

Abstract
Substantial evidence has been accumulated suggesting that branched‐chain amino acid (BCAA) supplementation or BCAA‐rich diets have a positive effect on the regulation of body weight, muscle protein synthesis, glucose homeostasis, the ageing process and extend healthspan. Despite these beneficial effects, epidemiological studies have shown that BCAA plasma concentrations and BCAA metabolism are altered in several metabolic disorders, including type 2 diabetes mellitus and cardiovascular diseases. In this review article, we present an overview of the current literature on the different effects of BCAAs in health and disease. We also highlight the results showing the most promising therapeutic effects of dietary BCAA supplementation and discuss how BCAAs can trigger different and even opposite effects, depending on the catabolic and anabolic states of the organisms. Moreover, we consider the effects of BCAAs when metabolism is abnormal, in the presence of a mixture of different anabolic and catabolic signals. These unique pharmacodynamic properties may partially explain some of the markedly different effects found in BCAA supplementation studies. To predict accurately these effects, the overall catabolic/anabolic status of patients should be carefully considered. In wider terms, a correct modulation of metabolic disorders would make nutraceutical interventions with BCAAs more effective.
Linked Articles
This article is part of a themed section on Principles of Pharmacological Research of Nutraceuticals. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.11/issuetoc
Abbreviations
- ALS
amyotrophic lateral sclerosis
- BCAA
branched‐chain amino acid
- BCAT
BCAA aminotransferase
- BCKA
branched‐chain α‐keto acid
- BCKDC
BCK dehydrogenase complex
- EAA
essential amino acid
- eNOS
endothelial NOS
- GATOR
GAP activity towards rags
- GCN2
general amino acid control non‐derepressible 2
- GLP‐1
glucagon‐like peptide‐1
- 3‐HIB
3‐hydroxyisobutyrate
- IRS‐1
insulin receptor substrate 1
- mTORC1
mammalian/mechanistic target of rapamycin complex 1
- PGC‐1α
PPARγ coactivator‐1α
- PDH
pyruvate dehydrogenase complex
- PP2Cm
mitochondrial‐targeted 2C‐type Ser/Thr protein phosphatase
- T2DM
type 2 diabetes mellitus
- tRNA
transfer RNA
- WAT
white adipose tissue
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes e |
| Adiponectin receptors | AMPK |
| TNF‐α | BCAT |
| GPCRs b | BCKDH kinase |
| GLP‐1 receptor | eNOS |
| Nuclear hormone receptors c | GCN2 |
| Oestrogen receptors | IKK |
| PPARγ | mTOR |
| Transporters d | Sirt1 |
| GLUT4 |
| LIGANDS | |
|---|---|
| 5‐HT | IL‐22 |
| Adiponectin | Insulin |
| Dopamine | Leptin |
| GABA | Noradrenaline |
| GLP‐1 | Nitric oxide (NO) |
| IGF1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,d,eAlexander et al., 2015a,b,c,d,e).
Introduction
Branched‐chain amino acids (BCAAs), that is isoleucine, leucine and valine, are proteinogenic essential amino acids (EAAs) with aliphatic‐branched side chains. In addition to their role as key building blocks for peptide synthesis, BCAAs are also important sources for the biosynthesis of sterol, ketone bodies and glucose. They account for about 21% of the total body protein content and 35% of the dietary essential amino acids in muscle proteins (Harper et al., 1984). The free BCAA pool not bound to a peptide chain accounts for an extremely small proportion (272 to 556‐fold less) of the body's total mass of BCAAs (Waterlow et al., 1978). Nevertheless, free BCAAs act as important nutrient signals and metabolic regulators. Skeletal muscle contains the largest amount of free BCAAs, corresponding to ~0.1 g kg−1 muscle (Shimomura et al., 2006).
Given their hydrophobic nature, BCAAs are involved in the formation of α‐helices and β‐sheets, secondary structural motifs of proteins. Therefore, they are present in a variety of coiled‐coil α‐helices of proteins such as myosin, fibrinogen and keratin. BCAAs form the helical zipper structures of transcription factors (Chou and Fasman, 1978; Glover and Harrison, 1995). Moreover, BCAAs are present in most of the non‐aqueous interior environment of water‐soluble globular proteins, such as the oxygen‐binding portion of myoglobin and haemoglobin (Chou and Fasman, 1973).
Branched‐chain amino acid pharmacokinetics
Skeletal muscles represent the largest protein pool and reservoir of BCAAs in the body. Although the molecular basis and the regulation of BCAA uptake remain poorly studied, l‐type amino‐acid transporters and bidirectional transporters for l‐glutamine and l‐leucine/EAAs in the enterocytes of proximal jejunum play a major role in the transport of BCAAs and activation of downstream signalling (Broer, 2008). BCAAs enter the blood circulation by absorption, largely escape the first‐pass hepatic metabolism and appear directly in the systemic circulation (Brosnan and Brosnan, 2006). About 95–99% of all circulating BCAAs are reabsorbed in the kidney nephrons, largely through the proximal convoluted tubules (Broer, 2008). Measurements of the arteriovenous exchanges have shown that muscles and the splanchnic bed extract over half and about one quarter of the circulating BCAAs, respectively, while the remainder is removed by the brain and other tissues (Fernstrom, 2005). Both plasma and cerebrospinal BCAA levels are rapidly elevated after ingestion of a BCAA‐containing meal, and brain access to BCAAs is mediated by facilitative transport, which involves both saturable and unsaturable processes (Smith et al., 1987).
Catabolism
BCAA catabolism is highly regulated by both allosteric and covalent mechanisms (Harris et al., 2005). This may occur not only because BCAAs are EAAs, obtained exclusively from external food sources, but also because they are the major regulators of protein synthesis, particularly leucine. The true biochemical reasons of the tight control of BCAA catabolism remain, however, largely unknown. The first steps in their catabolism are common to the three BCAAs and require the mitochondrial enzymes BCAA aminotransferase (BCAT) and branched‐chain α‐keto acid dehydrogenase complex (BCKDC). Remarkably in contrast with other amino acids, only a relatively small fraction of BCAA catabolism capacity resides in the liver. Most of the BCAA catabolism is indeed located in skeletal muscle and the brain, whereas a considerable proportion also resides in white adipose tissue (WAT) (Suryawan et al., 1998; Herman et al., 2010). In the first and fully reversible step of degradation, mitochondrial BCAT transfers the amino group from BCAAs to α‐ketoglutarate to form the corresponding branched‐chain α‐keto acids (BCKAs) and glutamate. Thereafter, BCKDC catalyses the decarboxylation of the carboxyl groups of BCKAs, to form the corresponding branched‐chain acyl‐CoA esters. This reaction is irreversible and, therefore, commits the BCAAs to degradation (Brosnan and Brosnan, 2006).
Recent findings have shown that this step is an important site of regulation (Lu et al., 2009). BCKDC activity is regulated by end‐product allosteric inhibition by NADH, α‐ketoisocaproate and branched‐chain acyl‐CoA esters, and can also be inhibited by phosphorylation and activated by dephosphorylation. The BCKD complex is genetically similar to the pyruvate dehydrogenase complex (PDH), with similar subunit composition and regulatory mechanisms. Like PDH, BCKDC activity is determined by the phosphorylation status of its regulatory subunit E1a. When the BCAA level is low, E1a is hyper‐phosphorylated by a BCKD kinase, leading to inhibition of BCKDC activity and preservation of free BCAA. When the BCAA level is high, E1a is dephosphorylated by a mitochondrial‐targeted 2C‐type Ser/Thr protein phosphatase (PP2Cm) named PP2C in mitochondria or protein phosphatase, Mg2 +/Mn2 + dependent 1K (PPM1K), leading to BCKDC activation and a reduction in total BCAAs (Lu et al., 2009).
BCKAs undergo further catabolic degradation to different end products, such as glucose and/or ketone bodies. In particular, leucine is a ketogenic amino acid, while valine is a gluconeogenic amino acid and isoleucine is both a gluconeogenic and ketogenic amino acid. However, muscles are not a gluconeogenic tissue; therefore, if valine and isoleucine are to be converted to glucose, they cannot be completely metabolized in this tissue. Accordingly, the intermediate valine metabolite 3‐hydroxyisobutyrate (3‐HIB), which lacks the covalent linkage to CoA, exits from myocytes and acts as a gluconeogenic substrate in both hepatocytes and renal cortical tubular cells (Letto et al., 1986). Interestingly, a novel inter‐organ communication property of 3‐HIB has been recently described as a paracrine regulator of trans‐endothelial fatty acid transport that promotes lipid accumulation in diabetic muscle (Jang et al., 2016).
Branched‐chain amino acid pharmacodynamics
Mechanism of action
BCAAs are evolutionary conserved essential components of the diet. BCAA metabolism can be directed either to catabolic (i.e. oxidation) or anabolic (i.e. protein synthesis) fate by signals initiated by specific cellular energy/nutrient sensors (Figure 1). In the case of starvation, autonomic activation, glucagon, adrenaline, noradrenaline, cortisol and growth hormone increase intracellular BCAA uptake and favour BCAA oxidation. At the cellular level, the molecular events underlying these signals are mainly regulated by AMP activated kinase (AMPK), a master sensor of energy balance (Yuan et al., 2013). In turn, high levels of amino acids are activated, through the Rag guanosine triphosphatases (GTPases), the mammalian/mechanistic target of rapamycin complex 1 (mTORC1) (Yuan et al., 2013). This multiprotein complex acts at different levels, cooperates with other anabolic hormones [insulin and insulin‐like growth factor 1 (IGF1)] to increase the intracellular BCAA uptake, promotes protein synthesis, reduces protein degradation and increases cell growth (Rennie et al., 2006). Intracellular BCAAs can synthesize new proteins, but can also be converted to glutamate, which detoxifies ammonia via glutamine synthesis in skeletal muscle (Kawaguchi et al., 2011). Importantly, in catabolic states, BCAAs are normally oxidized to generate ATP. Carbon originating from leucine enters the tricarboxylic acid (TCA) cycle as acetyl‐CoA for complete disposal as CO2, whereas isoleucine and valine mainly provide carbon for anaplerotic conversion of propionyl‐CoA to succinyl‐CoA (Harris et al., 2005).
Figure 1.
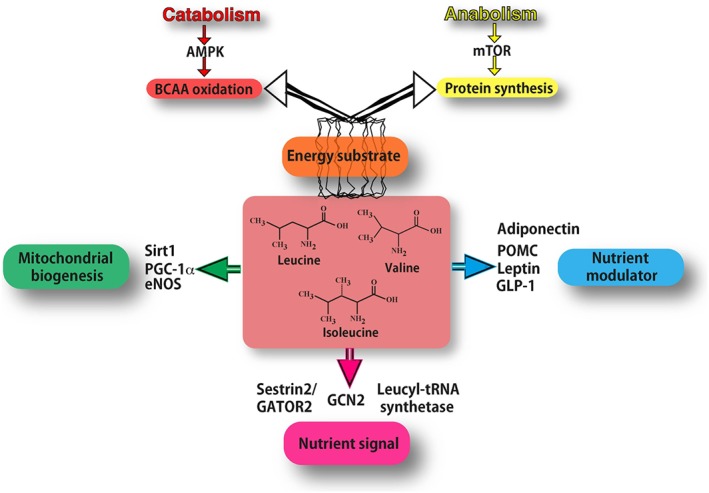
Mechanisms of action of branch‐chained amino acids (BCAAs). As energy substrate (orange box) BCAAs can be directed either to oxidation (through AMPK signalling in catabolic conditions) or to protein synthesis (through mTOR signalling in anabolic conditions). BCAAs act as nutrient signals to specific nutrient‐sensing systems (purple box). In particular, they may interact mostly with the general amino acid control non‐derepressible 2 (GCN2) pathway, specific leucyl‐tRNA synthetase and Sestrin2. Leucine inhibits the Sestrin2‐GATOR2 interaction and allows GATOR2 to activate mTOR signalling. BCAAs function also as nutrient modulators (blue box), by fine‐tuning the secretion of nutrient‐related hormones, such as leptin, adiponectin, GLP‐1 and pro‐opiomelanocortin (POMC). BCAAs increase the expression of PGC‐1α and sirtuin 1(Sirt1), thus promoting mitochondrial biogenesis (green box), partly through eNOS activity.
Nutrient signals
Whole body protein synthesis in humans (measured as g kg−1 of body weight day−1) drastically decreases with age, being 17.4 in new‐borns, 6.9 in infants, 3.0 in adults and 1.9 in elderly subjects. Similarly, protein catabolism also decreases with age. It has been estimated that in conditions of dietary nitrogen balance, the adult protein turnover (i.e. synthesis and degradation processes) accounts for 250 g day−1 (Waterlow et al., 1978). These parameters can largely change in conditions of nutrient deprivation and in disease states, for example in traumatized or septic subjects.
Cellular organisms have evolutionary conserved multiprotein complexes able to sense energy and nutrient levels, including BCAAs, and to coordinate a network of signalling cascades for cell growth (anabolism) or energy production (catabolism). Amino acid‐ and BCAA‐specific sensing systems seem to be redundant and act at different cellular localizations (Wolfson et al., 2016). Intracellular amino acids interact with the general amino acid control non‐derepressible 2 (GCN2) pathway. GCN2 senses the uncharged transfer RNAs (tRNAs) that accumulate when the amino acid concentration is low (Wek et al., 1995). Activated GCN2 decreases the translation of amino acids, reducing their consumption and the energy required for this process (Figure 1). Moreover, specific leucyl‐tRNA synthetase has been revealed as an intracellular leucine sensor in both yeast and mammals that is able to activate the mTORC1‐signalling pathway, but does not affect the AMPK pathway (Han et al., 2012). In the presence of leucine, leucyl‐tRNA synthetase translocates to the lysosome where it increases, in a dose‐dependent manner, the GTP hydrolysis of RagD required for mTORC1 activation (Figure 1). Furthermore, leucine, but not arginine, binds to Sestrin2, a known mTORC1 signalling inhibitor that acts on the mTORC1‐activator GAP activity towards rags 2 (GATOR2) (Saxton et al., 2016; Wolfson et al., 2016). Leucine concentrations able to activate mTORC1 can bind to Sestrin2, thus inhibiting the Sestrin2‐GATOR2 interaction and allowing GATOR2 to regulate positively mTOR activation (Figure 1). Interestingly, since Sestrin2 is a soluble protein, this leucine‐sensor system can sense the free leucine in the cytosol.
Brain leucine rapidly and strongly activates mTOR signaling and its downstream target p70 S6 kinase, within two sites adjacent to circumventricular organs, which have preferential access to blood borne nutrients (Cota et al., 2006). The leucine sensing structures are located in the mediobasal hypothalamus (including the arcuate and ventromedial nuclei) and the dorsal vagal complex of the caudal brainstem (including the nucleus of the solitary tract, the dorsal motor vagus and the area postrema). In these brain areas, leucine induces neurons to express the anorexigenic proopiomelanocortin (POMC) that reduces feeding and body weight gain (McAllan et al., 2013) (Figure 1).
Nutrient sensors
BCAAs have been found to increase the secretion of nutrient‐related hormones. Single EAAs have been infused in humans to determine their effects on plasma insulin. Arginine, lysine, phenylalanine and leucine resulted in the highest increases in plasma insulin levels, although of all of them arginine seems to be the most important (Floyd et al., 1966). Interestingly, only the BCAAs were able to concurrently inhibit glucagon secretion (Nair and Short, 2005), while leucine has been shown to increase the secretion of leptin (Lynch et al., 2006) and adiponectin from adipocytes (Blumer et al., 2008) (Figure 1). In obese mice, leucine supplementation, together with physical exercise, was found to increase adiponectin concentrations while reducing pro‐inflammatory adipokines (Torres‐Leal et al., 2011). BCAAs have been shown to induce a dose‐dependent increase in glucagon‐like peptide‐1 (GLP‐1) release from enterocytes in vitro (Chen and Reimer, 2009); however, this effect has not been confirmed in vivo (Steinert et al., 2015). Furthermore, a valine‐rich amino‐acid mixture (Colombel et al., 1988) or leucine alone (Steinert et al., 2015) directly infused into duodenum can increase cholecystokinin production and gallbladder contraction.
Effects on skeletal muscle
Circulating BCAAs, in particular leucine, have been shown to act as potent nutrient signals in muscle where they induce protein synthesis (Shimomura et al., 2006). A dietary supplementation of amino‐acid mixtures enriched in BCAAs (BCAAem) preserves muscle fibre size and improves physical endurance and motor coordination in middle‐aged mice (D'Antona et al., 2010). Moreover, BCAAem increases the expression of PPARγ coactivator‐1α (PGC‐1α) and sirtuin 1(SIRT1) and promotes mitochondrial biogenesis and function in cardiac and skeletal muscles through an mTORC1‐dependent effect (D'Antona et al., 2010). BCAAem‐activated mTOR signalling can enhance mitochondrial biogenesis partially through increasing of the NO generating system. Endothelial NOS (eNOS) gene silencing decreased the activation of mTOR by BCAAem in vitro and in vivo (D'Antona et al., 2010). Thus, a positive feedback mechanism between eNOS and mTOR pathways could promote the effects of BCAAs. Finally, exercise training further enhanced the BCAAem‐mediated improvement in muscle functional capacity.
Effects on the immune system
The link between metabolism and immunity has been clearly established from an evolutionary point of view. In fact, the functions of adipose tissue and bone marrow, specialized organs that regulate lipid stores and immune cell production in mammals, are sustained by a single organ in Drosophila, the fat body (Hotamisligil and Erbay, 2008). The activation of immune responses is a metabolically demanding process. It has been estimated that sepsis can increase the human metabolic rate by 30–60% and nitrogen excretion by two to threefold (Romanyukha et al., 2006). Catabolic states such as starvation and malnutrition can actually impair the functions of the immune system, but, on the other hand, most infections suppress the host's appetite, possibly by inducing the synthesis of leptin (Demas et al., 2003). Two crucial cell types of the immune and metabolic systems, macrophages and adipocytes, share similar functions and properties as far as they both secrete cytokines and can be activated by lipopolysaccharide. Nutrients may directly induce inflammation through activation of toll‐like receptor signalling by free fatty acids (Konner and Bruning, 2011). Among the better characterized intracellular points of crosstalk between nutrient sensing multiprotein complexes (i.e. AMPK, mTOR), anabolic hormones, inflammatory pathways, IκB kinase‐α and JNK play important roles (Lee et al., 2007). The anti‐inflammatory cytokine IL‐22 is able to regulate oxidative stress pathways and increase mouse and human insulin secretion from beta cells of the pancreas (Hasnain et al., 2014).
Protein malnutrition is one of the major causes of decreased immune function in the elderly (Lesourd, 1995). Although glutamine has been considered the most important amino acid for immune function (Roth, 2008), human immune cells express BCKDC and decarboxylase activities (Calder, 2006) and, thus, oxidize BCAAs. The BCAA uptake rate in lymphocytes varies according to the cell cycle phase, most likely reflecting the timing of protein synthetic activity (Glassy and Furlong, 1981). Moreover, dietary restriction of BCAAs impairs different aspects of the immune function, including the activity of cytotoxic T lymphocytes, natural killer cells and lymphocyte proliferation. The BCAA supplementation partially restored the immunosuppression occurring after intense long‐duration exercise (Bassit et al., 2002).
BCAA supplementation reduces by 30% the incidence of infections acquired in geriatric long‐term rehabilitation centres (Aquilani et al., 2011). Moreover, BCAAs can reduce the risk of bacterial and viral infection in patients with decompensated cirrhosis by restoring the impaired innate immune responses of these patients (Nakamura et al., 2004, 2007). Furthermore, the BCAA supplementation in haemodialysis patients on low‐protein diet has been found to be associated with a reduction in inflammatory markers and correction of nephropathy‐linked anaemia (Bolasco et al., 2011).
Effects on brain
BCAAs compete for large, neutral amino‐acid transport at the blood–brain barrier and can influence brain neurotransmitter synthesis (Fernstrom, 2005). Ingestion of BCAAs causes a rapid elevation of their plasma concentrations, increases their uptake into the brain and decreases the brain uptake and level of the aromatic amino acids tryptophan, phenylalanine and tyrosine. This interaction may interfere with the synthesis of the amine neurotransmitters 5‐HT, and the catecholamines dopamine and noradrenaline. Experimental studies show that BCAAs have favourable effects on cognitive functions. BCAA supplementation has been reported to improve cognitive performance in active dogs, with greater benefit to senior dogs (Fretwell et al., 2006). BCAA transamination plays an essential role in the synthesis of glutamate and subsequently of GABA. Mice subjected to traumatic brain injury had a significant reduction in BCAA concentration and neurotransmitter changes in the hippocampus (Cole et al., 2010). Dietary delivery of BCAAs to brain‐injured mice restored hippocampal BCAA levels, synaptic glutamate and GABA pools, and net synaptic efficacy, and eradicated injury‐induced cognitive impairments (Cole et al., 2010). Parenteral supplementation of BCAAs was shown to enhance the cognitive recovery of patients with traumatic brain injury (Aquilani et al., 2005), even when in a vegetative or minimally conscious state (Aquilani et al., 2008). BCAAs were administered to bipolar subjects during periods of mania for 7 days and produced a significant reduction in manic symptoms, possibly reducing brain tyrosine and phenylalanine uptake and, thus, catecholamine synthesis (Fernstrom, 2005).
Therapeutic windows and adverse effects
The recommended dietary allowance (RDA) of BCAAs is 19 mg kg−1 day−1 of isoleucine, 42 mg kg−1 day−1 of leucine and 24 mg kg−1 day−1 of valine. In humans, the daily BCAA requirement is estimated to be in a range between 10.3 and 22% required for the maintenance protein (Kamin, 1985). Extremely elevated blood concentrations of BCAAs (leucine greater than 1000 μmol L−1), as in the maple syrup urine disease, causes, within a few hours, a severe clinical condition that includes anorexia, vomiting, dehydration, lethargy, hypotonia, seizures, hypoglycaemia, ketoacidosis, pancreatitis, coma and cerebral oedema (Strauss et al., 1993).
BCAAs or leucine alone have been administered to humans in a variety of studies. The amounts of the BCAAs administered were typically double to triple the normal turnover of the BCAAs (Matthews, 2005). The administration periods ranged from hours to months. None of these studies reported any untoward effects of BCAA administration (Kamin, 1985). However, both BCAA and/or leucine administration significantly reduced the plasma concentration of several essential amino acids (Shimomura et al., 2006). This effect has been identified predominantly in muscle tissue (Shimomura et al., 2006). Both infusion of the BCAAs at three times the basal flux and dietary intake at six times the normal flux did not have any adverse effects. There are no reports of side effects associated with normal diets containing BCAAs nor with healthy subjects receiving single, infused supplemental BCAA doses as high as 9.75 g (Kamin, 1985).
The tolerable upper intake level of leucine, in both healthy young (20–35 years) and elderly (72.2 ± 3.5 years) subjects, has been shown to be similar at a dose of 500 mg kg−1 day−1 or ~35 g day−1 for an individual weighing 70 kg (Elango et al., 2012; Rasmussen et al., 2016). In a dose‐ranging study in normal volunteers, ingestion of 60 g BCAAs was well‐ tolerated and markedly raised the BCAA plasma concentration 300 min after administration (leucine went up to about 2000 nmol mL−1). The same BCAA dose was administered to bipolar subjects during periods of mania for 7 days and ameliorated their symptoms, without any side effects (Fernstrom, 2005).
BCAAs in pregnancy
The RDA of proteins in pregnancy increases by 24%. Dietary supplementation with an individual BCAA (2 g kg−1 day−1) in pregnant rats fed a low‐protein diet (6% casein) reduces fetal body weight and relative brain weights (Matsueda and Niiyama, 1982). When BCAA supplementation (2250 mg kg−1 day−1) was given 2 weeks before mating and continued through three generations, pup brain weights were reduced starting from the second generation (Thoemke and Huether, 1984). Moreover, concentrations of aspartate decreased in the brain of these animals, although the dose–response effect was not considered.
BCAAs and cancer
Leucine and isoleucine have been shown to promote bladder neoplasms originated by the oncogenic agent N‐butyl‐N‐(4‐hydroxybutyl) nitrosamine in rats at dietary levels of 2% and above (Nishio et al., 1986). However, there is no evidence that either of these amino acids could possibly be carcinogenic in the absence of an initiating agent, and no dose–response studies have actually identified any effective BCAA carcinogenic concentration. Positron emission tomography with 11C–leucine points to the high avidity of amino acid uptake of some tumours (Smith et al., 2005). On the other hand, BCAA supplementation improved the metabolic parameters, morbidity and quality of life in patients with hepatocellular carcinoma (Kawaguchi et al., 2011). The few data available on BCAAs in the tumour‐bearing state are not conclusive, and further work is needed to clarify the effects of BCAAs on cancer (Baracos and Mackenzie, 2006).
BCAAs and amyotrophic lateral sclerosis
Some epidemiological studies have correlated BCAA supplementation with a higher incidence of amyotrophic lateral sclerosis (ALS) among professional football players (Chio et al., 2005). Certain studies have shown that this effect could possibly be associated with BCAA‐induced hyperexcitability of the cortical motoneurons. In contrast, BCAA supplementation was given to ALS patients with the idea that BCAAs could activate the glutamate dehydrogenase enzyme, hence increase the catabolism of glutamate and reducing its harmful levels in the brain. A meta‐analysis concluded that BCAAs actually did not change the course of ALS (Parton et al., 2003). Although, to date, no scientific studies have clearly demonstrated that ALS is a direct consequence of BCAA supplementation, giving the popular usage of BCAAs among sportsman this risk should be taken into consideration. Pharmacovigilance studies assessing the risk of ALS incidence among cohorts of sportsman using BCAA supplementation are thus needed.
Branched‐chain amino acid supplementation as effective therapy of different disorders
Based on their mechanisms of action, BCAA mixtures have been successfully used in many disease conditions characterized by a catabolic state, including muscle sarcopenia, burn and trauma (De Bandt and Cynober, 2006). Long‐term supplementation of a specific BCAA‐enriched formula has been found to enhance mitochondrial biogenesis and increase healthy life span in middle‐aged mice (D'Antona et al., 2010). However, most of the pathological conditions are characterized by a combination of anabolic and catabolic signals that can variably activate even opposite molecular paths. In the next session, we will highlight studies showing the most promising therapeutic effects of dietary BCAA supplementation.
Muscle sarcopenia
Dietary BCAAem supplementation was found to preserve muscle fibre size, improve physical endurance and motor coordination in middle‐aged mice (D'Antona et al., 2010). Accordingly, BCAAs have been shown to improve sarcopenia, that is the age‐associated loss of muscle mass and function (Pansarasa et al., 2008), an effect possibly due to the recovery of the altered Akt/mTOR signalling in skeletal muscles of aged rats (Flati et al., 2010). BCAAem‐mediated improvement of muscle functional capacity was further enhanced by exercise training (D'Antona et al., 2010). Similarly, other groups have reported that BCAAs decrease protein breakdown and protect against dexamethasone‐induced soleus muscle atrophy in rats (Yamamoto et al., 2010). BCAAem has recently been reported to protect mice from rosuvastatin‐induced myopathy without impairing the ability of this drug to lower plasma cholesterol levels. These positive effects seem to be due to multiple mechanisms, including rescue of de‐novo protein synthesis and reduction of protein breakdown, improvement of mitochondrial dysfunction and strengthening of the anti‐ROS defence mechanisms of statin, with an effective prevention of oxidative stress in muscle (D'Antona et al., 2016). These findings may be important considering the high number of statin prescriptions over the world, also because they suggest that the modulation of different molecular and cellular processes, at the same time, can successfully affect degenerative myopathies.
Chronic renal failure
Plasma concentrations of BCAAs decrease during untreated chronic renal failure (CRF), as well as during dialysis (Cano et al., 2006). Metabolic acidosis is one of the major alterations of CRF contributing to impaired BCAA production and metabolism, eventually leading to a progressive depletion in muscle mass (Tizianello et al., 1983; Hara et al., 1987; Kooman et al., 1997). In muscle, metabolic acidosis induces protein breakdown via activation of both a cytosolic ATP‐ubiquitin‐dependent proteolytic pathway and BCKA dehydrogenase, responsible for the irreversible breakdown of BCAAs. Metabolic acidosis increases protein catabolism, BCAA breakdown and glutamine release in muscle and stimulates amino acid and glutamine metabolism towards ammonium excretion and bicarbonate generation in the kidney (Garibotto et al., 1994). Furthermore, the muscle BCAA uptake represents only 30% of total amino acid extraction in CRF, compared with 46% in control subjects (Garibotto et al., 1995). In dialysis patients, it has been reported that the normalization of plasma BCAAs by BCAA oral supplementation was associated with an improvement in appetite and nutritional status (Cano et al., 2006). Correction of the plasma amino acid profile during CRF, through EAA administration, has been found to improve protein status, avoid uraemic toxicity and delay the progression of renal disease (Cano et al., 2006). In patients with severe CRF, BCAA supplements are always associated with low‐protein diets. This nutritional intervention has been shown to improve insulin sensitivity and hyperparathyroidism and to reduce proteinuria in these patients (Jones et al., 1983; Mitch et al., 1984; Cano et al., 2006).
Liver cirrhosis
BCAAs largely escape the first‐pass hepatic metabolism and appear directly in the systemic circulation. In liver cirrhosis, there is a low ratio of plasma BCAAs to aromatic amino acids. Large‐scale, multicentre, randomized, double‐blinded, controlled trials have been performed on BCAA supplementation with the aim of normalizing amino acid profiles and nutritional status in patients with liver diseases (Muto et al., 2006). These studies demonstrated that the BCAA supplementation improves not only the nutritional status but also the prognosis and the quality of life in cirrhotic patients (Steigmann et al., 1984; Kawaguchi et al., 2011). Moreover, the favourable effects of BCAAs include an increase in albumin synthesis and an improvement in insulin resistance (Kawaguchi et al., 2008).
Anti‐ageing effects
Dietary supplementation of BCAAem at the beginning of rat senescence induced eNOS and vascular endothelial growth factor in the kidney, with increased vascularization and reduced kidney fibrosis (Corsetti et al., 2014). Topical application of BCAAs and other EAAs in aged rats improved vascularization, accompanied by an increase in collagen deposition and fibroblast proliferation, which also seem also to be involved in cutaneous wound healing (Corsetti et al., 2010).
Branched‐chain amino acid concentration in dysmetabolic conditions
The recent large‐scale analysis of the use of metabolites (i.e. metabolomics) in several conditions has allowed us to relate the plasma BCAA concentrations to disease states and progression. Diseases characterized by a prominent catabolic state showed reduced plasma BCAA levels (De Bandt and Cynober, 2006; Cole et al., 2010). In different conditions, however, metabolic deficits and altered hormonal and peripheral nervous system signals can induce changes in both BCAA absorption/distribution and catabolism (Batch et al., 2014).
Cardiovascular diseases
Altered body metabolism is a well‐known risk factor for CVDs, and it greatly affects cardiovascular‐related mortality (Long and Fox, 2016). In addition to alterations in lipid and glucose metabolism, amino acid metabolism has recently been considered as a potential factor involved in the onset and progression of heart diseases (Huang et al., 2011). BCAAs are mostly metabolized in non‐hepatic tissue, and the cardiac muscle represents an important site of BCAA catabolism (Shimomura et al., 2006). In the failing heart, BCAA utilization and catabolism are modified, and BCKAs accumulate in cardiac tissue (Sun et al., 2016). In a high cardiovascular risk population, higher concentrations of baseline BCAAs have been associated with increased risk of cardiovascular diseases (Huang et al., 2011; Yang et al., 2014). Mechanistically, it has been proposed that BCAAs, and in particular l‐leucine, could modulate the l‐arginine‐dependent NO production in endothelial cells, partially through an interaction with glutamine:fructose‐6‐phosphate aminotransferase (Yang et al., 2015).
After ischaemic stroke, plasma BCAA concentrations significantly decrease both in human and in mouse models (Kimberly et al., 2013). Low plasma BCAA levels have been related to the severity of acute ischaemic stroke and worse outcomes after stroke (Kimberly et al., 2013). Further investigations are need to fully clarify the relationship between BCAA plasma and tissue levels and the overall effect of BCAAs in specific CVDs.
Obesity and type 2 diabetes mellitus (T2DM)
Elevated circulating BCAA levels have been found to be predictive of TDM2 (Newgard et al., 2009; Yang et al., 2015; Lee et al., 2016; Yoon, 2016). So far, whether increased BCAA levels can cause insulin resistance and obesity or if they are only markers or consequences of loss of insulin action is unclear (Lynch and Adams, 2014; Giesbertz and Daniel, 2016). This doubt results from the fact that, in T2DM and obesity, a complex of several hormonal and metabolic adjustments influence most of the body metabolic processes (Cornier et al., 2008). In these conditions, an exaggerated activation of the sympathetic nervous system can indeed co‐exist with inflammation (a state named metaflammation) (Hotamisligil, 2006), reduced secretion of gut incretines (GLP‐1) (Cantini et al., 2016), inadequate suppression of counter‐regulatory hormones (i.e. glucagon, cortisol and growth hormone), dysfunction of adipose hormones (Fasshauer and Bluher, 2015), such as adiponectin, leptin, resistin and visfatin, or sex hormones, like oestrogens (Gupte et al., 2015) and testosterone (Rao et al., 2013), thyroid hormones (Iwen et al., 2013), dyslipidaemia (i.e. high blood triglycerides and low HDL‐cholesterol) and hyperuricaemia (Newgard, 2012).
Obesity, insulin resistance and T2DM have been associated with impaired BCAA catabolism that leads to the accumulation of these amino acids. The altered BCAA catabolism may be the result of different and opposing signals. Adiponectin, an adipokine predominantly synthesized in and secreted from adipose tissue (Yamauchi et al., 2002), has been shown to activate the BCKDC through PP2Cm. In obese and T2DM animal models, low circulating levels of adiponectin impair the BCAA catabolism through a down‐regulation of PP2Cm expression. Interestingly, this effect was mediated by the AMPK signaling pathway (Lian et al., 2015). Resistin and visfatin are adipokines highly expressed in visceral fat, and they induce insulin resistance. When resistin is overexpressed, BCAA uptake and protein synthesis are increased, while the insulin signal is impaired (Kang et al., 2011). Furthermore, BCAAs significantly inhibit visfatin‐induced signals (Ninomiya et al., 2011). Hyperinsulinaemic‐euglicemic clamp studies in diabetic patients showed that high‐blood levels of insulin can normalize the plasma BCAA concentration. Total pancreatectomized patients showed a better BCAA uptake, suggesting that a basal level of glucagon is important for regulating the plasma concentrations of BCAAs (Trevisan et al., 1989). Moreover, reduced levels of testosterone impaired leucine‐dependent stimulation of protein synthesis (Jiao et al., 2009).
In contrast, other studies have suggested a causal role of the circulating high levels of BCAAs in impairing insulin signalling (Adeva et al., 2012; Yoon, 2016). BCAAs in fact activate mTOR/p70S6 kinase, which phosphorylates the insulin receptor substrate‐1 (IRS‐1), thereby inhibiting phosphatidylinositol‐3‐kinase and insulin signalling (Tremblay and Marette, 2001). Phosphorylation can target IRS‐1 for proteolysis, via a proteasomal process (Tremblay et al., 2005). However, numerous observations indicate that the BCAA‐associated mTORC1 activation is not necessary or sufficient to trigger insulin resistance (Leibowitz et al., 2008). Also a BCAA metabolite (i.e. 3‐HIB) produced in diabetic muscles has been shown to increase endothelial fatty acid transportation to the muscle contributing to the insulin resistance becoming worse (Jang et al., 2016).
In an obese mouse model, BCAT expression and activity was reduced in liver and adipose tissue, but not in muscle, resulting in increased BCAA levels in blood (She et al., 2007a). However, deletion of BCAT in BCAT2−/− mice, in which circulating BCAA levels were very high, resulted in improved glycaemic control, insulin sensitivity, adiposity and lipid profiles, despite increased mTORC1 signalling (She et al., 2007b). The long‐chain fatty acids and their metabolites, present in blood of insulin‐resistant and T2DM subjects, can inhibit BCKDC activity, either by affecting redox state or acetyl‐CoA synthesis (Ruskovska and Bernlohr, 2013). Although high levels of BCAAs and inflammatory cytokines have been observed in obese‐insulin resistant patients, a high dose of TNF‐α and/or IL‐1β or IL‐1α (50 μg kg−1) has been shown to increase muscle BCKDC activity by two to threefold (Nawabi et al., 1990).
Despite the fact that BCAA concentrations are elevated in obese and TDM2 subjects, some studies have investigated the effects of BCAA supplementation in a selected subset of obese patients, and have shown beneficial effects on body weight and body fat accumulation. BCAA supplementation in a long‐term randomized study of elderly subjects with T2DM showed improvements in metabolic control [i.e. reduced glycated haemoglobin (HBA1c)] and insulin sensitivity (Solerte et al., 2008). Noteworthy, BCAAs effectively reduce insulin resistance in patients with chronic viral liver disease (Kawaguchi et al., 2008).
Conclusion
BCAAs have been shown to play an important role in the regulation of metabolism and energy balance by directly affecting peripheral tissues, such as WAT, liver and muscle. The effect of BCAAs drastically changes when they act in catabolic or anabolic conditions. In catabolic states, BCAAs can behave as energy substrate, which can be directly oxidized in the muscle or converted to gluconegenic‐chetogenic substrates. In contrast, in anabolic conditions, BCAAs stimulate protein synthesis and cell growth (Figure 2). In exclusively catabolic disorders, such as muscle sarcopenia, burn and trauma, BCAA supplementation improves muscle function and clinical outcomes (Figure 2). However, in metabolic disorders, in which different amounts of anabolic and catabolic signals coexist, the effects of BCAA supplementation are difficult to predict. This might be the case in obesity and insulin resistance, where BCAA supplementation seems to exert opposing effects depending on the prevalence of the catabolic or anabolic signals (Figure 2). Thus, a future challenge in this field will be to approach systemically the complex network of molecules and metabolites, beyond the environmental signals (i.e. foods, nutrient composition, calorie restriction, exercise, gut microbiome, etc.), that regulates BCAA metabolism and is regulated by BCAAs themselves. This will be possible only taking into account both the complexity and the peculiarity of single, specific diseases, which depend on the variable contributions of the nervous system – both sympathetic and parasympathetic – in addition to the immune and the endocrine systems, acting in the presence of particular nutritional states.
Figure 2.
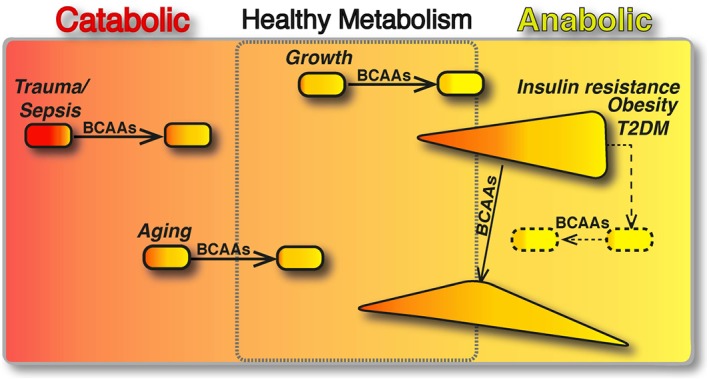
BCAAs differently modulate catabolic and anabolic states. The boxed area represents the catabolic (red, left) or anabolic (yellow, right) states of the organisms. In catabolic (sepsis, trauma and ageing) and anabolic (growth) conditions, BCAAs have been shown to restore the energy balance and to improve the clinical outcomes (arrows). In contrast, in metabolic disorders, different amounts of anabolic and catabolic signals coexist. Here, we present insulin resistance, obesity and T2DM, which are primarily anabolic conditions, in which several catabolic signals are co‐expressed with anabolic ones (e.g. exaggerated sympathetic nervous system activity, inadequate suppression of counter‐regulatory hormones, such as glucagon, cortisol and growth hormone). In these pathological conditions, the effects of BCAA supplementation seem (arrow) to exert opposing effects depending on the prevalence of the catabolic or anabolic signals. Multiple interventions capable of balancing the aberrant metabolic signals (dashed line) may be required to potentiate the healthy effects of BCAAs (dashed arrow).
To obtain significant data, it is vital to evaluate carefully the experimental conditions that are demonstrated to largely influence the outcomes of treatments with BCAAs. Also, given the potential discrepancy found in the treatments' results, it is advisable to establish the effective BCAA dose and toxicity in every single clinical setting. Multiple interventions capable of balancing any aberrant metabolic signals may be required to potentiate the healthy effects of BCAAs.
Author contributions
F.B. and E.N. contributed to article drafting and revision. F.B. and E.N. authorized the final version of the review.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This study is supported by the Ministero dell'Istruzione, dell'Università e della Ricerca (2009E48P9M_001).
Bifari, F. , and Nisoli, E. (2017) Branched‐chain amino acids differently modulate catabolic and anabolic states in mammals: a pharmacological point of view. British Journal of Pharmacology, 174: 1366–1377. doi: 10.1111/bph.13624.
References
- Adeva MM, Calvino J, Souto G, Donapetry C (2012). Insulin resistance and the metabolism of branched‐chain amino acids in humans. Amino Acids 43: 171–181. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015e). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilani R, Boselli M, Boschi F, Viglio S, Iadarola P, Dossena M et al. (2008). Branched‐chain amino acids may improve recovery from a vegetative or minimally conscious state in patients with traumatic brain injury: a pilot study. Arch Phys Med Rehabil 89: 1642–1647. [DOI] [PubMed] [Google Scholar]
- Aquilani R, Iadarola P, Contardi A, Boselli M, Verri M, Pastoris O et al. (2005). Branched‐chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil 86: 1729–1735. [DOI] [PubMed] [Google Scholar]
- Aquilani R, Zuccarelli GC, Dioguardi FS, Baiardi P, Frustaglia A, Rutili C et al. (2011). Effects of oral amino acid supplementation on long‐term‐care‐acquired infections in elderly patients. Arch Gerontol Geriatr 52: e123–e128. [DOI] [PubMed] [Google Scholar]
- Baracos VE, Mackenzie ML (2006). Investigations of branched‐chain amino acids and their metabolites in animal models of cancer. J Nutr 136: 237S–242S. [DOI] [PubMed] [Google Scholar]
- Bassit RA, Sawada LA, Bacurau RF, Navarro F, Martins E Jr, Santos RV et al. (2002). Branched‐chain amino acid supplementation and the immune response of long‐distance athletes. Nutrition 18: 376–379. [DOI] [PubMed] [Google Scholar]
- Batch BC, Hyland K, Svetkey LP (2014). Branched chain amino acids: biomarkers of health and disease. Curr Opin Clin Nutr Metab Care 17: 86–89. [DOI] [PubMed] [Google Scholar]
- Blumer RM, van Roomen CP, Meijer AJ, Houben‐Weerts JH, Sauerwein HP, Dubbelhuis PF (2008). Regulation of adiponectin secretion by insulin and amino acids in 3 T3‐L1 adipocytes. Metabolism 57: 1655–1662. [DOI] [PubMed] [Google Scholar]
- Bolasco P, Caria S, Cupisti A, Secci R, Saverio Dioguardi F (2011). A novel amino acids oral supplementation in hemodialysis patients: a pilot study. Ren Fail 33: 1–5. [DOI] [PubMed] [Google Scholar]
- Broer S (2008). Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev 88: 249–286. [DOI] [PubMed] [Google Scholar]
- Brosnan JT, Brosnan ME (2006). Branched‐chain amino acids: enzyme and substrate regulation. J Nutr 136: 207S–211S. [DOI] [PubMed] [Google Scholar]
- Calder PC (2006). Branched‐chain amino acids and immunity. J Nutr 136: 288S–293S. [DOI] [PubMed] [Google Scholar]
- Cano NJ, Fouque D, Leverve XM (2006). Application of branched‐chain amino acids in human pathological states: renal failure. J Nutr 136: 299S–307S. [DOI] [PubMed] [Google Scholar]
- Cantini G, Mannucci E, Luconi M (2016). Perspectives in GLP‐1 Research: New Targets, New Receptors. Trends Endocrinol Metab 27: 427–438. [DOI] [PubMed] [Google Scholar]
- Chen Q, Reimer RA (2009). Dairy protein and leucine alter GLP‐1 release and mRNA of genes involved in intestinal lipid metabolism in vitro. Nutrition 25: 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio A, Benzi G, Dossena M, Mutani R, Mora G (2005). Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain 128: 472–476. [DOI] [PubMed] [Google Scholar]
- Chou PY, Fasman GD (1973). Structural and functional role of leucine residues in proteins. J Mol Biol 74: 263–281. [DOI] [PubMed] [Google Scholar]
- Chou PY, Fasman GD (1978). Empirical predictions of protein conformation. Annu Rev Biochem 47: 251–276. [DOI] [PubMed] [Google Scholar]
- Cole JT, Mitala CM, Kundu S, Verma A, Elkind JA, Nissim I et al. (2010). Dietary branched chain amino acids ameliorate injury‐induced cognitive impairment. Proc Natl Acad Sci U S A 107: 366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombel JF, Sutton A, Chayvialle JA, Modigliani R (1988). Cholecystokinin release and biliopancreatic secretion in response to selective perfusion of the duodenal loop with aminoacids in man. Gut 29: 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR et al. (2008). The metabolic syndrome. Endocr Rev 29: 777–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsetti G, D'Antona G, Dioguardi FS, Rezzani R (2010). Topical application of dressing with amino acids improves cutaneous wound healing in aged rats. Acta Histochem 112: 497–507. [DOI] [PubMed] [Google Scholar]
- Corsetti G, D'Antona G, Ruocco C, Stacchiotti A, Romano C, Tedesco L et al. (2014). Dietary supplementation with essential amino acids boosts the beneficial effects of rosuvastatin on mouse kidney. Amino Acids 46: 2189–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC et al. (2006). Hypothalamic mTOR signaling regulates food intake. Science 312: 927–930. [DOI] [PubMed] [Google Scholar]
- D'Antona G, Ragni M, Cardile A, Tedesco L, Dossena M, Bruttini F et al. (2010). Branched‐chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle‐aged mice. Cell Metab 12: 362–372. [DOI] [PubMed] [Google Scholar]
- D'Antona G, Tedesco L, Ruocco C, Corsetti G, Ragni M, Fossati A et al. (2016). A peculiar formula of essential amino acids prevents rosuvastatin myopathy in mice. Antioxid Redox Signal 25: 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bandt JP, Cynober L (2006). Therapeutic use of branched‐chain amino acids in burn, trauma, and sepsis. J Nutr 136: 308S–313S. [DOI] [PubMed] [Google Scholar]
- Demas GE, Drazen DL, Nelson RJ (2003). Reductions in total body fat decrease humoral immunity. Proc Biol Sci 270: 905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango R, Chapman K, Rafii M, Ball RO, Pencharz PB (2012). Determination of the tolerable upper intake level of leucine in acute dietary studies in young men. Am J Clin Nutr 96: 759–767. [DOI] [PubMed] [Google Scholar]
- Fasshauer M, Bluher M (2015). Adipokines in health and disease. Trends Pharmacol Sci 36: 461–470. [DOI] [PubMed] [Google Scholar]
- Fernstrom JD (2005). Branched‐chain amino acids and brain function. J Nutr 135: 1539S–1546S. [DOI] [PubMed] [Google Scholar]
- Flati V, Caliaro F, Speca S, Corsetti G, Cardile A, Nisoli E et al. (2010). Essential amino acids improve insulin activation of AKT/MTOR signaling in soleus muscle of aged rats. Int J Immunopathol Pharmacol 23: 81–89. [DOI] [PubMed] [Google Scholar]
- Floyd JC Jr, Fajans SS, Conn JW, Knopf RF, Rull J (1966). Stimulation of insulin secretion by amino acids. J Clin Invest 45: 1487–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fretwell LK, McCune S, Fone JV, Yates DJ (2006). The effect of supplementation with branched‐chain amino acids on cognitive function in active dogs. J Nutr 136: 2069S–2071S. [DOI] [PubMed] [Google Scholar]
- Garibotto G, Deferrari G, Robaudo C, Saffioti S, Sofia A, Russo R et al. (1995). Disposal of exogenous amino acids by muscle in patients with chronic renal failure. Am J Clin Nutr 62: 136–142. [DOI] [PubMed] [Google Scholar]
- Garibotto G, Russo R, Sofia A, Sala MR, Robaudo C, Moscatelli P et al. (1994). Skeletal muscle protein synthesis and degradation in patients with chronic renal failure. Kidney Int 45: 1432–1439. [DOI] [PubMed] [Google Scholar]
- Giesbertz P, Daniel H (2016). Branched‐chain amino acids as biomarkers in diabetes. Curr Opin Clin Nutr Metab Care 19: 48–54. [DOI] [PubMed] [Google Scholar]
- Glassy MC, Furlong CE (1981). Neutral amino acid transport during the cell cycle of cultured human lymphocytes. J Cell Physiol 107: 69–74. [DOI] [PubMed] [Google Scholar]
- Glover JN, Harrison SC (1995). Crystal structure of the heterodimeric bZIP transcription factor c‐Fos‐c‐Jun bound to DNA. Nature 373: 257–261. [DOI] [PubMed] [Google Scholar]
- Gupte AA, Pownall HJ, Hamilton DJ (2015). Estrogen: an emerging regulator of insulin action and mitochondrial function. J Diabetes Res 2015: 916585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK et al. (2012). Leucyl‐tRNA synthetase is an intracellular leucine sensor for the mTORC1‐signaling pathway. Cell 149: 410–424. [DOI] [PubMed] [Google Scholar]
- Hara Y, May RC, Kelly RA, Mitch WE (1987). Acidosis, not azotemia, stimulates branched‐chain, amino acid catabolism in uremic rats. Kidney Int 32: 808–814. [DOI] [PubMed] [Google Scholar]
- Harper AE, Miller RH, Block KP (1984). Branched‐chain amino acid metabolism. Annu Rev Nutr 4: 409–454. [DOI] [PubMed] [Google Scholar]
- Harris RA, Joshi M, Jeoung NH, Obayashi M (2005). Overview of the molecular and biochemical basis of branched‐chain amino acid catabolism. J Nutr 135: 1527S–1530S. [DOI] [PubMed] [Google Scholar]
- Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP et al. (2014). Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med 20: 1417–1426. [DOI] [PubMed] [Google Scholar]
- Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB (2010). Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem 285: 11348–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2006). Inflammation and metabolic disorders. Nature 444: 860–867. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Erbay E (2008). Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 8: 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhou M, Sun H, Wang Y (2011). Branched‐chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res 90: 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwen KA, Schroder E, Brabant G (2013). Thyroid hormones and the metabolic syndrome. Eur Thyroid J 2: 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC et al. (2016). A branched‐chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med 22: 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Q, Pruznak AM, Huber D, Vary TC, Lang CH (2009). Castration differentially alters basal and leucine‐stimulated tissue protein synthesis in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab 297: E1222–E1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R, Dalton N, Turner C, Start K, Haycock G, Chantler C (1983). Oral essential aminoacid and ketoacid supplements in children with chronic renal failure. Kidney Int 24: 95–103. [DOI] [PubMed] [Google Scholar]
- Kamin H (1985). Status of the 10th edition of the Recommended Dietary Allowances‐‐prospects for the future. Am J Clin Nutr 41: 165–170. [DOI] [PubMed] [Google Scholar]
- Kang S, Chemaly ER, Hajjar RJ, Lebeche D (2011). Resistin promotes cardiac hypertrophy via the AMP‐activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c‐Jun N‐terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J Biol Chem 286: 18465–18473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T, Izumi N, Charlton MR, Sata M (2011). Branched‐chain amino acids as pharmacological nutrients in chronic liver disease. Hepatology 54: 1063–1070. [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Nagao Y, Matsuoka H, Ide T, Sata M (2008). Branched‐chain amino acid‐enriched supplementation improves insulin resistance in patients with chronic liver disease. Int J Mol Med 22: 105–112. [PubMed] [Google Scholar]
- Kimberly WT, Wang Y, Pham L, Furie KL, Gerszten RE (2013). Metabolite profiling identifies a branched chain amino acid signature in acute cardioembolic stroke. Stroke 44: 1389–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konner AC, Bruning JC (2011). Toll‐like receptors: linking inflammation to metabolism. Trends Endocrinol Metab 22: 16–23. [DOI] [PubMed] [Google Scholar]
- Kooman JP, Deutz NE, Zijlmans P, van den Wall Bake A, Gerlag PG, van Hooff JP et al. (1997). The influence of bicarbonate supplementation on plasma levels of branched‐chain amino acids in haemodialysis patients with metabolic acidosis. Nephrol Dial Transplant 12: 2397–2401. [DOI] [PubMed] [Google Scholar]
- Lee CC, Watkins SM, Lorenzo C, Wagenknecht LE, Il'yasova D, Chen YD et al. (2016). Branched‐Chain Amino Acids and Insulin Metabolism: The Insulin Resistance Atherosclerosis Study (IRAS). Diabetes Care 39: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y et al. (2007). IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130: 440–455. [DOI] [PubMed] [Google Scholar]
- Leibowitz G, Cerasi E, Ketzinel‐Gilad M (2008). The role of mTOR in the adaptation and failure of beta‐cells in type 2 diabetes. Diabetes Obes Metab 10 (Suppl. 4): 157–169. [DOI] [PubMed] [Google Scholar]
- Lesourd B (1995). Protein undernutrition as the major cause of decreased immune function in the elderly: clinical and functional implications. Nutr Rev 53: S86–91; discussion S92–84. [DOI] [PubMed] [Google Scholar]
- Letto J, Brosnan ME, Brosnan JT (1986). Valine metabolism. Gluconeogenesis from 3‐hydroxyisobutyrate. Biochem J 240: 909–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian K, Du C, Liu Y, Zhu D, Yan W, Zhang H et al. (2015). Impaired adiponectin signaling contributes to disturbed catabolism of branched‐chain amino acids in diabetic mice. Diabetes 64: 49–59. [DOI] [PubMed] [Google Scholar]
- Long MT, Fox CS (2016). The Framingham Heart Study‐‐67 years of discovery in metabolic disease. Nat Rev Endocrinol 12: 177–183. [DOI] [PubMed] [Google Scholar]
- Lu G, Sun H, She P, Youn JY, Warburton S, Ping P et al. (2009). Protein phosphatase 2Cm is a critical regulator of branched‐chain amino acid catabolism in mice and cultured cells. J Clin Invest 119: 1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch CJ, Adams SH (2014). Branched‐chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol 10: 723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch CJ, Gern B, Lloyd C, Hutson SM, Eicher R, Vary TC (2006). Leucine in food mediates some of the postprandial rise in plasma leptin concentrations. Am J Physiol Endocrinol Metab 291: E621–E630. [DOI] [PubMed] [Google Scholar]
- Matsueda S, Niiyama Y (1982). The effects of excess amino acids on maintenance of pregnancy and fetal growth in rats. J Nutr Sci Vitaminol (Tokyo) 28: 557–573. [DOI] [PubMed] [Google Scholar]
- Matthews DE (2005). Observations of branched‐chain amino acid administration in humans. J Nutr 135: 1580S–1584S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllan L, Cotter PD, Roche HM, Korpela R, Nilaweera KN (2013). Impact of leucine on energy balance. J Physiol Biochem 69: 155–163. [DOI] [PubMed] [Google Scholar]
- Mitch WE, Walser M, Steinman TI, Hill S, Zeger S, Tungsanga K (1984). The effect of a keto acid‐amino acid supplement to a restricted diet on the progression of chronic renal failure. N Engl J Med 311: 623–629. [DOI] [PubMed] [Google Scholar]
- Muto Y, Sato S, Watanabe A, Moriwaki H, Suzuki K, Kato A et al. (2006). Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long‐term oral supplementation with branched‐chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol Res 35: 204–214. [DOI] [PubMed] [Google Scholar]
- Nair KS, Short KR (2005). Hormonal and signaling role of branched‐chain amino acids. J Nutr 135: 1547S–1552S. [DOI] [PubMed] [Google Scholar]
- Nakamura I, Ochiai K, Imai Y, Moriyasu F, Imawari M (2007). Restoration of innate host defense responses by oral supplementation of branched‐chain amino acids in decompensated cirrhotic patients. Hepatol Res 37: 1062–1067. [DOI] [PubMed] [Google Scholar]
- Nakamura I, Ochiai K, Imawari M (2004). Phagocytic function of neutrophils of patients with decompensated liver cirrhosis is restored by oral supplementation of branched‐chain amino acids. Hepatol Res 29: 207–211. [DOI] [PubMed] [Google Scholar]
- Nawabi MD, Block KP, Chakrabarti MC, Buse MG (1990). Administration of endotoxin, tumor necrosis factor, or interleukin 1 to rats activates skeletal muscle branched‐chain alpha‐keto acid dehydrogenase. J Clin Invest 85: 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB (2012). Interplay between lipids and branched‐chain amino acids in development of insulin resistance. Cell Metab 15: 606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF et al. (2009). A branched‐chain amino acid‐related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya S, Shimizu M, Imai K, Takai K, Shiraki M, Hara T et al. (2011). Possible role of visfatin in hepatoma progression and the effects of branched‐chain amino acids on visfatin‐induced proliferation in human hepatoma cells. Cancer Prev Res (Phila) 4: 2092–2100. [DOI] [PubMed] [Google Scholar]
- Nishio Y, Kakizoe T, Ohtani M, Sato S, Sugimura T, Fukushima S (1986). L‐isoleucine and L‐leucine: tumor promoters of bladder cancer in rats. Science 231: 843–845. [DOI] [PubMed] [Google Scholar]
- Pansarasa O, Flati V, Corsetti G, Brocca L, Pasini E, D'Antona G (2008). Oral amino acid supplementation counteracts age‐induced sarcopenia in elderly rats. Am J Cardiol 101: 35E–41E. [DOI] [PubMed] [Google Scholar]
- Parton M, Mitsumoto H, Leigh PN (2003). Amino acids for amyotrophic lateral sclerosis / motor neuron disease. Cochrane Database Syst Rev 4: CD003457. [DOI] [PubMed] [Google Scholar]
- Rao PM, Kelly DM, Jones TH (2013). Testosterone and insulin resistance in the metabolic syndrome and T2DM in men. Nat Rev Endocrinol 9: 479–493. [DOI] [PubMed] [Google Scholar]
- Rasmussen B, Gilbert E, Turki A, Madden K, Elango R (2016). Determination of the safety of leucine supplementation in healthy elderly men. Amino Acids 48: 1707–1716. [DOI] [PubMed] [Google Scholar]
- Rennie MJ, Bohe J, Smith K, Wackerhage H, Greenhaff P (2006). Branched‐chain amino acids as fuels and anabolic signals in human muscle. J Nutr 136: 264S–268S. [DOI] [PubMed] [Google Scholar]
- Romanyukha AA, Rudnev SG, Sidorov IA (2006). Energy cost of infection burden: an approach to understanding the dynamics of host‐pathogen interactions. J Theor Biol 241: 1–13. [DOI] [PubMed] [Google Scholar]
- Roth E (2008). Nonnutritive effects of glutamine. J Nutr 138: 2025S–2031S. [DOI] [PubMed] [Google Scholar]
- Ruskovska T, Bernlohr DA (2013). Oxidative stress and protein carbonylation in adipose tissue ‐ implications for insulin resistance and diabetes mellitus. J Proteomics 92: 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T et al. (2016). Structural basis for leucine sensing by the Sestrin2‐mTORC1 pathway. Science 351: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ et al. (2007a). Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab 6: 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ (2007b). Obesity‐related elevations in plasma leucine are associated with alterations in enzymes involved in branched‐chain amino acid metabolism. Am J Physiol Endocrinol Metab 293: E1552–E1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura Y, Yamamoto Y, Bajotto G, Sato J, Murakami T, Shimomura N et al. (2006). Nutraceutical effects of branched‐chain amino acids on skeletal muscle. J Nutr 136: 529S–532S. [DOI] [PubMed] [Google Scholar]
- Smith CB, Schmidt KC, Qin M, Burlin TV, Cook MP, Kang J et al. (2005). Measurement of regional rates of cerebral protein synthesis with L‐[1‐11C]leucine and PET with correction for recycling of tissue amino acids: II. Validation in rhesus monkeys. J Cereb Blood Flow Metab 25: 629–640. [DOI] [PubMed] [Google Scholar]
- Smith QR, Momma S, Aoyagi M, Rapoport SI (1987). Kinetics of neutral amino acid transport across the blood–brain barrier. J Neurochem 49: 1651–1658. [DOI] [PubMed] [Google Scholar]
- Solerte SB, Fioravanti M, Locatelli E, Bonacasa R, Zamboni M, Basso C et al. (2008). Improvement of blood glucose control and insulin sensitivity during a long‐term (60 weeks) randomized study with amino acid dietary supplements in elderly subjects with type 2 diabetes mellitus. Am J Cardiol 101: 82E–88E. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigmann F, Szanto PB, Poulos A, Lim PE, Dubin A (1984). Significance of serum aminograms in diagnosis and prognosis of liver diseases. J Clin Gastroenterol 6: 453–460. [DOI] [PubMed] [Google Scholar]
- Steinert RE, Landrock MF, Ullrich SS, Standfield S, Otto B, Horowitz M et al. (2015). Effects of intraduodenal infusion of the branched‐chain amino acid leucine on ad libitum eating, gut motor and hormone functions, and glycemia in healthy men. Am J Clin Nutr 102: 820–827. [DOI] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Morton DH (1993). Maple Syrup Urine Disease In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH. et al. (eds). GeneReviews(R): Seattle (WA) Bookshelf ID: NBK1319PMID: 20301495 https://www.ncbi.nlm.nih.gov/books/NBK1319/?report=printable. [Google Scholar]
- Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z et al. (2016). Catabolic Defect of Branched‐Chain Amino Acids Promotes Heart Failure. Circulation 133: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE, Hutson SM (1998). A molecular model of human branched‐chain amino acid metabolism. Am J Clin Nutr 68: 72–81. [DOI] [PubMed] [Google Scholar]
- Thoemke F, Huether G (1984). Breeding rats on amino acid imbalanced diets for three consecutive generations affects the concentrations of putative amino acid transmitters in the developing brain. Int J Dev Neurosci 2: 567–574. [DOI] [PubMed] [Google Scholar]
- Tizianello A, Deferrari G, Garibotto G, Robaudo C, Lutman M, Passerone G et al. (1983). Branched‐chain amino acid metabolism in chronic renal failure. Kidney Int Suppl 16: S17–S22. [PubMed] [Google Scholar]
- Torres‐Leal FL, Fonseca‐Alaniz MH, Teodoro GF, de Capitani MD, Vianna D, Pantaleao LC et al. (2011). Leucine supplementation improves adiponectin and total cholesterol concentrations despite the lack of changes in adiposity or glucose homeostasis in rats previously exposed to a high‐fat diet. Nutr Metab (Lond) 8: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F, Marette A (2001). Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 276: 38052–38060. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Jacques H, Marette A (2005). Modulation of insulin action by dietary proteins and amino acids: role of the mammalian target of rapamycin nutrient sensing pathway. Curr Opin Clin Nutr Metab Care 8: 457–462. [DOI] [PubMed] [Google Scholar]
- Trevisan R, Marescotti C, Avogaro A, Tessari P, del Prato S, Tiengo A (1989). Effects of different insulin administrations on plasma amino acid profile in insulin‐dependent diabetic patients. Diabetes Res 12: 57–62. [PubMed] [Google Scholar]
- Waterlow JC, Golden MH, Garlick PJ (1978). Protein turnover in man measured with 15 N: comparison of end products and dose regimes. Am J Physiol 235: E165–E174. [DOI] [PubMed] [Google Scholar]
- Wek SA, Zhu S, Wek RC (1995). The histidyl‐tRNA synthetase‐related sequence in the eIF‐2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol 15: 4497–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR et al. (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351: 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto D, Maki T, Herningtyas EH, Ikeshita N, Shibahara H et al. (2010). Branched‐chain amino acids protect against dexamethasone‐induced soleus muscle atrophy in rats. Muscle Nerve 41: 819–827. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S et al. (2002). Adiponectin stimulates glucose utilization and fatty‐acid oxidation by activating AMP‐activated protein kinase. Nat Med 8: 1288–1295. [DOI] [PubMed] [Google Scholar]
- Yang R, Dong J, Zhao H, Li H, Guo H, Wang S et al. (2014). Association of branched‐chain amino acids with carotid intima‐media thickness and coronary artery disease risk factors. PLoS One 9: e99598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wu Z, Meininger CJ, Wu G (2015). L‐Leucine and NO‐mediated cardiovascular function. Amino Acids 47: 435–447. [DOI] [PubMed] [Google Scholar]
- Yoon MS (2016). The Emerging Role of Branched‐Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan HX, Xiong Y, Guan KL (2013). Nutrient sensing, metabolism, and cell growth control. Mol Cell 49: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]