

Abstract
Ca2+ release‐activated Ca2+ (CRAC) channels play an essential role in the immune system. The pore‐forming subunit, Orai1, is an important pharmacological target. Here, we summarize the recent discoveries on the structure–function relationship of Orai1, as well as its interaction with the native channel opener STIM1 and chemical modulator 2‐aminoethoxydiphenyl borate (2‐APB). We first introduce the critical structural elements of Orai1, which include a Ca2+ accumulating region, ion selectivity filter, hydrophobic centre, basic region, extended transmembrane Orai1 N‐terminal (ETON) region, transmembrane (TM) regions 2 and 3, P245 bend, 263SHK265 hinge linker and L273–L276 hydrophobic patch. We then hypothesize the possible mechanisms by which STIM1 triggers the conformational transitions of TM regions and exquisitely shapes the ion conduction pathway during generation of the CRAC current (I crac) with high Ca2+ selectivity. Finally, we propose mechanisms by which 2‐APB modulates I crac. On the STIM1‐activated Orai1 channel, a low dose of 2‐APB acts directly, dilating its extremely narrow pore diameter from 3.8 to 4.6 Å, increasing its unitary channel conductance, and potentiating the I crac. Further elucidation of the structure of the opened CRAC channel and a better understanding of structure–function relationship will benefit the future development of novel immune modulators.

Keywords: 2‐APB, channel gating, CRAC channel, ion channel modulation
Abbreviations
- CAD
CRAC activation domain
- CAR
Ca2+ accumulating region
- CC
coiled‐coil
- CRAC
Ca2+ release‐activated Ca2+
- dOrai
Drosophila Orai
- ETON
extended transmembrane Orai1 N‐terminal
- I–V
current–voltage
- Icrac
CRAC current
- Ip
potentiation current
- SCID
severe combined immunodeficiency
- SOAR
STIM1–Orai1 activation region
- 2‐APB
2‐aminoethoxydiphenyl borate
- TM
transmembrane
- Vrev
reversal potential
- WT
wild‐type
Introduction
CRAC channels, the major contributors of Ca2+ influx in immune cells, are composed of two essential components, the pore‐forming subunit, Orai1, and the channel activator, STIM1. CRAC channels have widespread tissue distribution and are involved in the function of immune cells, exocrine gland cells, platelets, skeletal muscle and brain (Gerasimenko et al. 2013, 2014; Prakriya & Lewis, 2015). The major symptoms of patients with loss‐of‐function Orai1 and STIM1 are life‐threatening severe combined immunodeficiency (SCID), as well as non‐life‐threatening muscular hypotonia, autoimmunity and anhydrotic ectodermal dysplasia (Lacruz & Feske, 2015). These patients require haematopoietic stem cell therapy at an early age to survive severe infections. Although STIM1 gates other channels such as TRPC and CaV1.2 (Zeng et al. 2008; Wang et al. 2010), the dominant phenotype of patients with loss‐of‐function STIM1 is SCID, which suggests the major role of STIM1 is in CRAC channels (Lacruz & Feske, 2015). Therefore, both Orai1 and STIM1 play an essential and relatively specific role in the immune system, acting as important pharmacological targets.
The sequence and structure of the Orai1 channel differ greatly from other ion channels (Hou et al. 2012), and determine its unique biophysical properties when activated by STIM1. These include the following: (1) an extraordinarily high selectivity for Ca2+ over Na+ (> 1000 times), accompanied by an inwardly rectified current–voltage (I–V) profile and a positive reversal potential (V rev) of ∼50 mV in Ca2+‐containing solutions; (2) an extremely low single‐channel conductance; (3) an unusually narrow pore diameter of 3.8 Å (Prakriya & Lewis, 2006; Yamashita et al. 2007; Xu et al. 2016); and (4) graded regulation of current density and ion selectivity by different amounts of STIM1 binding (Li et al. 2011; McNally et al. 2012). Notably, in divalent‐free solutions, CRAC channels readily conduct small monovalent ions such as Na+; however, micromolar levels of extracellular Ca2+ dose‐dependently block the Na+ current. A millimolar range of extracellular Ca2+ mediates the Ca2+ current, and 10 mm external Ca2+ produces a larger I crac than 2 mm external Ca2+.
Here, we focus on the structure–function relationships of Orai1, as well as its possible gating and modulation mechanisms by STIM1 and 2‐aminoethoxydiphenyl borate (2‐APB), to better understand its channel function for the development of pharmacological immune modulators.
Critical structural elements of Orai1
Each Orai1 subunit contains four TM α‐helices, TM1 to TM4, and two intracellular termini (the N‐ and C‐termini). The crystal structure of Drosophila Orai (dOrai) (Hou et al. 2012) shows that a single channel is formed by a hexamer in which all TM helices are assembled in three concentric rings. The inner ring formed of six TM1 helices, the Ca2+ accumulating region (CAR) (Frischauf et al. 2015) from the extracellular side and the extended transmembrane Orai1 N‐terminal (ETON) region (Derler et al. 2013) from the intracellular side together compose the entire ion conduction pathway (as shown in Fig. 1 A). Additional critical structural elements in the extracellular loop1, TM2‐4 and the C‐terminus are highlighted in Fig. 1 B. The properties of these highly conserved structural elements play important roles in the gating, modulation and determination of the unique biophysical properties of CRAC channels.
Figure 1. Schematic model for the ion conduction pathway and critical structural elements in Orai1.
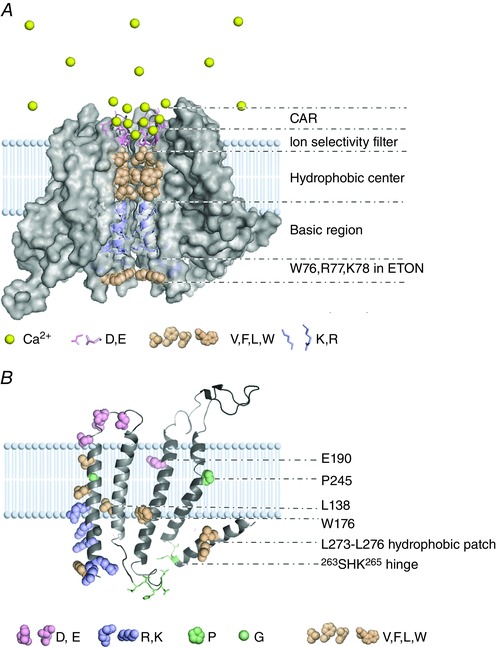
A, the ion conduction pathway of the Orai channel. Only four homologous dOrai monomers (Hou et al. 2012) are used to indicate the channel structure with the ion conduction pathway inside. Different colours of spheres and sticks denote important residues and the Ca2+ ion. The entire ion conduction pathway is composed of CAR, ion selectivity filter, hydrophobic centre, basic region and ETON region. B, critical structural elements on the homology‐modelled structure of human Orai1. To clearly show the locations of all critical structural elements, we have artificially enlarged the separation between TM2 and TM3 and denote critical residues or regions on TM2, TM3, TM4 and the C‐terminus.
CAR
Three acidic residues (D110, D112 and D114) are non‐pore lining (McNally et al. 2009) and located on external loop1 without a resolved structure. Using molecular dynamics simulation, Frischauf et al. revealed that D110/D112/D114 serve as a CAR, recruiting Ca2+ near the ion selectivity filter E106. Amazingly, they predicted that in a 10 mm external Ca2+ solution, the local Ca2+ concentration close to the pore entrance is concentrated by CAR and E106 up to 2.5 m within a 2 nm3 volume (Frischauf et al. 2015). The D110/112A mutant exhibits decreased ion selectivity (Vig et al. 2006) and current density at low concentrations of extracellular Ca2+, probably via impaired Ca2+ accumulation by increasing the distance between Ca2+ bound on CAR and E106 (Frischauf et al. 2015).
E106 as the ion selectivity filter
Early in 2006, E106 in the human Orai1 (Prakriya et al. 2006; Vig et al. 2006) and its homologous counterpart, E180 in Drosophila Orai (Yeromin et al. 2006), were identified as the most important determinant of Ca2+ selectivity on CRAC channels. The E106D mutation, which only shortens the side chain by one carbon atom, significantly impairs ion selectivity. Consistent with disulfide cross‐linking results (Zhou et al. 2010), the crystal structure of dOrai directly showed that a ring of negatively charged E106 residues is located on the extracellular mouth of the pore (Hou et al. 2012), serving as an ion selectivity filter. Non‐charged substitutions, such as human Orai1 E106A/Q and Drosophila Orai E180A mutation, result in non‐conducting channels (Prakriya et al. 2006; Vig et al. 2006; Yeromin et al. 2006), suggesting an additional role for E106 as part of the gating.
Hydrophobic centre
According to the structure of dOrai, V102, F99 and L95 are pore‐lining residues, with their hydrophobic side chains interacting with each other through van der Waals forces and forming a well‐packed hydrophobic centre (Hou et al. 2012). Consistent with this, the V102C and L95C mutants are effectively disulfide cross‐linked or highly accessible to Cd2+ (McNally et al. 2009; Zhou et al. 2010). This hydrophobic centre constitutes the relatively rigid core of the closed channel (Hou et al. 2012) and provides the major hindrance for ion permeation (Dong et al. 2013). Correspondingly, mildly hydrophobic and polar substitutions on V102 yield constitutively active currents in the absence of STIM1 (McNally et al. 2012), most likely by reducing the hydrophobic interaction and the energy barrier for ion conduction. In contrast to the published structure, G98 rather than F99 was suggested as a pore lining residue by cysteine accessibility mutagenesis (McNally et al. 2009). G98 was proposed as a gating hinge. Twisting the orientation of the α‐helix via the substitution of glycine into aspartic acid or proline, or elongating its long side chain by substitution into other residues except positively charged residues, results in constitutively active channels (Zhang et al. 2011; Zheng et al. 2013).
Basic region
It is unusual for three basic residues (R91, K87 and R83) to line the conduction pathway of cations. In the closed channel, this basic section is hypothesized to form a potential gate, to impede Ca2+ flow by binding anions or by forming an electrostatic barrier (Hou et al. 2012; Rothberg et al. 2013). When gated by STIM1, the positive charge provided by R91 is unnecessary because R91A/G/C mutants generate similar currents to wild‐type (WT), and R91K/D mutants produce even larger Ca2+ influx than WT (Zhang et al. 2011). R91W mutation results in non‐conducting channels and impedes the STIM1‐enhanced luminescence of Tb3+ (Gudlur et al. 2014), which is bound to negatively charged E106 and possibly also to CAR in the extracellular mouth of Orai1. It is likely that tryptophan substitution reinforces the hydrophobic barrier together with V102 and L95. This energy barrier is difficult for STIM1 to overcome and prevents STIM1 from triggering conformational rearrangement of the entire ion conduction pathway, including E106 and CAR. R83A–K87A double mutation substantially reduces STIM1‐dependent I crac and abolishes the constitutively active V102A current, whereas in the latter situation, the replacement of R83A–K87A with R83E–K87E mutation restores the V102A current (Derler et al. 2013), which suggests that in a channel opened by STIM1 or V102A, R83 and K87 residues may provide electrostatic repulsion forces to stabilize the elongated pore architecture, thereby facilitating the opening of Orai1.
ETON region
The truncation of the ETON (74–90) region significantly impairs STIM1‐dependent gating (Li et al. 2007; Muik et al. 2008), which emphasizes its functional necessity. It is likely that the ETON region is involved in channel gating by directly interacting with STIM1 because the N‐terminal fragment (48–91) directly binds to the CRAC activation domain (CAD) of STIM1 (Park et al. 2009), and mutations of hot spots such as hydrophobic (L74 and W76) and basic (R77, K78, R83 and K87) residues impair the coupling to STIM1 (Derler et al. 2013). In this region, the K85A/E mutation not only abolishes STIM1‐dependent I crac but also greatly impairs the STIM1‐independent V102A current (Lis et al. 2010; McNally et al. 2013). Additionally, its homologous mutation, K60E on Orai3, abolishes the direct gating mediated by 2‐APB (Lis et al. 2010). Although K85A/E impairs the STIM1–Orai1 association (McNally et al. 2013), the K85E or K60E mutation does not directly alter the binding affinity between CAD and the N‐terminus of Orai1 or that of Orai3 (Lis et al. 2010). Because of the universal impact of K85 on channel gating and its location on the pore‐averted side with the side chain protruding to TM2 and TM3, we propose that the ETON region may regulate channel gating by interacting with other regions of Orai1, in addition to interacting with STIM1 as discussed above.
TM2–TM3
In this region, the L138F mutation causes tubular aggregate myopathy and alters Ca2+ homeostasis in muscle cells via constitutive activation of CRAC channels (Endo et al. 2015). The W176C mutant is also constitutively active, and both W176C and E190Q substitutions dramatically impair ion selectivity (Prakriya et al. 2006; Vig et al. 2006; Srikanth et al. 2011). Therefore, TM2 and TM3 not only provide a structural surrounding for TM1 but also modulate channel gating and ion selectivity through unknown mechanisms.
P245 bend
P245 is located in the middle of the TM4 α‐helix (Hou et al. 2012). Interestingly, substitutions of almost any other amino acid for this proline lead to constitutively active channels (Palty et al. 2015), suggesting that the P245 bend stabilizes the closed state of the channel, probably by altering the orientation of the TM4 α‐helix.
Hinge linker
The 263SHK265 hinge links TM4 and the Orai1 C‐terminus. S263P mutation impairs the coupling between Orai1 and STIM1 (Tirado‐Lee et al. 2015) and abolishes I crac even when Orai1 is covalently linked to two STIM1 active domains (SS domains) (Palty et al. 2015), which suggests that the flexibility of this hinge plays a critical role in channel gating by STIM1.
L273–L276 hydrophobic patch
The C‐terminus (267–301) of Orai1 is a prerequisite for its coupling to STIM1 (Li et al. 2007; Muik et al. 2008; Park et al. 2009), most likely through the heteromeric interaction between the L273–L276 hydrophobic patch of the C‐terminus and STIM1. Orai1 with an L273S or L276D mutation fails to recruit and couple with STIM1 (Muik et al. 2008; Navarro‐Borelly et al. 2008; Frischauf et al. 2009). Interestingly, in STIM1‐unbound Orai1 channels, the L273–L276 hydrophobic patch mediates the homologous dimerization of two adjacent Orai1 C‐termini by forming an anti‐parallel coiled‐coil (CC) pair (Hou et al. 2012). Upon STIM1 binding, this interaction is disrupted to form the heteromeric interaction with STIM1, as demonstrated by the nuclear magnetic resonance structure of the complex formed by the Orai1 C‐terminal fragment (272–292) and STIM1 CC1–CC2 fragment (312–387). The former lies in the groove of the latter, with the L273–L276 hydrophobic patch contributing to this interaction (Stathopulos et al. 2013). Consistently, prior cross‐linking of the C‐termini of Orai1‐L273C or Orai1‐L276C prevents subsequent STIM1 binding to Orai1, and prior CAD binding to Orai1 impairs the diamide‐induced disulfide bridge formation of Orai1‐L273C/L276C (Tirado‐Lee et al. 2015).
Gating and regulation of the Orai1 channel by STIM1
The Orai1‐L273S mutant fails to couple with STIM1 and generate I crac. The N‐terminal truncated (△N1–80) Orai1 channel normally binds to STIM1 but is also non‐conducting. Tethered SS domains could restore I crac in the L273S mutant but not in the △N1–80 mutant. Therefore, Zheng et al. assumed that the Orai1 C‐terminus merely recruits and anchors STIM1, which subsequently engages the interaction between STIM1 and Orai1 N‐termini and triggers gating (Zheng et al. 2013). Recently, this assumption was revised. McNally et al. reported that N‐termini are also involved in coupling with STIM1 and that △N73–85 and K85E mutants exhibit diminished binding to CAD (McNally et al. 2013). Palty et al. revealed the critical role of C‐termini in channel gating because an Orai1 C‐terminal truncated (△C273–301) mutant and L273S–L276D double mutant fail to generate I crac even when tethered to the SS domains (Palty et al. 2015). When introducing mutation into V102A Orai1‐SS channels, V rev remains largely unaltered by the L273S or K85E single mutation, whereas V102A Orai1‐SS channels with L273S–K85E double mutation display greatly decreased V rev and non‐selective current (Palty & Isacoff, 2016). Taken together, both N‐ and C‐termini of Orai1 serve double roles in binding and gating, their cooperative interactions with STIM1 controlling channel activation and ion selectivity.
Based on the structure of the closed dOrai (Hou et al. 2012), it is presumed that the hydrophobic centre (V102, L95), P245 bend and W176 residue synergistically stabilize the channel in a closed state (Fig. 2 A). The P245 bend may force the intracellular end of TM4 to push TM3 inward, and W176 on the averted side of TM3 may further press TM1 inward through its bulky side chain to guarantee the tight packing of the hydrophobic centre and keep the channel closed. Then, what is the mechanism by which STIM1 opens the channel? First, upon STIM1 binding, the homologous anti‐parallel pair of L273–L276 hydrophobic patches are dissociated and the heteromeric interaction of this patch with STIM1 is reformed (Stathopulos et al. 2013). Then, as suggested in the hypothetical model of opened dOrai (Fig. 2 B), the P245 bend and hinge linker are pulled nearly straight by STIM1 (Hou et al. 2012), which is supported by the recently revealed roles of P245 and the hinge linker (Palty et al. 2015). We hypothesize the following key conformational transitions. The intracellular end of TM4 moves outward, releasing the steric restriction on TM3. Subsequently, the intracellular end of TM3 moves outward, removing the inward restriction effect of W176 on TM1. Based on the functions of E190 and L138 residues, TM2 may also contribute to gating of the channel and establishment of ion selectivity via interaction with TM1. Because the K85A/E mutation impairs STIM‐dependent gating and STIM1‐independent channel activation by 2‐APB and V102A mutation, we postulate that the ETON region interacts with STIM1, TM2 and/or TM2 extension in loop2 simultaneously and helps to exquisitely shape the ion conduction pathway, finishing the remaining gating process. The above discussed interactions and conformational changes, together with the intrinsic electrostatic repulsion formed by the charged residues lining the pore (E106, R91, K87 and R83), work together to dissociate the hydrophobic centre and open the channel. In the meantime, the gating rearrangement triggered by STIM1 coordinates the exquisite arrangement of E106 (Gudlur et al. 2014) and probably also E190 and CAR, establishing high ion selectivity.
Figure 2. Schematic model for Orai in the closed and opened states.

A, the closed model of Orai1. Two symmetrical dOrai monomers (Hou et al. 2012) are used to indicate the simplified channel architecture. TM1 and other TM α‐helices are labelled blue and grey, respectively. Different colours of spheres and sticks are used to denote critical residues and the Ca2+ ion. We postulate that the hydrophobic centre (represented by V102), W176 residue and P245 bend provide closing forces as indicated by black arrows, synergistically stabilizing the channel in a closed state. B, the hypothetical model of STIM1‐activated Orai1. The large yellow forks represent the STIM1–Orai1 activation region (SOAR) (Yang et al. 2012). Upon the binding of SOAR, the TM4–C‐terminus section may be pulled nearly straight as previously suggested (Hou et al. 2012). The intracellular end of TM4 may move outward, releasing the steric restriction on TM3. Subsequently, the intracellular end of TM3 may move outward, removing the inward restriction effect of W176 on TM1. Additionally, the ETON region may interact with STIM1, TM2 and/or TM2 extension in loop2 simultaneously. All of these interactions and conformational changes, together with the intrinsic electrostatic repulsion along the ion conduction pathway, work together to dissociate the hydrophobic centre and exquisitely shape the pore, resulting in a fully opened channel with high Ca2+ selectivity. Notably, the exact binding position for SOAR to Orai1 remain unclear and may differ from this schematic model.
It was proposed that STIM1 activates Orai1 not by following an ‘on–off’ mode but by regulating the current density and ion selectivity of the Orai1 channel in a dose‐dependent manner. When tethering one S domain to one Orai1, this unsaturated STIM1 binding results in decreased current density and V rev (Li et al. 2011; McNally et al. 2012).
How is the Ca2+ selectivity of CRAC channels established and modulated?
The exquisite Ca2+ selectivity of the CRAC channel is mainly determined by E106 and may be tuned by other factors. First, the ion is selected by its size because the extremely narrow pore diameter prevents large ions such as Cs+ from permeating and only allows small ions such as Ca2+ and Na+ to pass through. The variation of pore size by the V102 mutation results in an altered V rev and ion selectivity (Xu et al. 2016). Second, the competition of binding to E106/CAR helps to select Ca2+ over Na+. The Ca2+ ion has a similar size but a more positive charge; thus, it has higher binding affinity to E106/CAR compared to Na+. The external pH alters the ion selectivity (Scrimgeour et al. 2012; Beck et al. 2014), probably by affecting how much charge E106 and CAR carry, which determines their binding affinities for cations. Third, the high local concentration of Ca2+ close to the pore entrance, applied experimentally or concentrated by CAR, also contributes to the establishment of high Ca2+ selectivity. Fourth, the exquisite spatial arrangement of the six carbonyls of E106 contributes to high Ca2+ selectivity. The E106D mutation disturbs this arrangement by shortening one carbon atom of the side chain and results in decreased binding affinity to Ca2+ and dramatically impaired ion selectivity (Yamashita et al. 2007). The gating of STIM1 increases the luminescence of Tb3+ bound to E106 and triggers a spatial rearrangement of E106 (Gudlur et al. 2014), which sheds light on the mechanism by which STIM1 regulates ion selectivity.
Taken together, it is conceivable that the ion selectivity filter formed by E106, which is not a highly rigid ring with a fixed spatial arrangement of carbonyls, could be altered by STIM1 gating and 2‐APB binding (discussed below), as well as by E106D mutation, E190Q mutation (Prakriya et al. 2006; Vig et al. 2006) and G98D mutation (Zhang et al. 2011). The conformational change induced by these factors therefore modulates the ion selectivity of Orai1.
Gating and modulation of the Orai1 channel by 2‐APB
2‐APB exerts complex effects on CRAC channels, including potentiation of STIM1‐activated I crac at a low concentration, inhibition of STIM1‐activated I crac at a medium concentration, and direct gating of Orai3 and STIM1‐unbound Orai1 at a high concentration (Peinelt et al. 2008). Investigations of these multiple modes of action help to reveal the CRAC channel gating and modulation mechanisms, and benefit the development of novel immune enhancers and inhibitors.
Potentiation
The 2‐APB‐elicited potentiation current (I p) shares similar properties with I crac. It was proposed that 2‐APB generates I p by increasing channel number (N) at a constant open probability (Po) (Prakriya & Lewis, 2006), or by enhancing the interaction between Orai1 and STIM1 (Navarro‐Borelly et al. 2008; Wang et al. 2009; Wang et al. 2014), or by altering the pore architecture (Peinelt et al. 2008). Recently, we reported that the 2‐APB‐elicited I p is largely due to increased unitary conductance of the Orai1 channel (Xu et al. 2016). We revealed that I p is generated by the pore dilatation effect of 2‐APB and determined by the ratio of ion size/pore diameter. As shown in Fig. 3, when the ratio is high, such as when Cs+ ions are used to permeate the STIM1‐gated Orai1 channels (3.8 Å vs. 3.8 Å), the large hindrance could not be effectively overcome by the pore dilatation effect of 2‐APB and marginal Cs+‐conducting I p is detected. When the ratio is suitable (0.60–0.78), such as when Ca2+/Na+ ions pass through STIM1‐gated Orai1 channels (2.32/2.28 Å vs. 3.8 Å), or Cs+ ions through STIM1‐gated V102C Orai1 channels (3.8 Å vs. 4.9 Å), the pore dilatation effect of 2‐APB decreases hindrance and facilitates ion permeation and I p is generated. When the ratio is low (≤ 0.47), such as when Ca2+/Na+ ions pass through STIM1‐gated V102C/A Orai1 channels (2.32/2.28 Å vs. 4.9/6.8 Å), or Cs+ ions through STIM1‐gated V102G Orai1 channels (3.8 Å vs. 8.1 Å), no I p is elicited (Xu et al. 2016). The generation of I p requires the open state of Orai1, not STIM1 itself, suggesting that 2‐APB acts on Orai1 directly rather than on STIM1, although the exact binding site and pore dilatation mechanism remain to be revealed.
Figure 3. 2‐APB‐induced I p is determined by both the channel pore diameter and conducting ion size.

The estimated pore diameters of the STIM1‐activated Orai1 channels are 3.8 Å for WT, 4.9 Å for V102C, 6.8 Å for V102A and 8.1 Å for V102G. The naked ion sizes of Ca2+, Na+ and Cs+ are 2.28, 2.32 and 3.8 Å, respectively. A and B, bar graphs summarizing the potentiation ratios of Na+ (A) and Cs+ (B) currents on WT and V102X mutants (adapted from Xu et al. 2016). Ca2+ has a similar I p pattern to Na+. C–E, schematic model for the generation of 2‐APB‐induced I p on STIM1‐activated Orai1 WT (C), V102C mutant (D) and V102A/G mutant (E) channels (adapted from Xu et al. 2016). The 2‐APB‐induced I p is prominent on the WT channel when conducting small ions such as Na+ and Ca2+, as well as on the V102C mutant channel when conducting large ions such as Cs+.
The extremely narrow CRAC channel pore consists of the hydrophobic centre and basic region. Cations encounter steric hindrance and a large energy barrier formed by van der Waals forces and electrostatic repulsion along the permeation pathway. As shown in Fig. 3, our discovery emphasizes that the extremely narrow pore diameter limits the flow speed of cations. Additionally, the R91K/D mutation produces larger Ca2+ influx than WT (Zhang et al. 2011), and Na+ conducts a much larger current than Ca2+ on CRAC channels. In these two conditions, the increased ion permeation should be due to the decreased steric hindrance and abolished/reduced electrostatic repulsion between cations and R91 residue. Therefore, we hypothesize that the extremely narrow pore diameter and unusual conduction pathway lined with positively charged residues jointly lead to the extremely low single‐channel conductance. The newly developed single‐channel optical recording technique should help to clarify this hypothesis (Dynes et al. 2016).
Inhibition
It is unlikely that 2‐APB inhibits I crac by directly blocking the extremely narrow pore because even Cs+ ions with a diameter of 3.8 Å could not pass through it. Although 2‐APB acts on the STIM1–Orai coupling interface (Hendron et al. 2014), inhibition is not due to the dissociation of Orai1 from STIM1. By contrast, 2‐APB increases their association as measured by fluorescence resonance energy transfer (Navarro‐Borelly et al. 2008; Wang et al. 2009; Wang et al. 2014). 2‐APB impairs puncta formed by Orai1‐unbound STIM1 (DeHaven et al. 2008; Peinelt et al. 2008) via enhancing the interaction between STIM1‐CC1 and SOAR. However, this effect is not essential for its inhibition on I crac, because 2‐APB directly inhibits the constitutively active V102A/P245T currents in the absence of STIM1 (Wei et al. 2016). The inhibition is more effective when 2‐APB is applied in the extracellular solution than in the intracellular solution (Prakriya & Lewis, 2001), which suggests the extracellular location of the binding site on Orai1. Because of the important role of E106 and CAR on ion permeation, as well as evidence that Gd3+ inhibits I crac by tightly binding to E106 (Hou et al. 2012; Amcheslavsky et al. 2015) and possibly also CAR (Yeromin et al. 2006), it is possible that 2‐APB may inhibit I crac by modulating E106 and CAR too.
Direct gating
STIM1‐dependent gating and 2‐APB‐induced direct gating are different but mutually exclusive (Yamashita et al. 2011), suggesting that these two processes are competitive, probably because they share common structural elements in their gating processes. Indeed, they use the same ion conduction pathway (Amcheslavsky et al. 2014) and are abolished by the homologous sensitive residue (K85E on Orai1 and K60E on Orai3) (Lis et al. 2010).
However, the 2‐APB‐directly‐gated current is quite different from the STIM1‐activated I crac, as the former is characterized by the loss of high Ca2+ selectivity, altered I–V profile and enlarged pore diameter on Orai3 (Schindl et al. 2008; Yamashita & Prakriya, 2014). Consistently, many mutations differentially affect these two types of gating. On Orai3, the sensitive sites of 2‐APB were mapped to the TM2–TM3 region (Zhang et al. 2008). In this region, the C101/G158 linkage delays the development of 2‐APB directly gated current (Amcheslavsky et al. 2013). The Orai3‐E165A mutant, which is homologous to Orai1‐E190A, is normally activated by STIM1 but lacks 2‐APB direct gating (Amcheslavsky et al. 2014). G183A mutation on Orai1 confers 2‐APB sensitivity, whereas it impairs STIM1‐dependent gating. The constitutively active Orai1‐W176C mutant does not respond to 2‐APB but could be further activated by STIM1 (Srikanth et al. 2011). On the ETON region, K85Q mutation on Orai1 reduces STIM1‐gated I crac, whereas it results in a larger 2‐APB‐directly‐gated current than WT‐Orai1. R52E and R53E mutations on Orai3 decrease STIM1‐dependent gating but do not affect 2‐APB direct gating (Lis et al. 2010). Therefore, it is conceivable that these two types of gating elicit different conformational transitions in the TM2, TM3 and TM1 regions, which differentially shape the ion conduction pathway and result in two types of currents characterized by different ion selectivity and I–V profiles.
Conclusion
Here, we focus on the structure–function relationship of the Orai1 channel. The crystal structure of dOrai depicts the entire structure of this channel. Mutagenesis studies provided details on the possible mechanisms by which the critical structural elements, together with the binding of STIM1 and 2‐APB, orchestrate its gating, modulation and biophysical properties. Additional effort should focus on determining the structure of STIM1‐activated Orai1, constitutively active Orai1 mutants, and the Orai1–2‐APB complex, using techniques such as traditional crystallization, rapidly developed single‐particle cryo‐electron microscopy and computer simulation. These structures will then lead to subsequent site‐directed mutagenesis studies to clearly illustrate the exact gating and modulation mechanisms, which are essential for the future development of pharmacological immune modulators.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
S.A.: collection and assembly of data; manuscript writing. T.X.: conception and design; manuscript writing; administrative support. X.X.: data analysis and interpretation; conception and design; manuscript writing; financial support. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors research was supported by grants from the National Key laboratory of Biomacromolecules and the National Natural Science Foundation of China (81373422). This work was also sponsored by CAS‐TWAS President's Fellowship for International Students (to S.A.).
Acknowledgements
We thank Dr Fang Bai for the homology modelling of human Orai1.
Biographies
Sher Ali (left) is a PhD student in Tao Xu's Lab. He obtained a BS in Pharmaceutical Sciences from Hamdard University and an MS in Biotechnology from the COMSATS Institute of Information Technology.
Tao Xu (centre) received his PhD in Biophysics from Huazhong University of Science and Technology. From 1996 to 2000 he undertook postdoctoral work first with Erwin Neher at the Max‐Planck Institute for Biophysical Chemistry in Goettingen and then with Bertil Hille at the University of Washington in Seattle. In 2003, he moved to the Institute of Biophysics, Chinese Academy of Sciences, and was appointed the Director‐General in 2007.

Xiaolan Xu (right) received her PhD in Biochemistry from Peking University. She first started as a postdoctoral fellow and then continued as a research assistant in Tao Xu's lab. We share a common interest in gating of the CRAC channel and its modulation by STIM1 and 2‐APB.
This review was presented at “Advances and Breakthroughs in Calcium Signaling”, which took place in Honolulu, Hawaii, 7–9 April 2016.
References
- Amcheslavsky A, Safrina O & Cahalan MD (2013). Orai3 TM3 point mutation G158C alters kinetics of 2‐APB‐induced gating by disulfide bridge formation with TM2 C101. J Gen Physiol 142, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amcheslavsky A, Safrina O & Cahalan MD (2014). State‐dependent block of Orai3 TM1 and TM3 cysteine mutants: insights into 2‐APB activation. J Gen Physiol 143, 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amcheslavsky A, Wood ML, Yeromin AV, Parker I, Freites JA, Tobias DJ & Cahalan MD (2015). Molecular biophysics of Orai store‐operated Ca2+ channels. Biophys J 108, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A, Fleig A, Penner R & Peinelt C (2014). Regulation of endogenous and heterologous Ca2+ release‐activated Ca2+ currents by pH. Cell Calcium 56, 235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Bird GS & Putney JW Jr (2008). Complex actions of 2‐aminoethyldiphenyl borate on store‐operated calcium entry. J Biol Chem 283, 19265–19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Plenk P, Fahrner M, Muik M, Jardin I, Schindl R, Gruber HJ, Groschner K & Romanin C (2013). The extended transmembrane Orai1 N‐terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1. J Biol Chem 288, 29025–29034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Fiorin G, Carnevale V, Treptow W & Klein ML (2013). Pore waters regulate ion permeation in a calcium release‐activated calcium channel. Proc Natl Acad Sci USA 110, 17332–17337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynes JL, Amcheslavsky A & Cahalan MD (2016). Genetically targeted single‐channel optical recording reveals multiple Orai1 gating states and oscillations in calcium influx. Proc Natl Acad Sci USA 113, 440–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y, Noguchi S, Hara Y, Hayashi YK, Motomura K, Miyatake S, Murakami N, Tanaka S, Yamashita S, Kizu R, Bamba M, Goto Y, Matsumoto N, Nonaka I & Nishino I (2015). Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store‐operated Ca2+ channels. Hum Mol Genet 24, 637–648. [DOI] [PubMed] [Google Scholar]
- Frischauf I, Muik M, Derler I, Bergsmann J, Fahrner M, Schindl R, Groschner K & Romanin C (2009). Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1‐3 channels by a STIM1 coiled‐coil mutant. J Biol Chem 284, 21696–21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischauf I, Zayats V, Deix M, Hochreiter A, Jardin I, Muik M, Lackner B, Svobodova B, Pammer T, Litvinukova M, Sridhar AA, Derler I, Bogeski I, Romanin C, Ettrich RH & Schindl R (2015). A calcium‐accumulating region, CAR, in the channel Orai1 enhances Ca2+ permeation and SOCE‐induced gene transcription. Sci Signal 8, ra131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Gerasimenko OV & Petersen OH (2014). The role of Ca2+ in the pathophysiology of pancreatitis. J Physiol 592, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TO, Bychkova S, Peng S, Begg M, Gerasimenko OV & Petersen OH (2013). Ca2+ release‐activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci USA 110, 13186–13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudlur A, Quintana A, Zhou Y, Hirve N, Mahapatra S & Hogan PG (2014). STIM1 triggers a gating rearrangement at the extracellular mouth of the ORAI1 channel. Nat Commun 5, 5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendron E, Wang X, Zhou Y, Cai X, Goto J, Mikoshiba K, Baba Y, Kurosaki T, Wang Y & Gill DL (2014). Potent functional uncoupling between STIM1 and Orai1 by dimeric 2‐aminodiphenyl borinate analogs. Cell Calcium 56, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Pedi L, Diver MM & Long SB (2012). Crystal structure of the calcium release‐activated calcium channel Orai. Science 338, 1308–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS & Feske S (2015). Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci 1356, 45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu L, Deng Y, Ji W, Du W, Xu P, Chen L & Xu T (2011). Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res 21, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lu J, Xu P, Xie X, Chen L & Xu T (2007). Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release‐activated Ca2+ channel activation. J Biol Chem 282, 29448–29456. [DOI] [PubMed] [Google Scholar]
- Lis A, Zierler S, Peinelt C, Fleig A & Penner R (2010). A single lysine in the N‐terminal region of store‐operated channels is critical for STIM1‐mediated gating. J Gen Physiol 136, 673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Somasundaram A, Jairaman A, Yamashita M & Prakriya M (2013). The C‐ and N‐terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J Physiol 591, 2833–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Somasundaram A, Yamashita M & Prakriya M (2012). Gated regulation of CRAC channel ion selectivity by STIM1. Nature 482, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Yamashita M, Engh A & Prakriya M (2009). Structural determinants of ion permeation in CRAC channels. Proc Natl Acad Sci USA 106, 22516–22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, Kahr H, Madl J, Gruber H, Groschner K & Romanin C (2008). Dynamic coupling of the putative coiled‐coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem 283, 8014–8022. [DOI] [PubMed] [Google Scholar]
- Navarro‐Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ & Prakriya M (2008). STIM1‐Orai1 interactions and Orai1 conformational changes revealed by live‐cell FRET microscopy. J Physiol 586, 5383–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R & Isacoff EY (2016). Cooperative binding of stromal interaction molecule 1 (STIM1) to the N and C termini of calcium release‐activated calcium modulator 1 (Orai1). J Biol Chem 291, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Stanley C & Isacoff EY (2015). Critical role for Orai1 C‐terminal domain and TM4 in CRAC channel gating. Cell Res 25, 963–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE & Lewis RS (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinelt C, Lis A, Beck A, Fleig A & Penner R (2008). 2‐Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1‐dependent gating of CRAC channels. J Physiol 586, 3061–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A & Hogan PG (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. [DOI] [PubMed] [Google Scholar]
- Prakriya M & Lewis RS (2001). Potentiation and inhibition of Ca2+ release‐activated Ca2+ channels by 2‐aminoethyldiphenyl borate (2‐APB) occurs independently of IP3 receptors. J Physiol 536, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M & Lewis RS (2006). Regulation of CRAC channel activity by recruitment of silent channels to a high open‐probability gating mode. J Gen Physiol 128, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M & Lewis RS (2015). Store‐operated calcium channels. Physiol Rev 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg BS, Wang Y & Gill DL (2013). Orai channel pore properties and gating by STIM: implications from the Orai crystal structure. Sci Signal 6, pe9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindl R, Bergsmann J, Frischauf I, Derler I, Fahrner M, Muik M, Fritsch R, Groschner K & Romanin C (2008). 2‐Aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J Biol Chem 283, 20261–20267. [DOI] [PubMed] [Google Scholar]
- Scrimgeour NR, Wilson DP & Rychkov GY (2012). Glu106 in the Orai1 pore contributes to fast Ca2+‐dependent inactivation and pH dependence of Ca2+ release‐activated Ca2+ (CRAC) current. Biochem J 441, 743–753. [DOI] [PubMed] [Google Scholar]
- Srikanth S, Yee MK, Gwack Y & Ribalet B (2011). The third transmembrane segment of orai1 protein modulates Ca2+ release‐activated Ca2+ (CRAC) channel gating and permeation properties. J Biol Chem 286, 35318–35328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Schindl R, Fahrner M, Zheng L, Gasmi‐Seabrook GM, Muik M, Romanin C & Ikura M (2013). STIM1/Orai1 coiled‐coil interplay in the regulation of store‐operated calcium entry. Nat Commun 4, 2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirado‐Lee L, Yamashita M & Prakriya M (2015). Conformational changes in the Orai1 C‐terminus evoked by STIM1 binding. PLoS One 10, e0128622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP & Penner R (2006). CRACM1 multimers form the ion‐selective pore of the CRAC channel. Curr Biol 16, 2073–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J & Gill DL (2014). Distinct Orai‐coupling domains in STIM1 and STIM2 define the Orai‐activating site. Nat Commun 5, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD & Gill DL (2010). The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Deng X, Zhou Y, Hendron E, Mancarella S, Ritchie MF, Tang XD, Baba Y, Kurosaki T, Mori Y, Soboloff J & Gill DL (2009). STIM protein coupling in the activation of Orai channels. Proc Natl Acad Sci USA 106, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Zhou Y, Sun AM, Ma GL, He L, Zhou LJ, Zhang SC, Liu J, Zhang SL, Gill DL & Wang Y (2016). Molecular mechanisms underlying inhibition of STIM1‐Orai1 mediated Ca2+ entry induced by 2‐aminoethoxydiphenyl borate. Pflugers Arch 468, 2061–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Ali S, Li Y, Yu H, Zhang M, Lu J & Xu T (2016). 2‐Aminoethoxydiphenyl borate potentiates CRAC current by directly dilating the pore of open Orai1. Sci Rep 6, 29304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Navarro‐Borelly L, McNally BA & Prakriya M (2007). Orai1 mutations alter ion permeation and Ca2+‐dependent fast inactivation of CRAC channels: evidence for coupling of permeation and gating. J Gen Physiol 130, 525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M & Prakriya M (2014). Divergence of Ca2+ selectivity and equilibrium Ca2+ blockade in a Ca2+ release‐activated Ca2+ channel. J Gen Physiol 143, 325–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Somasundaram A & Prakriya M (2011). Competitive modulation of Ca2+ release‐activated Ca2+ channel gating by STIM1 and 2‐aminoethyldiphenyl borate. J Biol Chem 286, 9429–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Jin H, Cai X, Li S & Shen Y (2012). Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc Natl Acad Sci USA 109, 5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O & Cahalan MD (2006). Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF & Muallem S (2008). STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell 32, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Kozak JA, Jiang W, Yeromin AV, Chen J, Yu Y, Penna A, Shen W, Chi V & Cahalan MD (2008). Store‐dependent and ‐independent modes regulating Ca2+ release‐activated Ca2+ channel activity of human Orai1 and Orai3. J Biol Chem 283, 17662–17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Hu J, Amcheslavsky A, Zheng H & Cahalan MD (2011). Mutations in Orai1 transmembrane segment 1 cause STIM1‐independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc Natl Acad Sci USA 108, 17838–17843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Zhou MH, Hu C, Kuo E, Peng X, Hu J, Kuo L & Zhang SL (2013). Differential roles of the C and N termini of Orai1 protein in interacting with stromal interaction molecule 1 (STIM1) for Ca2+ release‐activated Ca2+ (CRAC) channel activation. J Biol Chem 288, 11263–11272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Ramachandran S, Oh‐Hora M, Rao A & Hogan PG (2010). Pore architecture of the ORAI1 store‐operated calcium channel. Proc Natl Acad Sci USA 107, 4896–4901. [DOI] [PMC free article] [PubMed] [Google Scholar]