

Abstract
The Human Gene Mutation Database (HGMD®) constitutes a comprehensive collection of published germline mutations in nuclear genes that underlie, or are closely associated with human inherited disease. At the time of writing (March 2017), the database contained in excess of 203,000 different gene lesions identified in over 8000 genes manually curated from over 2600 journals. With new mutation entries currently accumulating at a rate exceeding 17,000 per annum, HGMD represents de facto the central unified gene/disease-oriented repository of heritable mutations causing human genetic disease used worldwide by researchers, clinicians, diagnostic laboratories and genetic counsellors, and is an essential tool for the annotation of next-generation sequencing data. The public version of HGMD (http://www.hgmd.org) is freely available to registered users from academic institutions and non-profit organisations whilst the subscription version (HGMD Professional) is available to academic, clinical and commercial users under license via QIAGEN Inc.
Introduction
The Human Gene Mutation Database (HGMD®) represents an attempt to collate all known gene lesions underlying human inherited disease together with disease-associated/functional polymorphisms published in the peer-reviewed literature. The mutation data catalogued by HGMD (summarised by mutation type) are shown in Table 1.
Table 1.
Numbers of different mutations by mutation type present in HGMD Professional release 2017.1 and the publicly available version of the database (March 31st 2017)
| Mutation type | Numbers of mutations | ||
|---|---|---|---|
| HGMD Professional 2017.1 | Publicly available | ||
| Total (disease-associated/functional polymorphism sub-total) | With chromosomal coordinates and VCF data (GRCh38/hg38) | ||
| Missense substitutions | 92,331 (5132) | 91,671 | 62,759 |
| Nonsense substitutions | 22,372 (333) | 22,376 | 15,642 |
| Splicing substitutions (intronic and exonic) | 18,386 (632) | 18,083 | 13,087 |
| Regulatory substitutions (exonic, intronic, 5′- and 3′-untranslated regions) | 3801 (2499) | 3717 | 2764 |
| Micro-deletions ≤20 bp | 30,169 (292) | 29,540 | 21,744 |
| Micro-insertions/duplications ≤20 bp | 12,557 (175) | 12,227 | 8975 |
| Micro-indels ≤ 20 bp | 2866 (59) | 2770 | 2100 |
| Gross deletions >20 bp | 15,272 (147) | 0 | 10,337 |
| Gross insertions/duplications >20 bp | 3767 (84) | 0 | 2389 |
| Complex rearrangements (including inversions, translocations and complex indels) | 1857 (117) | 0 | 1417 |
| Repeat variations | 507 (306) | 0 | 421 |
| Totals | 203,885 (9776) | 180,386 | 141,635 |
HGMD has never sought to include either somatic or mitochondrial mutations, which are well covered by COSMIC (Forbes et al. 2015) and MitoMap (Lott et al. 2013), respectively. Nor does HGMD attempt to provide comprehensive coverage of pharmacological variants (except for those variants where evidence supporting a functional impairment has been provided); PharmGKB (https://www.pharmgkb.org/; Thorn et al. 2013) is a more comprehensive resource for these data. Finally, HGMD is not intended to be a general genetic variation database; users interested in such variants should visit dbSNP (http://www.ncbi.nlm.nih.gov/SNP/; Sherry et al. 2001), the NHLBI Exome Variant Server (http://evs.gs.washington.edu/EVS/) or the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/; Lek et al. 2016).
HGMD was originally established in 1996 for the scientific study of mutational mechanisms in human genes believed to cause inherited disease (Cooper et al. 2010; Stenson et al. 2014). However, over the last 20 years it has acquired a much broader utility as the central unified repository for disease-related functional genetic variation in the germline. It is now routinely accessed and utilised by next-generation sequencing (NGS) project researchers, human molecular geneticists, molecular biologists, clinicians and genetic counsellors as well as by those specialising in biopharmaceuticals, bioinformatics and personalised genomics.
The public version of HGMD (http://www.hgmd.org) is freely available to registered users from academic institutions/non-profit organisations. This version is, however, maintained in a basic form that is only updated twice annually, is permanently a minimum of 3.5 years out of date, and does not contain any of the additional annotations or extra features present in HGMD Professional (see below). The Professional version is available to both commercial and academic/non-profit users via subscription from QIAGEN (https://www.qiagenbioinformatics.com/) as either an online or a locally installed/downloadable version that is updated quarterly and includes a variety of additional annotations and extra features, such as GRCh38/hg38 and GRCh37/hg19 genomic chromosomal coordinates, HGVS nomenclature, Variant Call Format (VCF), additional literature reports, advanced search features, conservation data and functional predictions.
Source of mutation data
All HGMD mutation data have been obtained from the scientific literature and are manually curated on an ongoing basis. Identification of relevant literature reports is carried out via a combination of manual journal screening and automated text mining. The database currently contains >203,000 mutation entries obtained from over 57,000 primary literature reports (supported by 29,000 additional literature reports), which were published in more than 2600 different journals. The number of articles screened (both for novel mutations and additional annotations) appears to have reached a plateau, (Fig. 1); however, the number of mutations reported (per reference) continues to increase steadily. It is likely that the continuing development of high-throughput NGS methods will lead to an increased rate of deposition of disease-associated genetic variants in the published literature.
Fig. 1.
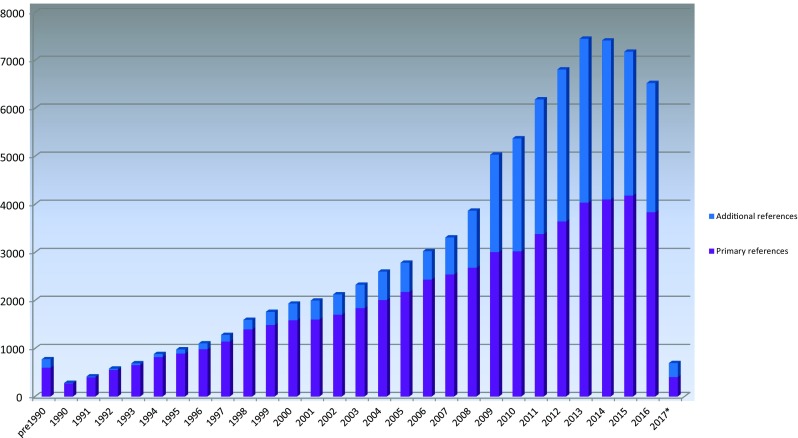
Annual numbers of cited literature references added to HGMD. *2017 figures not yet complete
Classes of variant listed in HGMD
There are six different classes of variant listed in HGMD (Fig. 2). Disease-causing mutations (DM) are entered into HGMD where the authors of the corresponding report(s) have established that the reported mutation(s) are involved (or very likely to be involved) in conferring the associated clinical phenotype upon the individuals concerned. The DM classification may, however, also appear with a question mark (DM?), denoting a probable/possible pathological mutation, reported as likely to be disease causing in the corresponding report, but where (i) the author has indicated that there may be some degree of doubt or uncertainty; (ii) the HGMD curators believe greater interpretational caution is warranted, or (iii) subsequent evidence has appeared in the literature which has called the initial putatively deleterious nature of the variant into question (e.g. a negative functional, case–control or population-scale sequencing study). The DM and DM? variant classes may include mutations that are believed to contribute to disease susceptibility in a multi-factorial manner (e.g. autism or schizophrenia), exhibit complex polygenic inheritance or possess an environmental trigger component to their pathogenicity. It can be seen from Fig. 2 that the proportion of reported mutations belonging to the DM? category has steadily increased over the last decade; we speculate that this is because authors, journal editors and referees (also database curators!) alike have become much more cautious than they used to be in ascribing pathogenicity to the putatively disease-associated variants that have been identified. This increase in caution appears to closely coincide with the advent of NGS and the consequent deluge of genetic variants that must be filtered and prioritised.
Fig. 2.
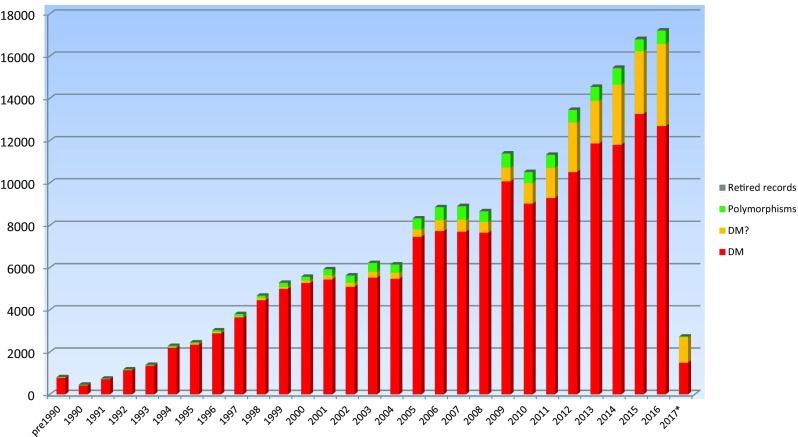
Annual mutation totals subdivided by variant class. *2017 figures not yet complete
Three categories of polymorphism are included in the database (combined into ‘polymorphisms’ in Fig. 2). Disease-associated polymorphisms (DP) are entered into HGMD where there is evidence for a significant association with a disease/clinical phenotype along with additional evidence that the polymorphism is itself likely to be of functional relevance (e.g. as a consequence of genic location, evolutionary conservation, transcription factor binding potential, etc.), although there may be no direct evidence (e.g. from an expression study) for a functional effect. The functional polymorphisms (FP) class includes those sequence changes for which a direct functional effect has been demonstrated (e.g. by means of an in vitro reporter gene assay or alternatively by protein structure, function or expression studies), but with no disease association reported as yet. Disease-associated polymorphisms with supporting functional evidence (DFP) must meet both of the above criteria in that the polymorphism should not only have been reported to be significantly associated with disease, but should also display direct evidence of being of functional relevance. The polymorphism data present in HGMD should be viewed with a degree of caution owing to (i) the possibility that an observed disease association may be simply due to a linkage disequilibrium effect and (ii) the fact that in vitro studies are not invariably accurate indicators of in vivo functionality (Cirulli and Goldstein 2007; Dimas et al. 2009). Retired records (R) are variants that have been removed from HGMD if found to have been erroneously included ab initio, or if the variant has been subject to retraction/correction in the literature resulting in the record becoming obsolete, merged or otherwise invalid.
The various HGMD variant classes described above should not be cross-correlated with the ‘benign to pathogenic’ 5-point classification system adopted by the ACMG consortium (Green et al. 2013). Although, by their very nature, there will be some overlap, these two classification systems are not directly interchangeable. The primary purpose of the ACMG guidelines appears to be to minimise false positives in a clinical setting, whereas HGMD aims to include mutation data based on the cogency and credibility of the associated literature, with a curation policy that opts to minimise false negatives by being broadly inclusive, whilst attempting to highlight potential false positives to users (e.g. via an allele frequency flag). Attempting to cross-correlate the two classification systems (e.g. by automatically considering HGMD DM to be equivalent to ACMG class 5) is likely to be potentially misleading at best, and may well lead to users drawing incorrect or inappropriate conclusions (Pinard et al. 2016).
Polymorphic copy number variations (CNVs) represent an important subset of potentially functional disease-associated variation (Mikhail 2014; Usher and McCarroll, 2015). While HGMD does not wish to replicate the excellent curatorial work of other resources (e.g. the Database of Genomic Variants http://dgv.tcag.ca/dgv/app/home, DECIPHER http://decipher.sanger.ac.uk/ and Copy Number Variation in Disease http://202.97.205.78/CNVD/), we do include such variants where they fulfil certain criteria. HGMD will include such variants if they have been shown to be of functional significance, associated with disease, and involve a single characterised gene or small group of genes that have been directly implicated in the disease association. Such variants would then be entered into the database under one of the above-mentioned polymorphism categories, depending upon the supporting evidence provided by the authors of the article in question.
The HGMD curators have adopted a policy of continual reassessment of the curated content within the database. If and when newly published information relevant to a specific mutation entry becomes available (e.g. additional case reports or alternate clinical or laboratory phenotypes, population frequency data or functional studies), the mutation entry may be revised or re-classified. Where new information becomes available which suggests that a given disease-causing mutation (DM) is likely to be of questionable pathological relevance or even a neutral polymorphism (on the basis of additional case reports, genome/population screening studies, negative case–control studies, etc.), it may be flagged with a question mark (DM?), re-categorised under one of the categories of polymorphism, or retired from the database altogether (R) if it turns out to have been erroneously included ab initio. The HGMD curators re-categorised or retired over 800 variants in 2015 with almost 26,000 existing records having at least one relevant additional reference added in the same year. Users of HGMD may utilise a feedback/comments function in order to inform the HGMD curators of relevant new or missing information, or to request the correction, recategorisation or removal of a listed variant.
Zygosity information (i.e. heterozygous, homozygous or compound heterozygous) for individual mutations in HGMD has not been recorded. Reasons for this include (i) the fact that this information is not always unequivocally provided in the corresponding literature reference; (ii) the possibility that a given mutation may be pathogenic irrespective of the zygosity in which it is found; (iii) the clinical consequences of zygosity may often be modified by other genetic variants either in cis or in trans; (iv) digenic or polygenic inheritance of other pathogenic variants or disease modifiers and (v) variable or reduced penetrance which ensures that the genotype is not invariably predictive of the clinical phenotype (Cooper et al. 2013). Sometimes the same mutation may be present in the heterozygous, compound heterozygous or homozygous states in different patients; in such cases, information on zygosity may not be easy to provide and may be even more difficult to interpret. Thus, information pertaining to zygosity would not always be helpful or informative with regard to ascertaining or predicting the clinical phenotype, and indeed might even prove inaccurate or misleading.
HGMD users should not assume that just because a sequence variant is labelled “DM”, it automatically follows that it is known or believed to be pathogenic in all individuals harbouring it (i.e. that the variant exhibits 100% penetrance). Indeed, many “disease-causing mutations” display reduced or variable penetrance for a variety of different reasons (reviewed by Cooper et al. 2013). Further, population sequencing programmes (such as the 1000 Genomes Project and ExAC) are now identifying considerable numbers of “DM” mutations in apparently healthy individuals (MacArthur et al. 2012; Xue et al. 2012; Lek et al. 2016). Such lesions should not be regarded automatically as being clinically irrelevant, even when they occur with significant frequency, because it is quite possible that these mutations either represent low-penetrance, mild or late onset, or more complex disease susceptibility alleles, as opposed to neutral variants (Cooper et al. 2013), or alternatively reside within transcripts that exhibit a degree of translational plasticity (Jagannathan and Bradley 2016).
It is HGMD curation policy to err on the side of inclusion and enter a variant into the database even if its pathological relevance may be questionable (while indicating this fact to our users wherever feasible), rather than run the risk of inadvertently excluding a variant that may be directly (or indirectly) relevant to disease. We have taken several different steps to highlight such equivocation in HGMD, viz. the DM? variant class, a dbSNP 1000 Genomes frequency flag (to highlight those HGMD variants that are also present in dbSNP, with allele frequency information included; see below) and the provision of additional literature citations which either support or cast doubt upon the pathogenicity of a particular variant (Fig. 3). This latter point is particularly pertinent in the clinical setting, where a greater burden of proof may be required as a prerequisite for use in diagnostic and predictive medicine, and when considering the return of incidental findings to patients after testing (Green et al. 2012, 2013; Ng et al. 2013; Gonsalves et al. 2013; Dewey et al. 2014; Tabor et al. 2014; Gambin et al. 2015; Jurgens et al. 2015).
Fig. 3.

Example of an HGMD Professional entry
Additional literature references are an important source of contextual information, and play a vital role in querying or confirming the pathogenicity of HGMD variants. Types of additional reference include functional studies, additional case reports, additional phenotypes and population case–control studies. The number of additional references in HGMD has grown steadily as a proportion of the total number of references and accounts for approximately 30–40% of the number of literature references screened and entered into HGMD over the last 3–5 years (Fig. 1). The number of literature references reporting novel variants appears to have reached a plateau over the last few years; however, the number of variants being reported per reference is still increasing, from 2.5 mutations per reference in the 1990s to over 4.0 in the last two years. We expect this trend to continue as ever larger numbers of patient population-scale sequencing studies are completed and published (Ellingford et al. 2016; Susswein et al. 2016; Lopes et al. 2015).
HGMD Professional
HGMD Professional serves as the subscription version of HGMD, and is available to both commercial and academic customers under license from QIAGEN Inc. HGMD Professional allows access to up-to-date mutation data with a quarterly release cycle; this version is therefore essential for checking the novelty of newly found mutations. HGMD Professional contains many features not available in the public version. More powerful search tools in the form of an expanded search engine with full text Boolean searching are provided. A batch search mode has been developed to allow users to query HGMD using gene (e.g. OMIM IDs, Entrez IDs), variant (e.g. dbSNP IDs, chromosomal coordinates, VCF format) and dataset (e.g. PubMed ID) oriented lists. Users can employ these tools to perform additional searches for gene-specific (e.g. chromosomal locations, gene names/aliases and gene ontology), mutation-specific (e.g. chromosomal coordinates, HGVS nomenclature, dbSNP ID) or citation-specific (e.g. first author, publication year, PubMed ID) information. Chromosomal coordinates (hg19/hg38) and HGVS nomenclature are provided for the vast majority of our nucleotide substitutions (99.8% coverage) and other micro-lesions (97.6% coverage). Provision of consistently accurate mutation descriptions is especially important in the era of NGS sequencing (Yen et al. 2017) and has helped to make HGMD an invaluable tool for the analysis of population-scale NGS datasets such as the 1000 Genomes Project (1000 Genomes Project Consortium 2015) and ExAC (Lek et al. 2016). Additional information is also provided on a mutation-specific basis (see Fig. 3) including curatorial comments (for example, if the mutation data presented in the original publication required in-house correction or author clarification [5–10% of all entries], or if the clinical phenotype is associated with a more complex, i.e. digenic or in-cis inheritance pattern), additional reports comprising functional characterisation, further phenotypic information, comparative biochemical parameters, evolutionary conservation and SIFT (Sim et al. 2012) and MutPred (Li et al. 2009) pathogenicity predictions. More recently, the functional predictions and nucleotide conservation data from dbNSFP2.0 (Liu et al. 2013), a database of all potential non-synonymous single-nucleotide variants in the human genome, have been included. These additional annotations are updated on a regular basis.
HGMD clinical phenotypes have been annotated against the Unified Medical Language System (UMLS) using a combination of manual curation and natural language processing. The UMLS is a compilation of biomedical ontologies and vocabularies catalogued into a single resource (e.g. OMIM phenotype data, Medical Subject Headings (MeSH) and other disease ontologies), and may be found at http://www.nlm.nih.gov/research/umls/. HGMD phenotype data have been mapped to approximately 18 different UMLS high-level concepts (Fig. 4). These UMLS mappings provide users with a more accurate and expanded phenotype search. Thus, searches using alternative disease names should return the same result-set, e.g. a search for “breast cancer” should yield identical results to a search for “malignant neoplasm of breast”. In addition, utilising the UMLS allows for powerful semantic searching (e.g. searches for all mutations linked to “blood disorders” or “immune disorders”). The UMLS ontology mappings have been utilised in a variety of different NGS sequencing studies (see below).
Fig. 4.

Overview of UMLS high-level disease concept mappings present in HGMD
Another feature involves the highlighting of HGMD entries where the pathogenicity of the variant may have been cast into doubt by virtue of its high allele frequency. HGMD Professional displays a frequency flag when a listed variant is to be found in dbSNP, and population frequency data from the 1000 Genomes Project are provided. In addition, HGMD will soon include allele frequencies derived from the more recent ExAC study (Lek et al. 2016). As well as searching and viewing mutation data, users of HGMD Professional may utilise a feedback facility to submit corrections to the database curators or to request additional features (see Fig. 3 to view a sample HGMD Professional variant entry).
HGMD Professional also includes an Advanced Search facility to enhance mutation searching, viewing and retrieval. Datasets may be combined (for example, micro-deletions, micro-insertions and indels) to enable powerful searching across comparable types of mutation. A variety of search parameters are available, including functional features [e.g. in vitro and/or in silico characterised transcription factor binding sites, post-translational modifications, microRNA binding sites, upstream open reading frames (ORFs), and catalytic residues] to search for the gain or loss of a specific feature as a consequence of mutation; type of amino acid substitution; nucleotide substitution; size and/or sequence composition of micro-deletions, micro-insertions or indels; pre- or user-defined sequence motifs (both those created and those abolished by the mutation in question); dbSNP number; keywords found in the article title or abstract. The Advanced Search also includes a batch mode termed “Mutation Mart” to query HGMD via multiple identifiers including dbSNP, Entrez gene (http://www.ncbi.nlm.nih.gov/gene) and PubMed. HGMD Professional is available to subscribers either as an online-only package or in downloadable form enabling users to incorporate HGMD data into their local variant analysis pipelines (https://www.qiagenbioinformatics.com/products/human-gene-mutation-database/).
Focus on NGS
HGMD data are available in VCF format allowing easy visualisation, for example by using the Integrative Genomics Viewer (Robinson et al. 2011), or incorporation into custom data analysis pipelines (Dorschner et al. 2013; Gambin et al. 2015; Johnston et al. 2015; Lek et al. 2016). This facility allows users to maximise their use of HGMD data in both a clinical diagnostic and research setting. The provision of disease UMLS concept mappings (including OMIM, SNOMED, MeSH and HPO) also greatly enhances both the web-based HGMD search facility and the downloadable package, allowing the stratification of variants according to recognised disease concepts.
When using HGMD Professional to annotate large NGS datasets, and depending on the context (e.g. an inherited disease screen), it is often useful to annotate the dataset with a subset of HGMD variants (e.g. those which fall into the DM and DM? categories). Any variants found concurrently in this subset and the dataset being tested may then be further prioritised by variant class; hence, DM variants could be ranked higher than DM? variants if so desired. We have plans to introduce a literature-based variant scoring system to allow NGS researchers and clinicians to improve their prioritisation of DM/DM? variants found in their result sets. This system will annotate additional references as being supportive, neutral or not supportive of the inclusion of the variant in HGMD, thereby allowing users to rank those variants that possess additional supporting literature evidence (e.g. those with a published functional study) more highly, in addition to de-prioritising variants that have additional literature evidence questioning their pathological relevance. This new information will be available in both the online and download versions of the next release of HGMD Professional (see Fig. 3).
One of the problems encountered by NGS researchers and clinicians is the mis-annotation of variants as pathogenic or disease-causing. A small number of literature reports have been published where common variants have not being properly filtered out at an early stage, thereby increasing the number of mis-categorised variants appearing in the literature. HGMD has instigated plans to mitigate this problem, including the pre-screening of entries against the population frequency data present in ExAC (in progress) and the introduction of a literature-based scoring system (see above).
Other variant databases
Several other databases are available that attempt to record disease-causing or disease-associated (i.e. pathogenic) variation. These include the Online Mendelian Inheritance in Man, OMIM (http://www.omim.org/; Amberger et al. 2015), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/; Landrum et al. 2016), dbSNP (http://www.ncbi.nlm.nih.gov/SNP/; Sherry et al. 2001), LOVD (http://grenada.lumc.nl/LSDB_list/lsdbs; Fokkema et al. 2011) and a variety of locus-specific mutation databases (LSDBs) (http://www.hgvs.org/dblist/glsdb.html). OMIM does not provide statistics for allelic variants on its website; however, 25,115 germline OMIM variants appear to have been added to ClinVar, which itself currently contains a total of 53,211 pathogenic or likely pathogenic germline variants, whereas dbSNP contains 49,675 pathogenic or likely pathogenic clinically significant variants (all databases accessed December 30th 2016). In comparison, HGMD currently contains 193,904 DM and DM? variant entries in 6770 genes. Owing to the highly dispersed nature of the LSDBs and the potential for duplication between databases, accurate statistics with regard to like-for-like bona fide germline disease-causing (i.e. not merely neutral) variation is difficult to obtain. Since OMIM only records a limited number of variants deemed newsworthy per gene, and ClinVar still lacks depth (in terms of variant and literature coverage) and obtains a significant proportion (~40% of the above-mentioned total) of its pathogenic variant data via direct submission from clinical testing laboratories, HGMD is the only database of inherited human pathological variants that can claim to approach comprehensive coverage of the peer-reviewed literature (Peterson et al. 2013). Since both ClinVar and the LSDBs contain unpublished (i.e. non-peer reviewed) mutation data, the question has arisen as to whether HGMD should also include these data (Patrinos et al. 2012). However, both ClinVar and the LSDBs have encountered problems pertaining to data quality, submission, provenance and consent. A recent study (Abouelhoda et al. 2016) found that a higher proportion (1.1% vs. 0.59%) of variants in ClinVar required reclassification when compared to HGMD Professional (Abouelhoda et al. 2016, Table 1). The reclassification data presented by the authors of this study have already been incorporated into HGMD Professional. At present, however, it does not appear that any revisions have been made to ClinVar as a result of this study. Therefore, we have opted not to include data from these databases at this time.
How HGMD is utilised
The registered users of the HGMD public website (>101,000 as of March 2017) performed more than 260,000 queries in 2016. HGMD data may not be downloaded in their entirety from the public website; however, data may be made available at the discretion of the curators for non-commercial research purposes. Potential collaborators who wish to access HGMD data in full are required to sign a confidentiality agreement.
HGMD data have been used to perform a series of meta-analyses on different types of gene mutation causing human inherited disease. These studies have helped to improve our understanding of mutational spectra and the molecular mechanisms underlying human inherited disease (Cooper et al. 2011). They have served to demonstrate not only that human gene mutation is an inherently non-random process, but also that the nature, location and frequency of different types of mutation are shaped in large part by the local DNA sequence environment (Cooper et al. 2011). Indeed, HGMD data have been instrumental in demonstrating that electron transfer reactions (Bacolla et al. 2013), base-pair flexibility (Bacolla et al. 2015) and non-B DNA forming sequences (Kamat et al. 2016) all contribute to sequence context-dependent mutagenesis causing inherited disease. HGMD mutation data were used to demonstrate that many in-frame pathogenic variations perturb protein–protein interactions (Das et al. 2014). HGMD mutations have also been used to demonstrate that proteins linked to autosomal dominant diseases exhibit more clustering of rare missense mutations than those linked to autosomal recessive diseases (Turner et al. 2015). Finally, HGMD mutations have been mapped to protein 3D structures in order to study the loss and gain of various types of functional attribute, thereby quantifying the impact of disease-causing amino acid substitutions on catalytic activity, metal binding, macromolecular binding, ligand binding, allosteric regulation and post-translational modification (Lugo-Martinez et al. 2016).
HGMD data have been used extensively in several international collaborative research projects including the Genotype-Tissue Expression (GTEx) project (Rivas et al. 2015), the ExAC project (Lek et al. 2016) and the 1000 Genomes project (Marth et al. 2011; MacArthur et al. 2012; 1000 Genomes Project Consortium 2015), where a surprising number of HGMD variants were found in apparently healthy individuals. They have also been used in the comparative analysis of orthologous sequences in model genomes including those of gorilla (Scally et al. 2012), mountain gorilla (Xue et al. 2015), cynomolgus and Chinese macaques (Yan et al. 2011), Rhesus macaque (Rhesus Macaque Genome Sequencing and Analysis Consortium 2007) and rat (Rat Genome Sequencing Project Consortium 2004), in which many apparently disease-causing mutations in human were found as wild-type (‘compensated mutations’) (Azevedo et al. 2015, 2016).
In a clinical setting, HGMD is widely utilised by many groups in ongoing NGS diagnostic (Bell et al. 2011; Johnston et al. 2012; Calvo et al. 2012; Makrythanasis et al. 2014; Karageorgos et al. 2015; Wilfert et al. 2016; Walsh et al. 2017) and human genome sequencing (Tong et al. 2010; Kim et al. 2009; Telenti et al. 2016) programmes. HGMD has also been used by a number of different groups to aid the development of a wide variety of post-NGS variant interpretation and exome prioritisation algorithms including MutPred (Li et al. 2009), MutPred Splice (Mort et al. 2014), PROVEAN (Choi et al. 2012), CAROL (Lopes et al. 2012), regSNPs (Teng et al. 2012), CRAVAT (Douville et al. 2013), NEST (Carter et al. 2013), FATHMM (Shihab et al. 2013), FATHMM-MKL (Shihab et al. 2015), PinPor (Zhang et al. 2014), MutationTaster2 (Schwarz et al. 2014), Phen-Gen (Javed et al. 2014), VEST-indel (Douville et al. 2016), Gene Damage Index (Itan et al. 2015), DDIG-in (Folkman et al. 2015), RSVP (Peterson et al. 2016), ExonImpact (Li et al. 2017), IntSplice (Shibata et al. 2016), snvForest (Wu et al. 2015), IMHOTEP (Knecht et al. 2017) and M-CAP (Jagadeesh et al. 2016). A list of some of the articles which have utilised HGMD data or expertise in their analyses can be found on the HGMD website (http://www.hgmd.cf.ac.uk/docs/articles.html).
Data sharing
A limited HGMD data set, containing both chromosomal coordinates and HGMD identifiers, has been made available via academic data exchange programmes to the European Bioinformatics Institute (EBI)/Ensembl (Flicek et al. 2013) and the University of California, Santa Cruz (UCSC) (Meyer et al. 2013) and may be viewed in these projects’ respective genome browsers. Data from HGMD Professional have additionally been made available to subscribers of Ingenuity Variant Analysis™ (QIAGEN) and Alamut (Interactive Biosoftware), but are also accessible as part of the HGMD Professional stand-alone package (QIAGEN). Allowing free access to the bulk of the mutation data present in HGMD, while generating sufficient income to support maintenance and development via commercial distribution, represents a business model that is intended to maximise the availability of HGMD at the same time as ensuring its long-term sustainability. Although we are obliged to be prudent with regard to data sharing with public data repositories, we have always taken the view that making as much data publicly available as possible is generally beneficial to HGMD as well as to its users worldwide.
Future plans
The provision of chromosomal coordinates (both GRCh37 and 38) for the vast majority of coding region micro-lesions in HGMD is now complete. Expanding this provision to include micro-lesions in non-coding regions and the gross (in progress) and complex lesion (where feasible) datasets is a high priority, We plan to add other commonly utilised NGS formats such as General Feature Format (GFF) (http://www.sanger.ac.uk/resources/software/gff/) and Browser Extensible Data (BED) format to complement the Variant Call Format (VCF) (Danecek et al. 2011) data currently available in HGMD Professional. The provision of allele frequency data from large-scale NGS projects such as ExAC (http://exac.broadinstitute.org/), more complete references (i.e. including article titles) and HGVS protein level descriptions for HGMD micro-lesions are also priorities. Provision of genomic reference sequences based on the NCBI RefSeqGene project (Pruitt et al. 2014), links to available protein structures and homology models, and expanding our coverage of secondary references (additional case reports and functional studies) are also regarded as priorities, as well as updating our set of functional predictions using the new dbNSFP v3.0 dataset (Liu et al. 2016).
HGMD provides the user with a unique resource that can be utilised not only to obtain evidence to support the pathological authenticity and/or novelty of detected gene lesions and to acquire an overview of the mutational spectra for specific genes, but also as a knowledgebase for use in the bioinformatics and whole genome screening projects that underpin personalised genomics, next-generation sequencing research and diagnostic medicine.
Compliance with ethical standards
Conflict of interest
The authors wish to declare an interest in so far as HGMD is financially supported by QIAGEN Inc. through a License agreement with Cardiff University.
Contributor Information
Peter D. Stenson, Phone: +44(0)29 2074 4062, Email: stensonPD@cardiff.ac.uk
David N. Cooper, Phone: +44(0)29 2074 4062, Email: cooperDN@cardiff.ac.uk
References
- 1000 Genomes Project Consortium. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abouelhoda M, Faquih T, El-Kalioby Alkuraya FS. Revisiting the morbid genome of Mendelian disorders. Genome Biol. 2016;17:235. doi: 10.1186/s13059-016-1102-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo L, Serrano C, Amorim A, Cooper DN. Trans-species polymorphism in humans and the great apes is generally maintained by balancing selection that modulates the host immune response. Hum Genom. 2015;9:21. doi: 10.1186/s40246-015-0043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo L, Mort M, Costa AC, Silva RM, Quelhas D, Amorim A, Cooper DN. Improving the in silico assessment of pathogenicity for compensated variants. Eur J Hum Genet. 2016;25:2–7. doi: 10.1038/ejhg.2016.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Temiz NA, Yi M, Ivanic J, Cer RZ, Donohue DE, Ball EV, Mudunuri US, Wang G, Jain A, Volfovsky N, Luke BT, Stephens RM, Cooper DN, Collins JR, Vasquez KM. Guanine holes are prominent targets for mutation in cancer and inherited disease. PLoS Genet. 2013;9:e1003816. doi: 10.1371/journal.pgen.1003816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Zhu X, Chen H, Howells K, Cooper DN, Vasquez KM. Local DNA dynamics shape mutational patterns of mononucleotide repeats in human genomes. Nucl Acids Res. 2015;43:5065–5080. doi: 10.1093/nar/gkv364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, Sheth V, Woodward JE, Peckham HE, Schroth GP, Kim RW, Kingsmore SF. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3:65ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, Laskowski A, Garone C, Liu S, Jaffe DB, Christodoulou J, Fletcher JM, Bruno DL, Goldblatt J, Dimauro S, Thorburn DR, Mootha VK. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Douville C, Stenson PD, Cooper DN, Karchin R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013;14(Suppl 3):S3. doi: 10.1186/1471-2164-14-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Goldstein DB. In vitro assays fail to predict in vivo effects of regulatory polymorphisms. Hum Mol Genet. 2007;16:1931–1939. doi: 10.1093/hmg/ddm140. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Chen JM, Ball EV, Howells K, Mort M, Phillips AD, Chuzhanova N, Krawczak M, Kehrer-Sawatzki H, Stenson PD. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum Mutat. 2010;31:631–655. doi: 10.1002/humu.21260. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Bacolla A, Férec C, Vasquez KM, Kehrer-Sawatzki H, Chen JM. On the sequence-directed nature of human gene mutation: the role of genomic architecture and the local DNA sequence environment in mediating gene mutations underlying human inherited disease. Hum Mutat. 2011;32:1075–1099. doi: 10.1002/humu.21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132:1077–1130. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J, Lee HR, Sagar A, Fragoza R, Liang J, Wei X, Wang X, Mort M, Stenson PD, Cooper DN, Yu H. Elucidating common structural features of human pathogenic variations using large-scale atomic-resolution protein networks. Hum Mutat. 2014;35:585–593. doi: 10.1002/humu.22534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, Merker JD, Goldfeder RL, Enns GM, David SP, Pakdaman N, Ormond KE, Caleshu C, Kingham K, Klein TE, Whirl-Carrillo M, Sakamoto K, Wheeler MT, Butte AJ, Ford JM, Boxer L, Ioannidis JP, Yeung AC, Altman RB, Assimes TL, Snyder M, Ashley EA, Quertermous T. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311:1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, Gagnebin M, Nisbett J, Deloukas P, Dermitzakis ET, Antonarakis SE. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, Bennett RL, Jones KL, Tokita MJ, Bennett JT, Kim JH, Rosenthal EA, Kim DS, National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project. Tabor HK, Bamshad MJ, Motulsky AG, Scott CR, Pritchard CC, Walsh T, Burke W, Raskind WH, Byers P, Hisama FM, Nickerson DA, Jarvik GP. Actionable, pathogenic incidental findings in 1000 participants’ exomes. Am J Hum Genet. 2013;93:631–640. doi: 10.1016/j.ajhg.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douville C, Carter H, Kim R, Niknafs N, Diekhans M, Stenson PD, Cooper DN, Ryan M, Karchin R. CRAVAT: cancer-related analysis of variants toolkit. Bioinformatics. 2013;29:647–648. doi: 10.1093/bioinformatics/btt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douville C, Masica DL, Stenson PD, Cooper DN, Gygax DM, Kim R, Ryan M, Karchin R. Assessing the pathogenicity of insertion and deletion variants with the variant effect scoring tool (VEST-Indel) Hum Mutat. 2016;37:28–35. doi: 10.1002/humu.22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingford JM, Barton S, Bhaskar S, O’Sullivan J, Williams SG, Lamb JA, Panda B, Sergouniotis PI, Gillespie RL, Daiger SP, Hall G, Gale T, Lloyd IC, Bishop PN, Ramsden SC, Black GC. Molecular findings from 537 individuals with inherited retinal disease. J Med Genet. 2016;53(11):761–767. doi: 10.1136/jmedgenet-2016-103837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, García-Girón C, Gordon L, Hourlier T, Hunt S, Juettemann T, Kähäri AK, Keenan S, Komorowska M, Kulesha E, Longden I, Maurel T, McLaren WM, Muffato M, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, Riat HS, Ritchie GR, Ruffier M, Schuster M, Sheppard D, Sobral D, Taylor K, Thormann A, Trevanion S, White S, Wilder SP, Aken BL, Birney E, Cunningham F, Dunham I, Harrow J, Herrero J, Hubbard TJ, Johnson N, Kinsella R, Parker A, Spudich G, Yates A, Zadissa A, Searle SM. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v. 2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–563. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- Folkman L, Yang Y, Li Z, Stantic B, Sattar A, Mort M, Cooper DN, Liu Y, Zhou Y. DDIG-in: detecting disease-causing genetic variations due to frameshifting indels and nonsense mutations employing sequence and structural properties at nucleotide and protein levels. Bioinformatics. 2015;31:1599–1606. doi: 10.1093/bioinformatics/btu862. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambin T, Jhangiani SN, Below JE, Campbell IM, Wiszniewski W, Muzny DM, Staples J, Morrison AC, Bainbridge MN, Penney S, McGuire AL, Gibbs RA, Lupski JR, Boerwinkle E. Secondary findings and carrier test frequencies in a large multiethnic sample. Genome Med. 2015;7:54. doi: 10.1186/s13073-015-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalves SG, Ng D, Johnston JJ, Teer JK, NISC Comparative Sequencing Program. Stenson PD, Cooper DN, Mullikin JC, Biesecker LG. Using exome data to identify malignant hyperthermia susceptibility mutations. Anesthesiology. 2013;119:1043–1053. doi: 10.1097/ALN.0b013e3182a8a8e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Berg JS, Berry GT, Biesecker LG, Dimmock DP, Evans JP, Grody WW, Hegde MR, Kalia S, Korf BR, Krantz I, McGuire AL, Miller DT, Murray MF, Nussbaum RL, Plon SE, Rehm HL, Jacob HJ. Exploring concordance and discordance for return of incidental findings from clinical sequencing. Genet Med. 2012;14:405–410. doi: 10.1038/gim.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itan Y, Shang L, Boisson B, Patin E, Bolze A, Moncada-Vélez M, Scott E, Ciancanelli MJ, Lafaille FG, Markle JG, Martinez-Barricarte R, de Jong SJ, Kong XF, Nitschke P, Belkadi A, Bustamante J, Puel A, Boisson-Dupuis S, Stenson PD, Gleeson JG, Cooper DN, Quintana-Murci L, Claverie JM, Zhang SY, Abel L, Casanova JL. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci USA. 2015;112:13615–13620. doi: 10.1073/pnas.1518646112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, Bejerano G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–1586. doi: 10.1038/ng.3703. [DOI] [PubMed] [Google Scholar]
- Jagannathan S, Bradley RK. Translational plasticity facilitates the accumulation of nonsense genetic variants in the human population. Genome Res. 2016;26:1639–1650. doi: 10.1101/gr.205070.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javed A, Agrawal S, Ng PC. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat Methods. 2014;11:935–937. doi: 10.1038/nmeth.3046. [DOI] [PubMed] [Google Scholar]
- Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK, Mullikin JC, Biesecker LG. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet. 2012;91:97–108. doi: 10.1016/j.ajhg.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Lewis KL, Ng D, Singh LN, Wynter J, Brewer C, Brooks BP, Brownell I, Candotti F, Gonsalves SG, Hart SP, Kong HH, Rother KI, Sokolic R, Solomon BD, Zein WM, Cooper DN, Stenson PD, Mullikin JC, Biesecker LG. Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations. Am J Hum Genet. 2015;96:913–925. doi: 10.1016/j.ajhg.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens J, Ling H, Hetrick K, Pugh E, Schiettecatte F, Doheny K, Hamosh A, Avramopoulos D, Valle D, Sobreira N. Assessment of incidental findings in 232 whole-exome sequences from the Baylor-Hopkins Center for Mendelian Genomics. Genet Med. 2015;17:782–788. doi: 10.1038/gim.2014.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat MA, Bacolla A, Cooper DN, Chuzhanova N. A role for non-B DNA forming sequences in mediating microlesions causing human inherited disease. Hum Mutat. 2016;37:65–73. doi: 10.1002/humu.22917. [DOI] [PubMed] [Google Scholar]
- Karageorgos I, Mizzi C, Giannopoulou E, Pavlidis C, Peters BA, Zagoriti Z, Stenson PD, Mitropoulos K, Borg J, Kalofonos HP, Drmanac R, Stubbs A, van der Spek P, Cooper DN, Katsila T, Patrinos GP (2015) Identification of cancer predisposition variants in apparently healthy individuals using a next-generation sequencing-based family genomics approach. Hum Genomics 9:12 [DOI] [PMC free article] [PubMed]
- Kim JI, Ju YS, Park H, Kim S, Lee S, Yi JH, Mudge J, Miller NA, Hong D, Bell CJ, Kim HS, Chung IS, Lee WC, Lee JS, Seo SH, Yun JY, Woo HN, Lee H, Suh D, Lee S, Kim HJ, Yavartanoo M, Kwak M, Zheng Y, Lee MK, Park H, Kim JY, Gokcumen O, Mills RE, Zaranek AW, Thakuria J, Wu X, Kim RW, Huntley JJ, Luo S, Schroth GP, Wu TD, Kim H, Yang KS, Park WY, Kim H, Church GM, Lee C, Kingsmore SF, Seo JS. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knecht C, Mort M, Junge O, Cooper DN, Krawczak M, Caliebe A. IMHOTEP-a composite score integrating popular tools for predicting the functional consequences of non-synonymous sequence variants. Nucleic Acids Res. 2017;45:e13. doi: 10.1093/nar/gkw886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Feng W, Zhang X, Yang Y, Wang K, Mort M, Cooper DN, Wang Y, Zhou Y, Liu Y. ExonImpact: prioritizing pathogenic alternative splicing events. Hum Mutat. 2017;38:16–24. doi: 10.1002/humu.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34(9):E2393–E2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes MC, Joyce C, Ritchie GR, John SL, Cunningham F, Asimit J, Zeggini E. A combined functional annotation score for non-synonymous variants. Hum Hered. 2012;73:47–51. doi: 10.1159/000334984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LR, Syrris P, Guttmann OP, O’Mahony C, Tang HC, Dalageorgou C, Jenkins S, Hubank M, Monserrat L, McKenna WJ, Plagnol V, Elliott PM. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart. 2015;101:294–301. doi: 10.1136/heartjnl-2014-306387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, Procaccio V, Wallace DC. mtDNA variation and analysis using Mitomap and Mitomaster. Curr Protoc Bioinform. 2013;44:1.23.1-26. doi: 10.1002/0471250953.bi0123s44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo-Martinez J, Pejaver V, Pagel KA, Jain S, Mort M, Cooper DN, Mooney SD, Radivojac P. The loss and gain of functional amino acid residues is a common mechanism causing human inherited disease. PLoS Comput Biol. 2016;12:e1005091. doi: 10.1371/journal.pcbi.1005091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZD, Conrad DF, Lunter G, Zheng H, Ayub Q, DePristo MA, Banks E, Hu M, Handsaker RE, Rosenfeld JA, Fromer M, Jin M, Mu XJ, Khurana E, Ye K, Kay M, Saunders GI, Suner MM, Hunt T, Barnes IH, Amid C, Carvalho-Silva DR, Bignell AH, Snow C, Yngvadottir B, Bumpstead S, Cooper DN, Xue Y, Romero IG, 1000 Genomes Project Consortium. Wang J, Li Y, Gibbs RA, McCarroll SA, Dermitzakis ET, Pritchard JK, Barrett JC, Harrow J, Hurles ME, Gerstein MB, Tyler-Smith C. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrythanasis P, Nelis M, Santoni FA, Guipponi M, Vannier A, Béna F, Gimelli S, Stathaki E, Temtamy S, Mégarbané A, Masri A, Aglan MS, Zaki MS, Bottani A, Fokstuen S, Gwanmesia L, Aliferis K, Bustamante Eduardo M, Stamoulis G, Psoni S, Kitsiou-Tzeli S, Fryssira H, Kanavakis E, Al-Allawi N, Sefiani A, Al Hait S, Elalaoui SC, Jalkh N, Al-Gazali L, Al-Jasmi F, Bouhamed HC, Abdalla E, Cooper DN, Hamamy H, Antonarakis SE. Diagnostic exome sequencing to elucidate the genetic basis of likely recessive disorders in consanguineous families. Hum Mutat. 2014;35:1203–1210. doi: 10.1002/humu.22617. [DOI] [PubMed] [Google Scholar]
- Marth GT, Yu F, Indap AR, Garimella K, Gravel S, Leong WF, Tyler-Smith C, Bainbridge M, Blackwell T, Zheng-Bradley X, Chen Y, Challis D, Clarke L, Ball EV, Cibulskis K, Cooper DN, Fulton B, Hartl C, Koboldt D, Muzny D, Smith R, Sougnez C, Stewart C, Ward A, Yu J, Xue Y, Altshuler D, Bustamante CD, Clark AG, Daly M, DePristo M, Flicek P, Gabriel S, Mardis E, Palotie A, Gibbs R, 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, Raney BJ, Pohl A, Malladi VS, Li CH, Lee BT, Learned K, Kirkup V, Hsu F, Heitner S, Harte RA, Haeussler M, Guruvadoo L, Goldman M, Giardine BM, Fujita PA, Dreszer TR, Diekhans M, Cline MS, Clawson H, Barber GP, Haussler D, Kent WJ. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41(Database issue):D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhail FM. Copy number variations and human genetic disease. Curr Opin Pediatr. 2014;26:646–652. doi: 10.1097/MOP.0000000000000142. [DOI] [PubMed] [Google Scholar]
- Mort M, Sterne-Weiler T, Li B, Ball EV, Cooper DN, Radivojac P, Sanford JR, Mooney SD. MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol. 2014;15:R19. doi: 10.1186/gb-2014-15-1-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS, Lewis KL, Cooper DN, Stenson PD, Mullikin JC, Biesecker LG. Interpreting secondary cardiac disease variants in an exome cohort. Circ Cardiovasc Genet. 2013;6:337–346. doi: 10.1161/CIRCGENETICS.113.000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrinos GP, Cooper DN, van Mulligen E, Gkantouna V, Tzimas G, Tatum Z, Schultes E, Roos M, Mons B. Microattribution and nanopublication as means to incentivize the placement of human genome variation data into the public domain. Hum Mutat. 2012;33:1503–1512. doi: 10.1002/humu.22144. [DOI] [PubMed] [Google Scholar]
- Peterson TA, Doughty E, Kann MG. Towards precision medicine: advances in computational approaches for analysis of human variants. J Mol Biol. 2013;425:4047–4063. doi: 10.1016/j.jmb.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TA, Mort M, Cooper DN, Radivojac P, Kann MG, Mooney SD. Regulatory single-nucleotide variant predictor increases predictive performance of functional regulatory variants. Hum Mutat. 2016;37:1137–1143. doi: 10.1002/humu.23049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinard A, Miltgen M, Blanchard A, Mathieu H, Desvignes JP, Salgado D, Fabre A, Arnaud P, Barré L, Krahn M, Grandval P, Olschwang S, Zaffran S, Boileau C, Béroud C, Collod-Béroud G. Actionable genes, core databases, and locus-specific databases. Hum Mutat. 2016;37:1299–1307. doi: 10.1002/humu.23112. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, Murphy MR, O’Leary NA, Pujar S, Rajput B, Rangwala SH, Riddick LD, Shkeda A, Sun H, Tamez P, Tully RE, Wallin C, Webb D, Weber J, Wu W, DiCuccio M, Kitts P, Maglott DR, Murphy TD, Ostell JM. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42(Database issue):D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rat Genome Sequencing Project Consortium Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Rhesus Macaque Genome Sequencing and Analysis Consortium Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Rivas MA, Pirinen M, Conrad DF, Lek M, Tsang EK, Karczewski KJ, Maller JB, Kukurba KR, DeLuca DS, Fromer M, Ferreira PG, Smith KS, Zhang R, Zhao F, Banks E, Poplin R, Ruderfer DM, Purcell SM, Tukiainen T, Minikel EV, Stenson PD, Cooper DN, Huang KH, Sullivan TJ, Nedzel J, GTEx Consortium. Geuvadis Consortium. Bustamante CD, Li JB, Daly MJ, Guigo R, Donnelly P, Ardlie K, Sammeth M, Dermitzakis ET, McCarthy MI, Montgomery SB, Lappalainen T, MacArthur DG. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348:666–669. doi: 10.1126/science.1261877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T, McCarthy S, Montgomery SH, Schwalie PC, Tang YA, Ward MC, Xue Y, Yngvadottir B, Alkan C, Andersen LN, Ayub Q, Ball EV, Beal K, Bradley BJ, Chen Y, Clee CM, Fitzgerald S, Graves TA, Gu Y, Heath P, Heger A, Karakoc E, Kolb-Kokocinski A, Laird GK, Lunter G, Meader S, Mort M, Mullikin JC, Munch K, O’Connor TD, Phillips AD, Prado-Martinez J, Rogers AS, Sajjadian S, Schmidt D, Shaw K, Simpson JT, Stenson PD, Turner DJ, Vigilant L, Vilella AJ, Whitener W, Zhu B, Cooper DN, de Jong P, Dermitzakis ET, Eichler EE, Flicek P, Goldman N, Mundy NI, Ning Z, Odom DT, Ponting CP, Quail MA, Ryder OA, Searle SM, Warren WC, Wilson RK, Schierup MH, Rogers J, Tyler-Smith C, Durbin R. Insights into hominid evolution from the gorilla genome sequence. Nature. 2012;483:169–175. doi: 10.1038/nature10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Okuno T, Rahman MA, Azuma Y, Takeda J, Masuda A, Selcen D, Engel AG, Ohno K. IntSplice: prediction of the splicing consequences of intronic single-nucleotide variations in the human genome. J Hum Genet. 2016;61:633–640. doi: 10.1038/jhg.2016.23. [DOI] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, Gaunt TR. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, Gaunt TR, Campbell C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31:1536–1543. doi: 10.1093/bioinformatics/btv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, Vaccari EM, Bissonnette J, Booker JK, Cremona ML, Gibellini F, Murphy PD, Pineda-Alvarez DE, Pollevick GD, Xu Z, Richard G, Bale S, Klein RT, Hruska KS, Chung WK. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2016;18:823–832. doi: 10.1038/gim.2015.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor HK, Auer PL, Jamal SM, Chong JX, Yu JH, Gordon AS, Graubert TA, O’Donnell CJ, Rich SS, Nickerson DA, NHLBI Exome Sequencing Project. Bamshad MJ. Pathogenic variants for Mendelian and complex traits in exomes of 6,517 European and African Americans: implications for the return of incidental results. Am J Hum Genet. 2014;95:183–193. doi: 10.1016/j.ajhg.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenti A, Pierce LC, Biggs WH, di Iulio J, Wong EH, Fabani MM, Kirkness EF, Moustafa A, Shah N, Xie C, Brewerton SC, Bulsara N, Garner C, Metzker G, Sandoval E, Perkins BA, Och FJ, Turpaz Y, Venter JC. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci USA. 2016;113:11901–11906. doi: 10.1073/pnas.1613365113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M, Ichikawa S, Padgett LR, Wang Y, Mort M, Cooper DN, Koller DL, Foroud T, Edenberg HJ, Econs MJ, Liu Y. regSNPs: a strategy for prioritizing regulatory single nucleotide substitutions. Bioinformatics. 2012;28:1879–1886. doi: 10.1093/bioinformatics/bts275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn CF, Klein TE, Altman RB. PharmGKB: the pharmacogenomics knowledge base. Methods Mol Biol. 2013;1015:311–320. doi: 10.1007/978-1-62703-435-7_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong P, Prendergast JG, Lohan AJ, Farrington SM, Cronin S, Friel N, Bradley DG, Hardiman O, Evans A, Wilson JF, Loftus B. Sequencing and analysis of an Irish human genome. Genome Biol. 2010;11:R91. doi: 10.1186/gb-2010-11-9-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Douville C, Kim D, Stenson PD, Cooper DN, Chakravarti A, Karchin R. Proteins linked to autosomal dominant and autosomal recessive disorders harbor characteristic rare missense mutation distribution patterns. Hum Mol Genet. 2015;24:5995–6002. doi: 10.1093/hmg/ddv309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usher CL, McCarroll SA. Complex and multi-allelic copy number variation in human disease. Brief Funct Genom. 2015;14:329–338. doi: 10.1093/bfgp/elv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, Exome Aggregation Consortium. MacArthur DG, Farrall M, Cook SA, Watkins H. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. doi: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfert AB, Chao KR, Kaushal M, Jain S, Zöllner S, Adams DR, Conrad DF. Genome-wide significance testing of variation from single case exomes. Nat Genet. 2016;48:1455–1461. doi: 10.1038/ng.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Wu J, Chen T, Jiang R. Prioritization of nonsynonymous single nucleotide variants for exome sequencing studies via integrative learning on multiple genomic data. Sci Rep. 2015;5:14955. doi: 10.1038/srep14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN, Tyler-Smith C, The 1000 Genomes Project Consortium Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet. 2012;91:1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Prado-Martinez J, Sudmant PH, Narasimhan V, Ayub Q, Szpak M, Frandsen P, Chen Y, Yngvadottir B, Cooper DN, de Manuel M, Hernandez-Rodriguez J, Lobon I, Siegismund HR, Pagani L, Quail MA, Hvilsom C, Mudakikwa A, Eichler EE, Cranfield MR, Marques-Bonet T, Tyler-Smith C, Scally A. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science. 2015;348:242–245. doi: 10.1126/science.aaa3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G, Zhang G, Fang X, Zhang Y, Li C, Ling F, Cooper DN, Li Q, Li Y, van Gool AJ, Du H, Chen J, Chen R, Zhang P, Huang Z, Thompson JR, Meng Y, Bai Y, Wang J, Zhuo M, Wang T, Huang Y, Wei L, Li J, Wang Z, Hu H, Yang P, Le L, Stenson PD, Li B, Liu X, Ball EV, An N, Huang Q, Zhang Y, Fan W, Zhang X, Li Y, Wang W, Katze MG, Su B, Nielsen R, Yang H, Wang J, Wang X, Wang J. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol. 2011;29:1019–1023. doi: 10.1038/nbt.1992. [DOI] [PubMed] [Google Scholar]
- Yen JL, Garcia S, Montana A, Harris J, Chervitz S, Morra M, West J, Chen R, Church DM. A variant by any name: quantifying annotation discordance across tools and clinical databases. Genome Med. 2017;9:7. doi: 10.1186/s13073-016-0396-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lin H, Zhao H, Hao Y, Mort M, Cooper DN, Zhou Y, Liu Y. Impact of human pathogenic micro-insertions and micro-deletions on post-transcriptional regulation. Hum Mol Genet. 2014;23:3024–3034. doi: 10.1093/hmg/ddu019. [DOI] [PMC free article] [PubMed] [Google Scholar]