

Abstract
Background
Shigellosis is the most common cause of gastrointestinal infections in developing countries. In China, the species most frequently responsible for shigellosis is Shigella flexneri. S. flexneri remains largely unexplored from a genomic standpoint and is still described using a vocabulary based on biochemical and serological properties. Moreover, increasing numbers of ESBL-producing Shigella strains have been isolated from clinical samples. Despite this, only a few cases of ESBL-producing Shigella have been described in China. Therefore, a better understanding of ESBL-producing Shigella from a genomic standpoint is required. In this study, a S. flexneri type 1a isolate SP1 harboring blaCTX-M-14, which was recovered from the patient with diarrhea, was subjected to whole genome sequencing.
Results
The draft genome assembly of S. flexneri strain SP1 consisted of 4,592,345 bp with a G+C content of 50.46%. RAST analysis revealed the genome contained 4798 coding sequences (CDSs) and 100 RNA-encoding genes. We detected one incomplete prophage and six candidate CRISPR loci in the genome. In vitro antimicrobial susceptibility testing demonstrated that strain SP1 is resistant to ampicillin, amoxicillin/clavulanic acid, cefazolin, ceftriaxone and trimethoprim. In silico analysis detected genes mediating resistance to aminoglycosides, β-lactams, phenicol, tetracycline, sulphonamides, and trimethoprim. The bla CTX-M-14 gene was located on an IncFII2 plasmid. A series of virulence factors were identified in the genome.
Conclusions
In this study, we report the whole genome sequence of a blaCTX-M-14-encoding S. flexneri strain SP1. Dozens of resistance determinants were detected in the genome and may be responsible for the multidrug-resistance of this strain, although further confirmation studies are warranted. Numerous virulence factors identified in the strain suggest that isolate SP1 is potential pathogenic. The availability of the genome sequence and comparative analysis with other S. flexneri strains provides the basis to further address the evolution of drug resistance mechanisms and pathogenicity in S. flexneri.
Electronic supplementary material
The online version of this article (doi:10.1186/s12941-017-0212-2) contains supplementary material, which is available to authorized users.
Keywords: Shigella flexneri, Extended-spectrum β-lactamase, IncFII2, Comparative genomic analysis
Background
Shigella species are a major causative cause of gastrointestinal infections throughout the world, especially in developing countries [1]. Globally, there are approximately 164.7 million cases per annum, of which 1.1 million people are estimated to die from Shigella infections [2]. Based on biochemical and serological properties, the genus Shigella comprises four serogroups: Shigella dysenteriae, Shigella flexneri, Shigella boydii and Shigella sonnei [2]. S. flexneri is endemic in many developing countries and causes more deaths than any other Shigella serotypes [3]. In China, shigellosis is the most common gastrointestinal infections [4] and most frequently isolated species responsible for shigellosis in mainland China is S. flexneri [5].
Antibiotic treatment is usually recommended for shigellosis as it reduces the duration and severity of symptoms, reduces the excretion of organisms and prevents potentially lethal complications [6]. However, the emergence of extended-spectrum β-lactamase (ESBL)-producing S. flexneri is a major public health problem in China, as these strains are associated with critical infections [7]. Most of the CTX-M, SHV, and TEM-type ESBLs genes are located on conjugative plasmids [4]. Moreover, co-existence with other antimicrobial resistance genes is frequently observed in ESBL-producers [8], which makes the choice of effective treatment extremely limited. Increasing instances of ESBL-producing Shigella strains isolated from Asia have been reported [1, 2]. So far, only a few cases of ESBL-producing Shigella have been described in China [4]. Therefore, a better understanding of ESBL-producing Shigella using a genomics approach is required.
The first genome sequence of S. flexneri was reported by Jin et al. in 2002 [9]. So far, more than 145 S. flexneri strains have been sequenced and analyzed [10]. To extend our understanding of the resistance mechanisms and pathogenesis of ESBL-producing S. flexneri, we performed sequencing and genomic analysis of the ESBL-harbouring S. flexneri SP1. Comparative genomics analysis of S. flexneri SP1 with other S. flexneri genomes may improve our understanding of the antibiotic resistance and virulence factors present in Shigella.
Methods
Strain information
Shigella flexneri 1a isolate SP1 was isolated from the stool sample of a 76-year-old female diarrhea patient. Standard biochemical tests were performed using the Vitek II system (BioMerieux, France) and species-specific 16S rRNA sequencing was used to confirm the identity of isolate SP1. Serotyping was performed with specific antiserum (Denka-Seiken). Genomic DNA of SP1 was extracted from a single colony of the pure bacterial culture. Possible contamination with other DNA and misassemblies were assessed by performing a BLAST search against the non-redundant database as described previously [11]. The whole genome of SP1 is in the expected size range for a Shigella genome and the coverage of the reads was consistent throughout the genome. The assembled draft genome sequence of SP1 was further verified by comparative analysis with the published complete genome sequences of S. flexneri strains.
Antimicrobial susceptibility testing
Susceptibility testing for ampicillin, amoxicillin/clavulanic acid, amikacin, aztreonam, piperacillin/tazobactam, cefazolin, cefoxitin, ciprofloxacin, ceftriaxone, cefepime, ertapenem, imipenem, gentamycin, tobramycin, levofloxacin, tigecycline, nitrofurantoin, and trimethoprim were performed by the microbroth dilution method and interpreted according to the CLSI guidelines [12]. Escherichia coli strain ATCC 25922 was used as the control strain for susceptibility studies. Late log phase cells were harvested, and genomic DNA was extracted from the pure culture using the DNeasy Blood & Tissue kit (Qiagen, Germany) according to the manufacturer’s instructions. For the purpose of bacterial identification, we amplified the 16S rRNA gene with a 16S rRNA universal primer set and the PCR product was sequenced. Ethical approval was granted by the Ethics Committee of the First Affiliated Hospital of Zhejiang University.
Genome sequencing and assembly
The extracted DNA was visualized by agarose gel electrophoresis and quantitated by Qubit 2.0. Whole-genome sequencing was performed on the Illumina HiSeq 4000-PE150 platform. DNA was tailed, ligated to paired-end adaptors and PCR amplified with a 500 bp insert size and a mate-pair libraries with an insert size of 5 kb were used for library construction at the Beijing Novogene Bioinformatics Technology Co., Ltd. Illumina PCR adapter reads and low quality reads from the paired-end and mate-pair library were filtered by the quality control step using Novogene pipeline. All high quality paired reads were assembled using Velvet 1.2.10 [13] into a number of scaffolds. The filtered reads were then passed handled by the next step of the gap-closing.
Genome annotation
Genome annotation included the prediction of coding genes, transfer RNAs, ribosomal RNA, prophage, and clustered regularly interspaced short palindromic repeat sequences (CRISPR). Open reading frames (ORFs) were identified and classified using the Rapid Annotation using Subsystem Technology (RAST) server [14]. Protein classification into functional groups was performed using the Clusters of Orthologous Groups of proteins (COGs) [15]. Transfer RNAs and ribosomal RNA genes rRNAs were detected by tRNAscan-SE [16] and RNAmmer 1.2 software [17], respectively. PHASTER [18] was used to identify prophage and putative phage-like elements and CRISPRFinder [19] was used to identify CRISPR sites. The plasmid replicon was predicted by the PlasmidFinder Tool [20]. ISfinder [21] was employed to search for IS sequences in the genome, with an e-value of 1E−3. plasmidSPAdes was used to produce plasmid sequences from the WGS data [22].
Antibiotic resistance genes prediction and virulence factors analysis
Antibiotic resistance genes were annotated using the comprehensive antibiotic resistance database (CARD) [23] and Resfinder [24] with default parameters. We further verified all putative antibiotic resistance genes (ARGs) through a BLAST search with cut-off e-value of 1E−0.5. Virulence factors were predicted by using BLAST to search against the VFDB database [25] with an e-value threshold of 1E−5 and also with VirulenceFinder 1.5 [26].
Plasmid characterization
PlasmidFinder 1.3 was used for identify the incompatibility group of the plasmid present in S. flexneri SP1 [20]. The plasmid sequence of bla CTX-M-14-harboring plasmid from isolate SP1 (named pSP1) was assembled with plasmidSPAdes [22]. Assignment of the plasmid to an incompatibility (Inc) group was performed by multiplex PCR. PCRs were performed as described previously [27].
Phylogenetic analysis and comparative genomic analysis
Comparative genomic analysis was performed by orthology identification method as previously described [11, 28]. Genome sequences of the following representative S. flexneri strains were downloaded from the NCBI genome database: S. flexneri 2a strain 981 (CP012137), S. flexneri S7737 (AMJY00000000), S. flexneri 5a strain M90T (CM001474), S. flexneri CDC 796.83 (AERO00000000), S. flexneri 4343.70 (AFHC00000000), S. flexneri NCTC1 (LM651928), S. flexneri 2a strain 301 (AE005674), S. flexneri G1663 (CP007037), S. flexneri Shi06HN006 (CP004057), S. flexneri 2003036 (CP004056), S. flexneri str 4S BJ10610 (JMRK00000000), S. flexneri 4c strain 1205 (CP012140), S. flexneri 2002017 (CP001383) and S. flexneri 1a strain 0228 (CP012735). Phylogenetic reconstruction and analysis was performed wih the phangorn package, written in the R language [29]. VennDiagram [30] was used to generate the Venn plots of S. flexneri SP1, S. flexneri str 4S BJ10610, S. flexneri 4c strain 1205 (CP012140), S. flexneri 2002017, and S. flexneri 1a strain 0228.
Results and discussion
General features
We performed whole genome sequencing using the Illumina HiSeq 4000 system with 2 × 150 bp paired-end reads. After quality control, we assembled the 1095 M bp filtered reads into contigs. The assembled genome of S. flexneri SP1 revealed a genome size of 4,592,345 bp with a G+C content of 50.46%. The largest contig consisted of 137,097 bp and the length of N50 contig was 33,394 bp. These scaffolds contain 4798 coding sequences (CDSs), and 100 RNA-encoding genes. The properties and the statistics of the genome are summarized in Additional file 1: Table S1. The resulting genomic size of strain SP1 was similar to previous studies within the range of 4.1–4.8 M bp [31, 32]. Similarly, CDSs numbers were close to the previous publication [31]. Gene functions were predicted using RAST and COG analysis. RAST server based annotation of the whole genome describes the distribution of subsystems in strain SP1 (Fig. 1a). Proteins responsible for carbohydrates (693 ORFs), amino acids and derivatives (384 ORFs), and cofactors, vitamins, prosthetic groups, pigments (292 ORFs) were abundant among the subsystem categories. The distribution of COGs is illustrated in Fig. 1b. The most abundant COG categories were R (general function prediction only), S (function unknown), E (amino acid transport and metabolism), G (carbohydrate transport and metabolism) and K (transcription). Furthermore, one incomplete prophage region was identified in the genome of SP1. It is a Salmonella ST64B-like phage (Acc-No. NC_004313) of 14.1 kb in length and a G+C content of 50.81%. Additionally, six questionable CRISPR loci were detected by CRISPERfinder.
Fig. 1.
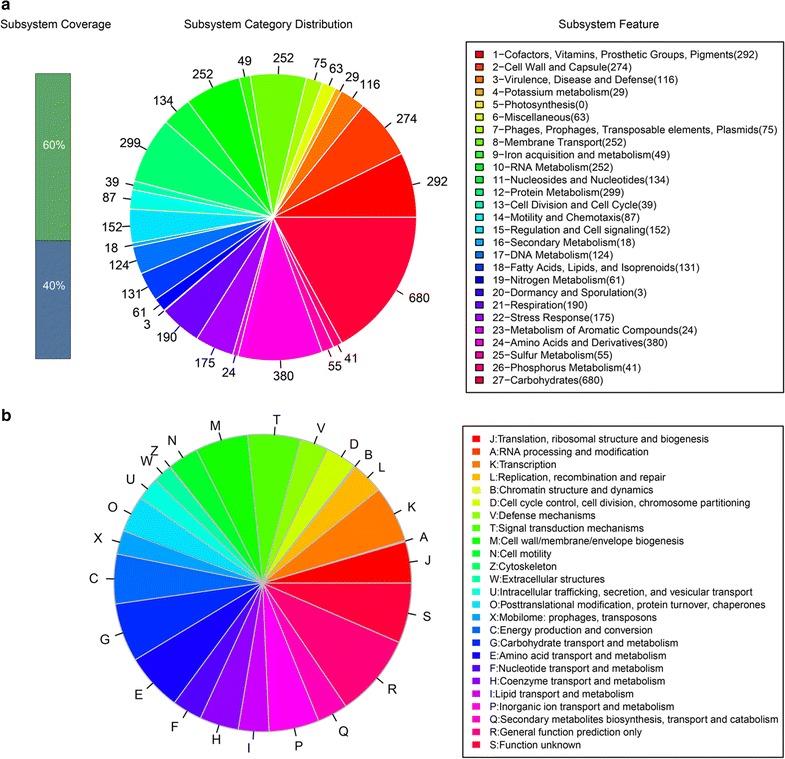
Analysis of annotated genes in S. flexneri strain SP1 based on the SEED and COG databases. a The green bar represents the percentage of proteins that were annotated by RAST server, while the blue bar indicated the proteins not annotated. The pie chart demonstrates the abundance of each subsystem category and the count of each subsystem feature is shown. b Distribution of COGs. Each bar indicates the number of annotated genes based on the COG database
Antimicrobial susceptibility profiles and antibiotic resistance genes
The in vitro antimicrobial susceptibility testing demonstrated that the strain SP1 was resistant to ampicillin, amoxicillin/clavulanic acid, cefazolin, ceftriaxone and trimethoprim, but susceptible to piperacillin/tazobactam, cefoxitin, cefepime, aztreonam, imipenem, amikacin, gentamicin, tobramycin, ciprofloxacin, levofloxacin, tigecycline and nitrofurantoin (Additional file 2: Table S2). We then screened the antibiotic resistance genes (ARGs) in the genome to further explore the genetic basis of multidrug resistance in this strain (Additional file 3: Table S3). In silico analysis revealed the presence of some putative ARGs for different drug classes. We detected genes mediating resistance to aminoglycosides (aadA24, strA and strB), β-lactams (bla CTX-M-14 and bla OXA-1), phenicol (catA1), tetracycline (tetD), sulphonamides (sul2), and trimethoprim (dfrA1). CTX-M-14 was the most frequent ESBL variant detected in Shigella isolates in China, followed by CTX-M-15 [4, 33]. Hitherto, only a few studies have reported the presence of ESBLs in S. flexneri [34]. Interestingly, all of the bla CTX-M-14-harbouring S. flexneri strains were isolated from China [4, 7, 34, 35]. A previous study reported a high prevalence of extended-spectrum cephalosporin resistance Shigella mediated mainly by bla CTX-M (mainly bla CTX-M-14, 14.1%) in Hangzhou City, Zhejiang Province, China [35]. Our study further indicates the existence of bla CTX-M-14-harbouring S. flexneri clone may responsible for these Shigella infections in this area. In addition, the insertion sequence (IS) elements were frequently detected in upstream of bla CTX-M genes [36]. ISfinder was thus employed to scan the blaCTX-M-14 flanking sequences in a range of 6-kb for IS sequences and junction associated proteins. ISEcp1 and IS903B were found upstream of blaCTX-M-14. ISEcp1 belongs to the IS1380 family, which may enhance the expression of bla CTX-M-14/-18, bla CTX-M-17 and bla CTX-M-19 β-lactamase genes [37].
Genetic context of blaCTX-M-14 gene
PlasmidFinder and plasmidSPAdes were used to detect the potential plasmids in the whole genome sequence. In silico analysis revealed that bla CTX-M-14 was located on an IncFII2 plasmid. To further explore the genetic environment of the bla CTX-M-14 gene in isolate SP1, using the bla CTX-M-14 carrying contig as a query against the nr/nt database revealed sequence homology to the ~74 kb annotated bla CTX-M-14-positive IncFII2 plasmid pAC2901 (GenBank: KU987452) from Citrobacter freundii strain AC2901 (Fig. 2). Multiple sequence alignments demonstrated that DNA sequences between pSP1 and pAC2901 share >99% identity. Successful dissemination of bla CTX-M-14 among Enterobacteriaceae isolates from humans, animals and the environment has mainly been associated with IncK, IncF and IncI1 plasmids [38], and there has been only 1 report of a bla CTX-M-14 located on an IncFII2 plasmid in the English literature [39]. Here we report for the first time an IncFII2 blaCTX-M-14-encoding plasmid in the genus Shigella.
Fig. 2.
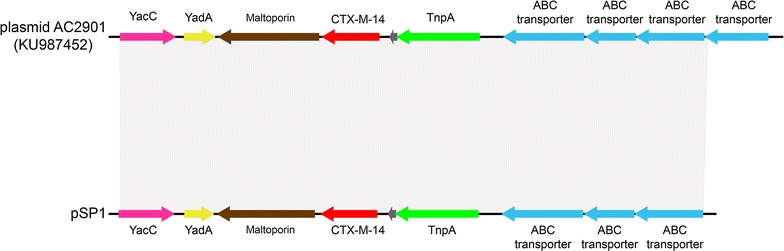
Genetic organization of scaffolds (portions of genome sequences reconstructed from the whole-genome sequence) containing bla CTX-M-14 harbored by plasmid pSP1 and structural comparison with plasmid pAC2901. Arrows indicate positions and direction of transcription of genes. Regions with >99% homology are shown in gray. Information in parentheses after isolates represents the GenBank accession number
Pathogenesis and virulence factors
Shigella flexneri remains a public health concern throughout the world and its pathogenesis should be further investigated. We performed a BLASTP search against the VRDB database and found several known virulence factors. These virulence factors include Shigella extracellular protein A (sepA), glutamate decarboxylase (gadA), invasion plasmid antigen (ipaH9.8), long polar fimbriae (lpfA), hexosyltransferase homolog (capU), invasion protein Shigella flexneri (ipaD), serine protease autotransporters of Enterobacteriaceae (pic), VirF transcriptional activator (virF), and Shigella IgA-like protease homologue (sigA). The ability to withstand an acid-challenge of pH 2.5 by S. flexneri is a necessary virulence trait, which requires acid-induction of a functional GdaA in the stationary-growth phase [40]. IpaH9.8 is a member of the IpaH family of Shigella, which are encoded on the 220 kb virulence plasmid or chromosome and has been shown to be secreted into the host cell where it is targeted to the nucleus [41]. Moreover, LpfA has been linked to virulence in enterohemorrhagic E. coli [42]. Earlier reports indicated the SigA is cytopathic for HEp-2 cells and contributes to the intestinal fluid accumulation associated with S. flexneri infections [43]. Translocation of effector proteins into host cells and the surrounding space is a common strategy used by S. flexneri to target signaling pathways in the host cell [44], and IpaD and VirF are required to facilitate bacterial invasion of host cells [45]. More importantly, Pic is secreted by pathogenic Gram-negative bacteria through the autotransporter pathway and targets a broad range of human leukocyte adhesion proteins, which represent unique immune-modulating bacterial virulence factors [46]. These data suggest that isolate SP1 is potential pathogenic, which is consistent with the isolation of SP1 from a diarrhea patient.
Comparative analysis with other S. flexneri strains
Based on genomes downloaded from the NCBI database, phylogenetic analysis was performed and the resulting tree topology was assessed to identify genetic relatedness between 14 S. flexneri isolates and strain SP1 (Fig. 3a). This revealed that SP1 is most closely related to S. flexneri str 4S BJ10610, which was also isolated from a severe diarrhea patient and was resistant to multiple drugs [47]. Other S. flexneri strains are also has highly similar, except for S. flexneri CDC 796.83, suggesting that S. flexneri strains show high similarity between different species. A previous study has highlighted that S. flexneri has a stable core genome that is equipped with a repertoire of virulence determinants that have enabled it to colonize, and persist, in multiple locations for hundreds of years [48]. A functional genomic comparison was performed between strain SP1 and its four most closely related neighbors: S. flexneri str 4S BJ10610 (JMRK00000000), S. flexneri 2002017 (CP001383), S. flexneri 4c str 1205 (CP012140), and S. flexneri 1a str 0228 (CP012735). The Venn diagram indicates the presence of 4449 core conserved genes present in the pan-genome of the analyzed S. flexneri isolates (Fig. 3b). Interestingly, a total of 178 strain-specific genes were identified in strain SP1.
Fig. 3.
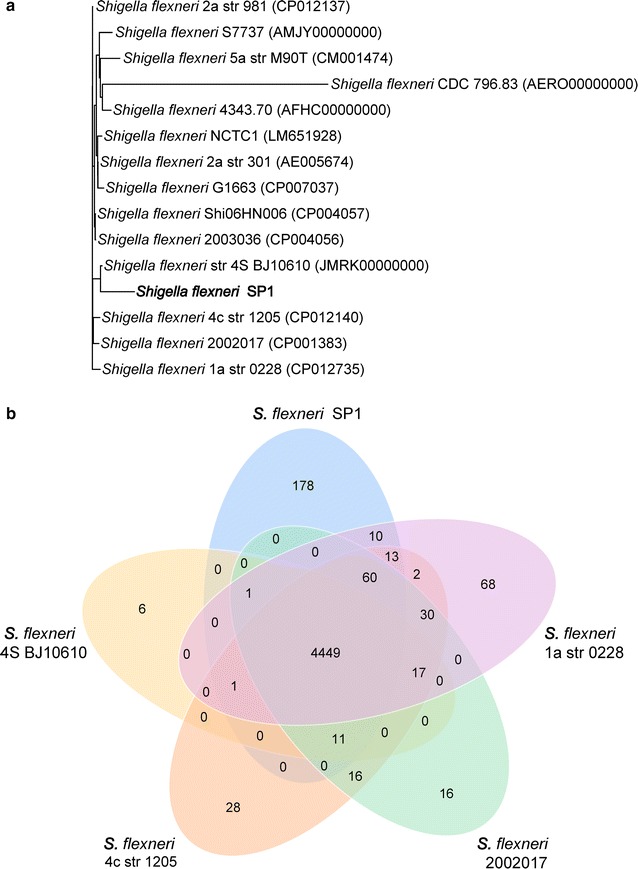
a Phylogenetic tree of S. flexneri strain SP1 with 14 other S. flexneri isolates. The tree was constructed based on based on alignments of orthologous genes. b Venn diagrams showing the orthologous groups in the five most closely related S. flexneri genomes. Numbers inside the Venn diagrams indicated the number of genes found to be shared among the indicated genomes
Conclusions
As more S. flexneri genomes have been sequenced in recent years, comparative genomic studies have progressed rapidly. So far, whole genome studies of S. flexneri have exclusively focused on the historical global spread and recent local persistence among these isolates. This work is the first description of the draft genome of a bla CTX-M-14-harbouring S. flexneri isolate and demonstrates the compares the genome of strain SP1 to other S. flexneri isolates. However, the data presented here is a preliminary report on the virulence profile and antibiotic resistance of S. flexneri strain SP1. Future studies involving more ESBL-encoding S. flexneri isolates from China are urgently needed to study the dynamics of the dissemination of ESBL genes, especially the complete sequence of plasmids carrying these ESBL genes.
Additional files
Additional file 1: Table S1. Summary of S. flexneri strain SP1 genome.
Additional file 2: Table S2. Antimicrobial susceptibility profile of S. flexneri SP1 recovered from a patient with diarrhea.
Additional file 3: Table S3. Antibiotic resistance genes predicted in the S. flexneri strain SP1 genome.
Authors’ contributions
BZ, PS and JF were involved in the overall experimental design. PS, JF, LG, JL, HX, JJ and CY performed microbiology and molecular biology experiments. BZ, LG and AL generated and analyzed the sequencing data. BZ participated in all discussions of data analysis and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Whole Genome Shotgun project of Shigella flexneri strain SP1 has been deposited at DDBJ/EMBL/GenBank under the accession MTPK00000000. The version described in this paper is version MTPK00000000.
Ethics approval and consent to participate
This research was approved by the Research Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81301460 and 81301461); National Key Research and Development Program of China (No. 2016YFD0501105).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12941-017-0212-2) contains supplementary material, which is available to authorized users.
Ping Shen and Jianzhong Fan contributed equally to this article
Contributor Information
Ping Shen, Email: shenping123451@126.com.
Jianzhong Fan, Email: assit500@163.com.
Lihua Guo, Email: glh171@126.com.
Jiahua Li, Email: zcyyljh@163.com.
Ang Li, Email: lia@zju.edu.cn.
Jing Zhang, Email: 21118028@zju.edu.cn.
Chaoqun Ying, Email: yingzi19910410@163.com.
Jinru Ji, Email: susu0572@126.com.
Hao Xu, Email: xuhao930523@163.com.
Beiwen Zheng, Email: zhengbw@zju.edu.cn.
Yonghong Xiao, Email: xiao-yonghong@163.com.
References
- 1.Taneja N, Mewara A, Kumar A, Verma G, Sharma M. Cephalosporin-resistant Shigella flexneri over 9 years (2001–2009) in India. J Antimicrob Chemother. 2012;67:1347–1353. doi: 10.1093/jac/dks061. [DOI] [PubMed] [Google Scholar]
- 2.Tajbakhsh M, Garcia Migura L, Rahbar M, Svendsen CA, Mohammadzadeh M, Zali MR, Aarestrup FM, Hendriksen RS. Antimicrobial-resistant Shigella infections from Iran: an overlooked problem? J Antimicrob Chemother. 2012;67:1128–1133. doi: 10.1093/jac/dks023. [DOI] [PubMed] [Google Scholar]
- 3.Bennish ML, Wojtyniak BJ. Mortality due to shigellosis: community and hospital data. Rev Infect Dis. 1991;13(Suppl 4):S245–S251. doi: 10.1093/clinids/13.Supplement_4.S245. [DOI] [PubMed] [Google Scholar]
- 4.Zhang R, Zhou HW, Cai JC, Zhang J, Chen GX, Nasu M, Xie XY. Serotypes and extended-spectrum beta-lactamase types of clinical isolates of Shigella spp. from the Zhejiang province of China. Diagn Microbiol Infect Dis. 2011;69:98–104. doi: 10.1016/j.diagmicrobio.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 5.Yang C, Li P, Zhang X, Ma Q, Cui X, Li H, Liu H, Wang J, Xie J, Wu F, et al. Molecular characterization and analysis of high-level multidrug-resistance of Shigella flexneri serotype 4 s strains from China. Sci Rep. 2016;6:29124. doi: 10.1038/srep29124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Y, Zhuang L, Kang H, Ma P, Xu T, Pan S, Gu B. Prevalence, resistance patterns, and characterization of integrons of Shigella flexneri isolated from Jiangsu Province in China, 2001–2011. Eur J Clin Microbiol Infect Dis. 2016;35:1347–1353. doi: 10.1007/s10096-016-2671-3. [DOI] [PubMed] [Google Scholar]
- 7.Zhang W, Luo Y, Li J, Lin L, Ma Y, Hu C, Jin S, Ran L, Cui S. Wide dissemination of multidrug-resistant Shigella isolates in China. J Antimicrob Chemother. 2011;66:2527–2535. doi: 10.1093/jac/dkr341. [DOI] [PubMed] [Google Scholar]
- 8.Zheng B, Dong H, Xu H, Lv J, Zhang J, Jiang X, Du Y, Xiao Y, Li L. Coexistence of MCR-1 and NDM-1 in clinical Escherichia coli isolates. Clin Infect Dis. 2016;63:1393–1395. doi: 10.1093/cid/ciw553. [DOI] [PubMed] [Google Scholar]
- 9.Jin Q, Yuan Z, Xu J, Wang Y, Shen Y, Lu W, Wang J, Liu H, Yang J, Yang F, et al. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 2002;30:4432–4441. doi: 10.1093/nar/gkf566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashton PM, Baker KS, Gentle A, Wooldridge DJ, Thomson NR, Dallman TJ, Jenkins C. Draft genome sequences of the type strains of Shigella flexneri held at Public Health England: comparison of classical phenotypic and novel molecular assays with whole genome sequence. Gut Pathog. 2014;6:7. doi: 10.1186/1757-4749-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang T, Jiang X, Feng C, Li A, Dong H, Wu S, Zheng B. Whole genome sequencing uncovers a novel IND-16 metallo-beta-lactamase from an extensively drug-resistant Chryseobacterium indologenes strain J31. Gut Pathog. 2016;8:47. doi: 10.1186/s13099-016-0130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.CLSI. Performance standards for antimicrobial susceptibility testing; 23rd informational supplement. Wayne: M100-S23: Clinical and Laboratory Standards Institute; 2013.
- 13.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST server: rapid annotations using subsystems technology. BMC Genom. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Natale DA, Shankavaram UT, Galperin MY, Wolf YI, Aravind L, Koonin EV. Towards understanding the first genome sequence of a crenarchaeon by genome annotation using clusters of orthologous groups of proteins (COGs) Genome Biol. 2000;1:RESEARCH0009. doi: 10.1186/gb-2000-1-5-research0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:0955–0964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carattoli A, Zankari E, Garcia-Fernandez A, Voldby Larsen M, Lund O, Villa L, Moller Aarestrup F, Hasman H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58:3895–3903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siguier P, Varani A, Perochon J, Chandler M. Exploring bacterial insertion sequences with ISfinder: objectives, uses, and future developments. Methods Mol Biol. 2012;859:91–103. doi: 10.1007/978-1-61779-603-6_5. [DOI] [PubMed] [Google Scholar]
- 22.Antipov D, Hartwick N, Shen M, Raiko M, Lapidus A, Pevzner PA. plasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics. 2016;32:3380–3387. doi: 10.1093/bioinformatics/btv688. [DOI] [PubMed] [Google Scholar]
- 23.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45:D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 2016;44:D694–D697. doi: 10.1093/nar/gkv1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinheinz KA, Joensen KG, Larsen MV. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage. 2014;4:e27943. doi: 10.4161/bact.27943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carattoli A, Bertini A, Villa L, Falbo V, Hopkins KL, Threlfall EJ. Identification of plasmids by PCR-based replicon typing. J Microbiol Methods. 2005;63:219–228. doi: 10.1016/j.mimet.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 28.Zheng B, Li A, Jiang X, Hu X, Yao J, Zhao L, Ji J, Ye M, Xiao Y, Li L. Genome sequencing and genomic characterization of a tigecycline-resistant Klebsiella pneumoniae strain isolated from the bile samples of a cholangiocarcinoma patient. Gut Pathog. 2014;6:40. doi: 10.1186/s13099-014-0040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lam F, Lalansingh CM, Babaran HE, Wang Z, Prokopec SD, Fox NS, Boutros PC. VennDiagramWeb: a web application for the generation of highly customizable Venn and Euler diagrams. BMC Bioinform. 2016;17:401. doi: 10.1186/s12859-016-1281-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rossi O, Baker KS, Phalipon A, Weill FX, Citiulo F, Sansonetti P, Gerke C, Thomson NR. Draft genomes of Shigella strains used by the STOPENTERICS consortium. Gut Pathog. 2015;7:14. doi: 10.1186/s13099-015-0061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang N, Lan R, Sun Q, Wang J, Wang Y, Zhang J, Yu D, Hu W, Hu S, Dai H, et al. Genomic portrait of the evolution and epidemic spread of a recently emerged multidrug-resistant Shigella flexneri clone in China. J Clin Microbiol. 2014;52:1119–1126. doi: 10.1128/JCM.02669-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Li B, Ni Y, Sun J. Molecular characterization of the extended-spectrum beta-lactamase (ESBL)-producing Shigella spp. in Shanghai. Eur J Clin Microbiol Infect Dis. 2015;34:447–451. doi: 10.1007/s10096-014-2244-2. [DOI] [PubMed] [Google Scholar]
- 34.Xia S, Xu B, Huang L, Zhao JY, Ran L, Zhang J, Chen H, Pulsrikarn C, Pornruangwong S, Aarestrup FM, Hendriksen RS. Prevalence and characterization of human Shigella infections in Henan Province, China, in 2006. J Clin Microbiol. 2011;49:232–242. doi: 10.1128/JCM.01508-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang CL, Liu QZ, Wang J, Chu X, Shen LM, Guo YY. Epidemic and virulence characteristic of Shigella spp. with extended-spectrum cephalosporin resistance in Xiaoshan District, Hangzhou, China. BMC Infect Dis. 2014;14:260. doi: 10.1186/1471-2334-14-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Song C, Duan G, Zhu J, Yang H, Xi Y, Fan Q. Transposition of ISEcp1 modulates blaCTX-M-55-mediated Shigella flexneri resistance to cefalothin. Int J Antimicrob Agents. 2013;42:507–512. doi: 10.1016/j.ijantimicag.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 37.Poirel L, Decousser JW, Nordmann P. Insertion sequence ISEcp1B is involved in expression and mobilization of a bla(CTX-M) beta-lactamase gene. Antimicrob Agents Chemother. 2003;47:2938–2945. doi: 10.1128/AAC.47.9.2938-2945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riccobono E, Di Pilato V, Villagran AL, Bartoloni A, Rossolini GM, Pallecchi L. Complete sequence of pV404, a novel IncI1 plasmid harbouring blaCTX-M-14 in an original genetic context. Int J Antimicrob Agents. 2014;44:374–376. doi: 10.1016/j.ijantimicag.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 39.Valat C, Forest K, Billet M, Polizzi C, Saras E, Madec JY, Haenni M. Absence of co-localization between pathovar-associated virulence factors and extended-spectrum beta-lactamase (blaCTX-M) genes on a single plasmid. Vet Microbiol. 2016;192:163–166. doi: 10.1016/j.vetmic.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 40.Bhagwat AA, Bhagwat M. Comparative analysis of transcriptional regulatory elements of glutamate-dependent acid-resistance systems of Shigella flexneri and Escherichia coli O157:H7. FEMS Microbiol Lett. 2004;234:139–147. doi: 10.1111/j.1574-6968.2004.tb09525.x. [DOI] [PubMed] [Google Scholar]
- 41.Edwards DJ, Streich FC, Jr, Ronchi VP, Todaro DR, Haas AL. Convergent evolution in the assembly of polyubiquitin degradation signals by the Shigella flexneri IpaH9.8 ligase. J Biol Chem. 2014;289:34114–34128. doi: 10.1074/jbc.M114.609164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, et al. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis. 2014;20:1919–1932. doi: 10.1097/MIB.0000000000000183. [DOI] [PubMed] [Google Scholar]
- 43.Al-Hasani K, Navarro-Garcia F, Huerta J, Sakellaris H, Adler B. The immunogenic SigA enterotoxin of Shigella flexneri 2a binds to HEp-2 cells and induces fodrin redistribution in intoxicated epithelial cells. PLoS ONE. 2009;4:e8223. doi: 10.1371/journal.pone.0008223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang F, Jiang Z, Li Y, He X, Zhao J, Yang X, Zhu L, Yin Z, Li X, Wang X, et al. Shigella flexneri T3SS effector IpaH4.5 modulates the host inflammatory response via interaction with NF-kappaB p65 protein. Cell Microbiol. 2013;15:474–485. doi: 10.1111/cmi.12052. [DOI] [PubMed] [Google Scholar]
- 45.Tobe T, Nagai S, Okada N, Adler B, Yoshikawa M, Sasakawa C. Temperature-regulated expression of invasion genes in Shigella flexneri is controlled through the transcriptional activation of the virB gene on the large plasmid. Mol Microbiol. 1991;5:887–893. doi: 10.1111/j.1365-2958.1991.tb00762.x. [DOI] [PubMed] [Google Scholar]
- 46.Ruiz-Perez F, Wahid R, Faherty CS, Kolappaswamy K, Rodriguez L, Santiago A, Murphy E, Cross A, Sztein MB, Nataro JP. Serine protease autotransporters from Shigella flexneri and pathogenic Escherichia coli target a broad range of leukocyte glycoproteins. Proc Natl Acad Sci USA. 2011;108:12881–12886. doi: 10.1073/pnas.1101006108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li P, Yang C, Wang J, Yi S, Li H, Wang Y, Qiu S, Song H. Draft genome sequence of a new Shigella flexneri subserotype, 4S BJ10610. Genome Announc. 2014;2:e00715. doi: 10.1128/genomeA.00715-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Connor TR, Barker CR, Baker KS, Weill FX, Talukder KA, Smith AM, Baker S, Gouali M, Pham Thanh D, Jahan Azmi I, et al. Species-wide whole genome sequencing reveals historical global spread and recent local persistence in Shigella flexneri. Elife. 2015;4:e07335. doi: 10.7554/eLife.07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Summary of S. flexneri strain SP1 genome.
Additional file 2: Table S2. Antimicrobial susceptibility profile of S. flexneri SP1 recovered from a patient with diarrhea.
Additional file 3: Table S3. Antibiotic resistance genes predicted in the S. flexneri strain SP1 genome.
Data Availability Statement
Whole Genome Shotgun project of Shigella flexneri strain SP1 has been deposited at DDBJ/EMBL/GenBank under the accession MTPK00000000. The version described in this paper is version MTPK00000000.