0 × 10−37 |
| Apolipoprotein B (top vs. bottom quintile) | 2.39 (2.03 – 2.82) | 9.0 × 10−26 |
| Apolipoprotein A1 (bottom vs. top quintile) | 1.87 (1.61 – 2.18) | 5.0 × 10−16 |
| Self-reported family history | 1.41 (1.29 – 1.53) | 2.9 × 10−15 |
| GRS (top vs. bottom quintile) | 1.85 (1.61 – 2.12) | 1.2 × 10−18 |
HR: hazard ratio, GRS: genetic risk score
Mendelian Randomization (Common and Rare)
To establish a causal relationship between a risk factor (usually a biomarker) and an outcome, randomized controlled trials (RCTs), which require a large amount of time and effort, are the gold standard. In contrast, the Mendelian randomization study is a technique that uses genotypes as instruments to assess a causal relationship between biomarkers and outcomes. In a Mendelian randomization study, a genetic variant associated with a particular biomarker is used as a proxy for the biomarker. Outcomes are compared between the group harboring the effect allele and a group with the reference allele. This approach can be considered a proxy for an RCT, in which the randomized groups have similar confounding variables. Accordingly, a Mendelian randomization study can be regarded as a natural RCT.
(1). Effect on Cardiovascular Diseases
A number of biomarkers have been shown to be associated with CAD from epidemiological studies. However, careful attention should be paid for the fact that those biomarkers are causal or not. In the case with LDL cholesterol, many epidemiological studies, as well as Mendelian randomization studies, using rare as well as common genetic variants consistently showed that the directions and magnitude by LDL cholesterol level and the odds for CAD were linearly correlated. Moreover, those odds induced by genetically determined LDL cholesterol levels were consistently larger than those induced by pharmacological interventions (Fig. 5). In contrast, common genetic variants in HDL-raising genes did not reduce the risk of CAD29, 30). Interestingly, plasma triglyceride and lipoprotein (a) (Lp(a)) have been shown to be associated with CAD in Mendelian randomization studies, suggesting their causal roles for the development of CAD31).
Fig. 5.
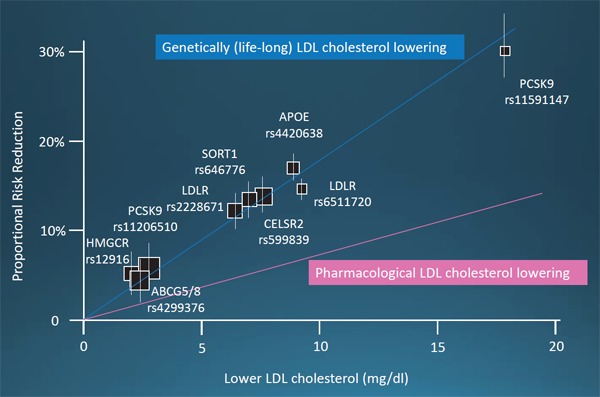
Relationship between coronary artery disease (CAD) risk reduction and LDL cholesterol lowering. Blue line indicates the relationship between CAD risk reduction and genetic (lifelong) LDL cholesterol lowering. Pink line indicates the relationship between CAD risk reduction and pharmacological lowering of LDL cholesterol.
Regarding other variables, a large-scale Mendelian randomization study has investigated the causal role of uric acid on CAD, heart failure, ischemic stroke, type 2 diabetes, and gout. They found that serum uric acid levels, increased by the common genetic backgrounds, were not associated with CAD, heart failure, ischemic stroke, or type 2 diabetes32). Moreover, the patients with Lesch–Nyhan syndrome exhibiting increased levels of uric acid caused by rare mutations in hypoxanthine phosphoribosyltransferase 1 gene manifests with severe hyperuricemia since birth, gout, renal stones, and neurological impairment but not CAD, heart failure, ischemic stroke, or type 2 diabetes. Those findings from common as well as rare variants suggest that uric acid does not seem to be causally associated with cardiovascular diseases33).
(2). Effect on Diabetes
A meta-analysis of RCT using statin showed consistent effectiveness of LDL cholesterol lowering therapy on reduction of CAD. However, it also revealed that LDL cholesterol lowering therapy using statin increased the risk of diabetes34). Soon after this report, a Mendelian randomization study focusing on the effects of LDL cholesterol lowering and development of diabetes by variants in HMG-coenzyme A reductase (HMGCR) gene, which is the target of statin. They found that loss of function variants in HMGCR gene is associated with reduced LDL cholesterol level as well as increased risk of diabetes, which is consistent with RCT35). Next critical questions are (1) how about other therapies, such as ezetimibe, ApoB, and PCSK9 and (2) how about rare variants with large effect size, such as the patients with FH. For the first question, a meta-analysis focusing on this point revealed that LDL cholesterol lowering alleles were associated with reduced risk of CAD as well as increased risk of diabetes regardless of gene. Such a notion could lead us to speculate that LDL cholesterol is inversely but weakly associated with diabetes36). For the second question, a large-scale, simple observational study has suggested that the prevalence of diabetes in the patients with FH is smaller than that of non-FH, suggesting that exposure of high LDL cholesterol level could be protective from diabetes37). Similarly, we can estimate any side effects by novel drugs inhibiting a particular molecule by investigating Mendelian diseases with large effect size or Mendelian randomization studies using common variants in the gene of interest.
Conclusion
It is well known that dyslipidemias and CAD are highly heritable diseases, motivating us to investigate their genetic backgrounds for a long time. Now, we have learned that common as well as rare genetic variants are contributing to such “heritability.” Systematic genetic analyses should be able to provide more clinically relevant information for the assessments of the patients with dyslipidemias and CAD in the near future.
Acknowledgments and Notice of Grant Support
A scientific research grant from the Ministry of Education, Science, and Culture of Japan (No. 16K19394).
Conflicts of Interest Disclosures
None.
References
- 1). Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D. eds. The metabolic and molecular bases of inherited disease, ed 8, vol 2. New York: McGraw-Hill; 2001: 2863-2913 [Google Scholar]
- 2). Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med, 2007; 4: 214-225 [DOI] [PubMed] [Google Scholar]
- 3). Tada H, Kawashiri MA, Ohtani R, Noguchi T, Nakanishi C, Konno T, Hayashi K, Nohara A, Inazu A, Kobayashi J, Mabuchi H, Yamagishi M. A novel type of familial hypercholesterolemia: double heterozygous mutations in LDL receptor and LDL receptor adaptor protein 1 gene. Atherosclerosis, 2011; 219: 663-636 [DOI] [PubMed] [Google Scholar]
- 4). Tada H, Kawashiri MA, Takata M, Matsunami K, Imamura A, Matsuyama M, Sawada H, Nunoi H, Konno T, Hayashi K, Nohara A, Inazu A, Kobayashi J, Mabuchi H, Yamagishi M. Infantile Cases of Sitosterolaemia with Novel Mutations in the ABCG5 Gene: Extreme Hypercholesterolaemia is Exacerbated by Breastfeeding. JIMD Rep, 2015; 21: 115-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Tada H, Kawashiri MA, Okada H, Endo S, Toyoshima Y, Konno T, Nohara A, Inazu A, Takao A, Mabuchi H, Yamagishi M, Hayashi K. A Rare Coincidence of Sitosterolemia and Familial Mediterranean Fever Identified by Whole Exome Sequencing. J Atheroscler Thromb, 2016; 23: 884-890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Tada H, Kawashiri MA, Ikewaki K, Terao Y, Noguchi T, Nakanishi C, Tsuchida M, Takata M, Miwa K, Konno T, Hayashi K, Nohara A, Inazu A, Kobayashi J, Mabuchi H, Yamagishi M. Altered metabolism of low-density lipoprotein and very-low-density lipoprotein remnant in autosomal recessive hypercholesterolemia: results from stable isotope kinetic study in vivo. Circ Cardiovasc Genet, 2012; 5: 35-41 [DOI] [PubMed] [Google Scholar]
- 7). Tada H, Kawashiri MA, Yamagishi M. Comprehensive genotyping in dyslipidemia: mendelian dyslipidemias caused by rare variants and Mendelian randomization studies using common variants. J Hum Genet. 2017. in press [DOI] [PubMed] [Google Scholar]
- 8). Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A, European Atherosclerosis Society Consensus Panel Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J, 2013; 34: 3478-3490a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Scientific Steering Committee on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ, 1991; 303: 893-896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Teramoto T, Sasaki J, Ishibashi S, Birou S, Daida H, Dohi S, Egusa G, Hiro T, Hirobe K, Iida M, Kihara S, Kinoshita M, Maruyama C, Ohta T, Okamura T, Yamashita S, Yokode M, Yokote K, Harada-Shiba M, Arai H, Bujo H, Nohara A, Ohta T, Oikawa S, Okada T, Wakatsuki A. Familial hypercholesterolemia. J Atheroscler Thromb, 2014; 21: 6-10 [DOI] [PubMed] [Google Scholar]
- 11). Tada H, Kawashiri MA, Nohara A, Inazu A, Mabuchi H, Yamagishi M. Impact of Clinical Signs and Genetic Diagnosis of Familial Hypercholesterolaemia on the Prevalence of Coronary Artery Disease in Patients with Severe Hypercholesterolaemia. Eur Heart J 2017. in press [DOI] [PubMed] [Google Scholar]
- 12). Tada H, Kawashiri MA, Nohara A, Saito R, Tanaka Y, Nomura A, Konno T, Sakata K, Fujino N, Takamura T, Inazu A, Mabuchi H, Yamagishi M, Hayashi K. Whole exome sequencing combined with integrated variant annotation prediction identifies asymptomatic Tangier disease with compound heterozygous mutations in ABCA1 gene. Atherosclerosis, 2015; 240: 324-329 [DOI] [PubMed] [Google Scholar]
- 13). Tada H, Kawashiri MA, Yamagishi M, Hayashi K. Whole Exome Sequencing in Monogenic Dyslipidemias. J Atheroscler Thromb, 2015; 22: 881-885 [DOI] [PubMed] [Google Scholar]
- 14). Tada H, Hosomichi K, Okada H, Kawashiri MA, Nohara A, Inazu A, Tomizawa S, Tajima A, Mabuchi H, Hayashi K. A de novo mutation of the LDL receptor gene as the cause of familial hypercholesterolemia identified using whole exome sequencing. Clin Chim Acta, 2016; 453: 194-196 [DOI] [PubMed] [Google Scholar]
- 15). Stitziel NO, Peloso GM, Abifadel M, Cefalu AB, Fouchier S, Motazacker MM, Tada H, Larach DB, Awan Z, Haller JF, Pullinger CR, Varret M, Rabès JP, Noto D, Tarugi P, Kawashiri MA, Nohara A, Yamagishi M, Risman M, Deo R, Ruel I, Shendure J, Nickerson DA, Wilson JG, Rich SS, Gupta N, Farlow DN, Neale BM, Daly MJ, Kane JP, Freeman MW, Genest J, Rader DJ, Mabuchi H, Kastelein JJ, Hovingh GK, Averna MR, Gabriel S, Boileau C, Kathiresan S. Exome sequencing in suspected monogenic dyslipidemias. Circ Cardiovasc Genet, 2015; 8: 343-350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, Harrison SC, Li K, Drenos F, Karpe F, Neil HA, Descamps OS, Langenberg C, Lench N, Kivimaki M, Whittaker J, Hingorani AD, Kumari M, Humphries SE. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet, 2013; 381: 1293-1301 [DOI] [PubMed] [Google Scholar]
- 17). Global Lipids Genetics Consortium. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A, Jackson AU, Johansson A, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikäinen LP, Magnusson PK, Mangino M, Mihailov E, Montasser ME, Müller-Nurasyid M, Nolte IM, O'Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney AS, Döring A, Elliott P, Epstein SE, Eyjolfsson GI, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJ, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimäki T, Lin SY, Lindström J, Loos RJ, Mach F, McArdle WL, Meisinger C, Mitchell BD, Müller G, Nagaraja R, Narisu N, Nieminen TV, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stancáková A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YD, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrières J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Järvelin MR, Jula A, Kähönen M, Kaprio J, Kesäniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, März W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njølstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PE, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BH, Ordovas JM, Boerwinkle E, Palmer CN, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Kathiresan S, Mohlke KL, Ingelsson E, Abecasis GR. Discovery and refinement of loci associated with lipid levels. Nat Genet, 2013; 45: 1274-1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18). Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikäinen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Huang J, Jalilzadeh S, Kessler T, König IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FU, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimäki T, Loos RJ, Melander O, Metspalu A, März W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O'Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ, Farrall M, CARDIoGRAMplusC4D Consortium A comprehensive 1,000 Genomes-based genome-wide association metaanalysis of coronary artery disease. Nat Genet, 2015; 47: 1121-1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19). Helgadottir A, Gretarsdottir S, Thorleifsson G, Hjartarson E, Sigurdsson A, Magnusdottir A, Jonasdottir A, Kristjansson H, Sulem P, Oddsson A, Sveinbjornsson G, Steinthorsdottir V, Rafnar T, Masson G, Jonsdottir I, Olafsson I, Eyjolfsson GI, Sigurdardottir O, Daneshpour MS, Khalili D, Azizi F, Swinkels DW, Kiemeney L, Quyyumi AA, Levey AI, Patel RS, Hayek SS, Gudmundsdottir IJ, Thorgeirsson G, Thorsteinsdottir U, Gudbjartsson DF, Holm H, Stefansson K. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet, 2016; 48: 634-639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Surakka I, Horikoshi M, Mägi R, Sarin AP, Mahajan A, Lagou V, Marullo L, Ferreira T, Miraglio B, Timonen S, Kettunen J, Pirinen M, Karjalainen J, Thorleifsson G, Hägg S, Hottenga JJ, Isaacs A, Ladenvall C, Beekman M, Esko T, Ried JS, Nelson CP, Willenborg C, Gustafsson S, Westra HJ, Blades M, de Craen AJ, de Geus EJ, Deelen J, Grallert H, Hamsten A, Havulinna AS, Hengstenberg C, Houwing-Duistermaat JJ, Hyppönen E, Karssen LC, Lehtimäki T, Lyssenko V, Magnusson PK, Mihailov E, Müller-Nurasyid M, Mpindi JP, Pedersen NL, Penninx BW, Perola M, Pers TH, Peters A, Rung J, Smit JH, Steinthorsdottir V, Tobin MD, Tsernikova N, van Leeuwen EM, Viikari JS, Willems SM, Willemsen G, Schunkert H, Erdmann J, Samani NJ, Kaprio J, Lind L, Gieger C, Metspalu A, Slagboom PE, Groop L, van Duijn CM, Eriksson JG, Jula A, Salomaa V, Boomsma DI, Power C, Raitakari OT, Ingelsson E, Järvelin MR, Thorsteinsdottir U, Franke L, Ikonen E, Kallioniemi O, Pietiäinen V, Lindgren CM, Stefansson K, Palotie A, McCarthy MI, Morris AP, Prokopenko I, Ripatti S, ENGAGE Consortium The impact of low-frequency and rare variants on lipid levels. Nat Genet, 2015; 47: 589-597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Peloso GM, Auer PL, Bis JC, Voorman A, Morrison AC, Stitziel NO, Brody JA, Khetarpal SA, Crosby JR, Fornage M, Isaacs A, Jakobsdottir J, Feitosa MF, Davies G, Huffman JE, Manichaikul A, Davis B, Lohman K, Joon AY, Smith AV, Grove ML, Zanoni P, Redon V, Demissie S, Lawson K, Peters U, Carlson C, Jackson RD, Ryckman KK, Mackey RH, Robinson JG, Siscovick DS, Schreiner PJ, Mychaleckyj JC, Pankow JS, Hofman A, Uitterlinden AG, Harris TB, Taylor KD, Stafford JM, Reynolds LM, Marioni RE, Dehghan A, Franco OH, Patel AP, Lu Y, Hindy G, Gottesman O, Bottinger EP, Melander O, Orho-Melander M, Loos RJ, Duga S, Merlini PA, Farrall M, Goel A, Asselta R, Girelli D, Martinelli N, Shah SH, Kraus WE, Li M, Rader DJ, Reilly MP, McPherson R, Watkins H, Ardissino D, NHLBI GO Exome Sequencing Project. Zhang Q, Wang J, Tsai MY, Taylor HA, Correa A, Griswold ME, Lange LA, Starr JM, Rudan I, Eiriksdottir G, Launer LJ, Ordovas JM, Levy D, Chen YD, Reiner AP, Hayward C, Polasek O, Deary IJ, Borecki IB, Liu Y, Gudnason V, Wilson JG, van Duijn CM, Kooperberg C, Rich SS, Psaty BM, Rotter JI, O'Donnell CJ, Rice K, Boerwinkle E, Kathiresan S, Cupples LA. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am J Hum Genet, 2014; 94: 223-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). Tada H, Won HH, Melander O, Yang J, Peloso GM, Kathiresan S. Multiple associated variants increase the heritability explained for plasma lipids and coronary artery disease. Circ Cardiovasc Genet, 2014; 7: 583-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Writing Group Members. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics Committee Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation, 2016; 133: e38-360 [DOI] [PubMed] [Google Scholar]
- 24). Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, Perola M, Jula A, Sinisalo J, Lokki ML, Nieminen MS, Melander O, Salomaa V, Peltonen L, Kathiresan S. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet, 2010; 376: 1393-1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Tada H, Melander O, Louie JZ, Catanese JJ, Rowland CM, Devlin JJ, Kathiresan S, Shiffman D. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J, 2016; 37: 561-567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26). Tada H, Shiffman D, Smith JG, Sjögren M, Lubitz SA, Ellinor PT, Louie JZ, Catanese JJ, Engström G, Devlin JJ, Kathiresan S, Melander O. Twelve-single nucleotide poly morphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke, 2014; 45: 2856-2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, Chasman DI, Baber U, Mehran R, Rader DJ, Fuster V, Boerwinkle E, Melander O, Orho-Melander M, Ridker PM, Kathiresan S. Genetic Risk Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med. 2016; 375: 2349-2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield MJ, Devlin JJ, Nordio F, Hyde CL, Cannon CP, Sacks FM, Poulter NR, Sever PS, Ridker PM, Braunwald E, Melander O, Kathiresan S, Sabatine MS. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet, 2015; 385: 2264-2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29). Haase CL1, Tybjærg-Hansen A, Grande P, Frikke-Schmidt R. Genetically elevated apolipoprotein A-I, highdensity lipoprotein cholesterol levels, and risk of ischemic heart disease. J Clin Endocrinol Metab, 2010; 95: E500-510 [DOI] [PubMed] [Google Scholar]
- 30). Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Hólm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, König IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schäfer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O'Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet, 2012; 380: 572-580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31). Jansen H, Samani NJ, Schunkert H. Mendelian randomization studies in coronary artery disease. Eur Heart J, 2014; 35: 1917-1924 [DOI] [PubMed] [Google Scholar]
- 32). Keenan T, Zhao W, Rasheed A, Ho WK, Malik R, Felix JF, Young R, Shah N, Samuel M, Sheikh N, Mucksavage ML, Shah O, Li J, Morley M, Laser A, Mallick NH, Zaman KS, Ishaq M, Rasheed SZ, Memon FU, Ahmed F, Hanif B, Lakhani MS, Fahim M, Ishaq M, Shardha NK, Ahmed N, Mahmood K, Iqbal W, Akhtar S, Raheel R, O'Donnell CJ, Hengstenberg C, März W, Kathiresan S, Samani N, Goel A, Hopewell JC, Chambers J, Cheng YC, Sharma P, Yang Q, Rosand J, Boncoraglio GB, Kazmi SU, Hakonarson H, Köttgen A, Kalogeropoulos A, Frossard P, Kamal A, Dichgans M, Cappola T, Reilly MP, Danesh J, Rader DJ, Voight BF, Saleheen D. Causal Assessment of Serum Urate Levels in Cardiometabolic Diseases Through a Mendelian Randomization Study. J Am Coll Cardiol, 2016; 67: 407-416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Marian AJ. Genetic Causality in Complex Traits: The Case of Uric Acid. J Am Coll Cardiol, 2016; 67: 417-419 [DOI] [PubMed] [Google Scholar]
- 34). Cholesterol Treatment Trialists' (CTT) Collaborators. Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet, 2012; 380: 581-590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, Sofat R, Stender S, Johnson PC, Scott RA, Leusink M, Verweij N, Sharp SJ, Guo Y, Giambartolomei C, Chung C, Peasey A, Amuzu A, Li K, Palmen J, Howard P, Cooper JA, Drenos F, Li YR, Lowe G, Gallacher J, Stewart MC, Tzoulaki I, Buxbaum SG, van der A DL, Forouhi NG, Onland-Moret NC, van der Schouw YT, Schnabel RB, Hubacek JA, Kubinova R, Baceviciene M, Tamosiunas A, Pajak A, Topor-Madry R, Stepaniak U, Malyutina S, Baldassarre D, Sennblad B, Tremoli E, de Faire U, Veglia F, Ford I, Jukema JW, Westendorp RG, de Borst GJ, de Jong PA, Algra A, Spiering W, Maitland-van der Zee AH, Klungel OH, de Boer A, Doevendans PA, Eaton CB, Robinson JG, Duggan D, DIAGRAM Consortium; MAGIC Consortium; InterAct Consortium. Kjekshus J, Downs JR, Gotto AM, Keech AC, Marchioli R, Tognoni G, Sever PS, Poulter NR, Waters DD, Pedersen TR, Amarenco P, Nakamura H, McMurray JJ, Lewsey JD, Chasman DI, Ridker PM, Maggioni AP, Tavazzi L, Ray KK, Seshasai SR, Manson JE, Price JF, Whincup PH, Morris RW, Lawlor DA, Smith GD, Ben-Shlomo Y, Schreiner PJ, Fornage M, Siscovick DS, Cushman M, Kumari M, Wareham NJ, Verschuren WM, Redline S, Patel SR, Whittaker JC, Hamsten A, Delaney JA, Dale C, Gaunt TR, Wong A, Kuh D, Hardy R, Kathiresan S, Castillo BA, van der Harst P, Brunner EJ, Tybjaerg-Hansen A, Marmot MG, Krauss RM, Tsai M, Coresh J, Hoogeveen RC, Psaty BM, Lange LA, Hakonarson H, Dudbridge F, Humphries SE, Talmud PJ, Kivimäki M, Timpson NJ, Langenberg C, Asselbergs FW, Voevoda M, Bobak M, Pikhart H, Wilson JG, Reiner AP, Keating BJ, Hingorani AD, Sattar N. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet, 2015; 385: 351-361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Lotta LA, Sharp SJ, Burgess S, Perry JR, Stewart ID, Willems SM, Luan J, Ardanaz E, Arriola L, Balkau B, Boeing H, Deloukas P, Forouhi NG, Franks PW, Grioni S, Kaaks R, Key TJ, Navarro C, Nilsson PM, Overvad K, Palli D, Panico S, Quirós JR, Riboli E, Rolandsson O, Sacerdote C, Salamanca-Fernandez E, Slimani N, Spijkerman AM, Tjonneland A, Tumino R, van der A DL, van der Schouw YT, McCarthy MI, Barroso I, O'Rahilly S, Savage DB, Sattar N, Langenberg C, Scott RA, Wareham NJ. Association Between Low-Density Lipoprotein Cholesterol-Lowering Genetic Variants and Risk of Type 2 Diabetes: A Meta-analysis. JAMA, 2016; 316: 1383-1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Besseling J, Kastelein JJ, Defesche JC, Hutten BA, Hovingh GK. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA, 2015; 313: 1029-1036 [DOI] [PubMed] [Google Scholar]