

Abstract
Key points
Pharmacological and molecular inhibition of transient receptor potential melastatin 7 (TRPM7) reduces store‐operated calcium entry (SOCE).
Overexpression of TRPM7 in TRPM7−/− cells restores SOCE.
TRPM7 is not a store‐operated calcium channel.
TRPM7 kinase rather than channel modulates SOCE.
TRPM7 channel activity contributes to the maintenance of store Ca2+ levels at rest.
Abstract
The transient receptor potential melastatin 7 (TRPM7) is a protein that combines an ion channel with an intrinsic kinase domain, enabling it to modulate cellular functions either by conducting ions through the pore or by phosphorylating downstream proteins via its kinase domain. In the present study, we report store‐operated calcium entry (SOCE) as a novel target of TRPM7 kinase activity. TRPM7‐deficient chicken DT40 B lymphocytes exhibit a strongly impaired SOCE compared to wild‐type cells as a result of reduced calcium release activated calcium currents, and independently of potassium channel regulation, membrane potential changes or changes in cell‐cycle distribution. Pharmacological blockade of TRPM7 with NS8593 or waixenicin A in wild‐type B lymphocytes results in a significant decrease in SOCE, confirming that TRPM7 activity is acutely linked to SOCE, without TRPM7 representing a store‐operated channel itself. Using kinase‐deficient mutants, we find that TRPM7 regulates SOCE through its kinase domain. Furthermore, Ca2+ influx through TRPM7 is essential for the maintenance of endoplasmic reticulum Ca2+ concentration in resting cells, and for the refilling of Ca2+ stores after a Ca2+ signalling event. We conclude that the channel kinase TRPM7 and SOCE are synergistic mechanisms regulating intracellular Ca2+ homeostasis.
Keywords: αKinase, SOCE, TRPM7
Key points
Pharmacological and molecular inhibition of transient receptor potential melastatin 7 (TRPM7) reduces store‐operated calcium entry (SOCE).
Overexpression of TRPM7 in TRPM7−/− cells restores SOCE.
TRPM7 is not a store‐operated calcium channel.
TRPM7 kinase rather than channel modulates SOCE.
TRPM7 channel activity contributes to the maintenance of store Ca2+ levels at rest.
Abbreviations
- CRAC
calcium release‐activated calcium channel
- CRACR2
CRAC‐regulatory protein 2
- cTRPM7
chicken TRPM7
- cWT
chicken wild‐type
- ∆Kinase
deleted kinase domain
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- HEK
human embryonic kidney
- hTRPM7
human TRPM7
- IP3
inositol trisphosphate
- KAB
kinase activity buffer
- KO
knockout
- PI
propidium Iodide
- PM
plasma membrane
- SARAF
SOCE‐associated regulatory factor
- SOCE
store‐operated calcium entry
- STIM
stromal interaction molecule
- Tg
thapsigargin
- TRP
transient receptor potential
- TRPM7
transient receptor potential melastatin 7
- WT
wild‐type
- WT par
wild‐type parental
Introduction
Changes in intracellular calcium concentration regulate a plethora of biological functions within a cell, including signal transduction, proliferation, motility, transcription, exocytosis and apoptosis. However, dysregulation of Ca2+ homeostasis may drive carcinogenesis by modulating cell migration and metastasis (Prevarskaya et al. 2014). Cells increase their intracellular Ca2+ concentration either by releasing Ca2+ from intracellular stores, such as the endoplasmic reticulum (ER) or sarcoplasmic reticulum or by opening Ca2+ conducting channels within the plasma membrane (PM). In many electrically non‐excitable cells, both processes are coupled, in that Ca2+ release from the ER engages calcium release‐activated calcium channels (CRAC) in the PM, a process that is known as store‐operated calcium entry (SOCE). This mechanism has two crucial functions: cellular signalling and store refilling.
Although SOCE and the corresponding CRAC channels have been described in the late 1980s and early 1990s, the molecular key players of SOCE remained elusive until extensive RNAi screens in Drosophila identified two proteins crucial for SOCE: the ER‐resident Ca2+ sensor stromal interaction molecule STIM1 (Liou et al. 2005; Roos et al. 2005) and the PM‐resident channel‐forming protein Orai1 (Feske et al. 2006; Prakriya et al. 2006; Vig et al. 2006a,b; Zhang et al. 2006). STIM1 binds to and activates Orai1 channels upon store depletion, providing a Ca2+ influx pathway that is essential for proper cell function in almost all eukaryotic cells. Careful titration of the protein concentration of STIM1 and Orai1 thus determines the amount of Ca2+ influx (Hoover & Lewis, 2011; Kilch et al. 2013) and its subsequent downstream effects. Mammalian species contain three homologous CRAC channel proteins (Orai1, Orai2 and Orai3), as well as two homologous STIM molecules (STIM1 and STIM2). All Orai homologues exhibit distinct selectivity profiles for Ca2+, Ba2+ and Na+, differential pharmacological effects in response to 2‐aminoethoxydiphenyl borate, and strikingly different feedback regulation by intracellular Ca2+. STIM1 is thoroughly characterized in the literature, however knowledge about STIM2 awaits further study. Although STIM2 exhibits higher expression levels in primary human melanocytes (Stanisz et al. 2012) and is the prominent STIM protein in neurons (Berna‐Erro et al. 2009) and dendritic cells (Bandyopadhyay et al. 2011), gene depletion studies of STIM2 exhibit minimal to no deleterious effects in most cell lines. A significant functional difference between STIM1 and STIM2 is the two‐fold higher Ca2+ sensitivity of the EF‐hand domain of STIM1 (Stathopulos et al. 2009; Zheng et al. 2011), which allows STIM2 to activate Ca2+ influx at smaller decreases in Ca2+ store content. STIM2 has been identified as the feedback regulator for basal Ca2+ concentration and ER store content (Brandman et al. 2007). STIM2 may be pre‐coupled to Orai channels in the PM, enabling store independent activation of Ca2+ influx and regulation of basal cytosolic Ca2+ concentration. STIM2 can also remain in the ER, where it serves to activate CRAC channels after mild store depletion (Parvez et al. 2008; Thiel et al. 2013).
The regulation of SOCE/CRAC is subjected to multiple molecular mechanisms, including post‐translational modifications such as phosphorylation (Parekh & Penner, 1995; Smyth et al. 2009, 2012; Kawasaki et al. 2010), ubiquitination (Keil et al. 2010; Eylenstein et al. 2011; Lang et al. 2012) and glycosylation (Kilch et al. 2013), or interaction partners such as CRAC‐regulatory protein 2 (CRACR2)A and B (Srikanth et al. 2010) and Junctate (Srikanth et al. 2012), as well as inhibitory proteins such as Golli (Feng et al. 2006) and SOCE‐associated regulatory factor (SARAF) (Palty et al. 2012).
Another Ca2+‐conducting channel of significant research interest in the field of cancer is transient receptor potential melastatin 7 (TRPM7), which contains an ion channel domain that transports Mg2+, Co2+, Mn2+, Ni2+ and Zi2+ and an active protein kinase domain (Fleig & Chubanov, 2014). Knockout (KO) studies of TRPM7 have established it as a key player in cell growth and proliferation, as well as developmental processes (Nadler et al. 2001; Jin et al. 2008, 2012; Ryazanova et al. 2010; Lee et al. 2011; Sah et al. 2013a). Although the TRPM7 kinase domain is not essential for channel function, there is a functional coupling that modulates channel gating (Schmitz et al. 2003; Jansen et al. 2016). It is known that TRPM7 kinase activity is regulated by intracellular Mg2+ (Ryazanova et al. 2004) and caspases can cleave the kinase from the channel without affecting its phosphotransferase activity (Desai et al. 2012). Besides the autophosphorylation of TRPM7 itself, several targets of the kinase have been identified, including the assembly domain of myosin IIA, IIB, IIC (Clark et al. 2006), annexin I (Dorovkov & Ryazanov, 2004), phospholipase Cγ2 (Deason‐Towne et al. 2012) and eEF2‐k (Perraud et al. 2011), as well as components of chromatin‐remodelling complexes (Krapivinsky et al. 2014).
As a result of the almost ubiquitous presence of these ion channels and their significance for cell physiology and the development of immune diseases and cancer, further research is needed to fully understand their regulatory mechanisms and downstream effects. In the present study, we investigated how TRPM7 contributes to and interacts with SOCE and how this affects the store refilling process and the store content in B lymphocytes. We found that, although TRPM7 itself is not a store‐operated ion channel, its activity is acutely linked to SOCE and its facilitating modulatory effect is mediated by the kinase domain of the protein. In addition, TRPM7 contributes to Ca2+ homeostasis by maintaining the filling state of intracellular Ca2+ stores in resting cells, and this effect is mediated by the ion channel domain of TRPM7. We hypothesize that TRPM7 channel activity is required to maintain the cellular Ca2+ store content and that TRPM7 kinase phosphorylates and thereby modulates STIM proteins and/or other SOCE components. This represents the first study showing a relationship between a TRPM channel, CRAC/SOCE and the ER store content and demonstrates a new facet of how SOCE can be regulated.
Methods
Cell culture
DT40 B lymphocytes, both wild‐type (WT) parental (WT par) and genetically modified, were maintained in RPMI 1640 medium (Clontech, Palo Alto, CA, USA) with 10% fetal bovine serum (FBS), 1% chicken serum, 10 U ml−1 penicillin/streptomycin and 2 mm glutamine. Next, 50 μg ml−1 blasticidin was added to chicken TRPM7‐KO (cTRPM7‐KO) cells. Then, 50 μg ml−1 blasticidin and 1 mg ml−1 zeocin was added to all inducible overexpressing cell lines. DT40 cTRPM7‐KO, chicken wild‐type (cWT), human TRPM7 (hTRPM7)‐deleted kinase domain (∆Kinase) and hTRPM7‐KR were maintained in culture medium supplemented with 15 mm MgCl2 when uninduced (Schmitz et al. 2003). Overexpression in DT40 B cell lines was induced by 1 μg ml−1 doxycycline for 48 h. KO of chicken TRPM7 in the DT40 V79.1 mutant was induced by 200 nm tamoxifen (Nadler et al. 2001).
Rat basophilic leukaemia‐1 (obtained from the American Type Culture Collection, Manassas, Virginia) and tetracycline‐inducible human embryonic kidney (HEK) hTRPM7 cells (Schmitz et al. 2003) were maintained in Dulbecco's modified Eagle's medium (Clontech) with 10% FBS and 10 U ml−1 penicillin/streptomycin. The medium for HEK hTRPM7 cells was supplemented with 5 μg ml−1 blasticidin and 200 μg ml−1 zeocin. The overexpression in HEK cells was induced by the addition of 1 μg ml−1 tetracycline for 24 h.
All cells were cultured under 5% CO2 at 37°C and 95% humidity.
Constructs and transfection
hStim1 and hStim2 constructs were subcloned into pCAGGS‐IRES‐GFP and pIRES‐Neo, respectively. For transfection, 2 μg of DNA/106 cells was electroporated into HEK‐293 cells overexpressing TRPM7 with Nucleofactor II electroporator and kit L (Lonza, Basel, Switzerland). The cells were transfected in accordance with the manufacturer's instructions and cultured for 48 h before protein extraction.
Electrophysiology
Patch clamp recordings were performed at room temperature in the tight‐seal whole‐cell configuration. Recording electrodes with a resistance of 3–4 MΩ were used. Pipette and cell capacitance were electronically compensated before each voltage ramp with an EPC‐10 patch clamp amplifier controlled using Patchmaster software (HEKA, Lambrecht, Germany). After establishing whole‐cell configuration, voltage ramps from −100 to +120 mV (200 ms duration) for the measurement of TRPM7 currents and from −150 to +100 mV (50 ms duration) for the measurement of CRAC currents were applied every 2 s from a holding potential of 0 mV. Potassium currents were measured using voltage ramps from −100 to +100 mV with a holding potential of −80 mV. Membrane currents were sampled at 10 kHz and filtered at 2.9 kHz. Voltages were corrected for a liquid junction potential of 10 mV in standard bath solution. For leak current correction, the ramp current before current activation was subtracted and the currents were normalized to whole cell capacitance.
The internal pipette solution contained (in mm): 140 Cs‐glutamate, 8 NaCl, 10 Cs‐Hepes, 3 MgCl2, 10 BAPTA and 0.02 inositol trisphosphate (IP3) for recording CRAC currents and 140 Cs‐glutamate, 8 NaCl, 10 Cs‐Hepes and 5 mm EDTA for TRPM7 currents. For K+ currents, we used (in mm): 140 K‐glutamate, 8 NaCl, 10 Hepes and 7.5 CaCl2, buffered with 10 BAPTA to 1 μm free internal calcium.
Standard bath solution contained (in mm): 120 NaCl, 2.8 KCl, 2 MgCl2, 10 CaCl2, 10 CsCl, 10 Na‐Hepes and 10 glucose for recording CRAC currents and 140 NaCl, 2.8 KCl, 2 MgCl2, 1 CaCl2, 10 Na‐Hepes and 10 glucose for both TRPM7 and K+ currents.
Flow cytometric analysis of DNA content and cell cycle analysis
DNA content and cell cycle analyses were carried out after fixation of cells and staining with propidium iodide (PI). Briefly, 2–3 × 106 cells of each condition were washed once with PBS, then fixed by adding 1 ml of ice‐cold 70% ethanol and stored at 4°C overnight. Cells were washed twice with PBS to remove the EtOH, treated with 20 μg ml−1 RNase A for 30 min and stained by adding 50 μg ml−1 PI for another 30–45 min in the dark at 4°C. Cells were analysed using a BD FACSCalibur Flow Cytometer (Becton‐Dickinson Biosciences, Franklin Lakes, NJ, USA) with a 488 laser and data were collected with a 585/42 filter. For cell cycle analysis, the singlets were separated by a gate and 25 000 events per experiment were counted. Analysis was performed using FlowJo software (FlowJo LLC, Ashland, OR, USA).
Fura‐2AM based Ca2+ imaging
Cells were pre‐plated for 30 min on glass coverslips and then loaded with 3.5 μm fura‐2AM in RPMI for 15–20 min at 37°C and 5% CO2 in a humidified incubator. For store refilling experiments, cells were loaded in suspension and washed afterwards with cell culture medium. To deplete the stores, cells were treated with 1 μm ionomycin for 5 min. Ionomycin was removed by washing the cells three times with cell culture medium, followed by plating the cells on glass coverslips for 20 min. Control cells were treated the same way, but without exposure to ionomycin. All experiments were performed in an open perfusion chamber with an upright microscope at room temperature. Images were analysed with TILLVision software (FEI Munich GmbH, Gräfelfing, Germany). The effect of high‐K‐induced depolarization on SOCE was assessed using a functional drug screening system (FDSS‐7000EX; Hamamatsu Photonics KK, Japan), a 96/384‐well fluorescent kinetic plate reader. Induced and uninduced DT40 V79.1 cells were loaded with 3.5 μm fura‐2AM then plated in 96‐well plate at a density of 60 000 cells well–1 before measurement. Bath solutions contained (in mm): 140 NaCl or 140 KCl, 0 or 0.5 or 2 CaCl2, 2 MgCl2, 10 glucose and 10 Hepes (pH 7.2 with NaOH). Stock solutions of thapsigargin (Tg) were prepared in DMSO at a concentration of 1 or 5 mm.
Quantitative real‐time PCR
Total RNA was isolated from 3 × 106 cells using a Qiagen RNeasy Kit in accordance with the manufacturer's instructions. RNA integrity was assessed using 1 μl of RNA on RNA Nano 6000 chips and a 2100 Agilent Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA, USA). Next, 1 μg of RNA was converted to 20 μl of cDNA using a High Capacity cDNA RT Kit with RNase Inhibitor (Invitrogen, Carlsbad, CA, USA). The kit utilizes random priming. cDNA was further diluted to 8 ng μl−1 (based on the starting RNA concentration) with nuclease‐free water. Quantitative real‐time PCR was performed in a 10 μl reaction volume on a 384‐well plate using 32 ng of cDNA in triplicate for the genes of interest and housekeeping genes.
The final concentration of the QuantiTect assay was 1× and for individual primers 625 nm for each F‐ and R‐primer with a Quanti Q19 Tect SYBR green kit (Qiagen, Valancia, CA, USA). PCR conditions were: 15 min at 95°C, 15 s at 94°C, 30 s at 55°C (40 cycles) and 30 s at 72°C. Relative expression was calculated in accordance with the ∆Cq method (2−∆Cq). The primers used were: GAPDH forward: GGCACGCCATCACTATC; reverse: CCTGCATCTGCCCATTT; UB forward: GGGATGCAGATCTTCGTGAAA; reverse: CTTGCCAGCAAAGATCAACCTT; TRPM7 (Qiagen).
Immunoblotting
For analysis of expression and phosphorylation levels, HEK293 hTRPM7 cells were induced with tetracycline. As shown in Fig. 6 A, cells were treated with or without 30 μm NS8593 and lysed after 24 h. As shown in Fig. 7, cells were transfected with Stim1 or Stim2 constructs for 48 h before protein extraction (for details, see above). Lysis buffer contained (in mm): 10 Tris‐HCl, 69 Na Cl, 5% glycerol, 0.5% Triton‐X, 1 EDTA, 1 × HALT phosphatase inhibitor cocktail mix (Thermo Scientific, Waltham, MA, USA) and protease inhibitor cocktail (Sigma‐Aldrich, St Louis, MO, USA) in accordance with the manufacturer's instructions. For all western blot experiments, 300 μg of total proteins was used to immunoprecipitate TRPM7, Stim1 and Stim2 proteins using anti‐HA, anti‐FLAG and anti‐Stim2 antibodies, respectively. The in vitro phosphorylation assay of TRPM7 was performed as described previously (Schmitz et al. 2003). Briefly, the immunoprecipitated TRPM7 proteins were treated alone with PBS or kinase activity buffer (KAB) or mixed with Stim1 or Stim2 then incubated with PBS or KAB. All samples were incubated for 30 min at 30°C on a rotating wheel. Samples were then denatured with β‐mercaptoethanol for 5 min at 65°C and supernatants were transferred on a 3–8% gradient Tris‐acetate gel, separated by electrophoresis, and electrotransferred on polyvinylidene fluoride membranes at a constant 15 V overnight. Immunoblots were probed with anti‐HA (dilution 1:500; Roche, Basel, Switzerland), anti‐Phospho‐Mix (dilution 1:1000; Cell Signaling Technology, Beverly, MA, USA), anti‐Flag (dilution 1:1000; Sigma‐Aldrich) and anti‐STIM2 (dilution 1:400; Anaspec, Inc., Fremont, CA, USA).
Figure 6. TRPM7 kinase domain may be targeting STIM proteins.
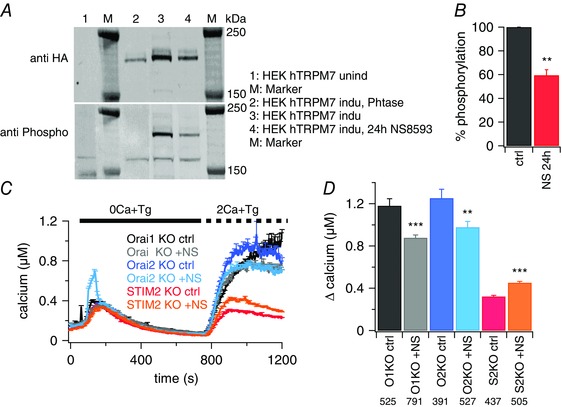
A, western blot analyses of TRPM7 expression and phosphorylation in tetracycline‐inducible HEK hTRPM7 cells. (1) uninduced cells (M, marker); (2) cells induced with tetracycline (24 h) and cell lysate treated with λ phosphatase; (3) cells induced with tetracycline (24 h); and (4) cells induced with tetracycline and treated with 30 μm NS8593 (24 h) (a representative blot for four independent experiments is shown) (M, marker). B, percentage of hTRPM7 phosphorylation from cells treated with or without NS8593; lane 3 vs. lane 4 in (A). Phosphorylation was normalized to total protein level (HA‐signal) and phosphorylation in untreated cells was set to 100%. C, average [Ca2+]i responses following store depletion by the addition of 5 μm Tg and readdition of 2 mm [Ca2+]o (SOCE) in DT40 B cells Orai1‐KO, Orai2‐KO and STIM2‐KO with 30 μm NS8593 (+NS) or without (ctrl) in the bath solution. Number of cells are shown in the corresponding bar graphs in (D). D, average of Ca2+ influx peaks assessed from baseline and obtained from SOCE measurements in (C). Asterisks indicate statistical significance: * P < 0.05; ** P < 0.01; *** P < 0.001.
Figure 7. No detectable phosphorylation of STIM proteins was observed by Western blot.
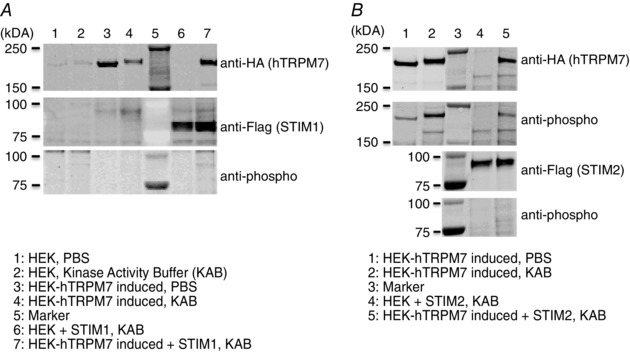
A, representative western blot of STIM1 co‐expression with TRPM7 and its phosphorylation in HEK hTRPM7 cells. B, representative western blot of STIM2 co‐expression with TRPM7 and its phosphorylation in HEK hTRPM7 cells.
Statistical analysis
All data are reported as the mean ± SEM. Currents were analysed with FitMaster, version 2.11 (HEKA) and IGOR Pro (Wavemetrics, Portland, OR, USA). CRAC current development was analysed at −130 mV and both potassium and TRPM7 currents were extracted at +80 mV for statistical analysis. Current amplitudes were normalized to cell size upon break‐in as current density (pA/pF). The amplitude of the reported SOCE response represents the difference in fluorescence before and after calcium readdition [(F max − F min)/F basal]. The absolute intracellular Ca2+ concentration was estimated from the relationship [Ca2+]i = K *(R − R min)/(R max − R) where the values of K, R min and R max were determined from an in situ calibration of fura‐2‐AM in DT40 cells as described by Grynkiewicz et al. (1985). Appropriate ANOVA or Student's t tests were performed to assess statistical significance. P < 0.05 was considered as statistically significant.
Results
TRPM7‐deficient B lymphocytes show impaired SOCE
To investigate the role of the Ca2+ conducting‐channels TRPM7 and CRAC in store‐operated calcium entry, as well as their potential interaction, we measured SOCE in different clones of DT40 Chicken B lymphocytes, either WT or TRPM7‐deficient, using a fura‐2‐based Ca2+ imaging approach with a Ca2+ re‐addition protocol. As shown in Fig. 1, SOCE was strongly reduced from a peak value of 1953 ± 134 nm Ca2+ in WT par cells to 756 ± 56 nm Ca2+ in stable DT40 cTRPM7‐KO cultured in medium supplemented with 15 mm Mg2+. Removal of the supplemented Mg2+ from the culture medium of cTRPM7‐KO cells for the indicated period of time reduced SOCE even more, probably as a result of a quiescent metabolic transition (Sahni et al. 2010) (Fig. 1 A). Similarly, the tamoxifen‐inducible DT40 cTRPM7‐KO cell line V79.1 also responded with reduced SOCE (Fig. 1 B and C). The maximal increase in [Ca2+]i after store depletion and external Ca2+ re‐addition was reduced from 515 ± 19 nm in uninduced V79.1 to 214 ± 19 nm in cells 4 days after induction of the KO, although the cells were supplemented with 15 mm Mg2+ in the culture medium.
Figure 1. Stable or inducible TRPM7‐KO decreases SOCE.
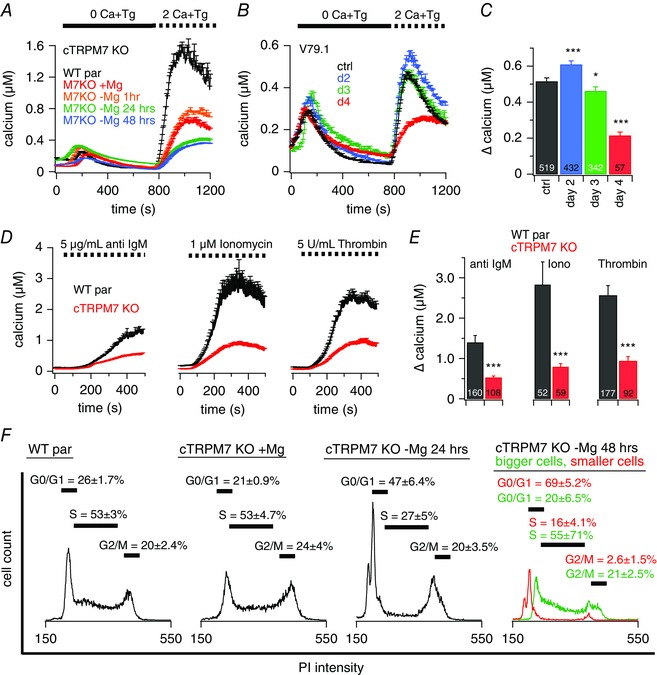
Ca2+ imaging experiments of intact DT40 chicken B lymphocytes. A–D, at the beginning of the experiment, cells were kept in extracellular standard solution supplemented with 0.5 mm Ca2+. A and B, average [Ca2+]i responses following store depletion by the addition of 5 μm Tg and readdition of 2 mm [Ca2+]o (SOCE) in DT40 WT par cells (n = 469) or TRPM7‐deficient cells (cTRPM7‐KO) and inducible V79.1 TRPM7‐KO DT40 cells (B). A, cTRPM7‐KO cells were kept in medium with or without supplemented 15 mm Mg2+ for the indicated time (+Mg2+, n = 546; –Mg2+ 1 h, n = 304; –Mg2+ 24 h, n = 129, –Mg2+ 48 h, n = 207). Data showing 24 h of Mg2+ exposure (in green) are shifted by +50 nm Ca2+ to make them visible behind the overlapping 48 h of data (in blue). B, KO was induced with tamoxifen and cells were kept in Mg2+ supplemented medium. C, average of Ca2+ influx peaks assessed from baseline and obtained from SOCE measurements in cells in (B). D, average [Ca2+]i responses following store depletion and Ca2+ influx by the addition of 2 mm [Ca2+]o (SOCE) triggered by different stimuli as indicated, in DT40 WT par cells (anti‐IgM n = 207, ionomycin n = 52, thrombin n = 177) or cTRPM7‐KO cells (anti‐IgM n = 108, ionomycin n = 59, thrombin n = 92). E, average Ca2+ influx peaks obtained from SOCE measurements in cells in (D). F, PI based cell cycle analysis of DT40 WT par and cTRPM7‐KO cells kept in medium with or without supplemented 15 mm Mg2+ for the indicated time (representative traces of six independent experiments are shown). Stars indicate statistical significance: * P < 0.05; ** P < 0.01; *** P < 0.001.
To test whether other stimuli of SOCE showed the same phenomenon, we used anti‐IgM antibodies, ionomycin and thrombin to release Ca2+ from the stores. The impaired SOCE in cTRPM7‐KO cells compared to WT par cells was also observed after induction of SOCE with these stimuli (Fig. 1 D and E).
Previous work has shown that the cell cycle state affects the amount of SOCE (Tani et al. 2007; Smyth et al. 2009). We therefore aimed to examine whether the reduced SOCE in TRPM7‐deficient cells was a result of the effect of TRPM7 on cell‐cycle distribution. Using PI‐based flow cytometry analyses, we measured the cell cycle distribution of B lymphocytes under all the experimental conditions described above. cTRPM7‐KO cells, cultured with an additional 15 mm Mg2+ in the cell culture medium, showed a similar cell cycle distribution as the WT par cells. The transition into the quiescent metabolic state occurs only upon removal of additional Mg2+ from the culture medium (Sahni et al. 2010) (Fig. 1 F). The cells then undergo apoptosis, resulting in an increase of the amount of cells in G0/G1 and hardly any cells in M‐phase. Forty‐eight hours after removal of additional Mg2+ from the culture medium, there are predominantly two populations of cells that differ in cell size and in cell cycle distribution (Fig. 1 F, right) and represent dying and living cells. Taken together, our results show that the SOCE phenotype in cTRPM7‐KO cells is a result of the absence of TRPM7 and not the subsequent changes in cell cycle distribution and cell growth.
TRPM7‐KO impaires SOCE by reducing ICRAC rather than affecting K+ conductances
To test whether TRPM7‐KO affects potassium (K+) conductances and thereby possibly changes membrane potential and calcium driving force, we used DT40 V79.1 cells cultured in the absence (uninduced) or presence of 200 nm tamoxifen (induced) to generate the cTRPM7‐KO. We first investigated whether impaired SOCE persists under completely potassium‐depolarized conditions. We performed fura‐2‐based Ca2+ imaging experiments under physiological condition using standard Ringer solution containing 140 mm NaCl, as well as under a depolarized condition where NaCl was equimolarly replaced by KCl and collapsing the membrane potential to 0 mV. We found that TRPM7‐KO reduced SOCE in a similar way under both physiological (∼45 % inhibition) and depolarizing conditions (∼40 % inhibition) (Fig. 2 A and B).
Figure 2. Impaired SOCE in TRPM7‐deficient B lymphocytes is not due to altered K+ currents.
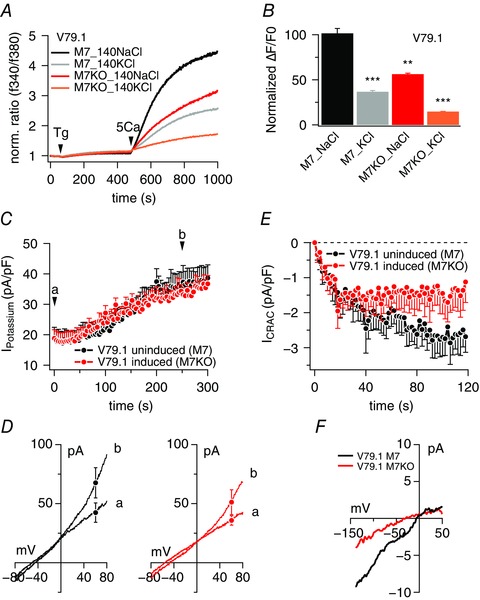
A, average [Ca2+]i responses following store depletion by the addition of 5 μm Tg and readdition of 5 mm [Ca2+]o (SOCE) in uninduced DT40 V79.1 or induced DT40 V79.1. Calcium signals were acquired on a 96‐well fluorescence kinetic plate reader and measured in standard Ringer solution with 140 mm NaCl or in depolarized condition by replacing NaCl with KCl. Data are averages of triplicates. B, average peak Ca2+ influx responses assessed from baseline and obtained in (A) (n = 3 each). Asterisks indicate statistical significance: * P < 0.05; ** P < 0.01; *** P < 0.001. C, time course of average K+ current development in uninduced DT40‐V79.1 cells (n = 9) and in induced DT40 V79.1 (n = 10). Tamoxifen (200 nm) was used to induce TRPM7‐KO and currents were recorded 4 days post‐induction. Cells were held at −80 mV and stimulated with a 50 ms voltage ramp from −100 mV to +100 mV at 0.5 Hz to activate voltage‐gated K+ channels. Cells were concomitantly perfused with 1 μm Ca2+ to activate any additional Ca2+‐sensitive K currents. Currents were extracted at +80 mV, normalized to cell size and plotted as current density in pA/pF over the time of the experiment. D, average I–V behaviour of K+ currents extracted at the two time points labelled (a) and (b) in (C) from uninduced (black I–V traces; n = 9) and induced‐KO cells (red I–V traces; n = 10). E, average CRAC currents (I CRAC) evoked by 20 μm IP3 in uninduced DT40 V79.1 (n = 12) or induced DT40 V79.1 (n = 20). Cells were held at 0 mV and stimulated with a 50 ms voltage ramp from −150 mV to +100 mV at 0.5 Hz. Cells were concomitantly perfused with 20 μm IP3 to activate I CRAC. Currents were extracted at −130 mV, normalized to cell size and plotted as current density in pA/pF over time of the experiment. F, average I–V behaviour of CRAC currents extracted from 116 to 120 s. The black trace represents average current from uninduced cells (n = 6) and the red trace represents average current from M7KO cells (n = 14).
To confirm these findings and more directly establish that K+ conductances are not affected by TRPM7‐KO, we next investigated whole‐cell K+ currents using experimental conditions that were designed to simultaneously reveal effects on both voltage‐ and calcium‐activated K+ currents, if present. Cells were held at −80 mV to keep voltage‐dependent K+ channels activatable and stimulated at 0.5 Hz with a 50 ms voltage ramp from –100 mV to +100 mV to reveal any voltage‐dependent currents. We have previously used this voltage protocol successfully in Jurkat T cells to assess Kv1.3 (Ren et al. 2008). In parallel, we supplemented the internal solution with 1 μm Ca2+ to concomitantly activate any Ca2+‐gated K+ channels. Under these conditions, we did not observe significant voltage‐dependent currents in DT40 cells but, instead, observed a constitutive and linear conductance that reversed at around −55 mV, probably reflecting a resting K+ conductance in both WT and cTRPM7‐KO cells (Fig. 2 C and D). Over time, we observed an increase in a slightly outwardly rectifying conductance that had a similar reversal potential and might have been a result of Ca2+‐dependent K+ channels (Fig. 2 C and D). We have not attempted to further characterize these currents because they were unaltered in WT and cTRPM7‐KO cells. These data further support the conclusions derived from the Ca2+ measurements in intact cells under fully depolarized conditions (Fig. 2 A and B), indicating that membrane potential effects are not responsible for the reduction in SOCE.
Next, we assessed the store‐operated CRAC current (I CRAC) in both WT and TRPM7‐deficient cells using a voltage ramp protocol from −150 to +100 mV over 50 ms and delivered at 0.5 Hz intervals from a holding membrane potential of 0 mV. Figure 2 E and F shows that I CRAC was significantly reduced in cTRPM7‐KO cells compared to cTRPM7 expressing cells. These results demonstrate that the mechanism by which TRPM7‐KO impairs SOCE appears to result directly from reduced CRAC currents rather than indirectly through effects on K+ currents and membrane potential changes.
Pharmacological blockade of TRPM7 shows that SOCE is acutely linked to TRPM7 function
To assess whether impaired SOCE in TRPM7‐deficient B lymphocytes is a long‐term adaptive effect as a result of the KO of TRPM7 protein or an acutely linked process, we performed fura‐2‐based Ca2+ imaging experiments using different known TRPM7 or CRAC inhibitors. As expected, Gd3+, which inhibits CRAC channels without affecting TRPM7 (Hermosura et al. 2002), decreased the maximal [Ca2+]i influx peak after store depletion from 1259 ± 41 nm in control cells to 34 ± 4 nm [Ca2+]i (Fig. 3 A). The TRPM7 blockers NS8593 (Chubanov et al. 2012) and waixenicin A (Zierler et al. 2011) were also able to strongly inhibit the SOCE response to 243 ± 22 nm and 191 ± 30 nm [Ca2+]i, respectively. This effect was specific because, in control experiments in B lymphocytes deficient for TRPM7 expression, the TRPM7 blocker NS8593 failed to impair SOCE (Fig. 3 B and D).
Figure 3. SOCE is acutely linked to TRPM7 function.
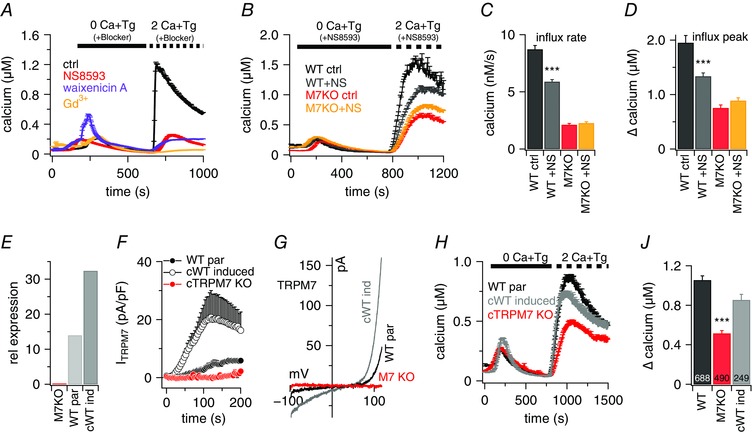
A and B, average [Ca2+]i responses following store depletion by the addition of 5 μm Tg and readdition of 2 mm [Ca2+]o (SOCE) in rat basophilic leukaemia cells (A) or DT40 B cells (B). A, bath solution was non‐supplemented (ctrl, n = 79) or supplemented with 1 μm Gd3+ (n = 22), 30 μm NS8593 (n = 29) or 10 μm waixenicin A (n = 17) at the time of application, as indicated by the black bar. B, bath solution was supplemented with 30 μm NS8593 or without (ctrl). Shown are traces for DT40 WT par cells with (n = 732) or without (n = 469) and cTRPM7‐KO cells with (n = 441) or without (n = 546) blocker. C and D, average of Ca2+ influx rates (C) and maximum Ca2+ influx peaks assessed from baseline of each cell (D) and obtained from SOCE measurements in (B). E, quantitative RT‐PCR analyses of TRPM7 expression [2−(∆Cq)] in DT40 B cells cTRPM7‐KO (M7KO), WT par and cWT induced (cWT ind) for hTRPM7 overexpression. The relative expression of TRPM7 was normalized to the average expression of GAPDH and UB house keeping genes. F, TRPM7 current densities in DT40 B cells cTRPM7‐KO (red, n = 5), WT par (n = 6) and cWT after induction (n = 6) as assessed at +80 mV. G, I–V relationships of currents evoked by 200 ms voltage ramps extracted from cells measured in (F). H, average [Ca2+]i responses following store depletion by the addition of 5 μm Tg and readdition of 2 mm [Ca2+]o (SOCE) in DT40 B cells cTRPM7‐KO (n = 490), WT par (n = 688) and cWT after induction (n = 249). I, average Ca2+ influx peaks assessed from baseline and quantified from SOCE measurements in (H). Asterisks indicate statistical significance: * P < 0.05; ** P < 0.01; *** P < 0.001.
Impaired SOCE in TRPM7‐KO B lymphocytes is restored by expression of TRPM7
Corroborating evidence linking TRPM7 functionality and SOCE is provided by the rescue experiments shown in Fig. 3 E and J. Inducing the overexpression of hTRPM7 in the cTRPM7‐KO DT40 cell line (cWT) (Fig. 3 E and F) restores SOCE to almost the same degree seen in DT40 WT par cells (WT par: 1055 ± 43 nm; cTRPM7‐KO: 515 ± 28 nm; cWT induced: 855 nm [Ca2+]i) (Fig. 3 H and J). The overexpression was confirmed by quantitative RT‐PCR (Fig. 3 E), as well as by patch clamp experiments (Fig. 3 F). Although the cTRPM7‐KO cells did not exhibit any TRPM7 current, the average maximal current density was 284 pA/pF in the WT par cells and 766 pA/pF in the overexpressing cells (cWT). The correspondent I–V relationships are shown in Fig. 3 G. These results demonstrate a clear interaction and a direct link between TRPM7 functionality and SOCE.
TRPM7 itself is not a store‐operated channel
To test whether the observed decrease of SOCE in TRPM7‐deficient cells stems from the contribution of Ca2+ entry through TRPM7 channels following store depletion, we performed fura‐2‐based imaging experiments where we inhibited CRAC currents either by the CRAC channel inhibitor Gd3+ or by using DT40 chicken B lymphocytes deficient in CRAC components (Wang et al. 2009). The addition of Gd3+ to the bath solution completely abolished SOCE in WT par, cTRPM7‐KO and hTRPM7‐overexpressing B lymphocytes (Fig. 4 A and B).
Figure 4. TRPM7 itself is not a store‐operated channel.
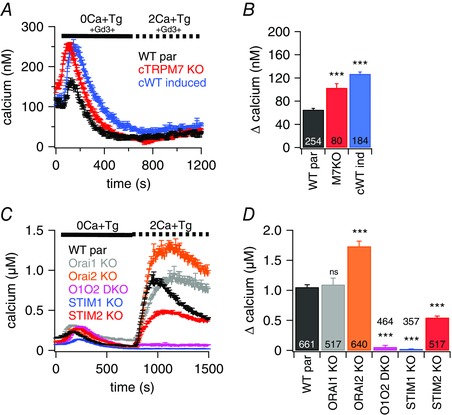
A and C, average [Ca2+]i responses following store depletion by the addition of 5 μm Tg and readdition of 2 mm [Ca2+]o (SOCE) in intact DT40 B cells. A, traces are shown for cTRPM7‐KO (n = 80), WT par (n = 254) and cWT after induction (n = 184). Bath solution was supplemented with 1 μm Gd3+. C, traces are shown for WT par (n = 661), Orai1‐KO (n = 517), Orai2‐KO (n = 640), Orai1/Orai2 double KO (O1O2 DKO; n = 464), STIM1‐KO (n = 357) and STIM2‐KO (n = 517). B and D, average of Ca2+ influx peaks obtained from SOCE measurements in cells in (A) or (C), respectively. The maximum Ca2+ release was measured for each individual cell from the resting baseline, averaged and plotted. Asterisks indicate statistical significance: * P < 0.05; ** P < 0.01; *** P < 0.001.
We next examined SOCE in B lymphocyte clones deficient in one or more CRAC components. Although the lack of Orai1 had little effect on SOCE, the absence of Orai2 actually increased the signal (Fig. 4 C and D), which is consistent with previous work (Holzmann et al. 2013). However, the double‐KO of Orai1/Orai2 almost completely eliminated SOCE. Similarly, STIM1‐KO cells exhibited no significant SOCE signal, whereas STIM2 reduced SOCE by 52%. Taken together, these results demonstrate that TRPM7 is not store‐operated and does not contribute significantly to SOCE‐mediated signals. Rather, it appears to play a regulatory role in support of SOCE.
The TRPM7 kinase domain modulates SOCE
Because the above experiments show that Ca2+ flux through TRPM7 does not contribute to SOCE, TRPM7 might shape SOCE through its kinase domain activity. To test this, we made use of two DT40 cell lines overexpressing kinase‐dead hTRPM7 mutants after induction with doxycycline, namely hTRPM7‐∆‐kinase and hTRPM7‐K1648R (Schmitz et al. 2003; Yu et al. 2013). Although both TRPM7 variants lack kinase activity, they exhibit different degrees of ion channel function. As illustrated in Fig. 5 A and B, the hTRPM7‐K1648R mutant conducts currents comparable to the hTRPM7‐WT current (cWT) (Fig. 3 G) and the ∆‐kinase mutation essentially eliminates conductivity through the pore of the channel. Fig. 5 C and D depicts the results of the fura‐2‐based Ca2+ imaging experiments. Although overexpression of hTRPM7‐WT (cWT) accomplishes the rescue of the cTRPM7‐KO phenotype in SOCE, neither the hTRPM7‐∆‐kinase, nor the hTRPM7‐K1648R proteins, despite their differences in ion transport abilities, are able to do the same. These results demonstrate that the TRPM7 kinase domain rather than the TRPM7 channel domain regulates SOCE in DT40 B‐lymphocytes.
Figure 5. TRPM7 kinase domain affects SOCE and TRPM7 conductivity is needed to refill Ca2+ stores.
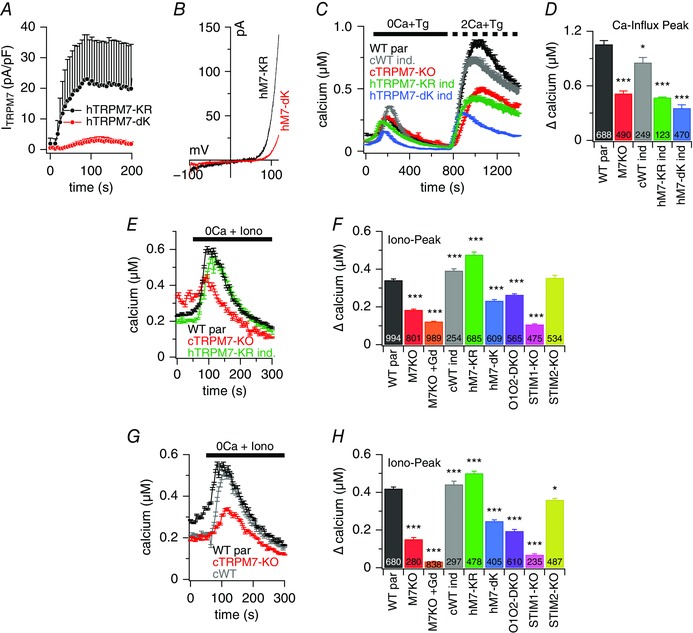
A, TRPM7 current densities in DT40 B cells overexpressing either hTRPM7‐∆Kinase (n = 6) or hTRPM7‐KR mutants (n = 6), 48 h after induction. B, I–V behaviour of currents evoked by 50 ms voltage ramps obtained from cells measured in (A). Holding potential was 0 mV. C, average [Ca2+]i responses following store depletion by the addition of 5 μm Tg and readdition of indicated [Ca2+]o (SOCE) in DT40 B cells WT par (n = 688) and cWT (n = 249), cTRPM7‐KO (n = 490), hTRPM7‐∆Kinase (n = 470) and hTRPM7‐KR mutant (n = 123) after induction. D, average of maximal Ca2+ influx assessed from baseline and obtained from SOCE measurements in (C). E, average [Ca2+]i responses following store depletion by application of 1 μm ionomycin (Iono) in DT40 B cells WT par and cTRPM7‐KO, and cTRPM7‐KR mutant after induction. Ionomycin was applied in the absence of extracellular Ca2+ in the solution. In one experiment, cTRPM7‐KO cells were analysed in the presence of 1 μm Gd3+ in the bath solution. F, average of Ca2+ release peaks assessed from baseline and obtained from measurements in (E), and additional [Ca2+]i responses not depicted in (E) but using the same experimental approach for cTRPM7‐KO with 1 μm Gd3++ in the bath, induced hTRPM7 KR mutant, induced hTRPM7‐∆Kinase mutant, Orai1/Orai2 double KO (O1O2 DKO), STIM1‐KO and STIM2‐KO. Numbers in black indicate number of cells averaged. G, for the refilling experiments, the experimental procedure was the same as shown for control cells in (E) but cells were treated with 1 μm ionomycin 20 min before the measurement (see Methods). Average [Ca2+]i responses following store depletion by the addition of 1 μm ionomycin (Iono) in DT40 B cells WT par, cTRPM7‐KO and cWT after induction. H, average of Ca2+ release peaks from baseline and obtained from measurements in (G) and additional DT40 mutant cell lines as investigated in (F). Asterisks indicate statistical significance: * P < 0.05; ** P < 0.01; *** P < 0.001.
TRPM7 currents are required to refill the internal Ca2+ stores after depletion
SOCE not only serves to translate signalling events from the PM into the cell, but also to refill the internal Ca2+ stores after signalling. Because we found that TRPM7 functionality affects SOCE, we next considered whether TRPM7 affects ER store content at rest and/or contributes to the refilling process after signalling. We examined the role of TRPM7 in regulating the resting Ca2+ store content (Fig. 5 E and F), as well as the refilling of the stores after depletion (Fig. 5 G and H), by performing fura‐2‐based intact‐cell imaging experiments and using ionomycin to probe ER store contents in various DT40 cell lines.
As illustrated in Fig. 5 E and F, cTRPM7‐KO cells had significantly less Ca2+ in their stores than WT par cells, as indicated by reduced Ca2+ release responses to ionomycin challenge (184 ± 4 nm vs. 341 ± 7 nm Ca2+). Interestingly, the phenotype could be rescued by overexpressing either hTRPM7‐WT (cWT) or hTRMP7‐K1648R (392 ± 10 nm and 476 ± 15 nm Ca2+) but not by the non‐conductive hTRPM7‐∆‐kinase protein (Fig. 5 E and F). Thus, overexpression of hTRMP7‐K1648R can rescue the store content phenotype of cTRPM7‐KO cells, even though it is unable to rescue the SOCE phenotype in those cells. This indicates that the constitutive activity of TRPM7 contributes to the filling state of resting cells. We further demonstrate that CRAC channels also appear to participate in this process because ionomycin responses in resting DT40 B cells are reduced in cells deficient in Orai1 and Orai2 (264 ± 6 nm) or STIM1 (107 ± 4 nm), whereas STIM2‐deficient cells have a similar store content as WT par cells, yielding similar ionomycin responses (354 ± 13 nm) (Fig. 5 E and F). These results show that TRPM7 and CRAC currents control the intracellular Ca2+ store content at rest. Surprisingly, however, and in contrast to other cell systems, where STIM2 is considered to regulate basal cytosolic and ER levels (Brandman et al. 2007; Várnai et al. 2009; Gruszczynska‐Biegala et al. 2011), it appears that, in the DT40 B cells, STIM1 may be more important for this resting activity of CRAC channels than STIM2.
In addition to contributing to Ca2+ store content under resting conditions, TRPM7 appears to also participate in the refilling process after depletion of Ca2+ stores. This is illustrated in Fig. 5 G and H, where experiments are depicted in which the transient application of ionomycin was used to fully deplete Ca2+ stores, followed by a 20 min waiting period that allowed cells to refill their stores. Some 20 min after depletion, the store content in cTRPM7‐KO cells was reduced by ∼64% compared to the WT par cells. In the additional presence of Gd3+ for blocking SOCE, there was no refilling of ER stores in cTRPM7‐KO cells at all. Cells overexpressing hTRPM7‐WT (cWT) or hTRMP7‐K1648R exhibited normal refilling levels comparable to WT par cells. The overexpression of hTRPM7‐∆‐kinase refilled the stores 41% less than the hTRPM7‐WT protein. The deficiency in SOCE components also affected the store refilling process because the Orai1/Orai2 double KO, which did not show any SOCE in the Ca2+ re‐addition measurements (Fig. 4), reduced the signal by ∼54%. Remarkably, the STIM1‐KO was more effective than the Orai1/Orai2 KO in that it reduced ionomycin‐induced Ca2+ responses by ∼84%, whereas STIM2‐KO achieved merely a 14% reduction. Taken together, these experiments demonstrate a major role of TRPM7 Ca2+ current in the refilling of intracellular Ca2+ stores. It appears that TRPM7 and SOCE work hand in hand to maintain the intracellular Ca2+ balance under resting conditions, as well as after Ca2+ signalling.
The TRPM7 kinase domain may be targeting STIM proteins
The results of the present study demonstrate that acute SOCE regulation by TRPM7 requires a functional kinase domain, irrespective of the remaining ion channel conductivity. To examine the target of the TRPM7 kinase domain, we made use of the TRPM7 channel blocker NS8593 to investigate whether the channel activity of TRPM7 is needed for efficient kinase activity. As a read out of kinase activity, we assessed the autophosphorylation of TRPM7 protein in immunoblot experiments. Tetracycline‐inducible HEK293 cells overexpressing hTRPM7 (Schmitz et al. 2003) and treated with NS8593 showed a 42% reduction in autophosphorylation of hTRPM7, illustrating the modulatory role of the TRPM7 current for kinase‐activity (Fig. 6 A and B). This result shows that NS8593 not only blocks the current through the TRPM7 pore, but also decreases the TRPM7 kinase activity.
Next, we used this pharmacological tool to examine which of the molecular components of SOCE might be the targets of the TRPM7 kinase. The rationale behind this approach was that blocking the ion flux through TRPM7 should affect the kinase activity, and removing the target of phosphorylation in B lymphocytes would render the SOCE of these cells resistant to NS8593 inhibition. Accordingly, we performed fura‐2‐based intact‐cell imaging experiments in which we examined the Ca2+ signals in the presence of the TRPM7 blocker NS8593 in different B lymphocyte clones deficient in one of the CRAC components. As depicted in Fig. 6 C and D, the TRPM7 blocker was able to reduce SOCE in Orai1‐KO (26% reduction) and Orai2‐KO (21% reduction) cells but not in STIM2‐KO cells. Because the STIM1‐KO cells did not show any SOCE, they could not be used for this type of experiment. These results suggest an effect of the TRPM7 kinase domain on STIM2 proteins, although we cannot rule out a possible effect on STIM1 as well.
Given our results indicating a possible mechanism involving STIM proteins, we assessed whether STIM1 and STIM2 are the direct targets of TRPM7 kinase. We transfected tetracycline‐inducible HEK‐293 overexpressing hTRPM7 with either STIM1 plasmid (Fig. 7 A) or STIM2 plasmid (Fig. 7 B). Although the anti‐phospho mix detected the in vitro phosphorylation of hTRPM7 (Fig. 7 B), we were unable to detect any phosphorylation of STIM1 and STIM2. The absence of STIM phosphorylation may be a result of the potential limiting factor of the sensitivity of the technique (detection limit) or the antibodies, or possibly because TRPM7 may affect STIM indirectly by engaging another protein partner of STIM or Orai proteins.
Discussion
In the present study, we report a new mechanistic target of the TRPM7 channel kinase: SOCE. The present study confirms that TRPM7 channel activity, although not directly contributing to SOCE, influences TRPM7 kinase function, thereby modifying the efficacy of Ca2+ entry via SOCE. In addition, constitutive TRPM7 ion channel activity serves to maintain Ca2+ store contents in resting unstimulated cells. Thus, our results suggest that TRPM7 and CRAC currents work hand in hand to regulate cytosolic Ca2+ signals as well as refill intracellular Ca2+ stores.
A previous study observed a quiescent metabolic transition of TRPM7‐deficient B lymphocytes (TRPM7‐KO) after removal of additional Mg2+ from the cell culture medium, causing a shift in the cell cycle distribution of the cells towards G0/G1 phase and resulting in a reduction in SOCE (Sahni et al. 2010). In the present study, we demonstrate that TRPM7‐KO cells, cultured with additional Mg2+, show a normal cell cycle distribution comparable to WT DT40 lymphocytes and nevertheless have impaired SOCE (Fig. 1). Furthermore, TRPM7 blockers such as NS8593 and waixenicin A also decrease SOCE (Fig. 3), indicating a direct and acute correlation between the two mechanisms. The TRPM7‐KO‐mediated phenotype can be rescued by overexpressing hTRPM7‐WT but not by the kinase dead mutant hTRPM7‐K1648R or hTRPM7‐∆Kinase, suggesting that kinase activity of the protein rather than its ion channel function imparts a regulatory effect on the molecular CRAC components Orai and/or STIM (Fig. 5).
Indeed, the literature reports several ways of regulating SOCE by phosphorylation. Previous work identified two serine residues in the STIM1 protein that are phosphorylated in mitotic cells and are capable of suppressing SOCE (Smyth et al. 2009). Another study showed that Orai1 is a direct target of the Ca2+‐dependent kinase PKCβ, and the phosphorylation of S27 and S30 abolishes SOCE (Kawasaki et al. 2010). However, phosphorylation can also augment SOCE. We have previously reported that Orai1 expression levels are regulated by serum‐ and glucocorticoid‐inducible kinase 1 (SGK1), a growth factor‐regulated kinase (Eylenstein et al. 2011). An enhancing effect has also been shown when STIM1 is phosphorylated by extracellular‐signal‐regulated kinases 1 or 2 on serine residues (Pozo‐Guisado et al. 2013). Our present data indicate that STIM2 might be the phosphorylation target of the endogenous kinase of TRPM7 rather than Orai (Fig. 6). Although one study has reported that Stim2 protein can be phosphorylated (Williams et al. 2001), we were unable to detect any phosphorylation in our current efforts (Fig. 7). The absence of STIM phosphorylation may be a result of the sensitivity of the technique (detection limit), the quality and sensitivity of antibodies, or possibly because TRPM7 may affect SOCE indirectly by engaging regulatory protein partners of STIM or ORAI. Possible candidates for such indirect modulatory effects include Septin, junctate, Golli, SARAF and CRACR2A, (Walsh et al. 2010; Srikanth et al. 2012; Hooper & Soboloff, 2015), warranting further investigation to determine the direct target of TRPM7 phosphorylation and its connection to SOCE.
There are numerous ways in which TRP channels and SOCE can interact in different cellular systems to generate a precisely adapted spatio‐temporal Ca2+ signalling (Saul et al. 2014). Our results depict for the first time an acute functional link between TRPM7 and store‐operated calcium entry. I CRAC is certainly the best described store‐operated calcium entry mechanism; however, other channels, especially members of the ‘canonical’ TRP subfamily, have also been reported to be store‐operated (Parekh & Putney, 2005; Pedersen et al. 2005; Ramsey et al. 2006). Taken together, these Ca2+ entry mechanisms are essential for signalling and store refilling. TRPM7 is constitutively active and can be modulated by internal Mg2+ or ATP concentrations (Nadler et al. 2001; Demeuse et al. 2006). We found that TRPM7 itself is not a store‐operated channel, at least not in DT40 B lymphocytes. However, the Ca2+ current through TRPM7, and thus the channel domain of the protein, contributes significantly in maintaining the filling state of internal Ca2+ stores under unstimulated resting conditions without modulating SOCE itself. To our knowledge, this is the first demonstration that the Ca2+ current through TRPM7 is linked to store refilling and maintenance of ER Ca2+ store contents.
Several studies discuss the effects of the TRPM7 knockdown or KO in vitro or in vivo, including inhibitory effects on T cell migration (Kuras et al. 2012), the requirement of TRPM7 expression to obtain cardiac automaticity (Sah et al. 2013b) or the propensity of TRPM7 expression levels to enhance the metastatic potential of invasive human breast cancer cells in vivo (Middelbeek et al. 2012). Furthermore, the TRPM7 kinase domain has been shown to regulate the Ca2+‐sensitivity of G‐protein induced histamine release in primary mouse mast cells (Zierler et al. 2016). Ca2+ signalling in B cells is fundamentally important for the development of adaptive immunity (Baba & Kurosaki, 2016). The apparent dependence of SOCE on functional TRPM7 kinase activity may represent a feedback loop for B‐cell receptor induced Ca2+ signalling that is in relation to the availability of intracellular Mg·ATP for the cell.
The connection between TRPM7 and SOCE should be considered when discussing TRPM7 effects because the possibility exists that these effects may be secondary to regulatory effects on SOCE. This may pertain to previous studies that have proposed a strong role for TRPM7 in mediating Ca2+ influx or Ca2+ oscillations and secondary physiological effects such as fibroblast proliferation (Du et al. 2010) or oocyte fertilization, egg activation and embryo development (Carvacho et al. 2016). Our data raise the distinct possibility, if not likelihood, that much of the TRPM7‐dependent Ca2+ entry attributed to TRPM7 might result from the additional secondary, indirect support of SOCE via the kinase activity of TRPM7 and the regulation of I CRAC. The dysregulation of Ca2+ homeostasis through either mechanism may drive the above‐mentioned cellular responses, as well as the expression of malignant phenotypes, such as proliferation, migration, invasion and metastasis. Therefore, it might not be coincidental that both channels, CRAC and TRPM7, have been linked to various types of cancer (Chen et al. 2013). For this reason, both CRAC and TRPM7 channels may be promising targets for developing therapeutic strategies for diseases in which these mechanisms are involved.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
RP conceived and co‐ordinated the study. TK and MF designed, performed and analysed the experiments and prepared the illustrations. AF performed and analysed experiments. FDH isolated and provided waixenicin A. RP, TK, MF and AF wrote and edited the paper. All authors reviewed the results and approved the final version of the manuscript submitted for publication.
Funding
This work was supported by DFG to TK (Ki 1821/1‐1) and partially supported by NIH/NCI U54CA143727 (RP) and NIH/NIGMS P20 GM103466 (FDH).
Acknowledgements
DT40 B lymphocyte cell lines WT par, cTRPM7‐KO, cWT, hTRPM7 △Kinase and inducible hTRPM7‐KO V79.1, hTRPM7 KR were kindly provided by Andrew Scharenberg (Seattle) and Carsten Schmitz (Denver), respectively. DT40 B lymphocyte cell lines Orai1 KO, Orai2 KO, Orai1Orai2 DKO, STIM1‐KO and STIM2‐KO were kindly provided by Tomohiro Kurosaki (Osaka) and Donald Gill (Philadelphia). The authors thank the University of Hawaii Cancer Centre Genomics Shared Resource (supported by NIH/NCI CA071789‐13) for performing the quantitative RT‐PCR assays for the present study. Some of the services for this research were provided with the help of the University of Hawaii John A. Burns School of Medicine Molecular and Cellular Immunology Core, which is supported in part by NIH/NIGMS P20GM103516 from the Centres of Biomedical Research Excellence (COBRE) program.
References
- Baba Y & Kurosaki T (2016). Role of calcium signalling in B cell activation and biology. Curr Top Microbiol Immunol 393, 143–174. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay BC, Pingle SC & Ahern GP (2011). Store‐operated Ca2+ signaling in dendritic cells occurs independently of STIM1. J Leukoc Biol 89, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berna‐Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G & Nieswandt B (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal 2, ra67. [DOI] [PubMed] [Google Scholar]
- Brandman O, Liou J, Park WS & Meyer T (2007). STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131, 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvacho I, Ardestani G, Lee HC, McGarvey K, Fissore RA & Lykke‐Hartmann K (2016). TRPM7‐like channels are functionally expressed in oocytes and modulate post‐fertilization embryo development in mouse. Sci Rep 6, 34236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y‐F, Chen Y‐T, Chiu W‐T & Shen M‐R (2013). Remodeling of calcium signaling in tumor progression. J Biomed Sci 20, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubanov V, Mederos Y, Schnitzler M, Meißner M, Schäfer S, Abstiens K, Hofmann T & Gudermann T (2012). Natural and synthetic modulators of SK (K(ca)2) potassium channels inhibit magnesium‐dependent activity of the kinase‐coupled cation channel TRPM7. Br J Pharmacol 166, 1357–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG, Figdor CG, Moolenaar WH, Jalink K & van Leeuwen FN (2006). TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J 25, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deason‐Towne F, Perraud A‐L & Schmitz C (2012). Identification of Ser/Thr phosphorylation sites in the C2‐domain of phospholipase C γ2 (PLCγ2) using TRPM7‐kinase. Cell Signal 24, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeuse P, Penner R & Fleig A (2006). TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol 127, 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai BN, Krapivinsky G, Navarro B, Krapivinsky L, Carter BC, Febvay S, Delling M, Penumaka A, Ramsey IS, Manasian Y & Clapham DE (2012). Cleavage of TRPM7 releases the kinase domain from the ion channel and regulates its participation in Fas‐induced apoptosis. Dev Cell 22, 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorovkov MV & Ryazanov AG (2004). Phosphorylation of annexin I by TRPM7 channel‐kinase. J Biol Chem 279, 50643–50646. [DOI] [PubMed] [Google Scholar]
- Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B & Yue L (2010). TRPM7‐mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res 106, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eylenstein A, Gehring E‐M, Heise N, Shumilina E, Schmidt S, Szteyn K, Münzer P, Nurbaeva MK, Eichenmüller M, Tyan L, Regel I, Föller M, Kuhl D, Soboloff J, Penner R & Lang F (2011). Stimulation of Ca2+‐channel Orai1/STIM1 by serum‐ and glucocorticoid‐inducible kinase 1 (SGK1). FASEB J 25, 2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J‐M, Hu YK, Xie L‐H, Colwell CS, Shao XM, Sun X‐P, Chen B, Tang H & Campagnoni AT (2006). Golli protein negatively regulates store depletion‐induced calcium influx in T cells. Immunity 24, 717–727. [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel S‐H, Tanasa B, Hogan PG, Lewis RS, Daly M & Rao A (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. [DOI] [PubMed] [Google Scholar]
- Fleig A & Chubanov V (2014). TRPM7. Handb Exp Pharmacol 222, 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszczynska‐Biegala J, Pomorski P, Wisniewska MB & Kuznicki J (2011). Differential roles for STIM1 and STIM2 in store‐operated calcium entry in rat neurons. PloS ONE 6, e19285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M & Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260, 3440–3450. [PubMed] [Google Scholar]
- Hermosura MC, Monteilh‐Zoller MK, Scharenberg AM, Penner R & Fleig A (2002). Dissociation of the store‐operated calcium current I(CRAC) and the Mg‐nucleotide‐regulated metal ion current MagNuM. J Physiol 539, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann C, Kilch T, Kappel S, Armbrüster A, Jung V, Stöckle M, Bogeski I, Schwarz EC & Peinelt C (2013). ICRAC controls the rapid androgen response in human primary prostate epithelial cells and is altered in prostate cancer. Oncotarget 4, 2096–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper R & Soboloff J (2015). STIMATE reveals a STIM1 transitional state. Nat Cell Biol 17, 1232–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover PJ & Lewis RS (2011). Stoichiometric requirements for trapping and gating of Ca2+ release‐activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1). Proc Natl Acad Sci USA 108, 13299–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen C, Sahni J, Suzuki S, Horgen FD, Penner R & Fleig A (2016). The coiled‐coil domain of zebrafish TRPM7 regulates Mg·nucleotide sensitivity. Sci Rep 6, 33459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Desai BN, Navarro B, Donovan A, Andrews NC & Clapham DE (2008). Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322, 756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Wu L‐J, Jun J, Cheng X, Xu H, Andrews NC & Clapham DE (2012). The channel kinase, TRPM7, is required for early embryonic development. Proc Natl Acad Sci USA 109, E225–E233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Ueyama T, Lange I, Feske S & Saito N (2010). Protein kinase C‐induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store‐operated Ca2+ channel. J Biol Chem 285, 25720–25730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil JM, Shen Z, Briggs SP & Patrick GN (2010). Regulation of STIM1 and SOCE by the ubiquitin‐proteasome system (UPS). PloS ONE 5, e13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilch T, Alansary D, Peglow M, Dörr K, Rychkov G, Rieger H, Peinelt C & Niemeyer BA (2013). Mutations of the Ca2+‐sensing stromal interaction molecule STIM1 regulate Ca2+ influx by altered oligomerization of STIM1 and by destabilization of the Ca2+ channel Orai1. J Biol Chem 288, 1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y & Clapham DE (2014). The TRPM7 chanzyme is cleaved to release a chromatin‐modifying kinase. Cell 157, 1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuras Z, Yun Y‐H, Chimote AA, Neumeier L & Conforti L (2012). KCa3.1 and TRPM7 channels at the uropod regulate migration of activated human T cells. PloS ONE 7, e43859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Eylenstein A & Shumilina E (2012). Regulation of Orai1/STIM1 by the kinases SGK1 and AMPK. Cell Calcium 52, 347–354. [DOI] [PubMed] [Google Scholar]
- Lee B‐C, Hong S‐E, Lim H‐H, Kim DH & Park C‐S (2011). Alteration of the transcriptional profile of human embryonic kidney cells by transient overexpression of mouse TRPM7 channels. Cell Physiol Biochem 27, 313–326. [DOI] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE & Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+‐store‐depletion‐triggered Ca2+ influx. Curr Biol CB 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelbeek J, Kuipers AJ, Henneman L, Visser D, Eidhof I, van Horssen R, Wieringa B, Canisius SV, Zwart W, Wessels LF, Sweep FCGJ, Bult P, Span PN, van Leeuwen FN & Jalink K (2012). TRPM7 is required for breast tumor cell metastasis. Cancer Res 72, 4250–4261. [DOI] [PubMed] [Google Scholar]
- Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM & Fleig A (2001). LTRPC7 is a Mg.ATP‐regulated divalent cation channel required for cell viability. Nature 411, 590–595. [DOI] [PubMed] [Google Scholar]
- Palty R, Raveh A, Kaminsky I, Meller R & Reuveny E (2012). SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 149, 425–438. [DOI] [PubMed] [Google Scholar]
- Parekh AB & Penner R (1995). Depletion‐activated calcium current is inhibited by protein kinase in RBL‐2H3 cells. Proc Natl Acad Sci USA 92, 7907–7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB & Putney JW (2005). Store‐operated calcium channels. Physiol Rev 85, 757–810. [DOI] [PubMed] [Google Scholar]
- Parvez S, Beck A, Peinelt C, Soboloff J, Lis A, Monteilh‐Zoller M, Gill DL, Fleig A & Penner R (2008). STIM2 protein mediates distinct store‐dependent and store‐independent modes of CRAC channel activation. FASEB J 22, 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SF, Owsianik G & Nilius B (2005). TRP channels: an overview. Cell Calcium 38, 233–252. [DOI] [PubMed] [Google Scholar]
- Perraud A‐L, Zhao X, Ryazanov AG & Schmitz C (2011). The channel‐kinase TRPM7 regulates phosphorylation of the translational factor eEF2 via eEF2‐k. Cell Signal 23, 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo‐Guisado E, Casas‐Rua V, Tomas‐Martin P, Lopez‐Guerrero AM, Alvarez‐Barrientos A & Martin‐Romero FJ (2013). Phosphorylation of STIM1 at ERK1/2 target sites regulates interaction with the microtubule plus‐end binding protein EB1. J Cell Sci 126, 3170–3180. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A & Hogan PG (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. [DOI] [PubMed] [Google Scholar]
- Prevarskaya N, Ouadid‐Ahidouch H, Skryma R & Shuba Y (2014). Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol Sci 369, 20130097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Delling M & Clapham DE (2006). An introduction to TRP channels. Annu Rev Physiol 68, 619–647. [DOI] [PubMed] [Google Scholar]
- Ren YR, Pan F, Parvez S, Fleig A, Chong CR, Xu J, Dang Y, Zhang J, Jiang H, Penner R & Liu JO (2008). Clofazimine inhibits human Kv1.3 potassium channel by perturbing calcium oscillation in T lymphocytes. PLoS ONE 3, e4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G & Stauderman KA (2005). STIM1, an essential and conserved component of store‐operated Ca2+ channel function. J Cell Biol 169, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazanova LV, Dorovkov MV, Ansari A & Ryazanov AG (2004). Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to the transient receptor potential ion channel. J Biol Chem 279, 3708–3716. [DOI] [PubMed] [Google Scholar]
- Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, Mazur A, Fleig A & Ryazanov AG (2010). TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat Commun 1, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni J, Tamura R, Sweet IR & Scharenberg AM (2010). TRPM7 regulates quiescent/proliferative metabolic transitions in lymphocytes. Cell Cycle Georget Tex 9, 3565–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R, Mesirca P, Mason X, Gibson W, Bates‐Withers C, Van den Boogert M, Chaudhuri D, Pu WT, Mangoni ME & (2013a). Timing of myocardial trpm7 deletion during cardiogenesis variably disrupts adult ventricular function, conduction, and repolarization. Circulation 128, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R, Mesirca P, Van den Boogert M, Rosen J, Mably J, Mangoni ME & Clapham DE (2013b). Ion channel‐kinase TRPM7 is required for maintaining cardiac automaticity. Proc Natl Acad Sci USA 110, E3037–E3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul S, Stanisz H, Backes CS, Schwarz EC & Hoth M (2014). How ORAI and TRP channels interfere with each other: Interaction models and examples from the immune system and the skin. Eur J Pharmacol 739C, 49–59. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Perraud A‐L, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A & Scharenberg AM (2003). Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 114, 191–200. [DOI] [PubMed] [Google Scholar]
- Smyth JT, Beg AM, Wu S, Putney JW & Rusan NM (2012). Phosphoregulation of STIM1 leads to exclusion of the endoplasmic reticulum from the mitotic spindle. Curr Biol CB 22, 1487–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, Petranka JG, Boyles RR, DeHaven WI, Fukushima M, Johnson KL, Williams JG & Putney JW (2009). Phosphorylation of STIM1 underlies suppression of store‐operated calcium entry during mitosis. Nat Cell Biol 11, 1465–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Jew M, Kim K‐D, Yee M‐K, Abramson J & Gwack Y (2012). Junctate is a Ca2+‐sensing structural component of Orai1 and stromal interaction molecule 1 (STIM1). Proc Natl Acad Sci USA 109, 8682–8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Jung H‐J, Kim K‐D, Souda P, Whitelegge J & Gwack Y (2010). A novel EF‐hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol 12, 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanisz H, Stark A, Kilch T, Schwarz EC, Müller CSL, Peinelt C, Hoth M, Niemeyer BA, Vogt T & Bogeski I (2012). ORAI1 Ca(2+) channels control endothelin‐1‐induced mitogenesis and melanogenesis in primary human melanocytes. J Invest Dermatol 132, 1443–1451. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Zheng L & Ikura M (2009). Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem 284, 728–732. [DOI] [PubMed] [Google Scholar]
- Tani D, Monteilh‐Zoller MK, Fleig A & Penner R (2007). Cell cycle‐dependent regulation of store‐operated I(CRAC) and Mg2+‐nucleotide‐regulated MagNuM (TRPM7) currents. Cell Calcium 41, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel M, Lis A & Penner R (2013). STIM2 drives Ca2+ oscillations through store‐operated Ca2+ entry caused by mild store depletion. J Physiol 591, 1433–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai P, Hunyady L & Balla T (2009). STIM and Orai: the long‐awaited constituents of store‐operated calcium entry. Trends Pharmacol Sci 30, 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet J‐P & Penner R (2006. a). CRACM1 multimers form the ion‐selective pore of the CRAC channel. Curr Biol CB 16, 2073–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan‐Huberson M, Kraft S, Turner H, Fleig A, Penner R & Kinet J‐P (2006b). CRACM1 is a plasma membrane protein essential for store‐operated Ca2+ entry. Science 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CM, Doherty MK, Tepikin AV & Burgoyne RD (2010). Evidence for an interaction between Golli and STIM1 in store‐operated calcium entry. Biochem J 430, 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Deng X, Zhou Y, Hendron E, Mancarella S, Ritchie MF, Tang XD, Baba Y, Kurosaki T, Mori Y, Soboloff J & Gill DL (2009). STIM protein coupling in the activation of Orai channels. Proc Natl Acad Sci USA 106, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ & Dziadek MA (2001). Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J 357, 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Zhang Z, Lis A, Penner R & Fleig A (2013). TRPM7 is regulated by halides through its kinase domain. Cell Mol Life Sci 70, 2757–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Zhang XH‐F, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA & Cahalan MD (2006). Genome‐wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release‐activated Ca(2+) channel activity. Proc Natl Acad Sci USA 103, 9357–9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Stathopulos PB, Schindl R, Li G‐Y, Romanin C & Ikura M (2011). Auto‐inhibitory role of the EF‐SAM domain of STIM proteins in store‐operated calcium entry. Proc Natl Acad Sci USA 108, 1337–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierler S, Sumoza‐Toledo A, Suzuki S, FÓ Dúill, Ryazanova LV, Penner R, Ryazanov AG & Fleig A (2016). TRPM7 kinase activity regulates murine mast cell degranulation. J Physiol 594, 2957–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierler S, Yao G, Zhang Z, Kuo WC, Pörzgen P, Penner R, Horgen FD & Fleig A (2011). Waixenicin A inhibits cell proliferation through magnesium‐dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J Biol Chem 286, 39328–39335. [DOI] [PMC free article] [PubMed] [Google Scholar]