

Abstract
In vivo two‐photon Ca2+ imaging has become an effective approach for the functional analysis of neuronal populations, individual neurons and subcellular neuronal compartments in the intact brain. When imaging individually labelled neurons, depth penetration can often reach up to 1 mm below the cortical surface. However, for densely labelled neuronal populations, imaging with single‐cell resolution is largely restricted to the upper cortical layers in the mouse brain. Here, we review recent advances of deep two‐photon Ca2+ imaging and the use of red‐shifted fluorescent Ca2+ indicators as a promising strategy to increase the imaging depth, which takes advantage of reduced photon scattering at their long excitation and emission wavelengths. We describe results showing that the newly introduced fluorescent Ca2+‐sensitive dye Cal‐590 can be used to record in vivo neuronal activity in isolated cortical neurons and in neurons within populations in depths of up to 900 μm below the pial surface. Thus, the new approach allows the comprehensive functional mapping of all six cortical layers of the mouse brain. Specific features of Cal‐590‐based in vivo Ca2+ two‐photon imaging include a good signal‐to‐noise ratio, fast kinetics and a linear dependence of the Ca2+ transients on the number of action potentials. Another area of application is dual‐colour imaging by combining Cal‐590 with other, shorter wavelength Ca2+ indicators such as OGB‐1. Overall, Cal‐590‐based two‐photon microscopy emerges as a promising tool for the recording of neuronal activity at depths that were previously inaccessible to functional imaging of neuronal circuits.
Keywords: calcium imaging, mouse cortical circuits, multicolor functional imaging, neuronal activity, red‐shifted calcium indicator
Abbreviations
- AM
acetoxymethyl
- GCaMP
genetically‐encoded calcium indicator protein containing circularly permutated green fluorescent protein, calmodulin and calmodulin‐binding domain from the myosin light chain kinase
- OGB‐1
Oregon Green® 488 BAPTA‐1
- RCaMP
red‐shifted GCaMP variant
Introduction
Two‐photon Ca2+ imaging has become an established tool to observe neuronal activity from the scale of individual dendritic spines (Chen et al. 2011) to entire cortical networks (Stosiek et al. 2003; Sofroniew et al. 2015; Stirman et al. 2016) in vivo. Advances of microscope and fluorophore technology continue to make two‐photon microscopy more accessible for neuroscientists and allow them to perform increasingly more sophisticated experiments in the intact brain of various model organisms. Combined with behavioural paradigms, spatially resolved neuronal network activity can now routinely be monitored with single‐cell resolution in different brain regions during specific tasks in, for example, Drosophila (Seelig et al. 2010), zebrafish (Brustein et al. 2003), mouse (Komiyama et al. 2010), rat (Sawinski et al. 2009) and even non‐human primates (Nauhaus et al. 2012; Sadakane et al. 2015).
While the brain size of small mammals, like mice or marmosets, pales in comparison with the much larger human brain, the thickness of their cortices already humbles experimenters when they try to observe neuronal activity with cellular resolution underneath the most superficial cortical layers of light‐scattering brain tissue (Helmchen & Denk, 2005). For example, two‐photon imaging of neuronal activity with the Ca2+‐sensitive dye Oregon Green 488 BAPTA‐1 acetoxymethyl ester (OGB‐1 AM) in the mouse brain typically provides optimal results in depths of not more than 300 μm (Stosiek et al. 2003) below the pia. Although recording neuronal activity with bright genetically encoded Ca2+ indicators such as GCaMP6 is possible in greater depths (Chen et al. 2013; Li et al. 2015), the deepest regions of the adult mouse cortex located in about 1 mm depth, not to mention brain structures lying underneath it, are out of reach for most current functional two‐photon microscopy approaches. An additional and major challenge is the simultaneous observation of activity in large neuronal populations. Even with the brightest fluorescent sensors that are currently available, good results are generally obtained only when recording from isolated neurons, as dense labelling of neurons in the upper layers can contribute to the generation of out‐of‐focus light and thereby reduces the optical contrast at larger imaging depths (Helmchen & Denk, 2005).
To address the limited imaging depth of two‐photon microscopy, a number of methods were developed. For example, regenerative amplifiers increase the efficiency of multiphoton excitation by increasing the energy of individual laser pulses, albeit at a reduced laser pulse repetition frequency (Mittmann et al. 2011). Another strategy involves the use of adaptive optics that were originally developed for astronomical observations to correct for aberrations introduced by the atmosphere. To correct for aberrations, the wavefront of the laser beam is deformed to correspond to the opposite of the phase aberrations introduced by the tissue, thus leading to a well‐focused laser spot in the tissue and extending the depth in which a detailed image of the desired structure can be recorded (Ji et al. 2012; Wang et al. 2015). An alternative approach involving the use of three‐photon excitation, which takes advantage of lower scattering at longer wavelengths (Kobat et al. 2009; Xu & Wise, 2013; Ouzounov et al. 2014), has shown promising potential for deep imaging applications. Finally, another, perhaps simpler, way to take advantage of reduced scattering of light at longer wavelengths is to use red‐shifted fluorophores with longer two‐photon excitation and emission wavelengths.
A direct comparison of the same sample excited with 775 and 1280 nm wavelengths shows the substantially increased imaging depth with longer wavelengths (Fig. 1 A). Importantly, the reduced scattering of light at longer wavelengths (Kobat et al. 2009; Smith et al. 2009; Xu & Wise, 2013) benefits both excitation and emission of a red‐shifted fluorophore. Excitation wavelengths in the range of 1000–1300 nm are well‐suited for imaging experiments in the brain, as they are less attenuated in biological tissue (Fig. 1 B) compared to the wavelengths typically used to excite OGB‐1, Cal‐520 or GCaMP6 ranging from 800 to 940 nm (Stosiek et al. 2003; Chen et al. 2013; Tada et al. 2014). From a practical point of view, an additional advantage of the wavelength range around 1050 nm is the possibility to equip two‐photon microscopes with ytterbium fibre lasers (Xu & Wise, 2013; Perillo et al. 2016; Pilz et al. 2016). These lasers are centred on wavelengths around 1050 nm and can be a convenient, cost‐effective alternative to the commonly used, broadly tuned Ti:Sapphire lasers, as they have a compact design, do not need water cooling and various available models provide femtosecond laser pulses combined with average power values exceeding 2 W.
Figure 1. Wavelength‐dependent attenuation length in brain tissue.
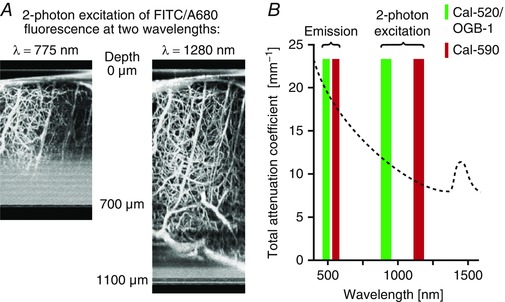
A, z‐stack projection of vasculature in the mouse cortex in vivo, recorded by two‐photon excitation at 750 or 1280 nm. The blood vessels are filled with fluorescein isothiocyanate‐dextran and Alexa680‐dextran. Figure adapted from Kobat et al. (2009). B, attenuation coefficient of biological tissue versus wavelength. Coloured bars illustrate the optimum emission and two‐photon excitation wavelength ranges for OGB‐1/Cal‐520 (green) and Cal‐590 (red). Figure adapted from Sordillo et al. (2014).
In this review, we focus on the last approach and show how a recently developed red‐shifted fluorescent Ca2+dye (Tischbirek et al. 2015; Zhao et al. 2015) was used for imaging of neuronal activity in previously unreachable depths within the intact brain.
Two‐photon Ca2+ imaging with Cal‐590
Since the development of the first membrane‐permeant Ca2+‐sensitive acetoxymethyl (AM) ester dyes (Tsien, 1981), AM dyes have been constantly improved and have become versatile tools for many applications, including in vivo two‐photon imaging to directly label a desired cell population in any part of the brain. However, widely used AM dyes such as OGB‐1 AM (Stosiek et al. 2003), fluo‐8 AM (Busche et al. 2012) and more recently Cal‐520 AM (Tada et al. 2014) are all excitable around 490 nm (or around 920–940 nm for two‐photon excitation) (Mutze et al. 2012) and emit around 520 nm. The use of Ca2+‐sensitive dyes excitable in a longer wavelength range was hindered by major limitations such as a low signal‐to‐noise ratio in the relevant range of Ca2+ concentrations, slow kinetics, low photostability, low solubility in aqueous solutions, or poor cellular dye retention (for a review see Oheim et al. 2014).
A recently developed fluorometric Ca2+ indicator dye with properties advantageous for in vivo imaging applications is Cal‐590 (Zhao et al. 2015), a red‐shifted variant of the fluorescent Ca2+ indicator Cal‐520 (Tada et al. 2014). The Ca2+ binding affinity of the dye is lower compared to, for example, OGB‐1 (K d = 561 nm for Cal‐590 compared to K d = 170 nm for OGB‐1). For a comparison with a wide range of other Ca2+ indicators, see Grienberger & Konnerth (2012). Cal‐590 has single‐photon excitation and emission peak wavelengths of 570 and 590 nm, respectively. We determined the maximum for two‐photon excitation to be at wavelengths around 1050 nm (Fig. 2 A) and made small modifications to our experimental set‐up for the optimization of the imaging performance at longer wavelengths (for details see Tischbirek et al. 2015).
Figure 2. Properties of the red‐shifted fluorescent Ca2+ indicator Cal‐590.
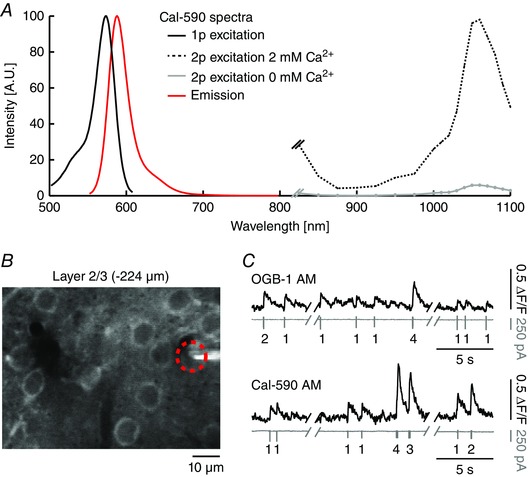
Figure adapted from Tischbirek et al. (2015). A, single‐photon, two‐photon excitation (with and without Ca2+) and emission spectrum of Cal‐590. Spectra were normalized for display purposes. Single‐photon spectrum adapted from AAT Bioquest (Sunnyvale, CA, USA). B, in vivo two‐photon image (average of 30 s recording time) of cortical layer 2/3 neurons bulk‐loaded with Cal‐590 AM. The red circle indicates a neuron measured with a patch‐pipette in cell‐attached mode and two‐photon imaging simultaneously. C, top, example of combined two‐photon Ca2+ imaging (upper traces) and cell‐attached recordings (lower traces) in a layer 2/3 neuron of the visual cortex in an anaesthetized mouse. Recordings were obtained after bulk‐loading with Cal‐590 AM. Numbers below the traces indicate the number of action potentials. Bottom, recordings obtained under similar recording conditions after bulk‐loading with OGB‐1 AM in another mouse.
We tested the suitability of Cal‐590 AM by performing in vivo two‐photon imaging experiments that were combined with cell‐attached recordings in layer 2/3 neurons of the mouse visual cortex (Tischbirek et al. 2015). Bulk‐loading the dye by pressure injection (Stosiek et al. 2003) into the cortex resulted in the ring‐shaped staining of neuronal somata, created by a brighter Cal‐590 staining in the neuronal cytosol compared to the nucleus (Fig. 2 B). Single action potential‐evoked Ca2+ transients had a good signal‐to‐noise ratio (Fig. 2 C), rapid rise (< 2 ms) and decay times. Despite the lower affinity for Ca2+ of Cal‐590 compared to OGB‐1, the recorded Ca2+ transients had a signal quality comparable to those obtained with OGB‐1 AM under the same recording conditions (Fig. 2 C), demonstrating that red‐shifted Cal‐590 AM is well suited for recordings of neuronal population activity with single cell resolution in vivo.
In order to test the performance of Cal‐590 also in dendrites, we labelled cortical layer 5 neurons via electroporation of the salt form of Cal‐590 using the shadow‐patching approach (Kitamura et al. 2008) in a headfixed, anaesthetized mouse (Tischbirek et al. 2015). An example of such an experiment is illustrated in Fig. 3 A. In the electroporated neuron we detected action potential‐associated Ca2+ transients with a much more rapid decay time course in the dendrites than in the cell bodies (Fig. 3 B), reflecting different surface‐to‐volume ratios of the two cellular compartments. Indeed, the kinetics of the Ca2+ transients in the dendrites were fast enough (Fig. 3 C) to distinguish peaks of individual Ca2+ transients even for high‐frequency trains (100 Hz) of action potentials (Fig. 3 D), a feature property that is particularly useful for highly active cells such as layer 5 neurons. Similarly to other small molecule Ca2+ indicator dyes like Cal‐520 (Tada et al. 2014), we observed a linear relationship between number of action potentials and corresponding fluorescence signal amplitudes (Fig. 3 E). The linearity of the indicator is beneficial for an accurate interpretation of neuronal activity levels when Ca2+ transients are recorded without additional electrophysiolgical validation.
Figure 3. Single‐cell imaging of a layer 5 neuron electroporated with Cal‐590.
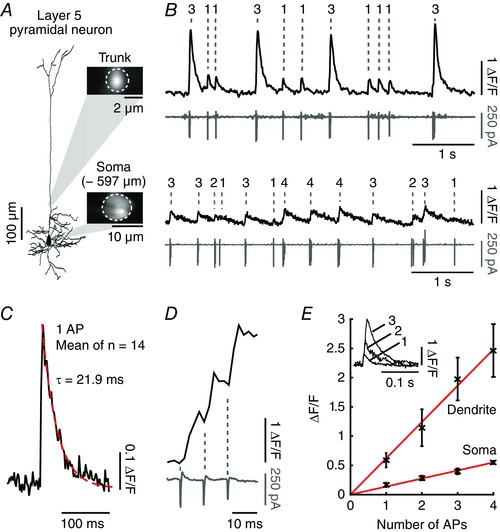
Figure adapted from Tischbirek et al. (2015). A, reconstruction of a pyramidal neuron in layer 5 of mouse visual cortex electroporated with Cal‐590. Insets: images of the focal planes showing two‐photon recordings of the dendritic trunk and the soma at 500 and 597 μm. The regions of interest used to obtain the fluorescence transients are indicated with white dotted lines. B, spontaneous activity recorded with two‐photon imaging (black traces) and simultaneous cell‐attached recordings (grey traces). The recordings were performed in the dendritic trunk close to the soma (upper traces) and in the soma (lower traces). C, average of 14 single action potential events recorded in the dendritic trunk indicated in A to evaluate the Cal‐590 dye kinetics. The red dotted line indicates a single exponential fit. D, example of a Ca2+ transient recorded in the dendritic trunk at 500 Hz and the corresponding action potential train measured in cell‐attached mode (bottom grey trace). E, evaluation of the linear relationship between dendritic and somatic fluorescence transients (y‐axis, mean ± SD) and the number of action potentials (x‐axis). Inset: representative fluorescence traces corresponding to one, two and three action potentials. AP, actiion potential.
Population Ca2+ imaging with cellular resolution in deep layers of the mouse neocortex
To explore the suitability of Cal‐590 AM‐based two‐photon microscopy for population imaging in deep layers with single cell resolution, we stained a small volume of brain tissue in the cortical layer of interest (Fig. 4 A). Such a localized staining is important because it helps to avoid out‐of‐focus fluorescence that is generated in regions located on top of the focal plane. Generally, pressure application of Cal‐590 AM is sufficiently accurate to allow for the labelling of spherical volumes with diameters of about 200 μm (Tischbirek et al. 2015). Figure 4 A–D illustrates two representative experiments in which Ca2+ transients were recorded in populations of neurons in layer 5 (–665 μm) and layer 6 (–870 μm), respectively. In cell‐attached patch‐clamp recordings combined with two‐photon Ca2+ imaging (Fig. 4 E), we evaluated the sensitivity of our measurements –740 μm below the pial surface and show that the Ca2+ transient amplitudes linearly report the number of action potentials. Staining with Cal‐590 AM in these depths allowed the high quality record neuronal activity for at least 2–3 h post‐staining.
Figure 4. Two‐photon Ca2+ imaging of deep cortical layers 5 and 6 stained with Cal‐590.
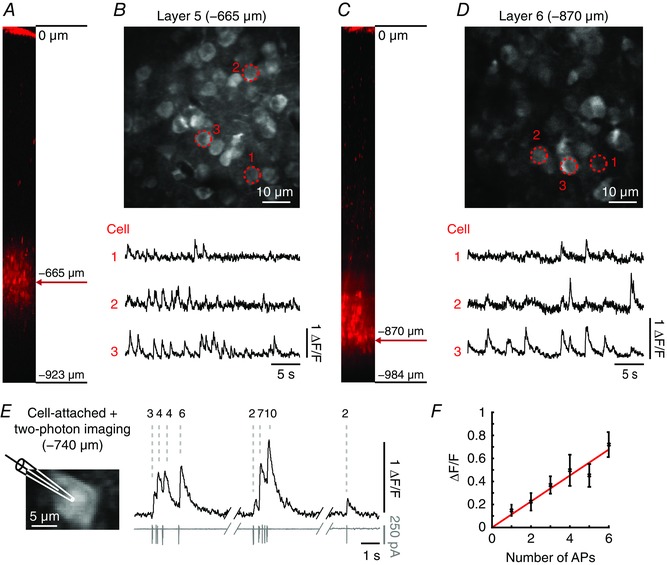
Figure adapted from Tischbirek et al. (2015). A, side view of the visual cortex showing layer 5 locally labelled with Cal‐590 AM and the depth from the pial surface (reconstruction from the z‐stack). B, neural population recorded in layer 5 at –665 μm from the pial surface. Bottom, spontaneous Ca2+ transients recorded from the cells marked with red dotted circles. C and D, neural population recorded in layer 6 at –870 μm from the pial surface with corresponding spontaneous activity with the same arrangement as in A and B for layer 5. E, left, image of a neuron in layer 5 at 740 μm below the pial surface stained with Cal‐590 AM with the recording patch‐pipette displayed schematically. Right, fluorescence traces (black, top) and corresponding action potential activity (grey, bottom) with the number of action potentials indicated on top. F, linear relationship between the number of action potentials (x‐axis) and Ca2+ transient amplitudes (y‐axis, mean ± SD). AP, action potential.
Dual‐colour functional imaging
An additional benefit of Cal‐590 is the possibility to combine it with another, spectrally well separated Ca2+ indicator for simultaneous dual‐fluorophore experiments. In the example shown in Fig. 5 A–C, layer 2/3 neurons were bulk‐labelled with Cal‐590 AM and individual neurons electroporated with OGB‐1 within the same field of view. As Cal‐590 and OGB‐1 are optimally excited for two‐photon imaging with wavelengths of 1050 and 920 nm, respectively, simultaneous imaging (with two separate detectors) was possible by using the intermediate wavelength of 1000 nm to excite both indicators at the same time (Fig. 5 D). The combination of two functional indicators allows for useful combinations of recordings in the intact tissue. For example, Ca2+ transients recorded from an individual dendrite labelled with OGB‐1 can be monitored together with the activity of the surrounding somata of neurons labelled with Cal‐590 (Fig. 5 E). To our knowledge, this is the first report of simultaneously recorded activity in distinct neurons with spectrally separated Ca2+ indicators in vivo (Tischbirek et al. 2015). A similar example – showing the possibility of simultaneously imaging a long‐wavelength Ca2+ indicator dye and a spectrally separable genetically encoded functional indicator for glutamate – used the new Ca2+ indicator CaRuby‐Nano (Collot et al. 2012; Collot et al. 2015) together with the extracellular glutamate sensor iGluSnFR (Marvin et al. 2013) to report activity in cerebellar Purkinje cells in vivo.
Figure 5. Simultaneous dual‐colour functional Ca2+ imaging.
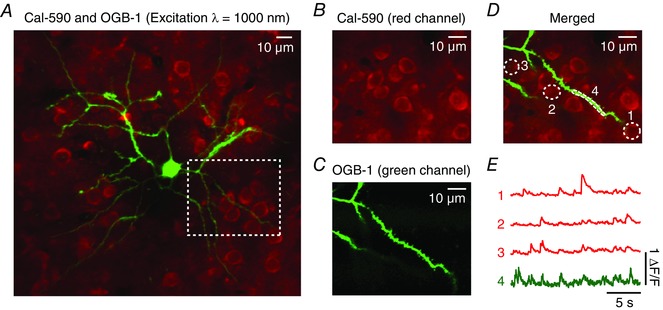
Figure adapted from Tischbirek et al. (2015). A, overlay of a single cell electroporated with OGB‐1 (green) and a cell population stained with Cal‐590 AM. The two colours were recorded simultaneously by exciting both dyes at a wavelength of 1000 nm. The emission light was separated by optical filters and detected with two photomultiplier tubes (red and green channel). B and C, images obtained from the two photomultiplier tubes separately. The fields of view in both images correspond to the magnified area indicated with a white dotted line in A). D, overlay of the magnified images in B and C with the regions of interest, which are used to determine the fluorescence traces, marked in white. E, Ca2+ transients recorded simultaneously from the somata of the neural population stained with Cal‐590 AM (red channel) and a dendrite of the single cell filled with OGB‐1 (green channel).
Discussion and conclusions
In summary, the red‐shifted fluorescent Ca2+ indicator Cal‐590 can be used for two‐photon imaging of neuronal activity with high signal‐to‐noise ratios in depths of up to 900 μm below the brain surface. It has the advantages inherent to many synthetic dyes such as rapid, linear response kinetics and the ability to readily stain any kind of neuronal tissue. However, it also has limitations such as a limited recording time and is suited therefore only for acute experiments. Additionally, it stains astrocytes along with neurons (Nimmerjahn et al. 2004; Tischbirek et al. 2015), which might cause the need for additional efforts to identify the recorded cell‐types in some experiments. As an alternative method, the use of genetically encoded Ca2+ indicators (Miyawaki et al. 1997; Nakai et al. 2001; Akerboom et al. 2013; Chen et al. 2013; Inoue et al. 2015) allows the targeted expression of a Ca2+ indicator protein in selected cell types of interest. Expression can also be targeted to specific brain areas, such as a single cortical layer, and controlled for density when, for example, a sparsely labelled neuronal network is necessary. Another benefit of genetically encoded Ca2+ indicators, which is important for experiments focusing on neuronal activity during behaviour, is that the recording time is not limited and can exceed multiple weeks.
In parallel to the recent development of the red‐shifted Cal‐590 dye, long‐wavelength variants of genetically encoded Ca2+ indicators with a high potential for deep two‐photon imaging were created and are being continuously improved. For example, jRCaMP1a was used to image sensory‐evoked Ca2+ transients in layer 6 neurons in the mouse visual cortex (Dana et al. 2016). The targeted expression of jRCaMP1a in a subpopulation of layer 6 neurons was achieved by using a neurotensin receptor 1 (NTSR1)‐Cre mouse (Gong et al. 2007). In another brain region, the red‐shifted genetically encoded Ca2+ indicator R‐CaMP1.07 (Ohkura et al. 2012) was used to record Ca2+ transients in dentate gyrus granule cells approximately 800 μm below the hippocampal surface (Pilz et al. 2016). To express R‐CaMP1.07, a Cre‐dependent adeno associated virus was injected into the dentate gyrus of transgenic mice expressing Cre‐recombinase in granule cells. The cortex overlying the hippocampus was removed and a chronic window implanted. It should be noted that in this particular study, similar results were obtained with the bright probe GCaMP6s (Chen et al. 2013). Chronic imaging experiments with RCaMP1.07 illustrate the possibility to use genetically encoded Ca2+ indicators to observe Ca2+ transients from deep brain structures over the course of multiple imaging sessions (Pilz et al. 2016). As the authors of the study note, the observed Ca2+ transients have not been calibrated for the underlying number of action potentials, so it cannot be excluded that some neuronal activity is below the detection threshold of the indicator.
Both types of indicators, synthetic dyes and genetically encoded Ca2+ indicators, have their own advantages and disadvantages and can be used depending on specific experimental needs (Grienberger & Konnerth, 2012). Regardless of their differences, the challenges for both types of Ca2+ indicators are similar: further improved probes would benefit from, among other parameters, an increased signal‐to‐noise ratio, higher photo‐stability, and dye kinetics that report neuronal Ca2+ transient time courses as accurately as possible. The experiments presented in this review are restricted to the mouse brain, although work on model organisms with different brain sizes, for example marmosets (Sadakane et al. 2015), will greatly benefit from the extended imaging depth. Additionally, further advances involving the use of long‐wavelength fluorophores will not only improve deep imaging applications, but also allow for multi‐colour experiments with spectrally separate indicators of neuronal activity (Collot et al. 2015; Inoue et al. 2015; Tischbirek et al. 2015) as well as the combination of imaging with the use of neuronal actuators, including various optogenetic probes. Finally, the maximum imaging depth reached with red‐shifted Ca2+ indicators may be extended when combined with hardware‐based deep‐imaging methods, such as adaptive optics (Ji et al. 2012), with the potential of eventually detecting neuronal network activity at the single‐cell level or even in small neuronal subcompartments (e.g. dendrites, spines, axons) in regions that were previously inaccessible to functional imaging studies in vivo.
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
C.H.T, A.B. and A.K. made the figures and wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (TRR 152 and SFB 870) and an ERC Advanced Grant (to A.K.).
Acknowledgements
We dedicate this paper to our colleague and friend Roger Tsien. His outstanding contributions were of decisive importance for this work.
Biographies
Carsten Tischbirek and Antje Birkner work in Prof. Konnerth's lab, using in vivo two‐photon microscopy and electrophysiology to study neuronal activity in various regions of the brain.
Arthur Konnerth is the Friedrich‐Schiedel‐Chair of Neuroscience and Director of the Institute for Neuroscience at the Technical University of Munich, Germany. His research is aimed at a better understanding of the cellular mechanisms underlying function and dysfunction of brain circuits. By using a variety of techniques, including optical and electrophysiological approaches, he is especially interested in the study of behaviour‐determined synaptic signalling and dendritic signal integration in neurons within the intact brain.
C. H. Tischbirek and A. Birkner contributed equally to this work. This review was presented at “Advances and Breakthroughs in Calcium Signaling”, which took place in Honolulu, Hawaii, 7–9 April 2016.
Contributor Information
Carsten H. Tischbirek, Email: carsten.tischbirek@tum.de.
Arthur Konnerth, Email: arthur.konnerth@tum.de.
References
- Akerboom J, Carreras Calderon N, Tian L, Wabnig S, Prigge M, Tolo J, Gordus A, Orger MB, Severi KE, Macklin JJ, Patel R, Pulver SR, Wardill TJ, Fischer E, Schuler C, Chen TW, Sarkisyan KS, Marvin JS, Bargmann CI, Kim DS, Kugler S, Lagnado L, Hegemann P, Gottschalk A, Schreiter ER & Looger LL (2013). Genetically encoded calcium indicators for multi‐color neural activity imaging and combination with optogenetics. Front Mol Neurosci 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustein E, Marandi N, Kovalchuk Y, Drapeau P & Konnerth A (2003). “In vivo” monitoring of neuronal network activity in zebrafish by two‐photon Ca2+ imaging. Pflugers Arch 446, 766–773. [DOI] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B & Konnerth A (2012). Critical role of soluble amyloid‐β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 109, 8740–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K & Kim DS (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Leischner U, Rochefort NL, Nelken I & Konnerth A (2011). Functional mapping of single spines in cortical neurons in vivo. Nature 475, 501–505. [DOI] [PubMed] [Google Scholar]
- Collot M, Loukou C, Yakovlev AV, Wilms CD, Li D, Evrard A, Zamaleeva A, Bourdieu L, Leger JF, Ropert N, Eilers J, Oheim M, Feltz A & Mallet JM (2012). Calcium rubies: a family of red‐emitting functionalizable indicators suitable for two‐photon Ca2+ imaging. J Am Chem Soc 134, 14923–14931. [DOI] [PubMed] [Google Scholar]
- Collot M, Wilms CD, Bentkhayet A, Marcaggi P, Couchman K, Charpak S, Dieudonne S, Hausser M, Feltz A & Mallet JM (2015). CaRuby‐Nano: a novel high affinity calcium probe for dual color imaging. Elife 4, e05808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, Patel R, Macklin JJ, Bargmann CI, Ahrens MB, Schreiter ER, Jayaraman V, Looger LL, Svoboda K & Kim DS (2016). Sensitive red protein calcium indicators for imaging neural activity. Elife 5, e12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N & Gerfen CR (2007). Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci 27, 9817–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienberger C & Konnerth A (2012). Imaging calcium in neurons. Neuron 73, 862–885. [DOI] [PubMed] [Google Scholar]
- Helmchen F & Denk W (2005). Deep tissue two‐photon microscopy. Nat Methods 2, 932–940. [DOI] [PubMed] [Google Scholar]
- Inoue M, Takeuchi A, Horigane S, Ohkura M, Gengyo‐Ando K, Fujii H, Kamijo S, Takemoto‐Kimura S, Kano M, Nakai J, Kitamura K & Bito H (2015). Rational design of a high‐affinity, fast, red calcium indicator R‐CaMP2. Nat Methods 12, 64–70. [DOI] [PubMed] [Google Scholar]
- Ji N, Sato TR & Betzig E (2012). Characterization and adaptive optical correction of aberrations during in vivo imaging in the mouse cortex. Proc Natl Acad Sci USA 109, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Judkewitz B, Kano M, Denk W & Hausser M (2008). Targeted patch‐clamp recordings and single‐cell electroporation of unlabeled neurons in vivo. Nat Methods 5, 61–67. [DOI] [PubMed] [Google Scholar]
- Kobat D, Durst ME, Nishimura N, Wong AW, Schaffer CB & Xu C (2009). Deep tissue multiphoton microscopy using longer wavelength excitation. Opt Express 17, 13354–13364. [DOI] [PubMed] [Google Scholar]
- Komiyama T, Sato TR, O'Connor DH, Zhang YX, Huber D, Hooks BM, Gabitto M & Svoboda K (2010). Learning‐related fine‐scale specificity imaged in motor cortex circuits of behaving mice. Nature 464, 1182–1186. [DOI] [PubMed] [Google Scholar]
- Li N, Chen TW, Guo ZV, Gerfen CR & Svoboda K (2015). A motor cortex circuit for motor planning and movement. Nature 519, 51–56. [DOI] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen TW, Bargmann CI, Orger MB, Schreiter ER, Demb JB, Gan WB, Hires SA & Looger LL (2013). An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods 10, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann W, Wallace DJ, Czubayko U, Herb JT, Schaefer AT, Looger LL, Denk W & Kerr JN (2011). Two‐photon calcium imaging of evoked activity from L5 somatosensory neurons in vivo. Nat Neurosci 14, 1089–1093. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M & Tsien RY (1997). Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388, 882–887. [DOI] [PubMed] [Google Scholar]
- Mutze J, Iyer V, Macklin JJ, Colonell J, Karsh B, Petrasek Z, Schwille P, Looger LL, Lavis LD & Harris TD (2012). Excitation spectra and brightness optimization of two‐photon excited probes. Biophys J 102, 934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M & Imoto K (2001). A high signal‐to‐noise Ca2+ probe composed of a single green fluorescent protein. Nat Biotechnol 19, 137–141. [DOI] [PubMed] [Google Scholar]
- Nauhaus I, Nielsen KJ, Disney AA & Callaway EM (2012). Orthogonal micro‐organization of orientation and spatial frequency in primate primary visual cortex. Nat Neurosci 15, 1683–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN & Helmchen F (2004). Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods 1, 31–37. [DOI] [PubMed] [Google Scholar]
- Oheim M, van ’t Hoff M, Feltz A, Zamaleeva A, Mallet JM & Collot M (2014). New red‐fluorescent calcium indicators for optogenetics, photoactivation and multi‐color imaging. Biochim Biophys Acta 1843, 2284–2306. [DOI] [PubMed] [Google Scholar]
- Ohkura M, Sasaki T, Kobayashi C, Ikegaya Y & Nakai J (2012). An improved genetically encoded red fluorescent Ca2+ indicator for detecting optically evoked action potentials. PLoS One 7, e39933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounov DG, Horton N, Wang T, Feng D, Nishimura N & Xu C (2014). In vivo three‐photon calcium imaging of brain activity from layer 6 neurons in mouse brain In CLEO: 2014 Postdeadline Paper Digest, STh5C.2 Optical Society of America, San Jose. [Google Scholar]
- Perillo EP, McCracken JE, Fernee DC, Goldak JR, Medina FA, Miller DR, Yeh HC & Dunn AK (2016). Deep in vivo two‐photon microscopy with a low cost custom built mode‐locked 1060 nm fiber laser. Biomed Opt Express 7, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilz GA, Carta S, Stauble A, Ayaz A, Jessberger S & Helmchen F (2016). Functional imaging of dentate granule cells in the adult mouse hippocampus. J Neurosci 36, 7407–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakane O, Masamizu Y, Watakabe A, Terada S, Ohtsuka M, Takaji M, Mizukami H, Ozawa K, Kawasaki H, Matsuzaki M & Yamamori T (2015). Long‐term two‐photon calcium imaging of neuronal populations with subcellular resolution in adult non‐human primates. Cell Rep 13, 1989–1999. [DOI] [PubMed] [Google Scholar]
- Sawinski J, Wallace DJ, Greenberg DS, Grossmann S, Denk W & Kerr JN (2009). Visually evoked activity in cortical cells imaged in freely moving animals. Proc Natl Acad Sci USA 106, 19557–19562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig JD, Chiappe ME, Lott GK, Dutta A, Osborne JE, Reiser MB & Jayaraman V (2010). Two‐photon calcium imaging from head‐fixed Drosophila during optomotor walking behavior. Nat Methods 7, 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Mancini MC & Nie S (2009). Bioimaging: second window for in vivo imaging. Nat Nanotechnol 4, 710–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew NJ, Vlasov YA, Andrew Hires S, Freeman J & Svoboda K (2015). Neural coding in barrel cortex during whisker‐guided locomotion. Elife 4, e12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordillo LA, Pu Y, Pratavieira S, Budansky Y & Alfano RR (2014). Deep optical imaging of tissue using the second and third near‐infrared spectral windows. J Biomed Opt 19, 056004. [DOI] [PubMed] [Google Scholar]
- Stirman JN, Smith IT, Kudenov MW & Smith SL (2016). Wide field‐of‐view, multi‐region, two‐photon imaging of neuronal activity in the mammalian brain. Nat Biotechnol 34, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K & Konnerth A (2003). In vivo two‐photon calcium imaging of neuronal networks. Proc Natl Acad Sci USA 100, 7319–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M, Takeuchi A, Hashizume M, Kitamura K & Kano M (2014). A highly sensitive fluorescent indicator dye for calcium imaging of neural activity in vitro and in vivo. Eur J Neurosci 39, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischbirek C, Birkner A, Jia H, Sakmann B & Konnerth A (2015). Deep two‐photon brain imaging with a red‐shifted fluorometric Ca2+ indicator. Proc Natl Acad Sci USA 112, 11377–11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY ( 1981). A non‐disruptive technique for loading calcium buffers and indicators into cells. Nature 290, 527–528. [DOI] [PubMed] [Google Scholar]
- Wang K, Sun W, Richie CT, Harvey BK, Betzig E & Ji N (2015). Direct wavefront sensing for high‐resolution in vivo imaging in scattering tissue. Nat Commun 6, 7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C & Wise FW (2013). Recent advances in fibre lasers for nonlinear microscopy. Nat Photon 7, 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Guo H, Yi G, Liao J & Diwu Z (2015). A novel red fluorescence calcium indicator for functional analysis of GPCRs and calcium channel targets. Biophys J 108, 109a–110a. [Google Scholar]