

Abstract
Aim: Previous studies demonstrated that endothelin-1 (ET-1) can significantly increase the cell size and stimulate adiponectin expression in cultured human cardiomyocytes (HCM). The aim of the present study was to investigate the effects of fenofibrate, a peroxisome proliferator-activated receptor-α (PPARα) activator, on cell hypertrophy and adiponectin expression in vitro and in a rat model of daunorubicin-induced cardiomyopathy.
Methods: The cultured human cardiomyocytes (HCM) were stimulated with or without ET-1. The cell size and the protein expressions of PPARα and adiponectin were tested by confocal Immunofluorescence study and Western blot, respectively. To study the effects of PPARα activation on ET-1-induced cell hypertrophy and adiponectin protein synthesis, HCM were pretreated with fenofibrate or small interfering RNA (siRNA) of PPARα. Echocardiographic parameters were measured and immunohistochemistry study of myocardial adiponectin expression was conducted in the in vivo study.
Results: ET-1 significantly increased the cell size, dose-dependently suppressed the expression of PPARα, and enhanced the expression of adiponectin; whereas, such an increase of cell size and enhancement of adiponectin expression were inhibited by the pre-treatment with fenofibrate. Addition of siRNA of PPARα abolished the effects of fenofibrate. Moreover, we found that fenofibrate treatment can significantly improve the left ventricular function and reverse the myocardial expression of adiponectin.
Conclusions: Our study shows that fenofibrate may protect against ET-1-induced cardiomyocyte hypertrophy and enhanced adiponectin expression through modulation of PPARα expression in vitro and limitation of daunorubicin cardiotoxicity in vivo, suggesting a novel mechanistic insight into the role of PPARα and adiponectin in cardiac hypertrophy and heart failure.
Keywords: Peroxisome proliferator-activated receptor, Fenofibrate, Endothelin-1, Adiponectin, Cardiomyocyte hypertrophy
Introduction
Cardiac hypertrophy is considered as a compensatory mechanism that temporarily maintains pump function in response to physiological or pathological stresses. However, unremitted hypertrophy can lead to heart failure and sudden death1). Among the neurohumoral factors activated in those conditions, endothelin-1 (ET-1) plays an important role in the genesis of myocyte hypertrophy2).
Peroxisome proliferator-activated receptor α (PPARα) deficiency can induce impaired functional capacity of the heart3–6). PPARα activator fenofibrate can reduce cardiac hypertrophy, decrease cardiac fibrosis, attenuate cardiac dysfunction, and improve survival through suppression of pro-inflammatory, pro-hypertrophic, and pro-fibrotic gene expression7–10). Fenofibrate was reported to suppress ET-1-induced cardiac hypertrophy by down-regulation of activator protein-1 (AP-1) binding capability, inhibition of p38 signaling, and modification of the nuclear factor of activated T-cells (NFAT)-related signaling systems11–14). Moreover, atorvastatin inhibited cardiac hypertrophy through inhibition of negative cross-talk between PPARα and nuclear factor-kappa B (NF-κB)15). Furthermore, the previous study has demonstrated that fenofibrate might also ameliorate cardiac hypertrophy via downregulation of myocardial lipid and glucose metabolism14). However, other possible molecular mechanisms of the inhibition of cardiomyocyte hypertrophy by activated PPARα remain to be elucidated.
Adiponectin, an adipokine, has also been reported to play an important role in the regulation of cardiac remodeling16–19). Adiponectin over-expression can reverse the cardiac dysfunction induced by various pathological factors, including ET-117–19). Moreover, adiponectin may significantly enhance PPARα activity, and the adiponectin-dependent PPARα activation may play a protective role against angiotensin II-induced cardiac hypertrophy and fibrosis18–20). In our previous studies, both ET-1 and angiotensin II have been discovered to acutely stimulate adiponectin expression and significantly increase cell size in cultured human cardiomyocytes (HCM)21, 22). As far as we know, no specific data concerning the functional relationship between fenofibrate, ET-1-induced adiponectin expression, and cell hypertrophy have yet been reported.
Aim
In the current study, we focused on surveying the effects of fenofibrate, a PPARα activator, on cell hypertrophy and adiponectin expression in vitro and on amelioration of daunorubicin cardiotoxicity in a rat model. The Ethical Committee of Cheng Hsin General Hospital has approved the experiments. Throughout the study, all animals were treated in accordance with the guidelines for animal experimentation of our institute.
Methods
Culture of HCM
HCM (Cat. No. 6200) were purchased from ScienCell Research Laboratories (Carlsbad, CA) and grown in Cardiac Myocyte Medium (CMM, Cat. No. 6201) supplemented with 5% of fetal bovine serum, 1% of cardiac myocyte growth supplement (CMGS, Cat. No. 6252), and 1% of penicillin/streptomycin solution (P/S, Cat. No. 0503). Cells were grown on poly-L-lysine-coated culture dish in a humidified incubator with 5% CO2 at 37°C. The culture medium was renewed every 3 to 4 days. With no exception, primary cells were used for experiments.
Western Blot Analysis
The cell lysate was prepared using cell lysis buffer (Cell Signaling, Beverly, MA) and then Western blot analysis was performed. The cell lysate (25 to 40 µg) was subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes blotted beforehand. Having been blocked with 5% skim milk in Tween-20/PBS, blots were incubated with various primary antibodies. Equal protein loading of the samples was verified by staining monoclonal α-tubulin or β-actin antibody (Chemicon, Temecula, CA). Blots were then incubated with the horseradish peroxidase-conjugated secondary antibodies. The signal was detected using Chemiluminescence Reagent Plus (NEN, Boston, MA). The intensity of each band was scanned and quantified using a densitometer linked to a computer software (ImageQuant, Amersham, UK).
Immunocytochemical Localization of β-Actin
Briefly, cells were cultured on glass coverslips coated with collagen IV (Sigma, Saint Louis, MO) and incubated with or without various agents. Cells were then washed with cold PBS and fixed with 4% paraformaldehyde in PBS at 4°C for 15 mins. The cells were treated with PBS containing 0.05% Triton X-100 for 5 mins and, then, blocked with PBS containing 5% BSA at room temperature for an hour. Mouse anti-human β-actin antibody (1:400; Santa Cruz Biotechnology, Santa Cruz, CA) was added and incubated at 37°C for 1.5 hrs. Having been rinsed with PBS, the cells were incubated with goat anti-mouse IgG antibody (1:400; Linex Technologies, Palm Beach Gardens, FL) for one hour. After washing with PBS, the cells were mounted using Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and examined with a Leica TCS SP2 confocal laser-scanning microscope (Leica Microsystems, Wetzlar, Germany).
Analysis of Cardiac Myocyte Hypertrophy23)
Phase contrast micrograph images of cardiac myocytes were captured using a camera (Nikon Europe B.V., Lijnden, Netherlands) and cell outlines were traced and digital image analyses were performed using NIS-Elements Imaging Software (Nikon Imaging, Japan). Myocytes were identified by immunofluorescence staining for β-actin. Cell surface areas calculated by planimetry were doubled to account for surfaces in contact with the plate. Cells were measured in each randomly selected field for cells treated under each condition. The identity of the samples was concealed from the observer during image analyses.
Small Interfering RNA of PPARα
Small interfering ribonucleic acid (siRNA), a specific double-stranded 21-nucleotide RNA sequence homologous to the target gene, was used to silence PPARα expression. Respectively, siRNA for PPARα and Negative Control (NC) siRNA were purchased from Ambion (Austin, TX) and Santa Cruz Technology (Santa Cruz, CA). Inhibition of PPARα protein expression was assessed by immunoblot analysis after transfection HCM with PPARα–siRNA. Briefly, cells were grown in 100-mm dishes and transiently transfected with 20 nM siRNA using 8 ml of siPORT Amine (Ambion Inc., Austin, TX) in a total transfection volume of 0.5 ml of medium. After incubation at 37°C in 5% CO2 for 5 hours, 1.5 ml of normal growth medium was added and incubated with cells for 48 hours.
Pharmacological Treatments with Fenofibrate
Fenofibrate was obtained from Laboratories Fournier S.A. (Fontaine Les Dijon, France) as a gift and subsequently dissolved in dimethyl sulfoxide as a stock solution. To test the probable inhibitory effect of fenofibrate on cardiomyocyte hypertrophy and adiponectin expression, the confluent HCM were pre-treated with the growth medium supplemented with 100 µM fenofibrate for 18 hrs, followed by ET-1 (150 nM) stimulation or control medium for 12 hrs at 37°C. The cardiomyocyte hypertrophy and the expression of adiponectin were next confirmed by Confocal Immunofluorescence study and Western blot, respectively. Since a daily dosage of 200 mg of fenofibrate produced plasma concentrations within the range of 5–35 mg/l (14–100 µM) in 12 dyslipidaemic patients receiving the drug over a 3-month period, the doses we studied in this experiment were clinically relevant24).
In vivo Study25)
An in vivo study using a rat model of daunorubicin (an anthracycline)-induced cardiomyopathy was designed to support the pathophysiological relevance between fenofibrate treatment, adiponectin, and heart failure. Briefly, four-week-old male Sprague-Dawley rats were treated with a cumulative dose of 9 mg/kg body weight daunorubicin (i.v.). These animals were then randomly assigned to fenofibrate-treated (100 mg/kg/day, p.o., the fenofibrate group, n = 6) or vehicle-treated (the HF group, n = 6) groups; age-matched normal rats were used as the control group (n = 6). Fenofibrate treatment was continued for 12 weeks. Echocardiographic parameters, such as left ventricular (LV) diastolic and systolic dimensions, and % fractional shortening (% FS) were measured at baseline and at the 4th, 6th, and 12th weeks. The rats were sacrificed in the 12th week and their blood samples were immediately collected and processed. The plasma was separated by centrifugation and then frozen under −20°C and stored at that temperature until analysis. Plasma concentrations of adiponectin were measured using commercial sandwich enzyme-linked immunosorbent assays (R & D Systems, Inc., Minneapolis, MN).
The heart specimens were dissected from the left ventricle, and the rat myocardial tissues were fixed in 4% buffered formalin solution overnight. They were then embedded in paraffin and sectioned in 5 µm thickness. The paraffin sections were de-paraffinized with xylene and stained with mouse anti-rat adiponectin antibodies (R&D Systems, Inc. Minneapolis, MN) overnight. After being washed in PBS, the sections were incubated for 1 hour at room temperature with horseradish peroxidase-conjugated rabbit anti-mouse IgG. Next, 0.1% DAB was mixed in Tris HCl and H2O2, and the mixture was dropped onto the slides; after 3–5 minutes, the stained tissues were observed with a microscope. Finally, the sections were washed with PBS again, counter-stained with Hematoxylin, mounted using mounting medium, and then examined with a microscope.
Statistics Analyses
All values were expressed as mean ± SEM. Comparisons between multiple groups were determined by means of a one-way analysis of variance (ANOVA) followed by Dunnett's test. A P value < 0.05 was considered statistically significant.
Results
Induction of Adiponectin on HCM by ET-1
The ET-1-induced adiponectin protein expressions were both time- and dose-dependent. ET-1 stimulation led to an increase in adiponectin protein expression (reaching a peak at 48 hrs.), compared to those incubated with control medium (Fig. 1A). Moreover, ET-1 at 100 nM showed the maximal effect in enhancing adiponectin protein expression in HCM; hence, the concentration of ET-1 used for the following experiments was 100 nM for 48 hrs (Fig. 1B).
Fig. 1.
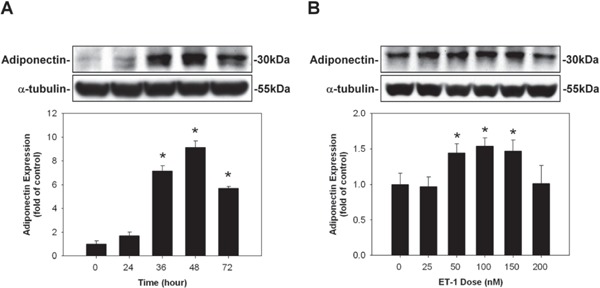
Induction of adiponectin on human cardiomyocytes (HCM) by endothelin-1 (ET-1). (A) ET-1 stimulation led to an increase, reaching a peak at 48 hrs, in adiponectin protein expression, compared to those incubated with control medium. (B) ET-1 at 100 nM (48 hrs) showed the maximal effect in enhancing adiponectin protein expression in HCM.
* denoted P < 0.05 vs. control. Results are representative of three independent experiments.
Suppression of the Expression of PPARα on HCM by ET-1
Incubation of HCM with ET-1 dose-dependently suppressed the expression of PPARα compared to those incubated with control medium (Fig. 2); however, incubation of HCM with ET-1 did not affect the expression of PPARβ/δ or PPARγ.
Fig. 2.
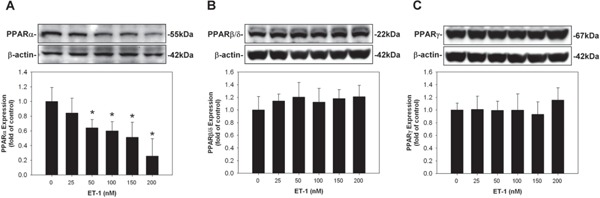
Suppression of the expression of peroxisome proliferator-activated receptor α (PPARα) on human cardiomyocytes (HCM) by endothelin-1 (ET-1). Incubation of HCM with ET-1 dose-dependently suppressed the expression of PPARα compared to those incubated with control medium; however, incubation of HCM with ET-1 did not affect the expression of PPARβ/δ or PPARγ.
* denoted P < 0.05 vs. control. Results are representative of three independent experiments.
Enhancement of Adiponectin Expression by ET-1 in HCM was Inhibited by Pre-Treatment with Fenofibrate
HCM were pre-treated with fenofibrate (a PPARα activator) at various indicated concentration levels, 30 mins before the addition of control medium and with 100 nM ET-1 for 48 hrs. The enhanced adiponectin protein expression in ET-1-stimulated HCM was significantly reduced dose-dependently, after the HCM had been pretreated with the growth medium supplemented with fenofibrate (Fig. 3A). Addition of siRNA of PPARα abolished the effects of fenofibrate (Fig. 3B).
Fig. 3.
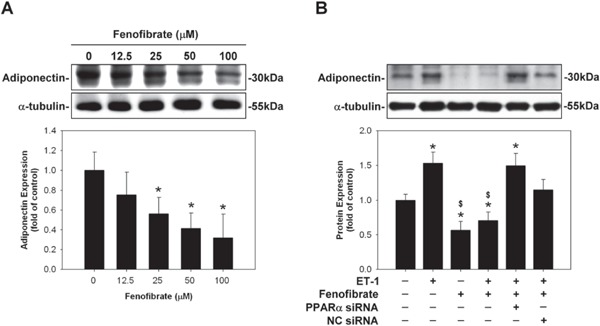
Enhancement of adiponectin expression by endothelin-1 (ET-1) in human cardiomyocytes (HCM) was inhibited by pre-treatment with fenofibrate. (A) Enhanced adiponectin protein expression in ET-1-stimulated HCM was significantly reduced dose-dependently, after the HCM had been pretreated with the growth medium supplemented with fenofibrate. (B) Addition of small interfering RNA (siRNA) of peroxisome proliferator-activated receptor α (PPARα) abolished the effects of fenofibrate.
* denoted P < 0.05 vs. control. Results are representative of three independent experiments.
ET-1 Induced Hypertrophy of Cardiomyocytes in vitro; Whereas, Such Increase of Cell Size was Inhibited by Pre-Treatment with Fenofibrate (Fig. 4)
Fig. 4.
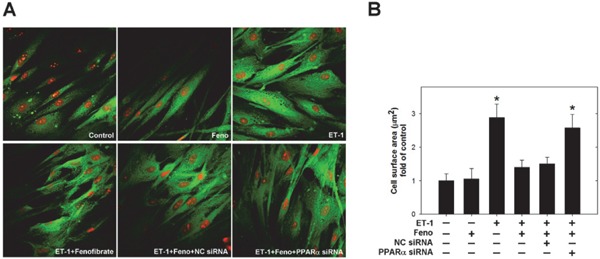
Cell hypertrophy of cardiomyocytes induced by endothelin-1 (ET-1) was inhibited by pre-treatment with fenofibrate. (A) To assess cellular hypertrophy, the cell size of cardiomyocytes was observed using Confocal Immunofluorescence study. ET-1 significantly increased the cell size of cardiomyocytes (upper right panel), compared to that of the control group (upper left panel); however, fenofibrate inhibited ET-1-induced cellular hypertrophy (lower left panel). In addition, the small interfering RNA (siRNA) of peroxisome proliferator-activated receptor α (PPARα), instead of Negative Contral (NC) siRNA, abolished the effects of fenofibrate (lower middle and right panels). (B) Quantitative results of the cell surface area of cardiomyocytes treated with fenofibrate, PPARα siRNA, or NC siRNA in response to ET-1 for 48 hours (n ≧ 10 cells per experimental group). Results are representative of three independent experiments.
To examine the effects of ET-1 in cardiomyocytes at the cellular level, we used in vitro cell culture studying cardiomyocyte hypertrophy with HCM. To assess cellular hypertrophy, the cell size of cardiomyocytes was observed using confocal immunofluorescence study. All cells were pretreated with fenofibrate 100 µM for 30 mins and, subsequently, stimulated with 100 nM ET-1 for 48 hrs. ET-1 significantly increased the cell size of cardiomyocytes (upper right panel), compared to that of the control group (upper left panel); but fenofibrate inhibited ET-1-induced cellular hypertrophy (lower left panel). In addition, the small interfering RNA (siRNA) of peroxisome proliferator-activated receptor α (PPARα), instead of the negative control (NC) siRNA, abolished the effects of fenofibrate (lower middle and right panels).
Results of the in vivo Study (Table 1 and Fig. 5)
Table 1. Demographic, echocardiographic data and circulating biomarkers of the rats in the in vivo study.
| Parameters | Control group | HF group | FENO group |
|---|---|---|---|
| (N = 6) | (N = 6) | (N = 6) | |
| Body weight (g) 5 week/17 week | 160.5 ± 0.5/280.3 ± 21.6 | 156.5 ± 1.5/331.7 ± 17.5* | 158.5 ± 0.7/352.0 ± 16.5* |
| Heart weight (g) | 0.76 ± 0.25 | 1.01 ± 0.10* | 0.98 ± 0.11* |
| HW/BW (%) | 0.27 ± 0.07 | 0.30 ± 0.02* | 0.28 ± 0.03 |
| Adiponectin (ng/mL) | 3.36 ± 2.5 | 11.3 ± 7.8* | 6.11 ± 5.5*, ** |
| Baseline FS (%) | 62 ± 3 | 59 ± 3 | 61 ± 2 |
| FS at 12-week (%) | 59 ± 3 | 52 ± 3* | 57 ± 2 |
P < 0.05 vs. Control group;
P < 0.05 vs. HF group;
(N) is the number of animals used to calculate the mean ± S.E.M. of the presented data in Control group (normal control rats), HF group (rats received daunorubicin followed by treatment with vehicle), and FENO group (rats received daunorubicin followed by treatment with fenofibrate).
HF, heart failure; FENO, fenofibrate; HW, heart weight; BW, body weight; FS, fractional shortening
Fig. 5.
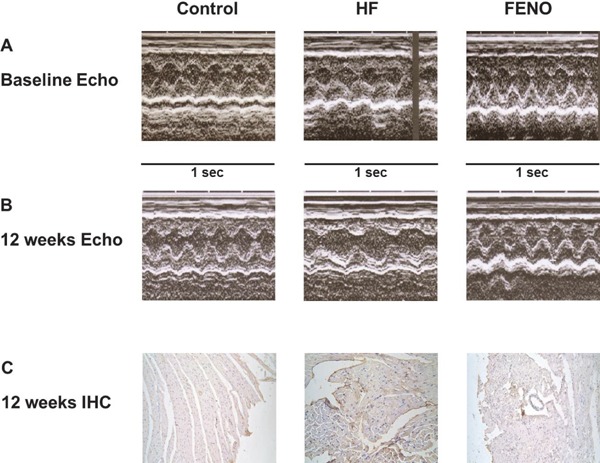
In the rat model of daunorubicin-induced cardiomyopathy, panels A and B show representative left ventricular (LV) M-mode echocardiographic recordings at baseline and at 12th week, respectively. Daunorubicin administration significantly increased LV dimensions (HF) and the increase in LV dimensions was not statistically significant with the treatment of fenofibrate. C. Immunohistochemistry study (IHC) demonstrates that the myocardial expression of adiponectin is significantly higher in the HF group, compared to those in the myocardium obtained from the rats in the fenofibrate group and normal controls.
In the in vivo study, heart weight/body weight (HW/BW) was significantly increased in HF group (P < 0.05 vs. Control group) and was reduced by fenofibrate treatment (as shown in Table 1), although the reduction did not reach statistical significance. Compared with those of the control group, circulating levels of adiponectin were significantly increased in HF group and fenofibrate group (P < 0.05). Moreover, they were significantly lower in fenofibrate group vs. HF group (P < 0.05).
Fig. 5A and 5B show representative left ventricular (LV) M-mode echocardiographic recordings. Daunorubicin administration significantly increased LV dimensions and reduced % FS (P < 0.05), relative to the control group. With fenofibrate treatment, the increase in LV dimensions and reduction of % FS were not statistically significant (Table 1, Fig. 5A and 5B).
To investigate whether adiponectin acts directly on cardiac tissues, we performed immunohistochemistry by staining heart sections with anti-adiponectin antibody. The presence of adiponectin proteins was observed in the representative rat myocardium specimens through immunohistochemistry study (Fig. 5C). The myocardial tissues obtained from the rats in the HF group expressed significantly higher amount of adiponectin, compared to the myocardium obtained from the rats in the fenofibrate group and normal controls.
Discussion
The study demonstrated that in the cultured HCM, ET-1 caused a significant increase in cardiomyocyte size, adiponectin protein expression, and down-regulation of PPARα; however, such increase of cell size and enhancement of adiponectin expression were reduced by pre-treatment with fenofibrate. The addition of siRNA of PPARα abolished the effects of fenofibrate, suggesting that the beneficial effects of fenofibrate on cardiomyocytes are exerted through PPARα modulation. Moreover, in a rat model of daunorubicin-induced cardiomyopathy, fenofibrate treatment improved left ventricular contraction, reduced the elevated circulating adiponectin levels, and ameliorated adiponectin protein expression in the myocardium.
ET-1 plays a critical role in the progression of myocyte hypertrophy2). The influence of ET-1 on the renin-angiotensin-aldosterone system also plays an important role in the structural, electrical, and neurohormonal remodeling of myocardium26). Down-regulation of PPARα is observed in diseased hearts, such as those with cardiac hypertrophy and failure3–6). ET-1 caused a significant down-regulation of PPARα in cultured HCM as observed in our study, and this is a novel finding and provides a strong support to the notion that PPARα deficiency can lead to impaired functional capacity of the heart8). PPARα activator fenofibrate reduces cardiac hypertrophy, decreases cardiac fibrosis, attenuates cardiac dysfunction and improves survival, and causes suppression of proinflammatory, prohypertrophic, and profibrotic gene expression7–10, 13, 14). Animal study has clearly demonstrated that myocardial fibrosis can be effectively inhibited by fenofibrate through suppression of AP-1-mediated ET-1 gene augmentation8). Our data also confirm that ET-1-induced cell hypertrophy and the decrease in the PPARα level in cardiomyocytes can be improved by fenofibrate, which is consistent with previous reports.
Another important and intriguing finding of our study is that ET-1 time- and dose-dependently enhanced the expression of adiponectin; whereas, such an enhancement of adiponectin expression was inhibited by pre-treatment with fenofibrate. Moreover, addition of siRNA of PPARα abolished the effects of fenofibrate. Recently, ET-1 has been shown as a regulatory factor in the secretion of different adipokines, including adiponectin27). Through the endothelin type A receptor, ET-1 stimulates adipocytes to secrete adiponectin, suggesting that ET-1 may play a significant role in the regulation of adiponectin in adipose tissue27). The reasons and detailed mechanisms as to why adiponectin expression increases in response to ET-1 stimulation in cultured HCM remain unclear. One possible explanation is that the elevated adiponectin is a physiological counter-regulating response to ET-1-induced downregulation of PPARα. One recent study has demonstrated that adiponectin significantly enhanced PPARα activity, and the adiponectin-dependent PPARα activation played a protective role against angiotensin II-induced cardiac fibrosis19). The other possible explanation is that acutely enhanced expression of adiponectin to ET-1 stimulation or accumulation of adiponectin in injured heart tissues may represent a compensatory response, and probably acts as an endogenous anti-hypertrophic and anti-fibrotic agent, which leads to protection against the progression of myocardial injury through regulation of cardiac remodeling17, 18), much like natriuretic peptides. Through activation of AMP-activated protein kinase (AMPK) signaling, adiponectin inhibits hypertrophic signaling in the myocardium, including that induced by endothelin stimulation17, 18, 20, 28). Moreover, supplementation of exogenous adiponectin may directly protect cardiomyocytes in adiponectin-deficient animals17, 18, 28).
However, adiponectin is best known to confer numerous cardioprotective effects, but it would be naive to assume that this has always been the case. Plasma adiponectin concentrations are increased in patients with overt heart failure and high adiponectin levels are independently predictive of clinical outcomes in heart failure29, 30). In our previous study, the myocardial tissue expression of adiponectin was related to the severity of heart failure, suggesting that adiponectin may play a role in the progression of heart failure and metabolism disturbance in advanced heart failure30). In fact, some of the effects of adiponectin may adversely affect the cardiovascular system or even the whole body while being chronically activated, such as in the case of chronic heart failure. For instance, adiponectin may activate NF-κB and AP-1, resulting in expression of proinflammatory genes and enhancement of angiotensin II-induced proliferation in cardiac fibroblasts, which may in turn play a role in heart failure progression31, 32). Moreover, adiponectin can increase energy expenditure and decrease body weight, which may participate in the development of cardiac cachexia33, 34). Therefore, the increase in plasma adiponectin in chronic heart failure can also represent shortterm beneficial adaptive responses on acute injury, which may eventually turn into harmful maladaptive signals on prolonged and chronic activation, like the sustained sympathetic hyperactivity in the advanced heart failure35). In actuality, greater cardiac disease severity can lead to greater salvage attempts; hence, a higher adiponectin concentration. If so, with successful anti-failure therapy, the plasma adiponectin concentrations should return to baseline conditions with improvement of HF. Supporters of this hypothesis have recently published reports to demonstrate that anti-failure treatment was associated with reduced plasma adiponectin concentration and the improvement of left ventricular ejection fraction in both acute and chronic heart failure patients35, 36). In our in vivo study, that also supports this hypothesis, we have discovered that fenofibrate treatment is able to significantly reduce circulating levels of adiponectin, improve the left ventricular function, and reverse the myocardial expression of adiponectin. However, whether the use of fenofibrate really works in the prevention of cardiac hypertrophy or treatment of heart failure patients, still awaits confirmation via further clinical trials. Whether adipokines, such as adiponectin, may become potential targets for the treatment, also calls for further investigations.
Our study does not exclude a role for the effects of PPARα activators on the expression of other factors involved in cardiomyocyte hypertrophy and adiponectin expression. Nor does it rule out the possibility that the molecules tested may act through unrelated mechanisms in addition to PPARα activation. For instance, fenofibrate can suppress ET-1-induced cardiac hypertrophy by down-regulation of AP-1 binding capability and inhibition of p38 signaling11, 12). In our previous study, we demonstrated that angiotensin II-enhanced adiponectin expression in cultured HCM can be inhibited by angiotensin receptor blocker through ERK, p38, and JNK pathways22). Therefore, it is reasonable to hypothesize that such a link may also exist between various mitogen-activated protein kinase signaling cascades, PPARα, adiponectin, and cardiac hypertrophy. However, it is only fair to emphasize here that the present study was designed to merely study the possible physiologic protective role of fenofibrate; hence, the study results proffer some clues to the research of the mechanisms of adiponectin in the pathophysiology of cardiac hypertrophy. No definite conclusion can be drawn regarding which mechanisms are more relevant from the present study. Further investigation into the pathophysiologic interaction among adiponectin, adiponectin receptors, together with their interplay in signal transduction, so as to gain additional insight into the precise regulatory mechanisms for the myocardial expression of adiponectin in cardiomyocyte hypertrophy and the complex interaction between ET-1, adiponectin, PPARα, and fenofibrate, will be most fascinating and has the potential to lead to the discovery of novel treatment strategies for cardiac hypertrophy and heart failure.
Conclusions
In conclusion, we have demonstrated that adiponectin, ET-1, and PPARα are closely associated with one another and partly contribute to the pathogenesis of cardiac hypertrophy and heart failure, but the precise mechanisms are not yet fully clear. However, this study may proffer some clues to the research of the mechanisms of adiponectin in the pathophysiology of cardiac hypertrophy and heart failure. Further clinical studies on a larger scale are also required to confirm whether fenofibrate may protect the heart against acute and chronic injuries.
Acknowledgments
The research team is most grateful for the generous subsidies provided by the Research Grant of Cheng Hsin General Hospital-National Yang-Ming University Cooperation Project (no. 98F117CY09) and Research Grant of Cheng Hsin General Hospital (no. 99-15). We also thank Ms. Chin-Feng Cheng for her excellent assistance in preparing and typing the manuscript.
Conflict of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- 1). Balakumar P, Jagadeesh G: Multifarious molecular signaling cascades of cardiac hypertrophy: can the muddy waters be cleared? Pharmacol Res, 2010; 62: 365-383 [DOI] [PubMed] [Google Scholar]
- 2). Drawnel FM, Archer CR, Roderick HL: The role of the paracrine/autocrine mediator endothelin-1 in regulation of cardiac contractility and growth. Br J Pharmacol, 2013; 168: 296-317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3). Schiffrin EL: Peroxisome proliferator-activated receptors and cardiovascular remodeling. Am J Physiol Heart Circ Physiol, 2005; 288: H1037-H1043 [DOI] [PubMed] [Google Scholar]
- 4). Smeets PJ, de Vogel-van den Bosch HM, Willemsen PH, Stassen AP, Ayoubi T, van der Vusse GJ, van Bilsen M: Transcriptomic analysis of PPARalpha-dependent alterations during cardiac hypertrophy. Physiol Genomics, 2008; 36: 15-23 [DOI] [PubMed] [Google Scholar]
- 5). Smeets PJ, Teunissen BE, Willemsen PH, van Nieuwenhoven FA, Brouns AE, Janssen BJ, Cleutjens JP, Staels B, van der Vusse GJ, van Bilsen M: Cardiac hypertrophy is enhanced in PPAR alpha−/− mice in response to chronic pressure overload. Cardiovasc Res, 2008; 78: 79-89 [DOI] [PubMed] [Google Scholar]
- 6). Oyekan A: PPARs and their effects on the cardiovascular system. Clin Exp Hypertens, 2011; 33: 287-293 [DOI] [PubMed] [Google Scholar]
- 7). Labinskyy V, Bellomo M, Chandler MP, Young ME, Lionetti V, Qanud K, Bigazzi F, Sampietro T, Stanley WC, Recchia FA: Chronic activation of peroxisome proliferator-activated receptor-alpha with fenofibrate prevents alterations in cardiac metabolic phenotype without changing the onset of decompensation in pacing-induced heart failure. J Pharmacol Exp Ther, 2007; 321: 165-171 [DOI] [PubMed] [Google Scholar]
- 8). Ogata T, Miyauchi T, Sakai S, Irukayama-Tomobe Y, Goto K, Yamaguchi I: Stimulation of peroxisome-proliferator-activated receptor alpha (PPAR alpha) attenuates cardiac fibrosis and endothelin-1 production in pressureoverloaded rat hearts. Clin Sci (Lond), 2002; 103 (Suppl 48): 284S-288S [DOI] [PubMed] [Google Scholar]
- 9). Ichihara S, Obata K, Yamada Y, Nagata K, Noda A, Ichihara G, Yamada A, Kato T, Izawa H, Murohara T, Yokota M: Attenuation of cardiac dysfunction by a PPAR-alpha agonist is associated with down-regulation of redox-regulated transcription factors. J Mol Cell Cardiol, 2006; 41: 318-329 [DOI] [PubMed] [Google Scholar]
- 10). Huang WP, Yin WH, Chen JW, Jen HL, Young MS, Lin SJ: Fenofibrate attenuates endothelial monocyte adhesion in chronic heart failure: an in vitro study. Eur J Clin Invest, 2009; 39: 775-783 [DOI] [PubMed] [Google Scholar]
- 11). Irukayama-Tomobe Y, Miyauchi T, Sakai S, Kasuya Y, Ogata T, Takanashi M, Iemitsu M, Sudo T, Goto K, Yamaguchi I: Endothelin-1-induced cardiac hypertrophy is inhibited by activation of peroxisome proliferator-activated receptor-alpha partly via blockade of c-Jun NH2-terminal kinase pathway. Circulation, 2004; 109: 904-910 [DOI] [PubMed] [Google Scholar]
- 12). Irukayama-Tomobe Y, Miyauchi T, Kasuya Y, Sakai S, Goto K, Yamaguchi I: Activation of peroxisome proliferator-activated receptor-alpha decreases endothelin-1-induced p38 mitogen-activated protein kinase activation in cardiomyocytes. J Cardiovasc Pharmacol, 2004; 44 (Suppl 1): S358-S361 [DOI] [PubMed] [Google Scholar]
- 13). Le K, Li R, Xu S, Wu X, Huang H, Bao Y, Cai Y, Lan T, Moss J, Li C, Zou J, Shen X, Liu P: PPARα activation inhibits endothelin-1-induced cardiomyocyte hypertrophy by prevention of NFATc4 binding to GATA-4. Arch Biochem Biophys, 2012; 518: 71-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14). Zou J, Le K, Xu S, Chen J, Liu Z, Chao X, Geng B, Luo J, Zeng S, Ye J, Liu P: Fenofibrate ameliorates cardiac hypertrophy by activation of peroxisome proliferator-activated receptor-α partly via preventing p65-NFκB binding to NFATc4. Mol Cell Endocrinol, 2013; 370: 103-112 [DOI] [PubMed] [Google Scholar]
- 15). Planavila A, Laguna JC, Vázquez-Carrera M: Atorvastatin improves peroxisome proliferator-activated receptor signaling in cardiac hypertrophy by preventing nuclear factor-kappa B activation. Biochim Biophys Acta, 2005; 1687: 76-83 [DOI] [PubMed] [Google Scholar]
- 16). Park M, Sweeney G: Direct effects of adipokines on the heart: focus on adiponectin. Heart Fail Rev, 2013; 18: 631-644 [DOI] [PubMed] [Google Scholar]
- 17). Goldstein BJ, Scalia RG, Ma XL: Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med, 2009; 6: 27-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18). Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K: Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med, 2004; 10: 1384-1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19). Fujita K, Maeda N, Sonoda M, Ohashi K, Hibuse T, Nishizawa H, Nishida M, Hiuge A, Kurata A, Kihara S, Shimomura I, Funahashi T: Adiponectin protects against angiotensin II-induced cardiac fibrosis through activation of PPAR-alpha. Arterioscler Thromb Vasc Biol, 2008; 28: 863-870 [DOI] [PubMed] [Google Scholar]
- 20). Fujioka D, Kawabata K, Saito Y, Kobayashi T, Nakamura T, Kodama Y, Takano H, Obata JE, Kitta Y, Umetani K, Kugiyama K: Role of adiponectin receptors in endothelin-induced cellular hypertrophy in cultured cardiomyocytes and their expression in infarcted heart. Am J Physiol Heart Circ Physiol, 2006; 290: H2409-H2416 [DOI] [PubMed] [Google Scholar]
- 21). Yin WH, Chen YH, Wei J, Jen HL, Huang WP, Young MS, Chen DC, Liu PL: Associations between endothelin-1 and adiponectin in chronic heart failure. Cardiology, 2011; 118: 207-216 [DOI] [PubMed] [Google Scholar]
- 22). Yin WH, Chen JW, Lin SJ, Young MS: Effect of irbesartan on angiotensin II-induced adiponectin expression in human cardiomyocytes. Acta Cardiologica Sinica, 2011: 27: 244-251 [Google Scholar]
- 23). Watkins SJ, Borthwick GM, Oakenfull R, Robson A, Arthur HM: Angiotensin II-induced cardiomyocyte hypertrophy in vitro is TAK1-dependent and Smad2/3-independent. Hypertens Res, 2012; 35: 393-398 [DOI] [PubMed] [Google Scholar]
- 24). Balfour JA, McTavish D, Heel RC: Fenofibrate. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in dyslipidaemia. Drugs, 1990; 40: 260-290 [DOI] [PubMed] [Google Scholar]
- 25). Soga M, Kamal FA, Watanabe K, Ma M, Palaniyandi S, Prakash P, Veeraveedu P, Mito S, Kunisaki M, Tachikawa H, Kodama M, Aizawa Y: Effects of angiotensin II receptor blocker (candesartan) in daunorubicin-induced cardiomyopathic rats. Int J Cardiol, 2006; 10: 378-385 [DOI] [PubMed] [Google Scholar]
- 26). Takahashi T, Saegusa S, Sumino H, Nakahashi T, Iwai K, Morimoto S, Nojima T, Kanda T: Adiponectin, T-cadherin and tumour necrosis factor-alpha in damaged cardiomyocytes from autopsy specimens. J Int Med Res, 2005; 33: 236-244 [DOI] [PubMed] [Google Scholar]
- 27). Clarke KJ, Zhong Q, Schwartz DD, Coleman ES, Kemppainen RJ, Judd RL: Regulation of adiponectin secretion by endothelin-1. Biochem Biophys Res Commun, 2003; 312: 945-949 [DOI] [PubMed] [Google Scholar]
- 28). Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, Hildebrandt P: Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure. Circulation, 2005; 112: 1756-1762 [DOI] [PubMed] [Google Scholar]
- 29). Yin WH, Wei J, Huang WP, Chen JW, Young MS, Lin SJ: Prognostic value of circulating adipokine levels and expressions of adipokines in the myocardium of patients with chronic heart failure. Circ J, 2012; 76: 2139-2147 [DOI] [PubMed] [Google Scholar]
- 30). Hattori Y, Hattori S, Kasai K: Globular adiponectin activates nuclear factor-kappaB in vascular endothelial cells, which in turn induces expression of proinflammatory and adhesion molecule genes. Diabetes Care, 2006; 29: 139-141 [DOI] [PubMed] [Google Scholar]
- 31). Hattori Y, Hattori S, Akimoto K, Nishikimi T, Suzuki K, Matsuoka H, Kasai K: Globular adiponectin activates nuclear factor-kappaB and activating protein-1 and enhances angiotensin II-induced proliferation in cardiac fibroblasts. Diabetes, 2007; 56: 804-808 [DOI] [PubMed] [Google Scholar]
- 32). Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF: Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A, 2001; 98: 2005-2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, Scherer PE, Ahima RS: Adiponectin acts in the brain to decrease body weight. Nat Med, 2004; 10: 524-529 [DOI] [PubMed] [Google Scholar]
- 34). Araújo JP, Lourenço P, Rocha-Gonçalves F, Ferreira A, Bettencourt P: Adiponectin is increased in cardiac cachexia irrespective of body mass index. Eur J Heart Fail, 2009; 11: 567-572 [DOI] [PubMed] [Google Scholar]
- 35). Yamaji M, Tsutamoto T, Tanaka T, Kawahara C, Nishiyama K, Yamamoto T, Fujii M, Horie M: Effect of carvedilol on plasma adiponectin concentration in patients with chronic heart failure. Circ J, 2009; 73: 1067-1073 [DOI] [PubMed] [Google Scholar]
- 36). Matsumoto M, Lee-Kawabata M, Tsujino T, Naito Y, Ezumi A, Sakoda T, Ohyanagi M, Shimomura I, Masuyama T: Decrease in serum adiponectin levels in response to treatment predicts good prognosis in acute decompensated heart failure. J Clin Hypertens (Greenwich), 2010; 12: 900-904 [DOI] [PMC free article] [PubMed] [Google Scholar]