

Abstract
C3 glomerulopathy is a potentially life-threatening disease of the kidney caused by dysregulated alternative pathway complement activation. The specific complement mediator(s) responsible for kidney injury in C3 glomerulopathy are yet to be defined and no specific therapy is currently available. We previously developed a mouse model of lethal C3 glomerulopathy with factor H and properdin gene double mutations. Therefore, we used this model to examine the role of C5 and C5a receptor (C5aR) in the pathogenesis of the disease. Disease severity in these factor H/properdin double mutant mice was found to be correlated with plasma C5 levels, and prophylactic anti-C5 mAb therapy was effective in preventing lethal C3 glomerulopathy. When given to these double mutant mice that had already developed active disease with severe proteinuria, anti-C5 mAb treatment also prevented death in half of the mice. Deficiency of C5aR significantly reduced disease severity, suggesting that C5aR-mediated inflammation contributed to C3 glomerulopathy. Thus, C5 and C5aR have a critical role in C3 glomerulopathy. Hence, early intervention targeting these pathways may be an effective therapeutic strategy for patients with C3 glomerulopathy.
Keywords: C3 glomerulopathy, complement, factor H, properdin, C5, C5a receptor
Introduction
C3 glomerulopathy (C3G) is a kidney disease caused by dysregulated alternative pathway (AP) complement activation. A pathological hallmark of the disease is prominent glomerular C3 deposition with no or scant immunoglobulin presence in the kidney glomeruli.1–3 C3G can be classified into several categories based on its pathological features, including dense deposit disease (DDD) and C3 glomerulonephritis (C3GN). DDD is characterized by prominent intramembranous dense deposits on electron microscopy, whereas the pattern of dense deposit in C3GN is more variable and can present as glomerulonephritis with subendothelial, subepithelial or mesangial immune deposits.4 Both genetic and acquired defects in the complement cascade can cause C3G.2, 3 In the former category, loss of function mutations in the fluid phase complement inhibitors factor H (FH) and factor I and in the cell surface inhibitor membrane cofactor protein (MCP), as well as gain of function mutations in the C3 gene, have been described.1 Acquired defects in C3G include the presence of C3 nephritic factors (C3Nef), IgG autoantibodies that bind to and stabilize the alternative pathway C3 convertase C3bBb, and autoantibodies against factor H and factor B (FB).1 Additionally, familial forms of C3G involving genetic abnormalities in complement factor H-related genes (CFHRs) have been identified and are referred to as CFHR nephropathy.1
Prognosis for C3G is generally poor and up to 50% of patient progress to end-stage renal failure within 10 years of diagnosis.5–7 Current treatments include plasma exchange, antiproteinuric and immunosuppressive reagents, and in some cases renal transplantation.6, 8 However, because most of these options focus on preserving renal function instead of addressing the underlying cause, they are largely ineffective and disease recurrence is common in transplanted tissue.6, 7, 9 Given the current understanding that C3G is a disease caused by abnormal complement activation and injury, it is expected that anti-complement therapy would be the most effective treatment. However, the pathogenic mechanisms of C3G and key complement effectors responsible for kidney injury remain to be fully elucidated. For example, despite the high frequency of C3Nef occurrence in C3G patients, its causal relationship with C3G is yet to be established.1, 5, 10–15 Also, whether upstream complement effectors originating from C3 cleavage or terminal complement activation products such as C5a and the membrane attack complex (MAC) are critical for C3G pathogenesis is uncertain. A better understanding of these issues will help the development of targeted anti-complement therapy for C3G that is safe and effective.
We previously created a lethal C3G mouse model by FH and properdin (P) gene double mutations. In this model, a C-terminal truncation mutation was introduced to FH to produce a FHm/m mouse which was found to have low plasma level of the truncated FH, resulting in extensive fluid phase AP complement activation with secondary C3 and FB insufficiency.16 FHm/m mice developed C3G with prominent glomerular C3 deposition and progressive renal pathology but without early mortality. Surprisingly, introduction of P gene mutation to the FHm/m mouse greatly exacerbated C3G such that FHm/m P−/− mice developed early onset proteinuria and rapidly progressive renal failure leading to premature death.16 In the present study, we have further examined FHm/m and FHm/m P−/− mice to understand the pathogenesis of C3G, specifically the underlying cause for the unexpected transition from mild C3G in FHm/m mice to lethal C3G in FHm/m P−/− mice. We also tested the effectiveness of blocking the terminal complement pathway in preventing lethal C3G of FHm/m P−/− mice. Our data show that plasma C5 level is a key determinant of C3G severity in FHm/m and FHm/m P−/− mice and that blocking C5 or C5aR was effective in treating lethal C3G.
Results
FHm/m/P−/− mouse has significantly higher plasma C5 level than FHm/m mouse
As we described before,16 FHm/m/P−/− mice developed more severe C3G than FHm/m mice. This outcome was unexpected and counterintuitive given that P is a positive regulator of AP complement activation. In the previous study, we found that P deficiency in FHm/m mice reduced C3 and FB consumption.16 This data suggested that partial restoration of AP complement may have contributed to exacerbated kidney injury in FHm/m/P−/− mice. To extend this earlier observation and examine the effect of P deficiency on C5 consumption in FHm/m mice, we used Western blotting and ELISA assays to compare plasma C5 levels in FHm/m and FHm/m/P−/− mice. Fig 1 shows that while WT and P−/− mice had similar levels of plasma C5, FHm/m mice had greatly reduced plasma C5, indicating excessive activation of the terminal complement pathway with C5 consumption. Interestingly, we found that concurrent P deficiency in FHm/m mice markedly increased plasma C5 with some fHm/m/P−/− mice showing C5 levels comparable to that of WT mice (Fig 1). Thus, P contributed significantly to C5 activation and consumption in FHm/m mice.
Figure 1. P deficiency partially restored plasma C5 levels.
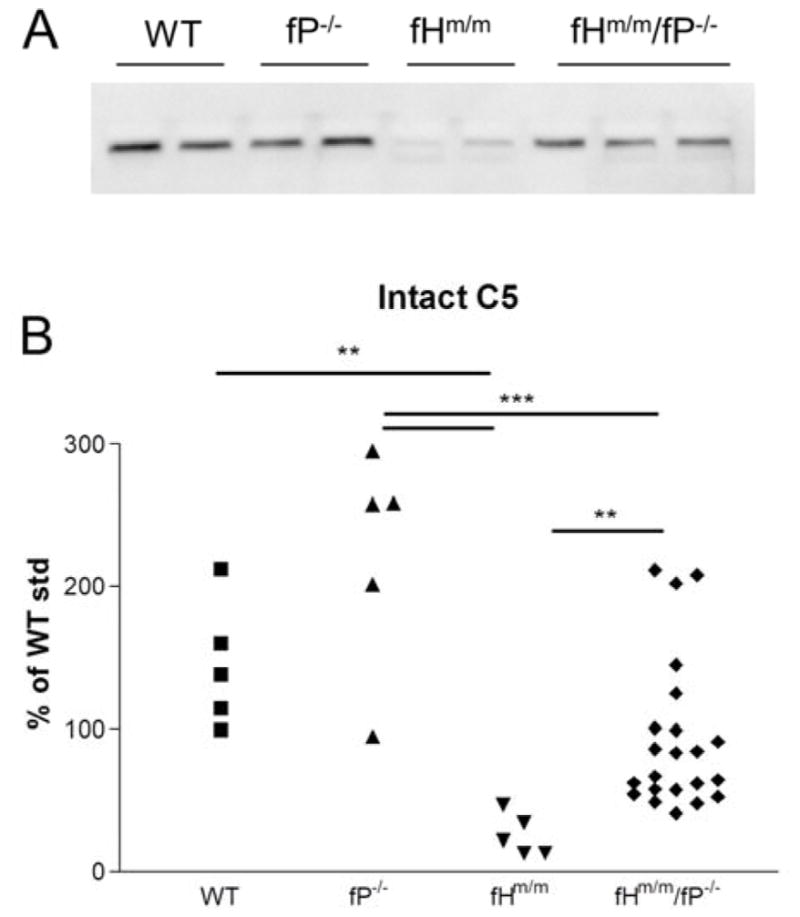
(A) Western blotting analysis of intact plasma C5 in WT, P−/−, fHm/m and fHm/m/P−/− mice. n=2 for WT, P−/−, and fHm/m samples and n=3 for fHm/m/P−/− mice. (B) Intact C5 levels in WT (n=5), P−/− (n=5), fHm/m (n=5) and fHm/m/P−/− mice (n=22). Results correlated with data in panel A, showing C5 levels were significantly decreased in fHm/m mice compared to WT and P−/− mice but were restored to near WT levels in most fHm/m/P−/− mice. ** p<0.01 and *** p<0.001, one-way ANOVA with Tukey’s test. An arbitrary WT mouse plasma sample was used as a reference against which all intact C5 values were normalized. Samples for A and B were from 2–3 month old mice.
C5 consumption in FHm/m mice does not occur in fluid phase
To determine if P-dependent C5 activation and consumption in FHm/m mice occurred in plasma or on the cell surface, we depleted FH from WT and P−/− mouse serum in vitro and assessed C5 activation. We previously observed in a similar experiment rapid and spontaneous C3 activation and consumption when FH was depleted, 16 confirming the key role of FH as a fluid phase AP C3 convertase inhibitor.17 While we again observed C3 consumption in the current experiment (data not shown), we detected no spontaneous C5 activation and consumption in either WT or P−/− mouse serum (Supplemental Fig 1). This result suggested that there is negligible fluid phase C5 convertase activity in the absence of FH, irrespective of whether P is present. Thus, P-dependent C5 activation and consumption in FHm/m mice likely occurred on the cell surface and not in plasma.
Prophylactic anti-C5 treatment of fHm/m/P−/− mice prevents lethal C3G
To determine if increased plasma C5 and terminal complement activation were responsible for lethal C3G in fHm/m/P−/− mice, we treated a group of fHm/m/P−/− mice (n=7) with a neutralizing anti-C5 (BB5.1) or an isotype control IgG (n=6). Mice were treated from 4 weeks of age before significant disease had developed. Fig 2A shows that fHm/m/P−/− mice treated with the control IgG all died or became moribund within 10 weeks of treatment, whereas all mice treated with anti-C5 mAb survived the entire 16-week treatment period. Additionally, progressive proteinuria and hematuria developed in control IgG-treated fHm/m/P−/− mice, but these symptoms were reduced and maintained at baseline levels in anti-C5-treated mice (Fig 2B–C). After 16 weeks of treatment, we euthanized 3 fHm/m/P−/− mice in the anti-C5 group and compared their renal pathology with that of 2 moribund control IgG-treated fHm/m/P−/− mice from which we were able to collect tissues. Mice given control IgG developed numerous glomerular crescents, interstitial inflammation and periglomerular fibrosis, as well as significant dense deposit formation and podocyte effacement (Fig 2D and Table 1). In contrast, anti-C5 treated mice showed largely normal kidney histology with no signs of interstitial fibrosis or glomerular crescents. EM analysis also illustrated that while glomerular dense deposits were still present, there was significant improvement in podocyte integrity in mice treated with anti-C5 (Fig 2D and Table 1). We also examined complement deposition and inflammatory cell infiltration in the glomeruli. C3 staining was observed in both groups at similar levels whereas C9 deposition was absent in anti-C5 mAb treated mice (Fig 2E and supplemental Fig 2). Anti-C5 treatment effectively prevented accumulation of CD11b+ cells in the glomeruli (Supplemental Fig 3). These data confirmed the efficacy and specificity of anti-C5 in blocking terminal complement activation and C5a-mediated glomerular inflammation. Of interest, glomerular IgG staining was increased in anti-C5 treated group (supplemental Fig 2), presumably reflecting trapping of the therapeutic mAb.
Figure 2. C5 inhibition rescued 4-week old fHm/m/P−/− mice from lethal C3G and preserved renal function.

(A) Survival curves of fHm/m/P−/− mice treated with anti-C5 mAb (C5 Ab, blue, n=7) and a control IgG mAb (ctrl IgG, red, n=6). All C5 mAb-treated mice survived the entire 4-month treatment period while mortality in ctrl IgG-treated mice was unaffected. ***p=0.0002, log rank test. (B–C) Urine testing over the course of mAb treatment showing improved renal condition in mice receiving anti-C5 but not the control mAb, as indicated by the absence of significant albuminuria (B) and hematuria (C). (D) PAS staining (i–ii) showed ctrl IgG-treated mice still developed fatal kidney damage with numerous cellular crescents, inflammation and tubulointerstitial injury. By EM, fHm/m/P−/− mice given ctrl IgG showed signs of podocyte injury and foot process effacement, intramembranous dense deposits and mesangial matrix expansion (iii). By contrast, anti-C5 mAb-treated mice demonstrated a significant reduction in inflammatory cell infiltration and lack of glomerular crescents and tubular injury (iv–v). Examination of anti-C5 mAb-treated mice by EM revealed improvement in podocyte foot process integrity, reduction in mesangial expansion and a largely subendothelial dense deposit pattern (vi). Scale bars: panel i and iv = 250 μm; panel ii and v = 50 μm; panel iii and vi = 2 μm. (E–F) Immunofluorescence staining of C3 (E) and C9 (F) in kidneys displayed significant C3 staining in both ctrl IgG and anti-C5 mAb-treated mice. In contrast, C9 deposition was present in ctrl IgG-treated mice but not detectable in anti-C5 mAb-treated mice. Scale bars = 25 μm. Sections in D–E were from moribund ctrl IgG-treated mice at 2–3 months of age whereas anti-C5 mAb-treated mice were studied at 5 months of age, 4 month after anti-C5 mAb treatment.
Table 1.
Summary of kidney pathology in fHm/m/fP−/− mice treated with anti-C5 or control IgG antibody starting at 4 weeks of age
| Genotype | n* | Glomerular Necrosis |
Mesangial Proliferation |
Glomerular Inflammation |
Endocapillary Proliferation |
Membranous Duplication |
Crescents | I/T Inflammation |
Glomerular Sclerosis |
Interstitial Fibrosis |
Tubular Atrophy |
Vascular Change |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4-month treatment | ||||||||||||
| fHm/m/fP−/− + ctrl IgG | 2 (of 6) | 10 | 0 | 50 | 0 | 0 | 90 | 0 | 60 | 10 | 30 | 0 |
| 10 | 0 | 50 | 30 | 50 | 90 | 0 | 40 | 10 | 30 | 0 | ||
| fHm/m/fP−/− + anti-C5 | 3 (of 3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 2-month extension/Crossover treatment | ||||||||||||
| fHm/m/fP−/− + anti-C5 → ctrl IgG | 2 (of 2) | 10 | 50 | 50 | 50 | 50 | 0 | 0 | 0 | 0 | 0 | 0 |
| 10 | 90 | 90 | 90 | 40 | 80 | 80 | 10 | 10 | 10 | 10 | ||
| fHm/m/fP−/− + anti-C5 → anti-C5 | 2 (of 2) | 0 | 20 | 100 | 20 | 0 | 20 | 0 | 10 | 0 | 0 | 0 |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
Data listed above (written as percentage of glomeruli and/or tissue with given characteristic) represent each mouse evaluated for given treatment schedule. n indicates number of kidney tissue samples analyzed (of total number started on treatment). 4 of 6 control IgG mice on 4-month treatment schedule died before tissue could be collected for analysis. Due to limited number of samples, statistical analysis was not performed.
n represents number of tissue samples available for histological analysis from total number of mice treated
To determine if continuous anti-C5 therapy is necessary to maintain viability of fHm/m/P−/− mice, we switched anti-C5 mAb to control IgG in two of the four remaining mice after 16 weeks of anti-C5 treatment. Over the next 8 weeks, both animals developed proteinuria, and one developed severe hematuria and died within 5 weeks of anti-C5 withdrawal (Fig 3, A–C). In contrast, the two fHm/m/P−/− mice that continued to receive anti-C5 treatment survived normally with only baseline proteinuria and no significant change in hematuria scores (Fig 3, A–C). Light microscopy revealed no detectible renal pathology in one of the mice continuing on anti-C5 treatment and only limited signs of renal disease in the other (Fig 3D and Table 1). In comparison, the two fHm/m/P−/− mice that were switched to control IgG developed severe renal pathology including mesangial and endocapilliary proliferations, glomerular inflammation, crescent formation and sclerosis, and membranous duplication (Fig 3D, Table 1 and data not shown). These data suggested that continuous prophylactic C5 inhibition is necessary to prevent lethal C3G in fHm/m/P−/− mice.
Figure 3. Renal disease returned in fHm/m/P−/− mice upon discontinuation of anti-C5 mAb treatment.

(A) Survival curves of fHm/m/P−/− mice treated with anti-C5 mAb for 4 months before switching to control mAb (C5 Ab → ctrl IgG, mouse 1–2, blue) or continuing on C5 mAb treatment (C5 Ab → C5 Ab, mouse 3–4, orange) for an additional 2 months. Black arrows indicate time of change in treatment. One ctrl IgG-treated mouse died while both anti-C5 mAb-treated mice survived. n=2 for each treatment. (B–C) Urine testing over the course of mAb treatment showed worsening renal condition in mice that were switched to control IgG compared to mice still receiving anti-C5 mAb, as illustrated by significant increases in proteinuria (B) and hematuria in the mouse that died (C). Each line in B–C represents one mouse. (D) Mice switched to control IgG (i–ii) showed development of glomerular crescents, inflammation, protein casts and tubulointerstitial injury. On the other hand, mice that continued on anti-C5 mAb treatment appeared to have some capillary wall thickening but did not show signs of significant crescents or tissue damage (iii–iv). Representative PAS staining for each of the 4 mice treated as indicated. Scale bars = 50 μm.
C5 inhibition is partially effective in fHm/m/P−/− mice with preexisting C3 glomerulopathy
We next tested if anti-C5 treatment was effective in fHm/m/P−/− mice that had already developed C3G and if there were any relevant biomarkers for disease severity or potential response to treatment. We prescreened 6-week old fHm/m/P−/− mice and selected those with detectible proteinuria for this experiment. Of 10 such mice that were treated with anti-C5 mAb, 5 (50%) survived a 10-week treatment period but the rest died within 6 weeks of treatment (Fig 4A). Notably, those that survived anti-C5 treatment showed dramatic improvement in kidney health with proteinuria dropping to baseline levels by end of the 10-week treatment period and hematuria maintained at low levels (Fig 4A). We observed an inverse correlation between the severity of preexisting proteinuria and anti-C5 treatment outcome. Mice that had albuminuria in the 0.9–10mg/16h range responded well to anti-C5 treatment with only 1/6 mice failed to survive, whereas all 4 mice with albuminuria in the 18–26mg/16h range died (Fig 4B). In the latter group, albuminuria but not hematuria was also noticeably reduced after anti-C5 treatment (Fig 4B).
Figure 4. Anti-C5 mAb treatment was partially effective in 6-week old fHm/m/P−/− mice with proteinuria.

(A) Survival curve and assessment of renal function in fHm/m/P−/− mice during 10-week treatment (n=10). Only 50% of mice survived entire treatment period, but surviving mice showed marked reduction in albuminuria and consistently low hematuria. (B) Correlation of treatment outcome with pre-treatment level of proteinuria in mice of (A). Mice with low starting albuminuria (0.9–10mg/16h, n=6) responded well whereas mice with high albuminuria (18–26mg/16h, n=4) responded poorly. (C–D) Analysis of intact plasma C5 and C3 as potential biomarkers for disease severity (using albuminuria as readout) in 6-week old naïve fHm/m/P−/− mice (n=13). Mice were stratified into high C5 or C3 (top 50%) and low C5 or C3 (bottom 50%) groups and their level of albuminuria was plotted. Albuminuria was significantly more severe in C5 high group than C5 low group (p=0.018, student’s t-test, panel C) but no difference in albuminuria was observed between C3 high and C3 low groups (panel D). For C5 and C3 measurement, an arbitrary WT mouse plasma sample was used as a reference against which all intact C3 and C5 values were normalized.
Given that P deficiency in fHm/m mice exacerbated C3G and restored plasma C5, and to a lesser extent plasma C3, we investigated if renal disease severity in fHm/m/P−/− mice is correlated with plasma C3 or C5. We measured relative plasma levels of intact (non-activated) C3 and C5 in a separate cohort of 6-week old fHm/m/P−/− mice (n=13) and stratified them into C3 or C5 high (top half) and C3 or C5 low (bottom half) groups. Urine albumin was simultaneously quantified and compared between C3 or C5 high and low groups. Fig 4C shows that there was no difference in albuminuria between C3 high and C3 low mice, but albuminuria was significantly more severe in mice with higher plasma C5 levels (Fig 4D). Thus, plasma C5 but not C3 appeared to predict severity of C3G in fHm/m/P−/− mice.
C5aR pathway plays a significant role in kidney injury in fHm/m/P−/− mice
The anti-C5 mAb BB5.1 prevents C5 cleavage and biological activities of both C5a and C5b.18 To determine if C5a receptor (C5aR)-mediated inflammation specifically contributed to exacerbated C3G in fHm/m/P−/− mice, we crossed the fHm/m mouse with a C5aR-deficient mouse (C5aR−/−) to generate fHm/m/C5aR−/− mice and blocked P in these mice using a neutralizing anti-P mAb. We previously demonstrated that anti-P mAb treatment could mimic P gene deficiency in fHm/m mice to cause exacerbated C3G.16 C5aR gene deficiency alone had no renal implications (data not shown).19 There was also no significant difference in disease severity between fHm/m/C5aR−/− and fHm/m mice at 8–12 week of age as assessed by light and electron microscopy, proteinuria, hematuria, plasma C3 and C5 levels (Suppl Figures 4, 5 and data not shown). We then treated 6-week old fHm/m/C5aR−/− mice and their fHm/m littermates with anti-P mAb for 22 weeks and assessed kidney disease. While two of the five fHm/m mice (40%) treated with anti-P mAb died during the course of treatment, all treated fHm/m/C5aR−/− mice (n=8) survived. Furthermore, anti-P mAb-treated fHm/m/C5aR−/− mice showed strikingly reduced albuminuria compared with similarly treated fHm/m mice (Fig 5A). Anti-P mAb-treated fHm/m mouse kidney sections also showed hallmarks of severe C3G, including glomerular inflammation, mesangial proliferation, crescent formation and electron dense deposit (Fig 5B and Table 2). In contrast, anti-P mAb-treated fHm/m/C5aR−/− mouse kidneys had significantly less pathology (Fig 5B and Table 2). Immunofluorescence staining showed no difference in glomerular C3 and C9 staining between the two groups of mice, but glomerular infiltration of CD11b+ cells was reduced in anti-P mAb-treated fHm/m/C5aR−/− mice (Fig 6 and supplemental Figs 6, 7), suggesting that C5aR deficiency protected mice from C3G by ameliorating glomerular inflammation.
Figure 5. C5aR deficiency ameliorated renal disease in fHm/m mice treated with anti-P mAb.

(A) Urine testing in fHm/m (n=5) and fHm/m/C5aR−/− (n=8) mice treated with anti-P mAb showed significantly lower proteinuria in fHm/m/C5aR−/− mice than in fHm/m mice. Hematuria remained relatively low and showed no significant difference between the two groups. Arrow indicates time of death of 2 fHm/m mice after anti-P mAb treatment was initiated. (B) Histological findings correlated with proteinuria (i/iv, PAS; ii/v H&E; iii/vi, EM). All fHm/m mice treated with anti-P mAb showed signs of C3G including mesangial proliferation (i), glomerular crescents and fibrosis (i–ii), glomerular inflammation, podocyte injury and glomerular dense deposits (iii). While GBM thickening and dense deposits persisted, fHm/m/C5aR−/− mice treated with anti-P mAb (iv–vi) showed better renal tissue integrity with reduced mesangial expansion (iv), reduced inflammation and absence of glomerular crescents (v), and improved podocyte integrity (vi). Dense deposits are marked on panel iii and vi with small arrows. Scale bars: panels i, ii, iv, v = 50 μm; panels iii, vi = 2 μm.
Table 2.
Summary of kidney pathology in fHm/m + anti-P and fHm/m/C5aR−/− + anti-P mice
| Genotype | n* | Glomerular Necrosis |
Mesangial Proliferation |
Glomerular Inflammation |
Endocapillary Proliferation |
Membranous Duplication |
Crescents | I/T Inflammation |
Glomerular Sclerosis |
Interstitial Fibrosis |
Tubular Atrophy |
Vascular Change |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| fHm/m + anti-P | 4 (of 5) | 50 | 50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 50 | 90 | 50 | 90 | 0 | 95 | 0 | 0 | 0 | 20 | 0 | ||
| 20 | 50 | 50 | 80 | 90 | 50 | 0 | 50 | 20 | 20 | 0 | ||
| 20 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| fHm/m/C5aR−/− + anti-P | 8 (of 8) | 0 | 0 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0 | 0 | 50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Data listed above (written as percentage of glomeruli and/or tissue with given characteristic) represent each mouse evaluated for given treatment schedule. n indicates number of kidney tissue samples analyzed (of total number started on treatment). 1 of 5 fHm/m + anti-P Ab treated mice died before tissue could be collected for analysis. Due to limited number of fHm/m + anti-P Ab samples, statistical analysis was not performed.
n represents number of tissue samples available for histological analysis from total number of mice treated
Figure 6. Immunofluorescence staining of inflammatory cells and complement deposition.

(A–B) Glomerular C3 (A) and C9 (B) staining in fHm/m and fHm/m/C5aR−/− mice treated with anti-P mAb were similar. Panels show representative images of C3 and C9 staining of kidney section from 2–5 mice. (C–D) CD11b+ inflammatory cells (green) were reduced both in glomeruli (C) and interstitium (D) of fHm/m/C5aR−/− mice treated with anti-P mAb compared to similarly treated fHm/m mice. Panels show representative images of immunofluorescence staining of kidney sections from 4–5 mice. Counterstain = DAPI (blue). Scale bars = 25 μm.
Discussion
We established in this study that a major consequence of P deficiency in fHm/m mice is reduced C5 consumption with marked recovery of plasma C5. Although in our earlier studies we observed also an increase in plasma C3 in fHm/m/P−/− mice compared with fHm/m mice,16 the extent of C5 recovery in the double mutant mice is much more pronounced. A stronger effect of P deficiency on plasma C5 was also independently noted in a separate FH and P double mutant mouse.20 The differential effect of P on C3 and C5 profiles in these models may be attributed to the following: 1) in the absence of FH, C5 was primarily activated and consumed on the cell surface whereas C3 was activated primarily in the fluid phase,16, 21 and 2) P is a stabilizer of surface-bound C3 and C5 convertases only with little effect on fluid phase C3 or C5 activation (Supplemental Fig 1).16, 21–26 Thus, C5 depletion in fHm/m mice depended on P whereas C3 depletion was largely independent of P. This may explain why P deficiency had a more dramatic effect on plasma C5 level in fHm/m/P−/− mice. The tissue(s) or organ(s) on which P-dependent C5 consumption occurred in fHm/m mice remains to be defined, but it is clear that such activation was less consequential as fHm/m mice were viable and did not show multi-organ pathologies.16 It is possible that C5 was activated on a wide range of tissue(s) or organ(s) in fHm/m mice such that its injurious potential was dissipated and damage to any give tissue or organ was limited.
We hypothesize that restoration of plasma C5 and a functional terminal complement pathway caused the transition from mild C3G in fHm/m mice to lethal C3G in fHm/m/P−/− mice. This theory was supported by the observation that plasma C5 level was correlated with severity of C3G in fHm/m/P−/− mice, and that prophylactic C5 inhibition prevented lethal C3G. That P was not required for C5 activation and kidney injury in fHm/m/P−/− mice may be explained by the unique structural characteristics of the kidney glomerular basement membrane (GBM). The GBM is an extracellular matrix that, unlike other cellular surfaces, lacks membrane complement regulators and this leaves FH as the critical complement inhibitor that protects the GBM from AP complement attack. Accordingly, in the context of FH insufficiency, AP and terminal complement activation on the GBM was unhindered, occurring spontaneously without the need of P stabilization of the convertases. In essence, the transition from mild C3G in fHm/m mice to lethal C3G in fHm/m/P−/− mice reflected a shunting from P-dependent C3/C5 activation and consumption on multiple tissues/organs to P-independent and selective C3/C5 activation on the kidney GBM, thus leading to exacerbated injury and lethal C3G.
The difference in efficacy between prophylactic and therapeutic anti-C5 treatment for lethal C3G in fHm/m/P−/− mice is notable. Blocking C5 in mice with no or less severe proteinuria (<10mg/16h) not only kept these mice alive but also prevented or ameliorated proteinuria and hematuria. On the other hand, anti-C5 treatment was not able to rescue fHm/m/P−/− mice with severe preexisting proteinuria (18–26 mg/16h). A possible explanation for the latter case, which remains to be investigated experimentally, is that loss of therapeutic mAb through proteinuria has occurred and this could have reduced the effective concentrations of the drug in plasma. It is also possible that in such mice kidney injury had reached a point of no-return and blocking terminal complement activation was no long able to reverse tissue injury and restore renal function. Regardless, our data suggest that if C5 and terminal complement pathway play a similar role in human C3G pathogenesis, disease stage and history may determine a patient’s response to anti-C5 therapy. Patients with early stages of C3G or newly transplanted kidneys may be more responsive to anti-C5 treatment.
An important question is whether terminal complement activation is critical to renal injury in human C3G patients as in fHm/m/P−/− mice. Eculizumab, a humanized anti-C5 mAb, has been used to treat human C3G in a number of case studies and in a small clinical trial involving six C3G patients with either native or transplanted kidneys.27–36 The outcome of these studies is mixed, with some patients responding positively and achieving significantly improved renal functions while other patients gained no benefit.37 Of interest, in some of these studies a correlation between serum sC5b-9 level and anti-C5 response has been noted but it is not yet established if serum sC5b-9 is a useful biomarker for anti-C5 response in general. Failure to respond to Eculizumab treatment by some C3G patients could be interpreted as an indication that C3-derived effectors rather than terminal complement activation plays a dominant role in their C3G pathogenesis.1, 28, 33 Based on the mouse studies presented here, an alternative explanation for Eculizumab non-responsiveness could be that renal injury in those patients had reached a point of no-return. Indeed, the partial response rate of anti-C5-treated fHm/m/P−/− mice with severe preexisting C3G was reminiscent of the mixed clinical outcome of Eculizumab-treated human C3G patients. While our data does not exclude the involvement of C3-derived effectors in C3G pathogenesis, it is notable that prophylactic anti-C5 treatment for 16 weeks effectively prevented lethal C3G in fHm/m/P−/− mice despite continuous plasma C3 activation and glomerular C3 deposition and GBM dense deposit (Fig 2). On the other hand, although fHm/m/P−/− mice on prolonged anti-C5 treatment survived and appeared to be grossly healthy, residual renal pathology was noted upon terminal examination (Table 2), suggesting that either anti-C5 therapy is losing efficacy or that C3 activation products contributed to renal injury in a longer time frame. Regardless, given that C3 activation is upstream of C5 activation, a therapeutic strategy targeting C3 would be expected to be effective even if the terminal complement pathway is the more critical pathogenic effector.
We showed that the detrimental effect of C5 and terminal complement is primarily mediated by the C5aR pathway and that complement-induced glomerular inflammation may be a major pathogenic mechanism in C3G. This finding is consistent with the notion and clinical observation that, if given early, conventional anti-inflammatory and immunosuppressive therapy could benefit C3G patients.38 Residual disease in fHm/m/C5aR−/− mice presumably reflected partial contribution of C5b-dependent membrane attack complexes and/or upstream C3-derived complement effectors. Given the significant effect of C5aR deficiency on disease development, anti-complement drugs that target the C5aR pathway may be expected to benefit human C3G patients.
In summary, our study of fHm/m and fHm/m/P−/− mice have revealed a key role of P in C5 activation and consumption in the context of fH insufficiency, and we demonstrated here that restoration of plasma C5 was responsible for exacerbated C3G in fHm/m/P−/− mice. Further, C5-dependent renal injury in fHm/m/P−/− mice was primarily mediated by the C5aR pathway. While in some of our experiments the striking data was based on limited number of mice and murine models do not always faithfully reproduce human diseases, when taken together our results suggest that early intervention targeting C5 or C5aR may be an effective therapeutic strategy for human C3G patients. Finally, in addition to sC5b-9 plasma C5 may be another useful biomarker for predicting the responsiveness of therapies targeting the terminal complement pathway.
Materials and Methods (supplemental Methods are available online)
Mice
The generation and source of fHm/m, P−/−, fHm/m/P−/− and C5aR−/− mice have been reported previously.16, 19, 39, 40 Except the C5aR−/− strain which are of C57BL/6 background, all mice were on a C57BL/6-129J mixed background. All experiments used age-matched littermates as controls and were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Measurement of proteinuria
Urine albumin was quantified using a mouse albumin ELISA kit according to manufacturer’s instructions (Bethyl Laboratories). Urine samples were collected in metabolic cages for 16h and volumes recorded. Total urine albumin was determined by multiplying albumin concentration (determined by ELISA) by total urine volume/16h.
C5 inhibition in fHm/m/P−/− mice
Three series of experiments were performed with the monoclonal mouse anti-mouse C5 Ab BB5.1, a function-blocking mouse anti-C5 IgG (anti-C5 Ab) that binds to intact C5 to prevent the cleavage of C5 into C5a and C5b.18 The mAb was purified from mouse ascites produced by Cocalico Biologicals, Inc. (Reamstown, PA, USA) and dialyzed against PBS.18, 41 In the first experiment, 4-week old fHm/m/P−/− mice were treated twice weekly (1 mg/mouse, i.p.) with mAb BB5.1. A control group of fHm/m/P−/− mice were given an irrelevant isotype control mouse IgG1 Ab (purified from MoPC 31C hybridoma, ATCC) on the same dosing schedule. Mice were treated for a total of 16 weeks, and survival was observed over the treatment period. To assess renal function, plasma and urine samples were collected every 2 weeks. Urine albumin was quantified as described above, and proteinuria, hematuria and leukocyturia were assessed semi-quantitatively using color-indicator strips (Uristix, Siemens). In the second experiment, a group of mice treated with anti-C5 Ab for 16 weeks continued to receive C5 mAb for 8 more weeks while a second group were switched to control mAb (MoPC 31C) for 8 weeks. In each case, the same treatment regimen, 1 mg/mouse twice weekly, was followed. Survival and renal function were assessed as described above. In the third experiment, 6-week old fHm/m/P−/− mice were screened for proteinuria by overnight (16h) urine collection and Uristix analysis. Mice with significant proteinuria, defined as a semi-quantitative score of 1.5–2 or higher by Uristix analysis, were treated with the anti-C5 mAb on the same dosing schedule as the previous two experiments for a total of 10 weeks. Survival and renal function were assessed as described above.
Properdin inhibition in fHm/m and fHm/m/C5aR−/− mice
Beginning at 6 weeks of age, fHm/m and fHm/m/C5aR−/− mice were treated weekly (1 mg/mouse, i.p.) with mAb 14E1, an inhibitory mouse anti-mouse P IgG (anti-P Ab).42 Mice were treated for a total of 22 weeks, and survival was assessed over the course of the experiment. To assess renal function, plasma and urine samples were collected every 2 weeks. Urine albumin was measured as described above, and hematuria and leukocyturia were assessed semi-quantitatively using color-indicator strips (Uristix, Siemens).
Grading of renal pathology
Renal sections (all stained with H&E and PAS, some also with trichrome) were graded for pathology in a blind fashion. The following characteristics were recorded: glomerular necrosis and sclerosis, mesangial proliferation, glomerular inflammation, endocapillary proliferation, membranous duplication, glomerular crescent formation, interstitial inflammation and fibrosis, tubular atrophy and vascular change. The severity of each change was graded as a percentage (0–100%). Approximately 50 glomeruli and surrounding tissue were examined for each sample.
Supplementary Material
Acknowledgments
Sources of support:
This work is supported by NIH grants AI085596, AI44970, AI49344, a grant from the Kidneeds Foundation (to WCS) and a pre-doctoral fellowship from the American Heart Association (to ALW).
Footnotes
Disclosures
All authors declared no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pickering MC, D’Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Vriese AS, Sethi S, Van Praet J, et al. Kidney Disease Caused by Dysregulation of the Complement Alternative Pathway: An Etiologic Approach. J Am Soc Nephrol. 2015;26:2917–2929. doi: 10.1681/ASN.2015020184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angioi A, Fervenza FC, Sethi S, et al. Diagnosis of complement alternative pathway disorders. Kidney Int. 2016;89:278–288. doi: 10.1016/j.kint.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Fakhouri F, Fremeaux-Bacchi V, Noel LH, et al. C3 glomerulopathy: a new classification. Nat Rev Nephrol. 2010;6:494–499. doi: 10.1038/nrneph.2010.85. [DOI] [PubMed] [Google Scholar]
- 5.Servais A, Noel LH, Roumenina LT, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 6.Barbour TD, Pickering MC, Cook HT. Recent insights into C3 glomerulopathy. Nephrol Dial Transplant. 2013;28:1685–1693. doi: 10.1093/ndt/gfs430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith RJ, Harris CL, Pickering MC. Dense deposit disease. Mol Immunol. 2011;48:1604–1610. doi: 10.1016/j.molimm.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith RJ, Alexander J, Barlow PN, et al. New approaches to the treatment of dense deposit disease. J Am Soc Nephrol. 2007;18:2447–2456. doi: 10.1681/ASN.2007030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nester CM, Smith RJ. Diagnosis and treatment of C3 glomerulopathy. Clin Nephrol. 2013;80:395–403. doi: 10.5414/CN108057. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Meyer NC, Wang K, et al. Causes of alternative pathway dysregulation in dense deposit disease. Clin J Am Soc Nephrol. 2012;7:265–274. doi: 10.2215/CJN.07900811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sethi S, Fervenza FC, Zhang Y, et al. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82:465–473. doi: 10.1038/ki.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwertz R, Rother U, Anders D, et al. Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: A long-term follow-up. Pediatr Allergy Immunol. 2001;12:166–172. doi: 10.1034/j.1399-3038.2001.012003166.x. [DOI] [PubMed] [Google Scholar]
- 13.Paixao-Cavalcante D, Lopez-Trascasa M, Skattum L, et al. Sensitive and specific assays for C3 nephritic factors clarify mechanisms underlying complement dysregulation. Kidney Int. 2012;82:1084–1092. doi: 10.1038/ki.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanuma Y, Ohi H, Hatano M. Two types of C3 nephritic factor: properdin-dependent C3NeF and properdin-independent C3NeF. Clin Immunol Immunopathol. 1990;56:226–238. doi: 10.1016/0090-1229(90)90144-f. [DOI] [PubMed] [Google Scholar]
- 15.Ohi H, Watanabe S, Fujita T, et al. Significance of C3 nephritic factor (C3NeF) in non-hypocomplementaemic serum with membranoproliferative glomerulonephritis (MPGN) Clin Exp Immunol. 1992;89:479–484. doi: 10.1111/j.1365-2249.1992.tb06984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesher AM, Zhou L, Kimura Y, et al. Combination of factor H mutation and properdin deficiency causes severe C3 glomerulopathy. J Am Soc Nephrol. 2013;24:53–65. doi: 10.1681/ASN.2012060570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Human Complement C3b Inactivator - Isolation, Characterization, and Demonstration of an Absolute Requirement for Serum-Protein Beta-1h for Cleavage of C3b and C4b in Solution. J Exp Med. 1977;146:257–270. doi: 10.1084/jem.146.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frei Y, Lambris JD, Stockinger B. Generation of a monoclonal antibody to mouse C5 application in an ELISA assay for detection of anti-C5 antibodies. Mol Cell Probes. 1987;1:141–149. doi: 10.1016/0890-8508(87)90022-3. [DOI] [PubMed] [Google Scholar]
- 19.Hopken UE, Lu B, Gerard NP, et al. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature. 1996;383:86–89. doi: 10.1038/383086a0. [DOI] [PubMed] [Google Scholar]
- 20.Ruseva MM, Vernon KA, Lesher AM, et al. Loss of properdin exacerbates C3 glomerulopathy resulting from factor H deficiency. J Am Soc Nephrol. 2013;24:43–52. doi: 10.1681/ASN.2012060571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Mol Immunol. 2013;56:191–198. doi: 10.1016/j.molimm.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farries TC, Lachmann PJ, Harrison RA. Analysis of the interactions between properdin, the third component of complement (C3), and its physiological activation products. Biochem J. 1988;252:47–54. doi: 10.1042/bj2520047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rawal N, Pangburn MK. Structure/function of C5 convertases of complement. Int Immunopharmacol. 2001;1:415–422. doi: 10.1016/s1567-5769(00)00039-4. [DOI] [PubMed] [Google Scholar]
- 24.Gotze O, Muller-Eberhard HJ. The role of properdin in the alternate pathway of complement activation. J Exp Med. 1974;139:44–57. doi: 10.1084/jem.139.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Medicus RG, Götze O, Müller-Eberhard HJ. Activation of Properdin (P) and Assembly and Regulation of the Alternative Pathway C5 Convertase. J Immunol. 1976;116:1741–1742. [Google Scholar]
- 26.Medicus RG, Gotze O, Muller-Eberhard HJ. Alternative pathway of complement: recruitment of precursor properdin by the labile C3/C5 convertase and the potentiation of the pathway. J Exp Med. 1976;144:1076–1093. doi: 10.1084/jem.144.4.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuber J, Fakhouri F, Roumenina LT, et al. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–657. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 28.Bomback AS, Smith RJ, Barile GR, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012;7:748–756. doi: 10.2215/CJN.12901211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herlitz LC, Bomback AS, Markowitz GS, et al. Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol. 2012;23:1229–1237. doi: 10.1681/ASN.2011121186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radhakrishnan S, Lunn A, Kirschfink M, et al. Eculizumab and refractory membranoproliferative glomerulonephritis. N Engl J Med. 2012;366:1165–1166. doi: 10.1056/NEJMc1106619. [DOI] [PubMed] [Google Scholar]
- 31.Vivarelli M, Pasini A, Emma F. Eculizumab for the treatment of dense-deposit disease. N Engl J Med. 2012;366:1163–1165. doi: 10.1056/NEJMc1111953. [DOI] [PubMed] [Google Scholar]
- 32.Daina E, Noris M, Remuzzi G. Eculizumab in a patient with dense-deposit disease. N Engl J Med. 2012;366:1161–1163. doi: 10.1056/NEJMc1112273. [DOI] [PubMed] [Google Scholar]
- 33.Gurkan S, Fyfe B, Weiss L, et al. Eculizumab and recurrent C3 glomerulonephritis. Pediatr Nephrol. 2013;28:1975–1981. doi: 10.1007/s00467-013-2503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCaughan JA, O’Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant. 2012;12:1046–1051. doi: 10.1111/j.1600-6143.2011.03923.x. [DOI] [PubMed] [Google Scholar]
- 35.Oosterveld MJ, Garrelfs MR, Hoppe B, et al. Eculizumab in Pediatric Dense Deposit Disease. Clin J Am Soc Nephrol. 2015;10:1773–1782. doi: 10.2215/CJN.01360215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Quintrec M, Lionet A, Kandel C, et al. Eculizumab for treatment of rapidly progressive C3 glomerulopathy. Am J Kidney Dis. 2015;65:484–489. doi: 10.1053/j.ajkd.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 37.Bomback AS. Eculizumab in the treatment of membranoproliferative glomerulonephritis. Nephron Clin Pract. 2014;128:270–276. doi: 10.1159/000368592. [DOI] [PubMed] [Google Scholar]
- 38.Rabasco C, Cavero T, Roman E, et al. Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int. 2015;88:1153–1160. doi: 10.1038/ki.2015.227. [DOI] [PubMed] [Google Scholar]
- 39.Kimura Y, Miwa T, Zhou L, et al. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood. 2008;111:732–740. doi: 10.1182/blood-2007-05-089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Kimura Y, Fang C, et al. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110:228–236. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, Miwa T, Hilliard B, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. 2005;201:567–577. doi: 10.1084/jem.20040863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miwa T, Sato S, Gullipalli D, et al. Blocking properdin, the alternative pathway, and anaphylatoxin receptors ameliorates renal ischemia-reperfusion injury in decay-accelerating factor and CD59 double-knockout mice. J Immunol. 2013;190:3552–3559. doi: 10.4049/jimmunol.1202275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.