

Abstract
Sporadic colorectal cancer (CRC) is classified into several molecular subtypes. We previously established two groups of DNA methylation markers through genome‐wide DNA methylation analysis to classify CRC into distinct subgroups: high‐, intermediate‐, and low‐methylation epigenotypes (HME, IME, and LME, respectively). HME CRC, also called CpG island methylator phenotype (CIMP)‐high CRC, shows methylation of both Group 1 markers (CIMP markers) and Group 2 markers, while IME/CIMP‐low CRC shows methylation of Group 2, but not of Group 1 markers, and LME CRC shows no methylation of either Group 1 or Group 2 markers. While BRAF‐ and KRAS‐mutation(+) CRC strongly correlated with HME and IME, respectively, clinicopathological features of NRAS‐mutation(+) CRC, including association with DNA methylation, remain unclear. To characterize NRAS‐mutation(+) CRC, the methylation levels of 19 methylation marker genes (6 Group 1 and 13 Group 2) were analyzed in 61 NRAS‐mutation(+) and 144 NRAS‐mutation(−) CRC cases by pyrosequencing, and their correlation with clinicopathological features was investigated. Different from KRAS‐mutation(+) CRC,NRAS‐mutation(+) CRC significantly correlated with LME. NRAS‐mutation(+) CRC showed significantly better prognosis than KRAS‐mutation(+) CRC (P = 3 × 10−4). NRAS‐mutation(+) CRC preferentially occurred in elder patients (P = 0.02) and at the distal colon (P = 0.006), showed significantly less lymph vessel invasion (P = 0.002), and correlated with LME (P = 8 × 10−5). DNA methylation significantly accumulated at the proximal colon. NRAS‐mutation(+) CRC may constitute a different subgroup from KRAS‐mutation(+) CRC, showing significant correlation with LME, older age, distal colon, and relatively better prognosis.
Keywords: BRAF mutation, colorectal cancer, DNA methylation, KRAS mutation, NRAS mutation
Introduction
Cancer arises through accumulation of genomic and epigenomic alternations 1, 2. Comprehensive genomic analyses of colorectal cancer (CRC) have been reported. Several important molecular aberrations are involved in CRC development, with disruption of critical signaling cascades such as RAS/RAF/ERK, WNT, TP53, and TGF‐β pathways 3, 4, 5. Epidermal growth factor receptor (EGFR) is activated in about 80% of CRC 6, leading to activation of the downstream RAS/RAF/ERK signaling and playing a significant role in tumor progression 6, 7. Cetuximab, a monoclonal antibody blocking the interaction between EGFR and its ligands, inhibits the downstream RAS signaling cascade and ERK activation in CRC therapy, and another EGFR‐neutralizing antibody, panitumumab, is available 8. Price et al. showed that these agents provided similar survival benefit, with more than 50% of participants having an overall survival longer than 10 months 9.
Downstream of EGFR, activating mutations of RAS and RAF also contribute to CRC development 10. BRAF mutation is observed in 5–10% of CRC and is mostly accompanied with frequent DNA hypermethylation and microsatellite instability (MSI) due to aberrant methylation of the MLH1 promoter 11, 12. KRAS mutation is more frequently observed in 35–40% of CRC 13, 14, 15, 16, 17. KRAS‐mutation(+) CRC reportedly shows worse prognosis, even under 5‐FU based chemotherapy 18, 19, 20. Targeting the EGFR using EGFR‐neutralizing antibodies is ineffective for treatment of CRC with mutation of these oncogenes because the RAS/RAF/ERK signaling cascade is downstream of EGFR 21, 22. NRAS mutation occurs infrequently at 2.6–4.2% 23, 24, 25, 26, 27, 28, 29, 30, 31, 32. NRAS‐mutation(+) CRC prognosis remains controversial, since only few studies on its prognosis, analyzing 4–73 cases, have been reported 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33. While Gavin et al. reported no significant difference in prognosis between 73 NRAS‐mutation(+) and 750 KRAS‐mutation(+) cases 26, other groups studied 4–35 NRAS‐mutation(+) cases and did neither reveal significant difference in prognosis nor conduct prognosis analysis 23, 24, 25, 27, 28, 29, 30, 31, 32.
We and others previously stratified CRC using comprehensive and quantitative DNA methylation data 18, 34. In 1999, Toyota et al. reported CRC subtypes with frequent CpG island hypermethylation, so‐called CpG island methylator phenotype (CIMP) 11. In 2010, Yagi et al. established two groups of methylation marker genes, Group 1 and Group 2 markers, to classify CRC into three distinct epigenotypes: high‐, intermediate‐, and low‐methylation epigenotypes (HME, IME, and LME, respectively) 18. While Group 1 markers are mostly equivalent to classical CIMP markers 11, 12, 35, 36, HME/CIMP‐high CRC shows methylation of both Group 1 and Group 2 markers, and strongly correlate with BRAF‐mutation(+). IME/CIMP‐low CRC shows methylation of Group 2, but not Group 1 markers, and strongly correlates with KRAS‐mutation(+). LME CRC shows no methylation of Group 1 and Group 2 markers, and do not correlate with mutations in these oncogenes 18, 37.
However, molecular features of NRAS‐mutation(+) CRC, including DNA methylation epigenotype and its clinicopathological features, are largely unknown. We therefore investigated DNA methylation levels in 61 NRAS‐mutation(+) CRC and 144 NRAS‐mutation(−) CRC samples using 6 Group 1 and 13 Group 2 methylation markers by pyrosequencing to characterize epigenetic features of NRAS‐mutation(+) CRC and analyzed its clinicopathological features.
Materials and Methods
Clinical samples
A total of 2045 CRC samples were obtained from CRC patients who underwent surgery at Saitama Cancer Center with written informed consent, and kept frozen until use. Among the 2045 CRC samples, 61 cases were positive for NRAS mutation, based on mutation analysis described below. In addition to these 61 NRAS‐mutation(+) CRC samples, 144 NRAS‐mutation(−) CRC samples, including 70 cases whose epigenotypes were already estimated in our previous study (10 HME, 30 IME, and 30 LME) 14, underwent subsequent analyses. CRC specimens were microscopically examined for determination of cancer cell contents by two independent pathologists. Samples that contained at least 40% of cancer cells were used for the subsequent analyses, and the specimens were dissected to enrich cancer cells when necessary. DNA was extracted using QIAamp DNA Micro Kit (Qiagen, Hilden, Germany). Their clinicopathological features, for example, age, gender, tumor location, mucinous component, and tumor stage based on American Joint Committee on Cancer (AJCC), lymph node metastasis, lymph vessel invasion, venous invasion, and microsatellite instability, are summarized in Table 1. This study was certified by the Ethics Committee of Chiba University and Saitama Cancer Center.
Table 1.
Comparison of clinicopathological features of CRC excluding stage I cases
| Clinical features | All cases | BRAF | KRAS | NRAS | No‐mut | P‐value (K vs. N vs. No) | P‐value (K vs. N) |
|---|---|---|---|---|---|---|---|
| Number of samples | 186 | 10 | 59 | 45 | 72 | ||
| Gender | |||||||
| Male | 110 | 9 | 33 | 20 | 48 | 0.2 | 0.7 |
| Female | 70 | 1 | 26 | 19 | 24 | ||
| Unknown | 6 | 0 | 0 | 6 | 0 | ||
| Age (y.o.) | |||||||
| Mean ± SD | 63.8 ± 9.4 | 72.0 ± 8.6 | 61.7 ± 9.4 | 66.0 ± 9.2 | 62.0 ± 9.1 | 0.04a | 0.02a |
| Tumor location | |||||||
| Proximal | 55 | 10 | 23 | 5 | 17 | 0.01a | 0.006a |
| Distal | 124 | 0 | 36 | 33 | 55 | ||
| Unknown | 7 | 0 | 0 | 7 | 0 | ||
| Mucinous component | |||||||
| (+) | 27 | 6 | 14 | 2 | 5 | 0.005a | 0.02a |
| (−) | 152 | 4 | 45 | 36 | 67 | ||
| Unknown | 7 | 0 | 0 | 7 | 0 | ||
| AJCC stage | |||||||
| I | 54 | 5 | 17 | 16 | 16 | 0.3 | 0.5 |
| III | 61 | 3 | 21 | 11 | 26 | ||
| IV | 65 | 2 | 21 | 12 | 30 | ||
| Unknown | 6 | 0 | 0 | 6 | 0 | ||
| Lymph node metastasis | |||||||
| (+) | 104 | 5 | 39 | 17 | 43 | 0.2 | 0.09 |
| (−) | 72 | 5 | 20 | 19 | 28 | ||
| Unknown | 10 | 0 | 0 | 9 | 1 | ||
| Lymph vessel invasion | |||||||
| (+) | 130 | 10 | 48 | 19 | 53 | 0.003a | 0.002a |
| (−) | 49 | 0 | 11 | 19 | 19 | ||
| Unknown | 7 | 0 | 0 | 7 | 0 | ||
| Venous invasion | |||||||
| (+) | 149 | 8 | 48 | 31 | 62 | 0.7 | 1.0 |
| (−) | 30 | 2 | 11 | 7 | 10 | ||
| Unknown | 7 | 0 | 0 | 7 | 0 | ||
| Microsatellite instability | |||||||
| MSI‐H | 14 | 8 | 3 | 0 | 3 | 0.4 | 0.3 |
| MSS | 164 | 2 | 56 | 37 | 69 | ||
| Unknown | 8 | 0 | 0 | 8 | 0 | ||
| Methylation epigenotype | |||||||
| HME | 10 | 6 | 2 | 0 | 2 | 2 × 10−4 a | 8 × 10−5 a |
| IME | 84 | 4 | 40 | 13 | 27 | ||
| LME | 92 | 0 | 17 | 32 | 43 | ||
No‐mut, no mutation; K vs. N vs. No, KRAS versus NRAS versus no mutation; K vs. N, KRAS versus NRAS; MSI‐H, microsatellite instability high; MSS, microsatellite stable; HME, high‐methylation epigenotype; IME, intermediate‐methylation epigenotype; LME, low‐methylation epigenotype.
P < 0.05
Mutation analysis
KRAS mutation (codons 12, 13, and 19) and NRAS mutation (codon 12, 13, 59, and 61) were analyzed as described previously 18, 38. BRAF mutation (V600E) at exon 15 was determined by direct sequencing using pyrosequencing as previously reported 39. The cut‐off value for positive result of mutation was set at 20% on the sequencer, considering the tumor cell content (≥40%).
Bisulfite treatment
Bisulfite conversion of 500 ng of genomic DNA from each tissue sample was performed using Zymo EZ DNA Methylation Kit (Zymo Research, Irvine, CA), and the DNA was eluted in 80 μL of 10 mEq Tris buffer. By bisulfite treatment, unmethylated cytosine is converted to uracil, that is, recognized as thymine (T) after PCR, but methylated cytosine is not converted, that is, cytosine (C) after PCR. Unmethylated DNA and methylated DNA are therefore distinguishable by detecting the difference of T and C in the sequence after bisulfite treatment.
Methylation control samples (0%, 25%, 50%, 75%, and 100%) were prepared as described previously 18. Briefly, human peripheral lymphocyte DNA was amplified using GenomiPhi v2 DNA amplification kit (GE Healthcare Life‐Science, Buckinghamshire, UK). The amplified DNA was not methylated in any CpG sites, and was used as unmethylated (0%) control. The amplified DNA was methylated by SssI methylase and used as fully methylated (100%) control. Other methylation control samples (25%, 50%, and 75%) were prepared by mixing 0% and 100% samples at a ratio of 3:1, 1:1, and 1:3. These control samples were also treated with bisulfite in the same manner.
Methylation analysis
Quantitative methylation analysis was performed by pyrosequencing as previously reported 37, 40. Briefly, the biotinylated PCR product was bound to Streptavidin Sepharose High Performance (Amersham Biosciences, Uppsala, Sweden), washed, and denatured using a 0.2 mol/L NaOH solution. After addition of 0.3 μmol/L sequencing primer to the single‐stranded PCR product, pyrosequencing was carried out according to the manufacturer's instructions. By using methylation control samples (0%, 25%, 50%, 75%, and 100%), it was confirmed in each pyrosequencing assay that methylation analysis for the 19 markers was done highly quantitatively. Primer sequences of the methylation markers are shown in Table S1.
Statistical analysis
The clinicopathological features were compared between BRAF‐mutation(+), KRAS‐mutation(+), NRAS‐mutation(+), and oncogene‐mutation(−) CRC groups. P‐value was calculated by the Student's t‐test for age and methylation level, by chi‐square test for AJCC stage, and by Fisher's exact test for gender, tumor location, mucinous component, lymph node metastasis, lymph vessel invasion, venous invasion, and microsatellite instability, using R software (https://www.r-project.org/). Unsupervised two‐way hierarchical clustering was performed based on the City‐block distance, the complete linkage‐clustering algorithm using Cluster 3.0 software. The heatmap was drawn using Java Tree View software. In survival analysis, Kaplan–Meier survival curve was drawn by GeneSpring 7.3.1 software, and P‐value was calculated by log‐rank test and Fisher's exact test. The end of the follow‐up period was 60 months from the primary surgery, and the death because of CRC was the primary endpoint; deaths by other causes were censored. Survival analysis by Cox proportional hazard model was also performed using R software. Correlation of the methylation level of each marker with tumor location and age was evaluated by linear single regression model using R software.
Result
Oncogene mutation analysis
Among 2045 CRC cases, 61 cases were positive for NRAS mutation. These and the additional 144 NRAS‐mutation(−) CRC cases were analyzed for BRAF and KRAS mutations. The 205 CRC cases included 13 cases with BRAF mutation, 59 with KRAS mutation only, 56 with NRAS‐mutation only, 5 with both KRAS and NRAS mutations, and 72 with no mutation of these oncogenes (Table S2).
Quantitative DNA methylation analysis
Methylation levels of six Group 1 and 13 Group 2 markers were quantitatively analyzed by pyrosequencing. Hierarchical clustering analysis was conducted using methylation data of these 19 markers to evaluate methylation epigenotype. First, we performed hierarchical clustering analysis using 70 CRC samples whose epigenotypes was previously evaluated (10 HME, 30 IME, and 30 LME) 18 (Fig. 1A). The 70 CRC samples were properly classified into three distinct clusters using the 19 markers. Second, we performed hierarchical clustering analysis of each CRC sample with the 70 CRC samples (Fig. 1B and C). Three CRC samples were clustered with 10 HME samples, 61 were clustered with 30 IME samples (Fig. 1B), and 71 were clustered with 30 LME samples (Fig. 1C).
Figure 1.
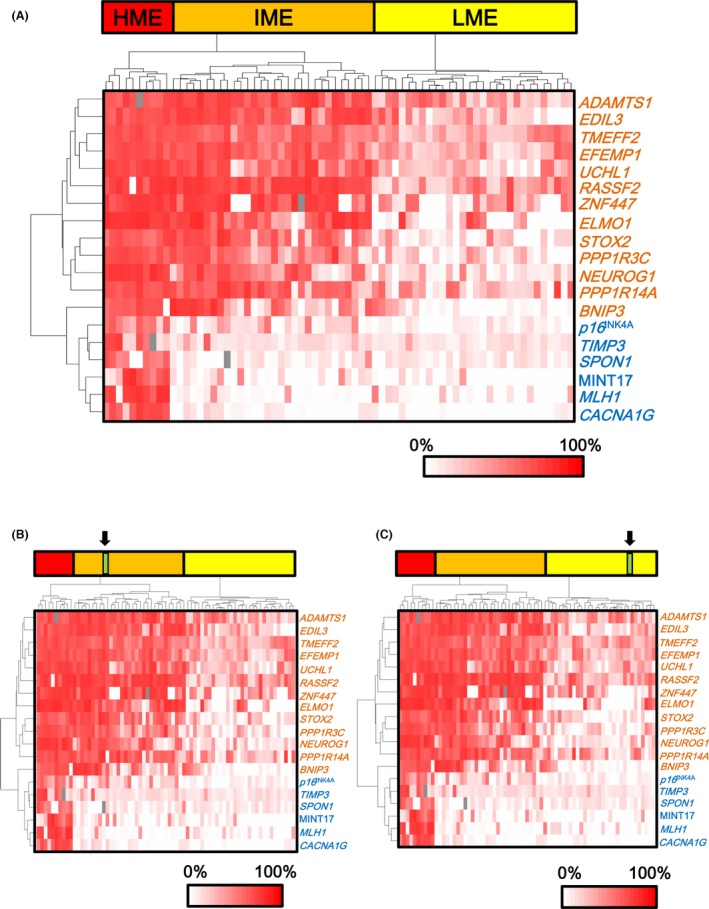
Hierarchical clustering of sporadic CRC samples. (A) Hierarchical clustering of 70 CRC samples. To evaluate the methylation epigenotype of CRC samples by comparing the previously established methylation epigenotypes of sporadic CRC, we used the cluster of 70 sporadic CRC samples, including 10 high‐, 30 intermediate‐, and 30 low‐methylation epigenotypes 18. Blue: Group 1 markers, including p16INK4A,TIMP3,SPON1, MINT17, MLH1, and CACNA1G. Orange: Group 2 markers, including ADAMTS1,TMEFF2,STOX2,COLA4A2,EDIL3,UCHL1,RASSF2,ELMO1,PPP1R3C,PPP1R14A,BNIP3,ZNF447, and NEUROG1. (B) One CRC sample belonging to IME. Arrow: One CRC sample. (C) One CRC sample belonging to LME. Arrow: One CRC sample.
The 61 NRAS‐mutation(+) CRC were classified into two major subtypes (Fig. 2). While none of the NRAS‐mutation(+) CRC was evaluated as HME, 20 NRAS‐mutation(+) CRC were evaluated as IME and 41 were LME. In contrast, KRAS‐mutation(+) cases were mostly IME (40 of 59), and BRAF‐mutation(+) cases were mostly HME (10 of 13) (P = 1 × 10−4, chi‐square test). These data indicated that NRAS‐mutation(+) CRC preferentially showed LME, while KRAS‐mutation(+) CRC strongly correlated with IME (Table S2).
Figure 2.
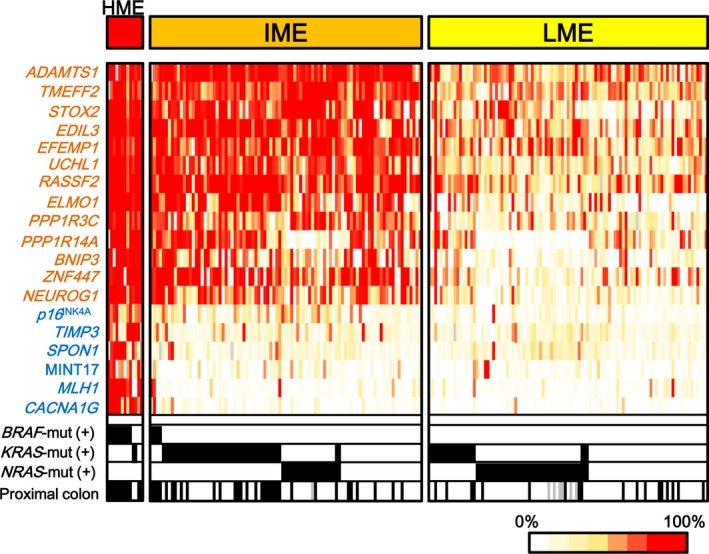
NRAS‐mutation(+) CRC showed two major subtypes. After evaluating methylation epigenotype of CRC samples, all CRC samples were divided into three groups. While none of NRAS‐mutation(+) cases was equivalent to HME, 20 NRAS‐mutation(+) CRC were equivalent to IME CRC and 41 cases were equivalent to LME CRC (P = 0.008). NRAS‐mut(+),KRAS‐mut(+), or BRAF‐mut(+): samples positive for NRAS‐mutation, KRAS‐mutation, or BRAF‐mutation are shown in black. Blue: Group 1 markers. Orange: Group 2 markers.
Comparison of the methylation level of each marker
To confirm that NRAS‐mutation(+) CRC preferentially showed LME, not IME, methylation levels of each marker were compared among the three epigenotypes, and among mutation types (Figs. 3 and 4).
Figure 3.
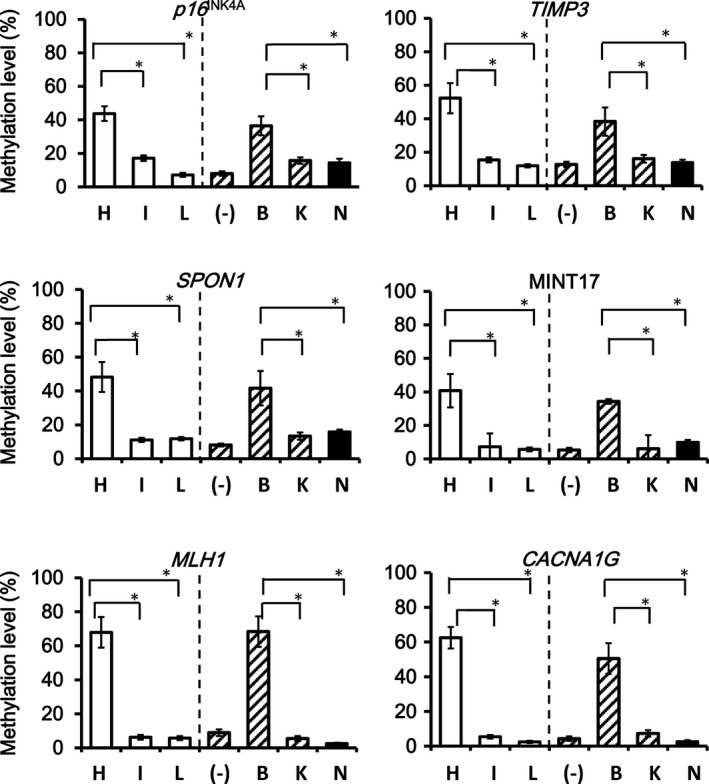
Comparison of the methylation level of Group 1 markers. All Group 1 markers showed that HME CRCs presented higher methylation levels than LME and IME CRC. Similarly, all the Group 1 markers showed significantly higher methylation levels in BRAF‐mutation(+) CRC than in KRAS‐mutation(+) and NRAS‐mutation(+) CRC (*P < 0.05).
Figure 4.
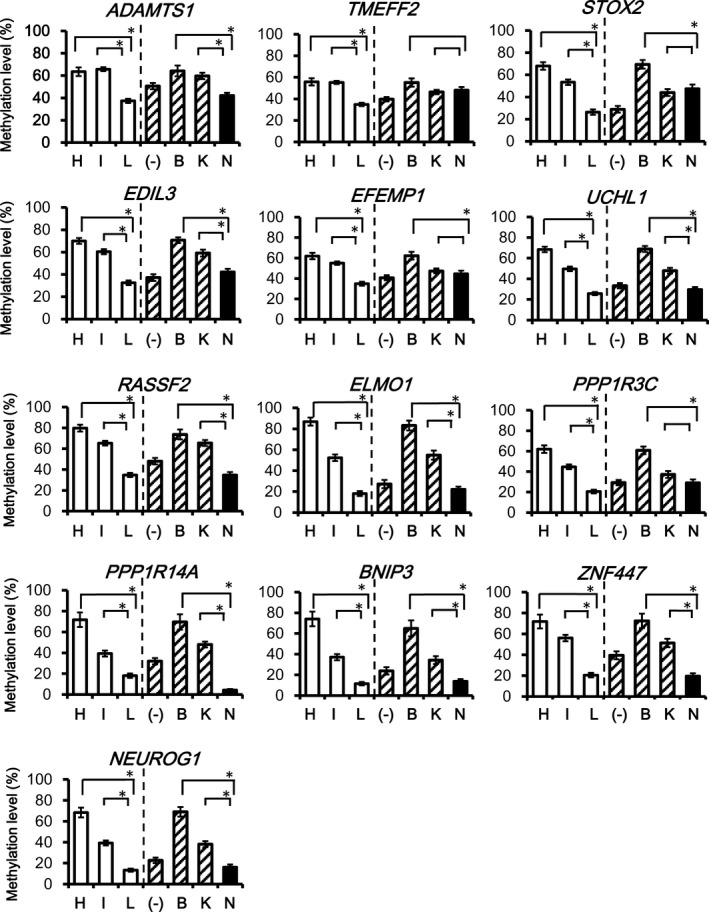
Comparison of the methylation level of Group 2 markers. While 12 of 13 Group 2 markers showed significantly lower methylation levels in NRAS‐mutation(+) CRC than in BRAF‐mutation(+) CRC, all Group 2 markers showed significantly lower methylation level in LME CRC than in HME CRC. Additionally, while 9 of 13 Group 2 markers presented low methylation in NRAS‐mutation(+) CRC than in KRAS‐mutation(+) CRC, all Group 2 markers showed significantly lower methylation levels in LME CRC than in IME CRC (*P < 0.05).
All six Group 1 markers showed significantly higher methylation levels in HME CRC than that in LME and IME CRC. Similarly, all Group 1 markers showed significantly higher methylation levels in BRAF‐mutation(+) CRC than KRAS‐mutation(+) and NRAS‐mutation(+) CRC (P < 0.05) (Fig. 3).
As for Group 2 markers, methylation levels in LME CRC were significantly lower than that in HME and IME in all genes (P < 0.05) (Fig. 4). Similarly, in all genes, except TMEFF2, methylation levels in NRAS‐mutation(+) CRC were significantly lower than that in BRAF‐mutation(+) CRC. For nine of 13 Group 2 markers, methylation levels in NRAS‐mutation(+) CRC were significantly lower than that in KRAS‐mutation(+) CRC.
Consistent with the clustering analysis, significantly lower levels of Group 1 and Group 2 markers indicated that NRAS‐mutation(+) CRC correlated with LME.
Comparison of clinocopathological features and mutation types
The clinicopathological data of the 205 analyzed CRC cases are summarized in Table S2. NRAS‐mutation(+) CRC significantly correlated with older age, distal colon, more mucinous component of the tumor, earlier AJCC stage, less lymph node metastasis, and less lymph vessel invasion (P = 0.01, 0.002, 0.02, 0.001, 0.003, and 8 × 10−6, respectively), compared with KRAS‐mutation(+) CRC. Since NRAS‐mutation(+) CRC included cases with earlier AJCC stage, we excluded stage I CRC cases and performed similar analyses using 186 cases with stage II–IV (Table 1). While NRAS‐mutation(+) CRC and KRAS‐mutation(+) CRC did not show significant difference in term of AJCC stage in this analysis (P = 0.5), NRAS‐mutation(+) CRC still significantly correlated with older age, distal colon, more mucinous component of the tumor, and less lymph vessel invasion (P = 0.02, 0.006, 0.02, and 0.002, respectively).
Survival analysis of CRC patients
We then conducted an overall survival analysis using these stage II–IV CRC cases. Kaplan–Meier survival analysis showed significant differences among NRAS‐mutation(+), KRAS‐mutation(+), and oncogene‐mutation(−) groups (P = 6 × 10−4, log‐rank test) (Fig. 5). Comparison of NRAS‐mutation(+) and KRAS‐mutation(+) groups, indicated a significantly better prognosis for NRAS‐mutation(+) CRC (P = 3 × 10−4, log‐rank test). When overall survival rates at 60 months were compared among NRAS‐mutation(+), KRAS‐mutation(+), and oncogene‐mutation(−) groups, NRAS‐mutation(+) and oncogene‐mutation(−) CRC also showed better survival rates than KRAS‐mutation(+) CRC (P = 0.02, chi‐square test) (Table 2). When analyzing the correlation of overall survival rates with other clinicopathological features (Fisher's exact test, t‐test, and chi‐square test), higher AJCC stage (P = 2 × 10−5), lymph node metastasis (P = 0.004), and venous invasion (P = 0.04) were also correlated with worse survival rate at 60 months. To identify independent prognosis factors, multivariate overall survival analysis was conducted by Cox proportional hazard model. RAS status and AJCC stage were statistically significant (P = 0.007 and 0.02, respectively), while other factors were not (Table 3).
Figure 5.
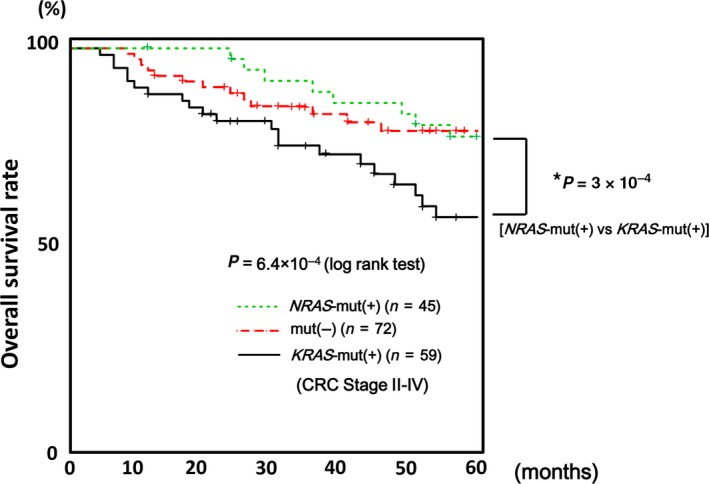
Kaplan–Meier analysis of survival in CRC, excluding stage I. Kaplan–Meier survival analysis showed significant differences (P = 6 × 10−4) among NRAS‐mutation(+), KRAS‐mutation(+), and oncogene‐mutation(−) groups. Compared with NRAS‐mutation(+) and KRAS‐mutation(+) groups, NRAS‐mutation(+) CRC did not show worse prognosis in overall survival (P = 3 × 10−4, log‐rank test).
Table 2.
Prognosis at 60 months and comparison with clinicopathological features
| Clinical features | Death (n = 42) | Alive (n = 65) | P‐value |
|---|---|---|---|
| RAS | |||
| NRAS | 8 | 26 | 0.02a |
| KRAS | 21 | 17 | |
| No‐mut | 13 | 22 | |
| Gender | |||
| Male | 26 | 35 | 0.4 |
| Female | 16 | 30 | |
| Age (y.o.) | 62.1 ± 9.6 | 64.0 ± 9.0 | 0.3 |
| Tumor location | |||
| Proximal | 11 | 15 | 0.8 |
| Distal | 31 | 50 | |
| AJCC stage | |||
| II | 5 | 21 | 2 × 10−5 a |
| III | 6 | 26 | |
| IV | 31 | 18 | |
| Mucinous component | |||
| (+) | 4 | 9 | 0.6 |
| (−) | 38 | 56 | |
| Lymph node metastasis | |||
| (+) | 34 | 34 | 0.004a |
| (−) | 8 | 31 | |
| Lymph vessel invasion | |||
| (+) | 33 | 53 | 0.8 |
| (−) | 9 | 12 | |
| Venous invasion | |||
| (+) | 40 | 53 | 0.04a |
| (−) | 2 | 12 | |
| Microsatellite instability | |||
| MSI‐H | 0 | 2 | 0.5 |
| MSS | 42 | 63 | |
| Methylation epigenotype | |||
| HME | 2 | 2 | 0.3 |
| IME | 23 | 27 | |
| LME | 17 | 36 | |
No‐mut, no mutation; MSI‐H, microsatellite instability high; MSS, microsatellite stable; HME, high‐methylation epigenotype; IME, intermediate‐methylation epigenotype; LME, low‐methylation epigenotype.
P < 0.05
Table 3.
Multivariate overall survival analysis by Cox proportional hazard model
| Clinicopathological features | P‐value |
|---|---|
| RAS | 0.007a |
| Gender | 0.4 |
| Age (y.o.) | 0.6 |
| Tumor location | 0.4 |
| AJCC stage | 0.02a |
| Mucinous component | 0.5 |
| Lymph node metastasis | 0.5 |
| Lymph vessel invasion | 0.3 |
| Venous invasion | 0.2 |
| Microsatellite instability | 0.9 |
| Methylation epigenotype | 0.9 |
P < 0.05.
Comparison using linear single regression
Correlation between DNA methylation levels and clinicopathological factors other than oncogene mutation status, for example, tumor location and age, was analyzed using linear single regression model (Fig. 6). A significant correlation was observed between higher methylation level and older age for three of six Group 1 markers, but not for any Group 2 markers (Figs. 6A and S1). A significant correlation was observed between higher methylation level and proximal colon for five of six Group 1 markers and 10 of 13 Group 2 markers (Figs. 5B and S2).
Figure 6.
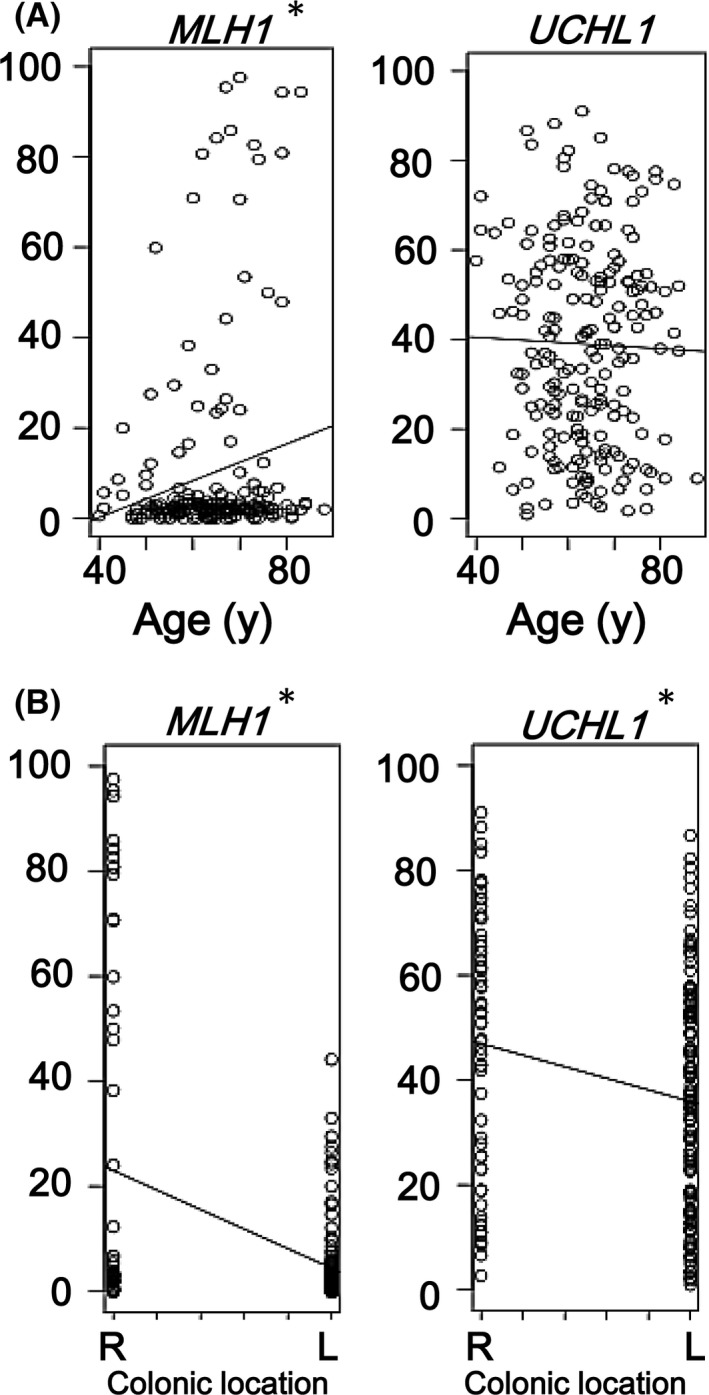
Comparison using linear single regression model. (A) Association of methylation accumulation with age. CRC samples of elder patients showed significantly higher methylation levels in Group 1 markers, for example., MLH1, than in Group 2 markers, for example., UCHL1 (see Fig. S1). (B) Association of methylation accumulation with location. Group 2 markers, for example., UCHL1, and Group 1 marker, for example., MLH1, showed significant correlation between higher methylation levels and proximal location (Fig. S2). Since 6 Group 1 markers and 13 Group 2 markers were evaluated for each factor, P < 0.008 (i.e., 0.05/6) and P < 0.004 (i.e., 0.05/13) were considered significant in the analysis of Group 1 and Group 2 markers, respectively, instead of P < 0.05 (*).
Discussion
The RAS pathway plays an important role in the development of various cancers 41, 42, 43. As one of the RAS family members, NRAS contains effector binding domains identical to those in KRAS. Thus, NRAS activating mutations yield effects similar to those observed after KRAS activation 44. Patients with KRAS and NRAS mutations are resistance to anti‐EGFR monoclonal antibody therapy. The PRIME trial showed a detrimental effect of adding panitumumab to first‐line FOLFOX in patients with RAS mutations 45. De Roock et al. evaluated the role of NRAS mutations in a large dataset of chemorefractory patients with CRC treated with cetuximab and chemotherapy in 11 centers in seven European countries. Only one RECIST response was reported among 13 patients with NRAS mutations in this retrospective series 46. Peeters et al. reported that none of the 11 patients with NRAS‐mutation(+) responded to panitumumab in a randomized phase III study compared to best supportive care 47. Considering the resistance to EGFR targeted therapy, the molecular basis and clinicopathological features of patients with NRAS‐mutation(+) CRC should be analyzed and clarified as previously performed for KRAS‐mutation(+) CRC.
However, NRAS mutations are observed infrequently in 2.6–4.2% CRC in sporadic CRC, while KRAS mutations are frequently observed in 35–40% CRC. The Cancer Genome Atlas conducted a comprehensive analysis of human CRC, but the study included only 20 NRAS‐mutation(+) CRC cases and revealed no correlation with CIMP or methylation of any genes 4. Here, we analyzed 61 NRAS‐mutation(+) and 144 NRAS‐mutation(−) CRC, and identified that, while BRAF mutation and KRAS mutation significantly correlated with HME and IME, NRAS mutation significantly correlated with LME, a different DNA methylation epigenotype.
In addition to epigenetic features, NRAS‐mutation(+) CRC also showed different clinicopathological features compared to KRAS‐mutation(+) CRC. NRAS‐mutation(+) CRC significantly correlated with older age, distal colon, more mucinous component of the tumor, and lower lymph vessel invasion when compared with KRAS‐mutation(+) CRC. The comparison of 73 NRAS‐mutation(+) and 750 KRAS‐mutation(+) cases by Gavin et al. 26 or that of 43 NRAS‐mutation(+) and 504 KRAS‐mutation(+) cases by Zhang et al. 30, revealed no significant difference in these parameters. Ogura et al. analyzed 35 NRAS‐mutation(+) CRC and found that NRAS‐mutation(+) CRC significantly correlated with older age comparing with KRAS‐mutation(+) CRC 23. While Schirripa et al. 33 reported lower prevalence of mucinous histology in 47 NRAS‐mutation(+) CRC compared with 393 KRAS‐mutation(+) CRC, this is the first report revealing the significant difference of the above clinicopathological features between NRAS‐mutation(+) CRC and KRAS‐mutation(+) CRC.
Although KRAS and BRAF mutations are both aberrations of the RAS/RAF/ERK pathway downstream of EGFR, they correlated with distinct DNA methylation epigenotypes and clinicopathological features, suggesting different molecular pathways of tumorigenesis 18, 48. In fact, BRAF‐mutation(+) CRC is mostly HME/CIMP‐high, MSI‐high CRC, whose precursor lesions are sessile serrated adenoma/polyps preferentially occurring at the proximal colon 49. Similarly, the different epigenetic and clinicopathological features of NRAS‐mutation(+) CRC may suggest a different tumorigenic pathway through different types of early lesions. While KRAS gene is located on the short (p) arm of chromosome 12 and NRAS is located on the chromosome 1 at position 13.1, the protein localization is different 50, 51. Oncogenic NRAS colocalizes with markers of the endoplasmic reticulum and Golgi 52. While the oncogenic NRAS is restricted to the endoplasmic reticulum with a transmembrane tether and retained transforming activity in a focus‐forming assay, NRAS restricted to the Golgi is unable to promote transformation 53. On the other hand, KRAS is targeted to the plasma membrane by an uncharacterized pathway and returns to endomembrane compartments following phosphorylation of the hypervariable region 54. Furthermore, NRAS and KRAS mutations may have different effects on cell biology. Haigis et al. found that activated KRAS affects cell proliferation and differentiation, whereas activated NRAS suppresses apoptosis 55. NRAS mutation does not seem to affect the early phases of tumor progression and the adenoma–carcinoma sequence, but it might inhibit epithelial cells’ stress‐induced apoptosis 55. A different effect of these mutations on downstream signaling cascade effectors has also been hypothesized 56. Wang et al. found that mutant NRAS strongly promotes tumorigenesis in the context of inflammation 56. In addition, mutations in different RAS genes are preferentially associated with distinct tumor types in human cancers. KRAS mutations are extremely common in cancer of the pancreas, colon, and lung, while NRAS mutations predominate in melanoma and hematopoietic cancers 57. To clarify whether NRAS‐mutation(+) CRC constitutes a unique CRC subtype occurring through distinct tumorigenic pathway, further analyses should be performed, including comprehensive analyses of genomic and epigenomic aberrations in precancerous NRAS‐mutation(+) colorectal lesions, for example, aberrant crypt foci and adenoma as well as NRAS‐mutation(+) CRC.
The prognosis and degree of malignancy of NRAS‐mutation(+) CRC is controversial. NRAS‐mutation(+) CRC shows a high degree of malignancy compared with oncogene‐mutation(−) CRC 33, and there is no significant difference in prognosis of NRAS‐mutation(+) and KRAS‐mutation(+) CRC 23, 26, 33. However, in this study, Kaplan–Meier survival analysis showed that, while KRAS‐mutation(+) CRC showed significantly worse prognosis, NRAS‐mutation(+) CRC and oncogene‐mutation(−) CRC showed relatively better prognosis. Survival analysis by Cox proportional hazard model showed that RAS status and AJCC stage were independent prognosis determining factors (P = 0.007 and 0.02, respectively).
To evaluate the possible association of methylation accumulation with tumor location and age, methylation levels were analyzed by a linear single regression model using all sporadic CRC samples (Figs. 6, S1, and S2). DNA methylation accumulated significantly more in the proximal colon than in the distal colon for most of Group 1 and Group 2 markers. Since sessile serrated adenoma/polyps are known as the precursor lesions of HME/CIMP‐high CRC and preferentially observed at the proximal colon 28, 49, we performed similar analyses using IME and LME CRC cases only (Fig. S3). A significant correlation was still observed between higher methylation level and proximal colon for 2 of 6 Group 1 markers and 7 of 13 Group 2 markers. Thus, DNA methylation significantly accumulated in the proximal colon, regardless of HME/CIMP‐high samples. Aging is known as an important factor causing DNA methylation accumulation 58. While three of six Group 1 markers (classical CIMP markers) showed a significant correlation between higher methylation level and older age, no correlation was observed in any Group 2 markers. Further analyses are necessary to identify factors that induce DNA methylation in Group 2 markers.
In summary, NRAS‐mutation(+) CRC showed distinct epigenetic and clinicopathological features. NRAS‐mutation(+) CRC significantly correlated with LME, while KRAS‐mutation(+) CRC correlated with IME. NRAS‐mutation(+) CRC significantly correlated with less lymph vessel invasion, occurred preferentially in elder patients and at the distal colon, and showed relatively better prognosis, compared with KRAS‐mutation(+) CRC.
Conflict of Interest
All authors have no potential conflict of interest to disclose.
Supporting information
Table S1. Methylation marker genes and printer sequences for pyrosequences.
Table S2. Comparison of clinicopathological features of all CRC cases.
Figure S1. Comparison between methylation levels and age using linear single regression model.
Figure S2. Comparison between methylation levels and tumor location using a linear single regression model.
Figure S3. Comparison between methylation levels and tumor location using a linear single regression model, excluding HME CRCs.
Acknowledgment
We thank Chihomi Sato for their technical help, and Editage (www.editage.jp) for English language editing.
Cancer Medicine 2017; 6(5):1023–1035
References
- 1. Toyota, M. , Sasaki Y., Satoh A., et al. 2003. Epigenetic inactivation of CHFR in human tumors. Proc. Natl Acad. Sci. USA 100:7818–7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akino, K. , Toyota M., Suzuki H., et al. 2005. The Ras effector RASSF2 is a novel tumor‐suppressor gene in human colorectal cancer. Gastroenterology 129:156–169. [DOI] [PubMed] [Google Scholar]
- 3. Suzuki, H. , Watkins D. N., Jair K. W., et al. 2004. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat. Genet. 36:417–422. [DOI] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas . 2012. N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Esteller, M. , Toyota M., Sanchez‐Cespedes M., et al. 2000. Inactivation of the DNA repair gene O6‐methylguanine‐DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K‐ras in colorectal tumorigenesis. Cancer Res. 60:2368–2371. [PubMed] [Google Scholar]
- 6. Spano, J. P. , Lagorce C., Atlan D., et al. 2005. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 16:102–108. [DOI] [PubMed] [Google Scholar]
- 7. Coffey, R. J. Jr , Goustin A. S., Soderquist A. M., et al. 1987. Transforming growth factor alpha and beta expression in human colon cancer lines: implications for an autocrine model. Cancer Res. 47:4590–4594. [PubMed] [Google Scholar]
- 8. Pozzi, C. , Cuomo A., Spadoni I., et al. 2016. The EGFR‐specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 22:624–631. [DOI] [PubMed] [Google Scholar]
- 9. Price, T. J. , Peeters M., Kim T. W., et al. 2014. Panitumumab versus cetuximab in patients with chemotherapy‐refractory wild‐type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open‐label, non‐inferiority phase 3 study. Lancet Oncol. 15:569–579. [DOI] [PubMed] [Google Scholar]
- 10. Yarden, Y. , and Sliwkowski M. X.. 2001. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2:127–137. [DOI] [PubMed] [Google Scholar]
- 11. Toyota, M. , Ahuja N., Ohe‐Toyota M., Herman J. G., Baylin S. B., and Issa J. P.. 1999. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 96:8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weisenberger, D. J. , Siegmund K. D., Campan M., et al. 2006. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 38:787–793. [DOI] [PubMed] [Google Scholar]
- 13. Roth, A. D. , Tejpar S., Delorenzi M., et al. 2010. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC‐3, EORTC 40993, SAKK 60‐00 trial. J. Clin. Oncol. 28:466–474. [DOI] [PubMed] [Google Scholar]
- 14. Perez‐Ruiz, E. , Rueda A., Pereda T., et al. 2012. Involvement of K‐RAS mutations and amino acid substitutions in the survival of metastatic colorectal cancer patients. Tumour Biol. 33:1829–1835. [DOI] [PubMed] [Google Scholar]
- 15. Cunningham, D. , Atkin W., Lenz H. J., et al. 2010. Colorectal cancer. Lancet 375:1030–1047. [DOI] [PubMed] [Google Scholar]
- 16. Naguib, A. , Mitrou P. N., Gay L. J., et al. 2010. Dietary, lifestyle and clinicopathological factors associated with BRAF and K‐ras mutations arising in distinct subsets of colorectal cancers in the EPIC Norfolk study. BMC Cancer 10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zlobec, I. , Bihl M. P., Schwarb H., Terracciano L., and Lugli A.. 2010. Clinicopathological and protein characterization of BRAF‐ and K‐RAS‐mutated colorectal cancer and implications for prognosis. Int. J. Cancer 127:367–380. [DOI] [PubMed] [Google Scholar]
- 18. Yagi, K. , Akagi K., Hayashi H., et al. 2010. Three DNA methylation epigenotypes in human colorectal cancer. Clin. Cancer Res. 16:21–33. [DOI] [PubMed] [Google Scholar]
- 19. Karapetis, C. S. , Khambata‐Ford S., Jonker D. J., et al. 2008. K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 359:1757–1765. [DOI] [PubMed] [Google Scholar]
- 20. Kadowaki, S. , Kakuta M., Takahashi S., et al. 2015. Prognostic value of KRAS and BRAF mutations in curatively resected colorectal cancer. World J. Gastroenterol. 21:1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Cutsem, E. , Kohne C. H., Hitre E., et al. 2009. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 360:1408–1417. [DOI] [PubMed] [Google Scholar]
- 22. Tol, J. , Koopman M., Cats A., et al. 2009. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N. Engl. J. Med. 360:563–572. [DOI] [PubMed] [Google Scholar]
- 23. Ogura, T. , Kakuta M., Yatsuoka T., et al. 2014. Clinicopathological characteristics and prognostic impact of colorectal cancers with NRAS mutations. Oncol. Rep. 32:50–56. [DOI] [PubMed] [Google Scholar]
- 24. Foltran, L. , De Maglio G., Pella N., et al. 2015. Prognostic role of KRAS, NRAS, BRAF and PIK3CA mutations in advanced colorectal cancer. Future Oncol. 11:629–640. [DOI] [PubMed] [Google Scholar]
- 25. Russo, A. L. , Borger D. R., Szymonifka J., et al. 2014. Mutational analysis and clinical correlation of metastatic colorectal cancer. Cancer 120:1482–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gavin, P. G. , Colangelo L. H., Fumagalli D., et al. 2012. Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin. Cancer Res. 18:6531–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oudejans, J. J. , Slebos R. J., Zoetmulder F. A., Mooi W. J., and Rodenhuis S.. 1991. Differential activation of ras genes by point mutation in human colon cancer with metastases to either lung or liver. Int. J. Cancer 49:875–879. [DOI] [PubMed] [Google Scholar]
- 28. Fernando, W. C. , Miranda M. S., Worthley D. L., et al. 2014. The CIMP Phenotype in BRAF Mutant Serrated Polyps from a Prospective Colonoscopy Patient Cohort. Gastroenterol Res. Pract. 2014:374926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen, Y. , Wang J., Han X., et al. 2013. Effectors of epidermal growth factor receptor pathway: the genetic profiling of KRAS, BRAF, PIK3CA, NRAS mutations in colorectal cancer characteristics and personalized medicine. PLoS ONE 8:e81628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang, J. , Zheng J., Yang Y., et al. 2015. Molecular spectrum of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancer patients: analysis of 1,110 cases. Sci. Rep. 5:18678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Osumi, H. , Shinozaki E., Suenaga M., et al. 2016. RAS mutation is a prognostic biomarker in colorectal cancer patients with metastasectomy. Int. J. Cancer 139:803–811. [DOI] [PubMed] [Google Scholar]
- 32. Irahara, N. , Baba Y., Nosho K., et al. 2010. NRAS mutations are rare in colorectal cancer. Diagn. Mol. Pathol. 19:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schirripa, M. , Cremolini C., Loupakis F., et al. 2015. Role of NRAS mutations as prognostic and predictive markers in metastatic colorectal cancer. Int. J. Cancer 136:83–90. [DOI] [PubMed] [Google Scholar]
- 34. Hinoue, T. , Weisenberger D. J., Lange C. P., et al. 2012. Genome‐scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 22:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen, L. , Toyota M., Kondo Y., et al. 2007. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 104:18654–18659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Toyota, M. , Ho C., Ohe‐Toyota M., Baylin S. B., and Issa J. P.. 1999. Inactivation of CACNA1G, a T‐type calcium channel gene, by aberrant methylation of its 5′ CpG island in human tumors. Cancer Res. 59:4535–4541. [PubMed] [Google Scholar]
- 37. Sakai, E. , Ohata K., Chiba H., et al. 2014. Methylation epigenotypes and genetic features in colorectal laterally spreading tumors. Int. J. Cancer 135:1586–1595. [DOI] [PubMed] [Google Scholar]
- 38. Akagi, K. , Uchibori R., Yamaguchi K., Kurosawa K., Tanaka Y., and Kozu T.. 2007. Characterization of a novel oncogenic K‐ras mutation in colon cancer. Biochem. Biophys. Res. Commun. 352:728–732. [DOI] [PubMed] [Google Scholar]
- 39. Seymour, M. T. , Brown S. R., Middleton G., et al. 2013. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild‐type, fluorouracil‐resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 14:749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsusaka, K. , Kaneda A., Nagae G., et al. 2011. Classification of Epstein‐Barr virus‐positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 71:7187–7197. [DOI] [PubMed] [Google Scholar]
- 41. Jaiswal, B. S. , Janakiraman V., Kljavin N. M., et al. 2009. Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors. PLoS ONE 4:e5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lea, I. A. , Jackson M. A., and Dunnick J. K.. 2009. Genetic pathways to colorectal cancer. Mutat. Res. 670:96–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quinlan, M. P. , and Settleman J.. 2009. Isoform‐specific ras functions in development and cancer. Future Oncol. 5:105–116. [DOI] [PubMed] [Google Scholar]
- 44. Downward, J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3:11–22. [DOI] [PubMed] [Google Scholar]
- 45. Douillard, J. Y. , Oliner K. S., Siena S., et al. 2013. Panitumumab‐FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 369:1023–1034. [DOI] [PubMed] [Google Scholar]
- 46. De Roock, W. , Claes B., Bernasconi D., et al. 2010. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy‐refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 11:753–762. [DOI] [PubMed] [Google Scholar]
- 47. Peeters, M. , Oliner K. S., Parker A., et al. 2013. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin. Cancer Res. 19:1902–1912. [DOI] [PubMed] [Google Scholar]
- 48. Kaneda, A. , and Yagi K.. 2011. Two groups of DNA methylation markers to classify colorectal cancer into three epigenotypes. Cancer Sci. 102:18–24. [DOI] [PubMed] [Google Scholar]
- 49. Sakai, E. , Fukuyo M., Ohata K., et al. 2016. Genetic and epigenetic aberrations occurring in colorectal tumors associated with serrated pathway. Int. J. Cancer 138:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alshahid, M. , Wakil S. M., Al‐Najai M., et al. 2013. New susceptibility locus for obesity and dyslipidaemia on chromosome 3q22.3. Hum. Genomics. 7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Stefano, A. , and Carlomagno C.. 2014. Beyond KRAS: Predictive factors of the efficacy of anti‐EGFR monoclonal antibodies in the treatment of metastatic colorectal cancer. World J. Gastroenterol. 20:9732–9743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chiu, V. K. , Bivona T., Hach A., et al. 2002. Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 4:343–350. [DOI] [PubMed] [Google Scholar]
- 53. Matallanas, D. , Sanz‐Moreno V., Arozarena I., et al. 2006. Distinct utilization of effectors and biological outcomes resulting from site‐specific Ras activation: Ras functions in lipid rafts and Golgi complex are dispensable for proliferation and transformation. Mol. Cell. Biol. 26:100–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bivona, T. G. , Quatela S. E., Bodemann B. O., et al. 2006. PKC regulates a farnesyl‐electrostatic switch on K‐Ras that promotes its association with Bcl‐XL on mitochondria and induces apoptosis. Mol. Cell 21:481–493. [DOI] [PubMed] [Google Scholar]
- 55. Haigis, K. M. , Kendall K. R., Wang Y., et al. 2008. Differential effects of oncogenic K‐Ras and N‐Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 40:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang, Y. , Velho S., Vakiani E., et al. 2013. Mutant N‐RAS protects colorectal cancer cells from stress‐induced apoptosis and contributes to cancer development and progression. Cancer Discov. 3:294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lau, K. S. , and Haigis K. M.. 2009. Non‐redundancy within the RAS oncogene family: insights into mutational disparities in cancer. Mol. Cells 28:315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ottaviano, Y. L. , Issa J. P., Parl F. F., Smith H. S., Baylin S. B., and Davidson N. E.. 1994. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 54:2552–2555. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Methylation marker genes and printer sequences for pyrosequences.
Table S2. Comparison of clinicopathological features of all CRC cases.
Figure S1. Comparison between methylation levels and age using linear single regression model.
Figure S2. Comparison between methylation levels and tumor location using a linear single regression model.
Figure S3. Comparison between methylation levels and tumor location using a linear single regression model, excluding HME CRCs.