

Abstract
Calcific aortic valve disease (CAVD) is common among the elderly, and aortic valve interstitial cells (AVICs) exhibit unique inflammatory and osteogenic responses to proinflammatory stimulation, which plays an important role in valvular fibrosis and calcification. Thus, suppression of AVIC proinflammatory response may have therapeutic utility for the prevention of CAVD progression. Interleukin (IL)-37, an antiinflammatory cytokine, reduces tissue inflammation. This study aimed to test the hypothesis that IL-37 suppresses human AVIC inflammatory responses to Toll-like receptor (TLR) agonists. Human AVICs were exposed to Pam3CSK4, poly(I:C) and lipopolysaccharide in the presence and absence of recombinant human IL-37. Stimulation of TLR4 increased the production of intercellular adhesion molecule-1, IL-6, IL-8 and monocyte chemoattractant protein-1. Knockdown of myeloid differentiation factor 88 (MyD88) or TIR-domain–containing adaptor-inducing interferon-β differentially affected inflammatory mediator production following TLR4 stimulation. IL-37 reduced production of these inflammatory mediators induced by TLR4. Moreover, knockdown of IL-37 enhanced induction of these mediators by TLR4. IL-37 also suppressed inflammatory mediator production induced by the MyD88-dependent TLR2, but had no effect on the inflammatory responses to the TRIF-dependent TLR3. Furthermore, IL-37 inhibited NF-κB activation induced by TLR2 or TLR4 through a mechanism dependent of IL-18 receptor α-chain. Activation of TLR2, TLR3 or TLR4 upregulates the production of inflammatory mediators in human AVICs. IL-37 suppresses MyD88-mediated responses to reduce inflammatory mediator production following stimulation of TLR2 and TLR4. This antiinflammatory cytokine may be useful for suppression of aortic valve inflammation elicited by MyD88-dependent TLR signaling.
INTRODUCTION
Calcific aortic valve disease (CAVD) is a major clinical problem and a significant cause of morbidity in the elderly. Furthermore, severe cases of this disease account for more than 10,000 deaths per year in the United States alone (1). Due to greater lifespan, the prevalence of CAVD is increasing. However, the pathogenic mechanism responsible for CAVD remains incompletely understood, and there is currently no pharmacological treatment capable of preventing progression of this disease (2).
CAVD is known to be a chronic inflammatory disease (3). The early events in CAVD development appear to include endothelial damage, lipid deposition, inflammatory cell accumulation and resultant production of proinflammatory mediators (4). In affected aortic valves, the cusps become progressively thickened and calcified (5). A hallmark of CAVD is the presence of inflammatory cells, such as macrophages and T cells, within and surrounding calcified nodules (6,7). Atherogenic factors and mechanical stress are known to activate valvular endothelial cells, causing them to express adhesion molecules that recruit monocytes and macrophages to the valvular tissue (8). Proinflammatory mediators produced by activated inflammatory cells are believed to modulate valvular cell pro-osteogenic activity (9).
Aortic valve interstitial cells (AVICs), the major cellular component of the valve tissue, maintain the microarchitecture of valvular leaflets and regulate valvular function. These cells can differentiate into osteoblast-like cells and thus contribute to valvular calcification (10,11). However, we previously observed that stimulation of Toll-like receptor (TLR) 2 or 4 in human AVICs induces osteogenic responses (12,13). Thus, the inflammatory and osteogenic mechanisms are interrelated in human AVICs, and suppression of AVIC responses to proinflammatory stimuli may have therapeutic potential for prevention of CAVD progression.
TLRs play a key role in innate and adaptive immunity (14). While functioning as pattern recognition receptors, several TLRs can be activated by damaged-associated molecular patterns (15). TLR signaling is controlled by four intracellular adaptor proteins: myeloid differentiation factor 88 (MyD88), MyD88 adaptor-like, TIR-domain– containing adaptor-inducing interferon-β (TRIF) and TRIF-related adaptor molecule (16). MyD88 mediates signaling in all TLRs except TLR3, whereas TRIF is involved in TLR3 and TLR4 signaling (14). Thus, TLR2 utilizes the MyD88 -dependent pathway and TLR3 utilizes the TRIF-dependent pathway. TLR4 signals through both the MyD88 -dependent and TRIF-dependent pathways. The MyD88-dependent pathway activates NF-κB and AP-1; the two transcription factors collectively regulate the expression of inflammatory mediators in most cell types (17). It has been demonstrated that the MyD88-mediated signaling pathway plays an important role in mediating tissue inflammation in many inflammation conditions, including myocardial ischemia-reperfusion injury (18), atherosclerosis (19) and cardiovascular calcification (20).
Interleukin (IL)-37, a member of the IL-1 family, functions as an inhibitor of broad inflammatory responses (21). The antiinflammatory effect of IL-37 has been observed in vitro as well as in vivo (22). IL-37 mRNA and protein are present in tissues of patients with inflammatory and autoimmune diseases, such as rheumatoid arthritis, atherosclerosis, Crohn’s disease and lupus (21). In cells that overexpress IL-37, the inflammatory responses to a variety of proinflammatory stimuli are markedly attenuated (21,23). More important, mice carrying the human IL-37 gene (IL-37 transgenic mice) are protected against aortic valve lesions induced by proinflammatory stimuli (23). However, it remains unclear whether IL-37 has an effect on the inflammatory responses to TLRs in human AVICs.
The present study determined the effect of IL-37 on suppression of TLR -mediated inflammatory responses in human AVICs and the TLR signaling pathway interfered by IL-37.
MATERIALS AND METHODS
Materials
Antibodies against intercellular adhesion molecule-1 (ICAM-1), MyD88, TRIF and β-actin as well as specific small interfering RNA (siRNA) for human IL-37 and scrambled siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to phospho-NF-κB, total NF-κB, specific siRNA for MyD88 and TRIF, and scrambled siRNA were purchased from Life Technologies (Grand Island, NY, USA). Pam3CSK4 and poly(I:C) were purchased from InvivoGen (San Diego, CA, USA). Antibody against IL-37, recombinant human IL-37b, neutralizing antibody against human IL-18 receptor α-chain (IL-18Rα) and enzyme-linked immunosorbent assay (ELISA) kits for IL-6, IL-8 and monocyte chemoattractant protein-1 (MCP-1) were purchased from R&D Systems (Minneapolis, MN, USA). TransAM NF-κB activity (p65 DNA-binding) assay kit was purchased from Active Motif (Carlsbad, CA, USA). Medium 199 was purchased from Lonza (Walkersville, MD, USA). Lipopolysaccharide (LPS, E. coli 0111:B4) and all other chemicals and reagents were purchased from Sigma -Aldrich Chemical (St Louis, MO, USA).
Cell Isolation, Culture and Treatment
Normal aortic valve leaflets were collected from explanted hearts of six patients (four male and two female, mean age 59 ± 8.1 years) undergoing heart transplantation due to cardiomyopathy at the University of Colorado Hospital. All patients gave informed consent for use of their aortic valves for this study. This study was approved by the Colorado Multiple Institutional Review Board.
AVICs were isolated and cultured using a previously described method (24) with modifications (12). Briefly, valve leaflets were subjected to sequential digestions with collagenase, and cells were collected by centrifugation. Cells were cultured in M199 growth medium containing penicillin G, streptomycin, amphotericin B and 10% fetal bovine serum. Cells of passages 3–6 were used for this study. Cells were subcultured on plates and treated when they reached 80–90% confluence.
To determine the effect of TLR stimulation on inflammatory mediator production, cells were treated with TLR4 agonist LPS (0.1 or 0.2 μg/mL), TLR2 agonist Pam3CSK4 (0.1 μg/mL) and TLR3 agonist poly(I:C) (2.5 μg/mL) for 24 h. To determine the effect of TLR stimulation on NF-κB activation, cells were treated with TLR ligands for 1–8 h. The effect of IL-37 on NF-κB activation and inflammatory responses was evaluated in cells preincubated with recombinant IL-37 (0.1 or 1.0 ng/mL) 1 h prior to treatment with TLR ligands. To determine the role of endogenous IL-37, MyD88 and TRIF in the inflammatory responses, knockdown of IL-37, TRIF and MyD88 with specific siRNA was performed. The role of IL-18Rα in mediating the effect of IL-37 on NF-κB activation was evaluated in cells treated with IL-18Rα–neutralizing antibody.
Gene Knockdown
To knock down IL-37, cells (60–70% confluent) in 24-well plates were incubated with a mixture of IL-37 siRNA (60 nmol/L) and transfection reagent (6 μL/mL of medium) for 48 h and then stimulated with LPS. Control cells were treated with scrambled siRNA and transfection reagent. To knock down MyD88 and TRIF, cells were separately incubated with a mixture of MyD88 siRNA (100 nmol/L) or TRIF siRNA (100 nmol/L) and transfection reagent (6 μL/mL of medium) for 72 h and then stimulated with LPS. Control cells were treated with scrambled siRNA and transfection reagent.
Immunoblotting
Immunoblotting was done to analyze ICAM-1, MyD88, TRIF and IL-37 with β-actin as a loading control. Phospho-NF-κB p65 levels were also analyzed by immunoblotting, using total NF-κB p65 for loading normalization. After the experiments, human AVICs were lysed in a sample buffer (100 mmol/L Tris-HCl, pH 6.8; 2% SDS; 0.02% bromophenol blue; and 10% glycerol). Protein samples were separated on gradient (4–20%) mini-gels and transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked with 5% skim milk solution for 1 h at room temperature. The blocked membranes were then incubated with a primary antibody. After washing with phosphate-buffered saline containing 0.05% Tween 20, the membrane was incubated with a peroxidase-linked secondary antibody specific to the primary antibody. After further washes, the membrane was treated with enhanced chemiluminescence reagents. Then the membrane was exposed on x-ray film. Band density was analyzed using Image J software (National Institutes of Health, Bethesda, MD, USA).
NF-κB Activity Assay
NF-κB activity (p65 DNA-binding) was analyzed using the TransAM NF-κB assay kit following the manufacturer’s instruction. Absorbance of samples was determined spectrophotometrically at 450 nm using a microplate reader (Bio-Rad Laboratories).
Cytokine ELISA
Cell culture supernatants were collected. Levels of IL-6, IL-8 and MCP-1 were analyzed using ELISA kits following the manufacturer’s protocol.
Statistical Analysis
Data are presented as mean ± standard error (SE). Statistical analysis was performed using StatView software (Abacus Concepts, Calabasas, CA, USA). Student t test was used for comparison between two groups. Analysis of variance with post hoc Bonferroni/Dunn test was used to analyze differences between multiple groups. Statistical significance was defined as P ≤0.05. Nonparametric Mann-Whitney U test was performed to confirm the difference of the two-group comparison. For multiple-group comparisons, nonparametric Kruskal-Wallis test was performed to confirm the differences.
RESULTS
IL-37 Suppresses the Inflammatory Responses Induced by TLR4 Stimulation in Human AVICs
We analyzed the levels of IL-6, IL-8, MCP-1 and ICAM-1 in human AVICs exposed to TLR4 agonist LPS and determined the effect of recombinant IL-37 on the production of these inflammatory mediators. As shown in Figure 1A, ICAM-1 levels were increased in cells exposed to LPS (0.2 μg/mL) for 24 h. Recombinant IL-37 suppressed the upregulation of ICAM-1 production by LPS. The data in Figure 1B show that LPS increased the levels of IL-6, IL-8 and MCP-1 in the cell culture supernatants. Recombinant IL-37 markedly reduced production of these cytokines. The data demonstrate that IL-37 is antiinflammatory for human AVICs.
Figure 1.

IL-37 attenuates the inflammatory responses induced by TLR4 stimulation in human AVICs. AVICs were treated with lipopolysaccharide (LPS, 0.2 μg/mL) for 24 h in the presence and absence of recombinant IL-37 (0.1 and 1.0 ng/mL). (A) Representative immunoblots and densitometric data show that IL-37 reduced ICAM-1 production induced by LPS. (B) Cytokine data show that IL-37 suppressed production of IL-6, IL-8 and MCP-1 following stimulation with LPS, and the higher concentration of IL-37 had a greater effect. Data are mean ± SE. n = 6 experiments using cell isolates from distinct donor valves; *P <0.05 versus untreated control; #P <0.05 versus LPS alone.
IL-37 Knockdown Enhances the Inflammatory Responses to TLR4 Stimulation in Human AVICs
To determine the role of endogenous IL-37 in modulating the inflammatory responses, we treated human AVICs with specific human IL-37 siRNA prior to exposure to LPS. The immunoblot in Figure 2A shows that LPS moderately upregulated IL-37 levels, and cells pretreated with IL-37 siRNA had lower levels of IL-37 protein following LPS stimulation compared to cells treated with scrambled siRNA before LPS stimulation. The data in Figures 2A and B show that treatment with IL-37 siRNA enhanced the production of ICAM-1, IL-6 and IL-8 after exposure to a lower concentration of LPS (0.1 μg/mL). Although MCP-1 levels increased by 12% (680 ± 22 in IL-37 siRNA + LPS group versus 605 ± 43 in scrambled siRNA + LPS group, Figure 2B), the effect of IL-37 knockdown on this cytokine is insignificant. The data confirm that IL-37 is present in human AVICs and provide evidence that endogenous IL-37 is antiinflammatory.
Figure 2.

Knockdown of IL-37 enhances the inflammatory responses to TLR4 stimulation in human AVICs. AVICs were pretreated with IL-37 siRNA (60 nmol/L) or scrambled siRNA (60 nmol/L) for 48 h and then treated with LPS (0.1 μg/mL) for 24 h. (A) A representative immunoblot shows that cells treated with IL-37 siRNA + LPS had lower levels of IL-37 protein compared to cells treated with LPS alone or scrambled siRNA + LPS. Densitometric data and representative immunoblot show that cells treated with IL-37 siRNA + LPS produced higher levels of ICAM-1. (B) Cytokine data show that knockdown of IL-37 enhanced the production of IL-6 and IL-8, but not MCP-1, induced by LPS. Data are mean ± SE. n = 5 experiments using cell isolates from distinct donor valves; *P <0.05 versus control; #P <0.05 versus LPS alone; ⊗P< 0.05 versus scrambled siRNA + LPS.
MyD88 and TRIF Have Differential Roles in TLR4-Mediated Inflammatory Mediator Production in Human AVICs
To determine the relative roles of the MyD88 and TRIF signaling pathways in mediating the inflammatory responses to TLR4 stimulation in human AVICs, we applied specific siRNAs to knock down MyD88 and TRIF. The data in Figure 3A show that treatment with specific siRNA effectively reduced MyD88 and TRIF levels in human AVICs. Knockdown of MyD88 markedly reduced the production of ICAM-1, IL-6, IL-8 and MCP-1 induced by LPS (Figures 3B, C). However, knockdown of TRIF reduced ICAM-1 and IL-8, but not IL-6 and MCP-1. It appears that the MyD88 pathway plays a broad role in controlling inflammatory mediator production elicited by TLR4 in human AVICs.
Figure 3.

MyD88 and TRIF have differential roles in mediating the inflammatory responses to TLR4 stimulation. AVICs were treated with MyD88 siRNA (100 nmol/L) or TRIF siRNA (100 nmol/L) for 48 h prior to LPS (0.2 μg/mL) stimulation. (A) Representative immunoblots show effective knockdown of MyD88 and TRIF. (B) Representative immunoblots and densitometric data show that knockdown of either MyD88 or TRIF reduced production of ICAM-1 following LPS stimulation, and knockdown of MyD88 had a bigger effect. (C) Cytokine data show that knockdown of MyD88 reduced production of all cytokines following LPS stimulation. Knockdown of TRIF markedly reduced IL-8 and moderately reduced MCP-1, but had no effect on IL-6. Data are mean ± SE. n = 6 experiments using cell isolates from distinct donor valves; *P <0.05 versus control; #P <0.05 versus LPS alone; ⊗P <0.05 versus scrambled siRNA + LPS.
IL-37 Suppresses MyD88-Mediated but Not TRIF-Mediated Inflammatory Responses
TLR3 utilizes the TRIF-dependent signaling pathway and TLR2 utilizes the MyD88-dependent signaling pathway. To understand which pathway is inhibited by IL-37 to suppress the inflammatory responses, we stimulated cells with TLR3 agonist poly(I:C) and TLR2 agonist Pam3CSK4. As shown in Figure 4, poly(I:C) also induced production of the inflammatory mediators. However, IL-37 had no effect on the inflammatory responses to poly(I:C), which utilizes the TLR3-TRIF pathway (25). In contrast, IL-37 markedly reduced production of inflammatory mediators induced by TLR2 agonist Pam3CSK4 (Figure 4). Thus, IL-37 selectively inhibits MyD88-mediated proinflammatory signaling in human AVICs.
Figure 4.

IL-37 attenuates the inflammatory responses to TLR2 stimulation, but has no effect on the responses to TLR3 stimulation. AVICs were treated with poly(I:C) (2.5 μg/mL) or Pam3CSK4 (0.1 μg/mL) for 24 h in the presence and absence of IL-37 (0.1 and 1.0 ng/mL). (A) Representative immunoblots and densitometric data show that poly(I:C) and Pam3CSK4 upregulated ICAM-1 production. Whereas IL-37 reduced ICAM-1 production induced by Pam3CSK4, it failed to inhibit the effect of poly(I:C) on ICAM-1 production. (B) Cytokine data show that cells produced higher levels of cytokines following stimulation with either poly(I:C) or Pam3CSK4. IL-37 only reduced cytokine production in cells exposed to Pam3CSK4. Data are mean ± SE. n = 6 experiments using cell isolates from distinct donor valves; *P <0.05 versus control; #P <0.05 versus Pam3CSK4 alone.
IL-37 Inhibits NF-κB Activation Mediated by TLR2 and TLR4
MyD88 mediates the proinflammatory signaling of TLR2 and TLR4, and signaling through either of these two innate immunoreceptors leads to activation of NF-κB, which turns on the proinflammatory genes (14). NF-κB p65 phosphorylation was upregulated in human AVICs exposed to LPS or PAM3CSK4 (Figure 5A). Elevated levels of phosphorylated NF-κB p65 was observed at 1 h and lasted to 8 h after stimulation. IL-37 markedly attenuated NF-κB p65 phosphorylation induced by LPS and essentially abrogated NF-κB p65 phosphorylation induced by PAM3CSK4 (Figure 5A). To confirm the effect of IL-37 on NF-κB activation, we examined NF-κB DNA-binding activity 4 h after exposing cells to TLR2 and TLR4 agonists in the presence or absence of recombinant IL-37. The data in Figure 5A show that IL-37 markedly reduced NF-κB activation in human AVICs following simulation of TLR4 and essentially abrogated NF-κB activation following TLR2 stimulation.
Figure 5.

IL-37 inhibits NF-κB activation induced by stimulation of TLR2 and TLR4. (A) AVICs were treated with LPS (0.2 μg/mL) or Pam3CSK4 (0.1 μg/mL) for 1–8 h in the presence and absence of IL-37 (1.0 ng/mL). NF-κB p65 phosphorylation was examined using immunoblotting, and NF-κB DNA-binding activity at 4 h was assessed using enzyme-linked immunosorbent assay. IL-37 reduced NF-κB phosphorylation induced by LPS and abrogated NF-κB phosphorylation induced by PAM3CSK4. The experiments were repeated using three cell isolates from distinct donor valves. IL-37 also reduced NF-κB DNA-binding activity in cells exposed to LPS and abrogated the increase in NF-κB DNA-binding activity in cells exposed to Pam3CSK4. Data are mean ± SE. n = 5 experiments using cell isolates from distinct donor valves; *P <0.05 versus control; #P <0.05 versus LPS alone or Pam3CSK4 alone. (B) AVICs were treated with IL-18Rα–neutralizing antibody (2.65 μg/mL) prior to treatment with IL-37 (1.0 ng/mL). Neutralization of IL-18Rα abolished the effect of IL-37 on NF-κB phosphorylation examined at 4 h after stimulation with LPS or Pam3CSK4. The experiments were repeated three times using cell isolates from distinct donor valves.
Extracellular IL-37 utilizes IL-18Rα to exert an effect on cells (26). To ascertain whether the effect of IL-37 on NF-κB activation in human AVICs is direct or indirect, we treated cells with a neutralizing antibody to the IL-18Rα. Figure 5B shows that the effect of IL-37 on NF-κB p65 phosphorylation was abrogated by blocking of IL-18Rα.
Figure 6.
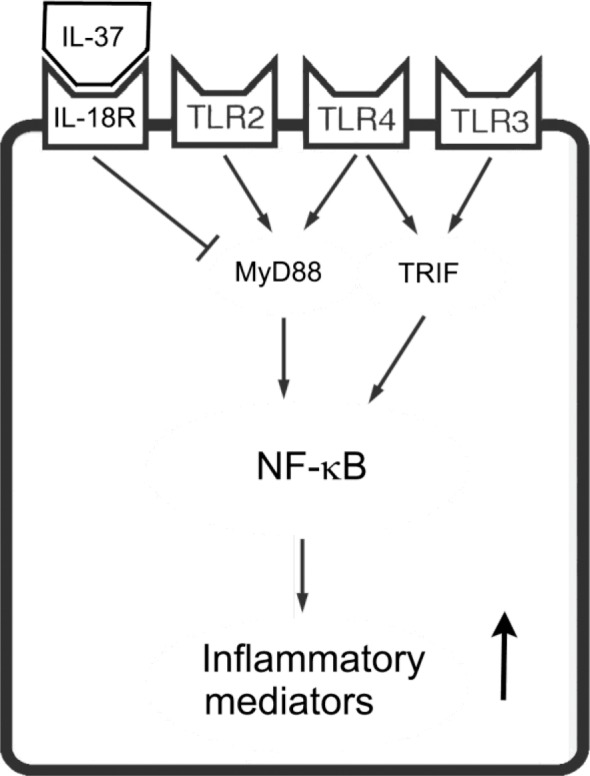
Schematic description of the suppression of human AVIC inflammatory responses to stimulation of TLR2 and TLR4. Both TLR2 and TLR4 utilize the MyD88 pathway to induce inflammatory mediator production in human AVICs. IL-37 inhibits NF-κB through an IL-18Rα–dependent mechanism to down-regulate inflammatory mediator production mediated by the MyD88 pathway.
Discussion
CAVD affects a significant number of elderly people and leads to heart failure. With the incidence of this disease increasing in the aging population, pharmacological intervention targeting its progression remains unavailable. Chronic inflammation in valvular tissue is linked to progression of this disease (27). Whereas AVICs respond to TLR stimulation and are involved in the overall inflammatory process in valvular tissue, the approach that modulates AVIC inflammatory responses to TLR stimulation is lacking. In this study, we observed that IL-37 suppresses the MyD88-depedent signaling pathway to reduce the production of multiple inflammatory mediators in human AVICs exposed to TLR2 and TLR4 ligands.
Inflammatory mediators promote the fibrotic and calcific processes associated with CAVD progression (28). In this regard, our previous studies demonstrated an important role of proinflammatory signaling pathways in mediating the osteogenic responses in human AVICs (13,20,29). Particularly, stimulation of TLR2 and TLR4 upregulates the expression of runt-related transcription factor, alkaline phosphatase and bone morphogenic protein-2 in human AVICs (29). In addition, several endogenous proteins are capable of inducing these osteogenic biomarkers (13,30). It appears that human AVICs elevate their pro-osteogenic activity in response to a variety of proinflammatory stimuli and that proinflammatory pathways modulate pro-osteogenic activity in this cell type. Thus, suppression of AVIC responses to proinflammatory stimuli may prevent the progression of valvular calcification.
We previously reported that stimulation of TLR4 induces inflammatory responses in human AVICs (12,31). TLR4 agonist LPS is capable of upregulating the expression of proinflammatory genes in MyD88-deficient mice, and such MyD88-independent activity of LPS is abolished in TRIF-deficient mice (32). In addition, TRIF-deficient mice were profoundly impaired for TLR4-mediated expression of IFN-inducible genes (33). In the present study, we show that knockdown of MyD88 reduces the production of ICAM-1, IL-6, IL-8 and MCP-1 in human AVICs exposed to LPS. However, knockdown of TRIF reduces the production of ICAM-1 and IL-8, but not IL-6 and MCP-1. Moreover, knockdown of MyD88 or TRIF alone fails to abrogate the inflammatory responses to TLR4 stimulation. Thus, both the MyD88-dependent and TRIF-dependent signaling pathways are required to mediate the TLR4-induced inflammatory responses in human AVICs, and the TRIF signaling pathway appears to be involved in upregulation of selected inflammatory mediators.
In the present study, we observed that recombinant IL-37 suppresses the production of ICAM-1, IL-6, IL-8 and MCP-1 induced by TLR4 ligand LPS in human AVICs. In addition, cells pretreated with IL-37 siRNA produce higher levels of ICAM-1, IL-6 and IL-8 in response to TLR4 stimulation, which is associated with lower levels of IL-37 protein following TLR4 stimulation. These findings demonstrate that IL-37 is antiinflammatory in human AVICs and recombinant IL-37 has the potential to suppress the inflammatory responses to certain TLR ligands. MCP-1, acting through its receptor CCR2, is critical for the recruitment of leukocytes to the sites of inflammation (34). ICAM-1 facilitates leukocyte filtration to inflammatory tissue (35). IL-6 and IL-8 released by leukocytes and AVICs may promote AVIC pro-osteogenic activity (36). These inflammatory mediators are present in the early aortic valve lesions associated with CAVD (37). Thus, the effect of IL-37 on the production of these inflammatory mediators has clinical significance.
TLRs utilize specific signaling pathways to elaborate cellular inflammatory responses. In this regard, TLR4 signaling involves both MyD88 and TRIF (38,39). However, TLR2 only utilizes the MyD88-dependent signaling pathway and TLR3 only utilizes the TRIF-dependent signaling pathway (33). Whereas production of ICAM-1, IL-6, IL-8 and MCP-1 is upregulated in human AVICs by stimulation of TLR2, TLR3 or TLR4, IL-37 selectively suppresses the inflammatory responses mediated by TLR2 and TLR4 that share the MyD88 signaling pathway. Thus, IL-37 suppresses the MyD88 -dependent responses but not the TRIF-dependent responses.
Inhibition of NF-κB activation by IL-37 attenuates the MyD88-dependent inflammatory responses. Furthermore, inhibition of NF-κB activation by IL-37 is downstream from IL-37 binding to IL-18R. This observation is consistent with previous reports that extracellular IL-37 binds to IL-18Rα to initiate signal transduction (26). Thus, recombinant IL-37 exerts an effect on human AVICs extracellularly.
In a recent study, we observed that recombinant IL-37 inhibits the production of pro-osteogenic mediators (bone morphogenic protein 2 and alkaline phosphatase) in human AVICs exposed to TLR2 and TLR4 agonists, and that the antiosteogenic effect of IL-37 is due to inhibition of ERK1/2 and NF-κB (23). The antiinflammatory effect of IL-37 observed in the present study is correlated with its inhibitory effect on NF-κB activation. It appears that in human AVICs, the pro-osteogenic and proinflammatory responses to TLR2 and TLR4 agonists are mediated by an overlapping signaling mechanism involving NF-κB. IL-37 inhibits NF-κB activation to suppress the production of both pro-osteogenic and proinflammatory mediators in human AVICs exposed to TLR2 and TLR4 agonists. However, it should be noted that IL-37 can increase the activities of several antiinflammatory signaling pathways, such as PTEN and AMPK (21). These pathways may also affect NF-κB activation observed in human AVICs.
It is known that dsRNA activates NF-κB through TLR3 (40). Previous work demonstrated that TLR3 stimulation also activates NF-κB in human AVICs (41,42). However, we recently found that the TLR3-TRIF cascade activates noncanonical NF-κB in human AVICs, whereas LPS activates canonical NF-κB in this cell type (43). It is likely that IL-37 selectively inhibits MyD88-mediated activation of canonical NF-κB.
CONCLUSION
Stimulation of TLR2, TLR3 or TLR4 in human AVICs upregulates the production of ICAM-1, IL-6, IL-8 and MCP-1. IL-37 suppresses MyD88-mediated upregulation of these inflammatory mediators. These findings suggest that IL-37 has therapeutic potential for suppression of valvular inflammation mediated by TLRs that utilize the MyD88 pathway to upregulate AVIC inflammatory responses.
ACKNOWLEDGMENTS
The authors thank Benjamin Posorske and Lihua Ao for their technical support.
This study was supported in part by National Institutes of Heart, Lung and Blood grants HL121776 and HL106582 (to XM), National Institute of Allergy and Infectious Disease Grant AI15414 (to CAD) and National Natural Sciences Foundation of China grant 81570352 (to QZ). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Zhan Q, et al. (2017) IL-37 suppresses MyD88-mediated inflammatory responses in human aortic valve interstitial cells. Mol. Med. 23:83–91.
References
- 1.Go AS, et al. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hutcheson JD, Aikawa E, Merryman WD. Potential drug targets for calcific aortic valve disease. Nat. Rev. Cardiol. 2014;11:218–31. doi: 10.1038/nrcardio.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajamannan NM, et al. Calcific Aortic Valve Disease: Not Simply a Degenerative Process. A Review and Agenda for Research From the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive Summary: Calcific Aortic Valve Disease-2011 Update. Circulation. 2011;124:1783–91. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ. Res. 2011;108:1392–1412. doi: 10.1161/CIRCRESAHA.110.234138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boström KI, Rajamannan NM, Towler DA. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ. Res. 2011;109:564–77. doi: 10.1161/CIRCRESAHA.110.234278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dweck MR, et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation. 2012;125:76–86. doi: 10.1161/CIRCULATIONAHA.111.051052. [DOI] [PubMed] [Google Scholar]
- 7.Hjortnaes J, et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur. Heart J. 2010;31:1975–84. doi: 10.1093/eurheartj/ehq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aikawa E, et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–86. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 9.Weiss RM, Miller JD, Heistad DD. Fibrocalcific Aortic Valve Disease: Opportunity to Understand Disease Mechanisms Using Mouse Models. Circ. Res. 2013;113:209–22. doi: 10.1161/CIRCRESAHA.113.300153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexopoulos A, et al. Bone regulatory factors NFATc1 and Osterix in human calcific aortic valves. Int. J. Cardiol. 2010;139:142–9. doi: 10.1016/j.ijcard.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 11.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J. Heart Valve Dis. 2004;13:841–7. [PubMed] [Google Scholar]
- 12.Meng X, et al. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. Am. J. Physiol. Cell Physiol. 2008;294:C29–C35. doi: 10.1152/ajpcell.00137.2007. [DOI] [PubMed] [Google Scholar]
- 13.Song R, et al. Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via Toll-like receptor-2. Arterioscler. Thromb. Vasc. Biol. 2012;32:2711–20. doi: 10.1161/ATVBAHA.112.300116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akira S, Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 15.Piccinini A, Midwood K. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010:672395. doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 17.Kaisho T, Akira S. Toll-like receptor function and signaling. J. Allergy Clin. Immunol. 2006;117:979–87. doi: 10.1016/j.jaci.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 18.Fang Y, Hu J. Toll-like receptor and its roles in myocardial ischemic/reperfusion injury. Med. Sci. Monit. 2011;1(7):RA100–09. doi: 10.12659/MSM.881709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Björkbacka H. Multiple roles of Toll-like receptor signaling in atherosclerosis. Curr. Opin. Lipidol. 2006;17:527–33. doi: 10.1097/01.mol.0000245258.25387.ec. [DOI] [PubMed] [Google Scholar]
- 20.Zeng Q, et al. Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of ERK1/2 and nuclear factor-κB activation. Arterioscler. Thromb. Vasc. Biol. 2013;33:1580–90. doi: 10.1161/ATVBAHA.112.300912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dinarello CA, et al. Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 2016;46:1067–81. doi: 10.1002/eji.201545828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boraschi D, et al. IL-37: a new anti -inflammatory cytokine of the IL-1 family. Eur. Cytokine Netw. 2011;22:127–47. doi: 10.1684/ecn.2011.0288. [DOI] [PubMed] [Google Scholar]
- 23.Zeng Q, et al. Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc. Natl. Acad. Sci. USA. 2017;114:1631–6. doi: 10.1073/pnas.1619667114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Messier RH, et al. Dual structural and functional phenotypes of the porcine aortic valve interstitial population: characteristics of the leaflet myofibroblast. J. Surg. Res. 1994;57:1–21. doi: 10.1006/jsre.1994.1102. [DOI] [PubMed] [Google Scholar]
- 25.Pimentel MM, Fuck RA. Neoproterozoic crustal accretion in central Brazil. Geology. 1992;20:375–9. [Google Scholar]
- 26.Nold-Petry CA, et al. IL-37 requires the receptors IL-18R [alpha] and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015;16:354–65. doi: 10.1038/ni.3103. [DOI] [PubMed] [Google Scholar]
- 27.Mohler ER, et al. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–28. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 28.New SE, Aikawa E. Cardiovascular calcification: an inflammatory disease. Circ. J. 2011;75:1305–13. doi: 10.1253/circj.cj-11-0395. [DOI] [PubMed] [Google Scholar]
- 29.Yang X, Fullerton DA, Su X, Ao L, Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J. Am. Coll. Cardiol. 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 30.Zeng Q, et al. Augmented osteogenic responses in human aortic valve cells exposed to oxLDL and TLR4 agonist: a mechanistic role of Notch1 and NF-κB interaction. PLoS One. 2014;9:e95400.. doi: 10.1371/journal.pone.0095400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng Q, et al. Cross-talk between the toll-like receptor 4 and notch1 pathways augments the inflammatory response in the interstitial cells of stenotic human aortic valves. Circulation. 2012;126:S222–30. doi: 10.1161/CIRCULATIONAHA.111.083675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K, Akira S. TLR signaling pathways. Semin. Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Schober A. Chemokines in vascular dysfunction and remodeling. Arterioscler. Thromb. Vasc. Biol. 2008;28:1950–9. doi: 10.1161/ATVBAHA.107.161224. [DOI] [PubMed] [Google Scholar]
- 35.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007;27:2292–2301. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 36.El Husseini D, et al. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: implication for calcific aortic valve disease. J. Mol. Cell. Cardiol. 2014;72:146–56. doi: 10.1016/j.yjmcc.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 37.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of “degenerative” valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–53. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 38.Akira S. Toll-like receptors: lessons from knockout mice. Biochem. Soc. Trans. 2000;28:551–56. doi: 10.1042/bst0280551. [DOI] [PubMed] [Google Scholar]
- 39.Kawai T, Akira S. TLR signaling. Semin. Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Alexopoulou L, Holt A, Medzhitov R, Flavell R. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 41.Zhan Q, et al. Activation of TLR3 induces osteogenic responses in human aortic valve interstitial cells through the NF-κB and ERK1/2 pathways. Int. J. Biol. Sci. 2015;11:482. doi: 10.7150/ijbs.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.López J, et al. Viral and bacterial patterns induce TLR-mediated sustained inflammation and calcification in aortic valve interstitial cells. Int. J. Cardiol. 2012;158:18–25. doi: 10.1016/j.ijcard.2010.12.089. [DOI] [PubMed] [Google Scholar]
- 43.Zhan Q, et al. Double-stranded RNA up-regulates the expression of inflammatory mediators in human aortic valve cells through the TLR3-TRIF-non-canonical NF-κB pathway. Am. J. Physiol. Cell Physiol. 2017;31(2):C407–17. doi: 10.1152/ajpcell.00230.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]