

Abstract
Ca2+ signalling is perhaps the most universal and versatile mechanism regulating a wide range of cellular processes. Because of the many different calcium‐binding proteins distributed throughout cells, signalling precision requires localized rises in the cytosolic Ca2+ concentration. In electrically non‐excitable cells, for example epithelial cells, this is achieved by primary release of Ca2+ from the endoplasmic reticulum via Ca2+ release channels placed close to the physiological target. Because any rise in the cytosolic Ca2+ concentration activates Ca2+ extrusion, and in order for cells not to run out of Ca2+, there is a need for compensatory Ca2+ uptake from the extracellular fluid. This Ca2+ uptake occurs through a process known as store‐operated Ca2+ entry. Ideally Ca2+ entering the cell should not diffuse to the target site through the cytosol, as this would potentially activate undesirable processes. Ca2+ tunnelling through the lumen of the endoplasmic reticulum is a mechanism for delivering Ca2+ entering via store‐operated Ca2+ channels to specific target sites, and this process has been described in considerable detail in pancreatic acinar cells and oocytes. Here we review the most important evidence and present a generalized concept.
Keywords: calcium activated chloride current, calcium entry, calcium signalling
Abbreviations
- ACh
acetylcholine
- ANO1
anoctamin‐1
- CaCC
Ca2+‐activated Cl− channel
- CCK
cholecystokinin
- CRAC channel
Ca2+ release‐activated Ca2+ channel
- ER
endoplasmic reticulum
- IP3
inositol trisphosphate
- IP3R
IP3 receptor
- MCU
mitochondrial Ca2+ uniporter
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NFAT
nuclear factor of activated T‐cells
- PIP2
phosphatidyl inositol bisphosphate
- PM
plasma membrane
- PMCA
plasma membrane Ca2+‐activated ATPase
- RyR
ryanodine receptor
- SERCA
sarco/endoplasmic reticulum Ca2+ activated ATPase
- SOAR
STIM1 Orai1‐activating region
- SOCE
store‐operated Ca2+ entry
- STIM
stromal interaction molecule
- TMEM16A
transmembrane member 16A
- TRP
transient receptor potential
Introduction
Effective and precise intracellular Ca2+ signalling depends on specific Ca2+ sensors and transport proteins expressed differentially on organelle and plasma membranes, as well as Ca2+ buffers with different affinities and kinetics in different cellular compartments (Petersen et al. 1994; Berridge, 2016). Because Ca2+ can interact with many potential cellular targets, signalling precision requires localized rises of the cytosolic [Ca2+] ([Ca2+]i) (Petersen et al. 1994; Petersen & Verkhratsky, 2016). In the nervous system, the extremely precise control of presynaptic neurotransmitter secretion depends on close co‐localization of voltage‐gated Ca2+ channels and the exocytotic machinery (Südhof, 2013). However, the target for the action of Ca2+ cannot always be very close to the site of Ca2+ entry. A prime example of such a scenario comes from the physiology of the pancreatic acinar cells, where Ca2+ entry occurs at the base of the cell, whereas the control of secretion has to take place at the opposite end of the cell at the apical pole (Petersen et al. 1994; Petersen & Tepikin, 2008). In these and many other electrically non‐excitable cells, which do not have voltage‐gated Ca2+ channels and do not fire action potentials (Petersen, 1992), the primary Ca2+ movement is from the endoplasmic reticulum (ER) into the cytosol (Nielsen & Petersen, 1972; Berridge, 2016) and this, in turn, triggers store‐operated Ca2+ entry (Putney, 1986; Petersen & Tepikin, 2008; Parekh, 2010). Diffusion of Ca2+ through the cytosol, from an entry site to a distant target, would potentially activate many inappropriate processes and a mechanism that could avoid this path would therefore be advantageous. Transport through an organelle, moving Ca2+ from its entry point to its target, would solve this problem (Fig. 1). The process of Ca2+ tunnelling through the ER was discovered in studies on pancreatic acinar cells carried out 20 years ago (Mogami et al. 1997). A similar process was later described in dopamine neurons (Choi et al. 2006) and the whole concept has more recently been generalized, based on experiments in oocytes (Courjaret & Machaca, 2014; Courjaret et al. 2016a). Furthermore, in a recent study (Kar et al. 2016), it has been shown that Ca2+ refilling of the nuclear envelope, after inositol trisphosphate (IP3)‐evoked Ca2+ release into the nucleoplasm through the inner nuclear membrane (Gerasimenko et al. 1995), depends on Ca2+ (entering via store‐operated Ca2+ channels) being tunnelled through the ER lumen directly into the nuclear envelope. In this article, we describe and review the most important evidence for Ca2+ tunnelling, primarily based on studies of pancreatic acinar cells and oocytes.
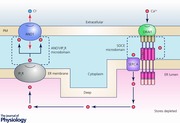
Figure 1. Schematic drawing of Ca2+ movement through pancreatic acinar cells.
Ca2+ moves from the extracellular solution (extreme right) via store‐operated CRAC channels, SERCA pumps, ER, IP3Rs and PMCA pumps into the acinar lumen (extreme left). Diffusion of Ca2+ from the base of the cell to the apical pole occurs quantitatively inside the ER rather than in the cytosol because of the much higher Ca2+ binding capacity in the cytosol than in the ER lumen. Ca2+ released from the ER in the apical part of the cell via IP3Rs activates Cl− channels (ANO1) in the luminal acinar plasma membrane. ANO1, anoctamin‐1; CRAC channel, Ca2+ release‐activated Ca2+ channel; IP3R, inositol trisphosphate receptor; PMCA, plasma membrane Ca2+‐activated ATPase; SERCA, sarco/endoplasmic reticulum Ca2+ activated ATPase.
Spatial and temporal features of Ca2+ signals
Specificity in Ca2+ signals is encoded in their spatial, temporal and amplitude features. These Ca2+ dynamics combine to activate a defined subset of Ca2+‐dependent downstream effectors to transduce the cellular response (Berridge et al. 2000, 2003). Spatially, Ca2+ signals are tightly regulated and are typically initiated by elementary events due to the opening of Ca2+ channels (intracellular or at the cell membrane). These elementary events due to the opening of one or a few channels can either remain localized resulting in Ca2+ signals in the microdomain around the channel(s), or coalesce through complex mechanisms into more global Ca2+ events that often encompass the entire cell (Berridge, 1997).
The extent and speed of Ca2+ movement is heavily influenced by the concentration and characteristics of the available Ca2+ buffers. It was shown many years ago, that adding a low affinity mobile Ca2+ buffer to the cytosol can profoundly change the timing and spatial extension of agonist‐elicited cytosolic Ca2+ signals (Petersen et al. 1991). The cytosolic Ca2+ buffering characteristics vary markedly between different cell types with, for example, a high level of relatively low mobility buffer in pancreatic acinar cells (Mogami et al. 1999), and a less restricted environment for Ca2+ diffusion in oocytes (Allbritton et al. 1992) and some nerve cells (Lin et al. 2017). Inevitably, [Ca2+] measurements using Ca2+‐sensitive fluorescent probes will be influenced by the Ca2+‐binding properties of the probes, so unless a careful analysis of the Ca2+ buffering situation has been carried out, as recently described by Lin et al. (2017), some caution with regard to interpreting quantitative results is called for.
In general, the diffusion of Ca2+ in the cytosol is always severely limited, as compared to movement in water, due to the relatively high buffering capacity. Some estimates indicate that free Ca2+ diffuses < 0.1 μm, and is free for ∼0.5 μs before it is buffered (Allbritton et al. 1992; Kasai & Petersen, 1994). At the mouth of an open Ca2+ channel and given the great concentration gradients across both the ER and plasma membrane, Ca2+ flow rapidly overwhelms the local buffering capacity resulting in a microdomain of high Ca2+ concentration in the order of 20–200 μm (Rizzuto & Pozzan, 2006). The spatial spread of these high Ca2+ microdomains is thus very tightly controlled, and is predicted based on theoretical modelling to be maintained within 20 nm of the channel (Simon & Llinas, 1985; Neher, 1998). Beyond the immediate point source of Ca2+ entry at the mouth of the channel, Ca2+ diffusion creates a downward gradient away from the channels that is thought to dissipate to the submicromolar range within 200 nm of the channel (Neher, 1998; Shuai & Parker, 2005; Demuro & Parker, 2006). This provides for an elegant mechanism to activate Ca2+‐dependent effectors that localize within the spatial spread of Ca2+ signals generated due to elementary Ca2+ events (Rizzuto & Pozzan, 2006; Parekh, 2008).
Global Ca2+ signals, in contrast, have a significantly broader spread on the order of 10–100 μm. This highlights a spatial gap between elementary and global Ca2+ signals, as there could be a physiological need to activate effectors that are not in the immediate vicinity of a Ca2+ channel without inducing a global Ca2+ rise that would activate a multitude of other signalling pathways. Although not discussed in details here, cells could maintain specificity in their Ca2+ signals despite the extent of their spatial spread by controlling their amplitude and frequency for example. Nonetheless, there are several examples that argue that Ca2+ signals activate effectors in the mid‐range, between the microdomain and global spatial extremes, with exquisite specificity. Nuclear factor of activated T‐cells (NFAT) activation in T‐cells in response to antigen stimulation (Dolmetsch et al. 1997; Rao et al. 1997), Ca2+‐activated K+ channels (Liu et al. 1998), and Ca2+‐activated Cl− channels (Courjaret & Machaca, 2014) are examples of effectors activated specifically without localizing to the Ca2+ channel microdomain. It is, however, not clear mechanistically how cells would mediate such Ca2+ signals in the mid‐range without inducing a global Ca2+ rise. One possible mechanism is to induce a higher level or a more sustained Ca2+ flux through open channels to saturate the local buffering capacity, thus allowing the Ca2+ signal to spread further. This would be a perilous path, though, given the Ca2+ dependence of intracellular Ca2+ release channels, IP3 and ryanodine receptors, which could lead to Ca2+‐induced Ca2+ release and a global Ca2+ rise (Osipchuk et al. 1990; Bootman et al. 2002).
Ca2+ signalling in electrically non‐excitable cells is typically initiated downstream of agonist stimulation through the activation of a phospholipase C that hydrolyses phosphatidyl inositol bisphosphate (PIP2) at the plasma membrane and results in the production of IP3 and diacylglycerol. IP3 diffuses and gates open IP3 receptors at the ER membrane, releasing store Ca2+ to mediate the first phase of the Ca2+ signal. Should the Ca2+ release phase result in significant store depletion, it leads to the activation of Ca2+ influx at the plasma membrane through store‐operated Ca2+ entry (SOCE). SOCE is mediated by members of the stromal interaction molecule (STIM) and Orai family (Prakriya & Lewis, 2015). STIM1 is a single‐pass ER membrane protein with lumenal EF hands allowing it to sense the ER Ca2+ concentration (Liou et al. 2005; Roos et al. 2005). Store depletion results in STIM1 losing its lumenal bound Ca2+ leading to a conformational change in the protein and its clustering and translocation to ER–plasma membrane (PM) junctions that are 20 nm apart (Luik et al. 2006; Prakriya et al. 2006; Stathopulos et al. 2006; Vig et al. 2006; Wu et al. 2006; Yeromin et al. 2006; Liou et al. 2007). The close proximity of the ER and PM at these junctions allows STIM1 to span the distance and physically interact with Orai1 at the PM. Orai1 is a four transmembrane domain protein that forms a hexameric channel that is exquisitely Ca2+ selective (Hou et al. 2012). STIM1 clusters stabilize at ER–PM junctions initially through interaction of the poly‐lysine domain at the C‐terminal end of STIM1 with PIP2 in the PM. Activated STIM1 in response to store depletion exposes the STIM1 Orai1‐activating region (SOAR)/CRAC‐activating domain, which interacts with Orai1, traps it within the STIM1‐defined ER–PM junctions, and gates it open, thus allowing Ca2+ flow into the cell. SOCE activation not only results in store refilling but also shapes Ca2+ signal dynamics. There is therefore a tight functional link between IP3‐dependent Ca2+ release in response to agonist stimulation and Ca2+ influx through SOCE.
Ca2+ tunnelling in pancreatic acinar cells
The function of the acinar cells
The principal function of the exocrine pancreas is to deliver digestive enzymes to the intestine in order to break down food products, so that they can be absorbed into the circulation. The most important secretory cell in the exocrine pancreas is the acinar cell, which manufactures the inactive pro‐enzymes and stores them in zymogen granules. When enzyme delivery is required, the acinar cells receive a signal in the form of either the neurotransmitter acetylcholine (ACh; released from parasympathetic nerve endings) and/or the hormone cholecystokinin (CCK). Interaction with specific surface membrane receptors activates signal transduction mechanisms that generate intracellular Ca2+, liberating messengers – IP3 in the case of ACh and cyclic ADP‐ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) in the case of CCK stimulation – thereby releasing Ca2+ from intracellular stores (Petersen & Tepikin, 2008). As a consequence of the depletion of intracellular Ca2+ stores, Ca2+‐permeable channels in the plasma membrane are opened allowing Ca2+ entry from the extracellular solution (Petersen & Tepikin, 2008).
Under physiological conditions the cytosolic Ca2+ concentration ([Ca2+]i) changes evoked by ACh or CCK consist of repetitive short‐lasting elevations mostly confined to the apical region (Kasai et al. 1993; Thorn et al. 1993), where the zymogen granules are concentrated. This local rise in [Ca2+]i triggers exocytosis of the granule content (Maruyama & Petersen, 1994) as well as opening up Cl− channels in the apical membrane and K+ channels in the apical part of the lateral membrane (Petersen & Maruyama, 1984; Petersen, 1992; Park et al. 2001b). These channel openings enable operation of the Na+/K+/2Cl− co‐transporter as well as increased turnover of the Na+/K+ pump across the basolateral membrane (Petersen, 1992). The net result is uptake of Cl− across the basolateral membrane and secretion of Cl− across the apical membrane with Na+ following via a paracellular pathway through the leaky tight junctions. Water moves along with the salt both through the cell and through the so‐called tight junctions (Petersen, 1992). The Ca2+‐activated acinar fluid secretion is a vehicle for transport of the pro‐enzymes into the duct system, where an additional ductal fluid secretion of a secretin‐stimulated bicarbonate‐rich solution (Scratcherd et al. 1981; Lee et al. 2012) will help wash the pro‐enzymes into the gut where they become active digestive enzymes.
The polarity of the acinar cells
The acinar cells secrete enzymes and fluid in one direction, namely into the lumen of the acinar unit, which is directly connected to the duct system and, therefore, these cells are highly polarized. The zymogen granules are in the apical part of the cells, whereas the nucleus – surrounded by densely packed rough ER – occupies the basolateral region. The apical membrane area is much smaller than the basolateral membrane area, but the final stage of secretion occurs exclusively through the apical membrane. The tight junctions, which are leaky in the case of the acinar epithelium, are placed close to the acinar lumen. Although the bulk of the ER is in the basolateral region, ER elements penetrate into the apical zymogen granule‐rich region all the way to the apical membrane (Gerasimenko et al. 2002).
With regard to the localization of the principal Ca2+‐activated ion channels in the plasma membrane of the acinar cells, we know that the Ca2+‐activated Cl− channels, transmembrane member 16A (TMEM16A)/anoctamin‐1 (ANO1) (Lee et al. 2012), are exclusively present in the apical membrane (Park et al. 2001b), whereas the high‐conductance and voltage‐sensitive Ca2+‐activated K+ channels (present in the pig and human acinar cells) are found in the basolateral membrane (Maruyama et al. 1983; Petersen et al. 1985). Simultaneous patch clamp recording of Cl− conductance and capacitance indicate that during normal physiological stimulus–secretion coupling, each local apical Ca2+ spike causes near‐synchronous (but see below) opening of Cl− channels and exocytosis (Maruyama & Petersen, 1994).
With regard to Ca2+ transport across the plasma membrane, we know that Ca2+‐ATPase‐driven Ca2+ extrusion occurs mostly through the apical membrane (Belan et al. 1996), whereas store‐operated Ca2+ entry occurs through the basolateral membrane (Mogami et al. 1997; Park et al. 2001a; Lur et al. 2009).
How Ca2+ entering through the basolateral plasma membrane allows Ca2+ signal generation near the apical membrane without passing through the cytosol
Although it has been known from the earliest days of work on stimulus–secretion coupling in pancreatic acinar cells that the initial Ca2+ signal generation evoked by stimulation with either ACh or CCK is due to release of Ca2+ from internal stores (Matthews et al. 1973; Petersen & Ueda, 1976), it has also been clear that supply of Ca2+ from the extracellular solution is essential for continuation of secretion (Petersen & Ueda, 1976). The reason for the extracellular Ca2+ requirement is that every rise in the cytosolic Ca2+ concentration ([Ca2+]i) inevitably activates Ca2+ pumps in the plasma membrane (plasma membrane Ca2+‐activated ATPase; PMCA) resulting in extrusion of Ca2+ (Tepikin et al. 1992), which then has to be compensated by Ca2+ entry, as otherwise the cell would gradually run out of Ca2+.
Agonist‐elicited cytosolic Ca2+ spiking, which is the normal physiological signal for secretion, requires a relatively high [Ca2+] in the lumen of the ER (Park et al. 2000). In experiments on isolated acinar cells where [Ca2+] changes in both the cytosol and the lumen of the ER were measured simultaneously, it could be shown that ACh evokes cytosolic Ca2+ spiking for several minutes in the absence of external Ca2+. However, spiking subsequently stops after only a relatively modest reduction of [Ca2+]ER (Park et al. 2000). Thus, Ca2+ entry through store‐operated Ca2+ channels, refilling the ER store, is essential for the normal physiological function of the acinar cells.
The original concept of Ca2+ tunnelling through the ER lumen, from entry at the base of the cell to release near the apical membrane (Fig. 1), was based on experiments in which isolated acinar cells were kept in a Ca2+‐free solution with a patch pipette attached to the basal surface (Mogami et al. 1997). The patch pipette was filled with a Ca2+‐containing solution and Ca2+ entry across the membrane covered by the pipette tip could be regulated by controlling the pipette potential. After supra‐maximal ACh stimulation had emptied the intracellular stores during a period without Ca2+ entry (negative – retaining – potential in the pipette), ACh stimulation was discontinued and a period of Ca2+ entry was enabled by switching the pipette potential from negative to positive. No change was observed in [Ca2+]i during this Ca2+ entry period, but after discontinuation of Ca2+ entry (switching the pipette voltage back to negative), a new period of ACh stimulation caused a local rise of [Ca2+]i in the apical pole near the apical membrane, exactly as under normal conditions. A rise in [Ca2+]i near the cell‐attached pipette during Ca2+ entry could only be observed when the ER Ca2+ pumps (sarco/endoplasmic reticulum Ca2+‐activated ATPase; SERCA) were arrested by thapsigargin (Thastrup et al. 1989). However, in this situation there was no sign of transfer of Ca2+ from the base to the apex, as ACh stimulation after a period of Ca2+ entry failed to elicit any Ca2+ release in the apical region (Mogami et al. 1997). As thapsigargin is a very selective blocker of SERCA pumps (Thastrup et al. 1989), the simplest explanation for the phenomenon observed, namely the non‐cytosolic transfer of Ca2+ across the cell from base to lumen, is movement through the ER lumen.
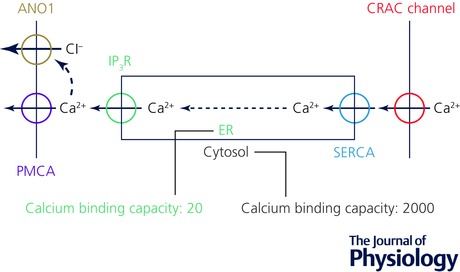
The ER Ca2+ tunnelling concept (Mogami et al. 1997) assumed that Ca2+ would move more easily within the lumen of the ER than in the cytosol (Fig. 1). The relatively low mobility of Ca2+ in the cytosol was demonstrated in the classical experiments of Baker & Crawford (1972) on axoplasm, in which it could be shown that radioactive Mg2+ moved much more quickly than radioactive Ca2+, and later confirmed by Allbritton et al. (1992). In the acinar cells, based on measurements of absolute calcium movements and changes in [Ca2+] in the cytosol and the ER, we estimated that the calcium binding capacity in the ER lumen is about 20 whereas in the cytosol it is about 1500–2000 (Mogami et al. 1999). Thus the mobility of Ca2+ in the ER lumen is very much higher than in the cytosol (Fig. 1).
The high mobility of Ca2+ in the ER lumen was demonstrated directly by experiments in which changes in [Ca2+]ER at various locations in the ER could be monitored after a highly localized uncaging of caged Ca2+ in the ER lumen (Park et al. 2000). These experiments showed that after a local Ca2+ uncaging event, rises in [Ca2+]ER were observed quickly over considerable distances (more than 10 μm away from the site of uncaging) and that the whole of the ER was re‐equilibrated with regard to [Ca2+]ER within a few seconds (less than the time interval between cytosolic Ca2+ spikes during physiological Ca2+ signalling) (Park et al. 2000; Petersen et al. 2001).
Movement of Ca2+ from the extracellular fluid into the ER lumen at the base
Early work on perfused submandibular glands showed that ACh‐evoked intracellular Ca2+ release was followed, after a delay, by Ca2+ influx into the gland cells from the perfusion fluid (Nielsen & Petersen, 1972). Later, work on isolated pancreatic acinar cells showed more precisely that the Ca2+ entry, following the ACh‐evoked immediate (< 0.5 s) apical Ca2+ release, occurred through the basolateral membrane after a delay of about 6–7 s (Toescu & Petersen, 1995). A few years later it was shown that it is possible to refill the emptied ER with Ca2+ flowing into the cell from a point source at the base of the cell (Mogami et al. 1997). Like in many other cell types, store‐operated Ca2+ entry is mediated by translocation of STIM to puncta near the plasma membrane, which in the pancreatic acinar cells are specifically located at the basolateral part of the cell (Lur et al. 2009). However, in these cells there is a specific challenge for this process, as the ER is of the rough type due to the presence of ribosomes. The size of ribosomes is such that it would not allow the close molecular interaction between STIM in the ER membrane and Ca2+ channels in the plasma membrane that is necessary for channel activation. However, it turns out that there are small areas of the otherwise rough ER that are devoid of ribosomes, allowing these parts to come very close to the plasma membrane (Lur et al. 2009) (Fig. 2).
Figure 2. Location of key organelles and molecules involved in Ca2+ uptake at the base of pancreatic acinar cells.
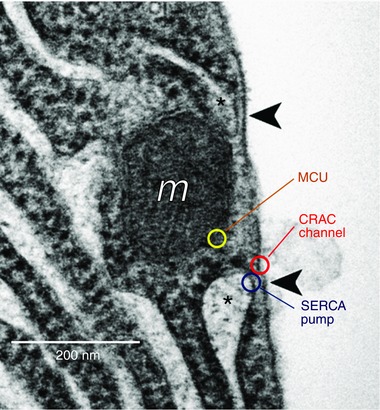
EM picture of basal area of a pancreatic acinar cell showing areas in which the ER is devoid of ribosomes and comes very close to the plasma membrane (indicated with asterisk). This arrangement allows molecular interaction between STIM in the ER membrane and CRAC channels in the plasma membrane. A mitochondrion (m) is also seen near the plasma membrane. Arrowheads signpost ER–plasma membrane junctions where Ca2+ entry can take place. Indicative locations of the three key Ca2+ transporters at the base of the cell are shown. MCU, mitochondrial Ca2+ uniporter. Adapted from Lur et al. (2009).
The biophysical nature of the Ca2+ entry process in the pancreatic acinar cells was not clarified until recently (Gerasimenko et al. 2013), when patch clamp whole‐cell current recording studies showed that the inward Ca2+ current evoked by ER store Ca2+ depletion has characteristics very similar to the Ca2+ release activated Ca2+ (CRAC) current previously discovered in immune cells (Hoth & Penner, 1992; Feske, 2007; Parekh, 2010), and could be blocked by a specific CRAC channel inhibitor (Gerasimenko et al. 2013). During Ca2+ refilling of the ER, for example after ACh‐evoked emptying of the store, there is no measurable increase in the cytosolic Ca2+ concentration near the Ca2+ entry channels although, as mentioned above, it is possible to observe a rise in the cytosolic [Ca2+] during Ca2+ entry if the SERCA pumps in the ER have been arrested by thapsigargin (Mogami et al. 1997). In that case there is also a clear increase in the [Ca2+] in the mitochondria (mitochondrial Ca2+ uptake being mediated by the mitochondrial Ca2+ uniporter; MCU; De Stefani et al., 2011, 2016) placed very close to the basolateral membrane (Park et al. 2001a) (Fig. 2). Ca2+ uptake into these peripheral mitochondria is functionally important as it will increase ATP production (De Stefani et al. 2016) locally, thereby fuelling the SERCA pumps. It would therefore appear that the crucial molecules, involved in the process of moving Ca2+ from the extracellular fluid into the ER, namely CRAC channels, SERCA pumps and the MCU, are localized very close together in the basolateral part of the cell during SOCE (Fig. 2).
Movement of Ca2+ from the ER into the apical cytosol where activation of exocytosis and Cl− channels occurs
The rise in [Ca2+]i, evoked by either ACh or CCK stimulation, always starts in the apical part of the cell, close to the apical membrane (Kasai & Augustine, 1990; Kasai et al. 1993; Thorn et al. 1993; Cancela et al. 2000) and, at near‐physiological intensities of stimulation, the rise is mostly confined to the apical region (Kasai et al. 1993; Thorn et al. 1993), due to the perigranular mitochondrial firewall (Tinel et al. 1999; Park et al. 2001a). Even under conditions where muscarinic receptor activation occurs exclusively within a small region at the base of the cell (uncaging of caged carbachol in a cell‐attached patch pipette), the rise in [Ca2+]i always starts near the apical membrane (Ashby et al. 2003). Close comparison of the time course of the increases in Cl− conductance and capacitance (indicative of fusion between zymogen granules and apical plasma membrane) during individual apical Ca2+ spikes shows (Fig. 3) that Cl− channels are activated slightly earlier than the start of exocytosis and that the Cl− conductance increase slightly outlasts the period of increased capacitance (Maruyama & Petersen, 1994). This could be explained either by the Cl− channels being located closer to the ER Ca2+ release channels than the sites of exocytosis, or by the Cl− channels being more sensitive to the local [Ca2+]i changes than the exocytosis machinery.
Figure 3. Ca2+‐activated secretory events at the apical pole of pancreatic acinar cells.
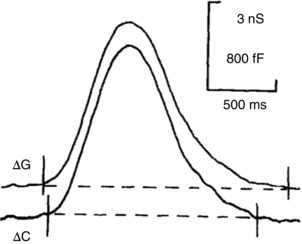
Simultaneous recording of changes in Cl− conductance (ΔG) and capacitance (ΔC) during a single IP3‐elicited Ca2+ spike (part of a train of spikes evoked by continuous intracellular IP3 infusion) shows the similar timing and trend of both events with a slight delay in the capacitance increase as compared to the rise of the Cl− conductance. It is also seen that the Cl− conductance increase outlasts the period of increased capacitance. From Maruyama & Petersen (1994).
Ca2+ spiking, induced by stimulation with either ACh or CCK, is abolished by blockade of IP3 receptors (IP3Rs) (Wakui et al. 1990), but also by blockade of ryanodine receptors (RyRs) (Cancela et al. 2000). Since Ca2+ spiking can also be elicited by intracellular Ca2+ infusion (Osipchuk et al. 1990; Wakui et al. 1990), it is probably due to interactive Ca2+‐induced Ca2+ release, involving both IP3Rs and RyRs. The mechanisms by which ACh and CCK initiate apical Ca2+ signal generation are different. In the case of ACh stimulation, there is IP3 generation due to phospholipase C activation whereas in the case of physiological CCK stimulation (low picomolar CCK concentration), there is primary generation of NAADP (Yamasaki et al. 2005). Thus blockade of NAADP receptors inhibits CCK‐ but not ACh‐elicited Ca2+ spiking (Cancela et al. 2000; Gerasimenko et al. 2015). In spite of these mechanistic differences, the measurable Ca2+ signal progression from the initiation site near the apical membrane towards the perigranular mitochondrial belt is quantitatively very similar in both cases (Cancela et al. 2000). This indicates that the initial trigger Ca2+ release is so small and so local that it is not observable with current technology. The local apical Ca2+ release that actually activates the Cl− channels in the apical membrane and the exocytotic enzyme release through the apical plasma membrane is therefore most likely the result of the final co‐activation of IP3Rs and RyRs triggered by the initial Ca2+ release from either IP3Rs or NAADP‐sensitive two‐pore channels (Gerasimenko et al. 2015).
Ca2+ tunnelling supports mid‐range Ca2+ signalling in the Xenopus oocyte
The Xenopus oocyte as an experimental model system to study Ca2+ signalling
The frog oocyte has long been a favoured model system to study Ca2+ signalling and has contributed significantly to our understanding of basic Ca2+ signalling mechanisms, including elementary Ca2+ release events, Ca2+ waves, fertilization‐specific Ca2+ signals, biophysical properties of the IP3 receptor, and remodelling of Ca2+ signalling during the cell cycle (Lechleiter & Clapham, 1992; Sun et al. 1998; Bugrim et al. 2003; Foskett et al. 2007; Machaca, 2007). Several features make the oocyte an attractive model system to study these various aspects of Ca2+ signalling. The oocyte is large (∼1.2 mm in diameter) allowing for easy spatial resolution of Ca2+ dynamics, which becomes particularly important for studies focused on the generation and propagation of Ca2+ waves, and elementary Ca2+ release events because their large spatial footprint in the oocyte makes them more amenable to investigation. The size of the oocyte also favours biochemical analyses and importantly linking them directly to Ca2+ signalling and other cell physiological processes at the single cell level (Machaca & Haun, 2002). Another unique advantage of the oocyte is the stage of the cell cycle oocytes transition through with two physiologically defined arrest points at prophase I and metaphase II of meiosis. Oocytes are arrested in prophase I at the G2/M transition of the cell cycle in a G2‐like state, which is the typical stage in which they have been used as an experimental model. However, physiologically oocytes transition to metaphase of meiosis II in preparation for fertilization. This well‐regulated progression through M‐phase provides a distinctive window into the cell division phase of the cell cycle that is transient and asynchronized in other systems such as mitosis in mammalian cells, making it more difficult to study. Another additional benefit of the oocyte is the relative simplicity of its Ca2+ signalling toolkit compared to other cells. The frog oocyte has a limited well defined complement of Ca2+ channels and transporters, significantly less complex than most mammalian cells (Machaca, 2007).
Further simplifying Ca2+ signalling studies in the frog oocyte is the endogenous expression of Ca2+‐activated Cl− channels (CaCCs), which are critical for oocyte biology and fertilization as they contribute significantly to the maintenance and regulation of the membrane potential. The Ca2+ transient generated at fertilization is initially localized at the site of sperm entry but gradually sweeps across the entire egg in the form of a Ca2+ release wave, which activates CaCCs and depolarizes the cell to prevent polyspermy (Machaca et al. 2001). This so called ‘fast electrical block’ to polyspermy in Xenopus is due to the fact that sperm–egg fusion is voltage sensitive in this species (Jaffe et al. 1983; Goul‐Somero & Jaffe, 1984). The molecular entity underlying the CaCC in the frog oocyte has been identified as anoctamin 1 (Ano1) or TMEM16A (Schroeder et al. 2008; Yang et al. 2008). The biophysical properties of the Xenopus oocyte CaCCs have been well characterized both for the endogenous current (Kuruma & Hartzell, 1998, 2000; Machaca & Hartzell, 1998, 1999; Callamaras & Parker, 2000), overexpressed Ano1 in the oocyte (Courjaret et al. 2016b), and heterologously expressed Xenopus Ano1 in the axolotl oocyte (Schroeder et al. 2008). In the oocyte, CaCC senses sub‐cell membrane changes in Ca2+ concentration in real time and with high fidelity, whether this Ca2+ is released from the ER or flows from the extracellular space through channels in the plasma membrane (Machaca & Hartzell, 1999). As such multiple studies have used the endogenous CaCC to monitor complex Ca2+ dynamics mediated by endogenous or heterologously expressed Ca2+‐permeable channels, such as ionotropic receptors (Kuruma & Hartzell, 1998), voltage‐gated Ca2+ channels (Zhou et al. 2004), transient receptor potential (TRP) channels (Courjaret et al. 2013) and SOCE channels (Courjaret & Machaca, 2016).
Mid‐range Ca2+ signalling
While studying the activation properties of CaCCs in the frog oocyte in response to various Ca2+ mobilizing agents, we noticed that CaCCs are stimulated to significantly higher levels when stores are depleted with IP3 as compared to other mobilizing agents that deplete Ca2+ stores by distinct mechanisms of action, including ionomycin, (N,N,N,N‐tetrakis(2‐pyridylmethyl)‐ethylenediamine (TPEN) and thapsigargin (Fig. 4) (Courjaret & Machaca, 2014). While IP3 replicates the physiological situation, ionomycin, a Ca2+ ionophore with preferential insertion in the ER membrane (Morgan & Jacob, 1994), empties Ca2+ stores rapidly and induces SOCE in the absence of activation of IP3 receptors (IP3R). TPEN is a transition metal chelator with low Ca2+ affinity that is freely membrane permeant and chelates high lumenal ER Ca2+, thus simulating Ca2+ store depletion and inducing SOCE (Hofer et al. 1998), again in the absence of IP3R activation. Thapsigargin is an irreversible, specific blocker of the endoplasmic reticulum Ca2+‐ATPase (SERCA) (Thastrup et al. 1989) that leads to store depletion due to an inherent constitutive Ca2+ leak pathway from the ER. Therefore, blocking SERCA, the primary ER Ca2+ refilling pathway leads to store depletion, although with slower kinetics than other aforementioned Ca2+ mobilizing agents. Given the differing mechanisms by which these agents induce SOCE, it was not clear why IP3 leads to a significantly higher induction of current through CaCCs. We ruled out an increased Ca2+ influx through SOCE under these various conditions (Courjaret & Machaca, 2014). Furthermore, we showed that Ca2+ transients in the cortical region of the oocyte were of similar amplitude when SOCE was stimulated with IP3 or thapsigargin, but of much smaller amplitude with ionomycin (Fig. 4 B) (Courjaret & Machaca, 2014). Given that measuring SOCE current reveals similar flow of Ca2+ into the cell, these data show that when SERCA is active, Ca2+ flowing through SOCE channels is immediately taken up by SERCA into the ER lumen, thus preventing its diffusion into the cortical region. When SERCA is blocked with thapsigargin, this pathway is inhibited allowing SOCE to flood the cell cortex with Ca2+. However, this does not explain why cortical Ca2+ is high during the Ca2+ influx phase when stores are depleted with IP3 (Fig. 4 B). Various approaches to block the IP3R or SERCA were used to show that the high CaCC current in response to SOCE when IP3 is present requires active IP3Rs (Courjaret & Machaca, 2014). This led to the model outlined in Fig. 5, where Ca2+ flowing through SOCE channels is taken up within the SOCE microdomain by SERCA into the ER lumen and then released through open IP3Rs at a distal site to specifically activate CaCCs, thus leading to high current levels specifically in response to SOCE (Fig. 5). This model matches the Ca2+ tunnelling mechanism discussed above in pancreatic acinar cells although the timing and functional links between the various components of the pathway diverge to meet the cell's physiological needs.
Figure 4. Differential response of Ca2+‐activated Cl− channels to modes of store depletion in the oocyte.
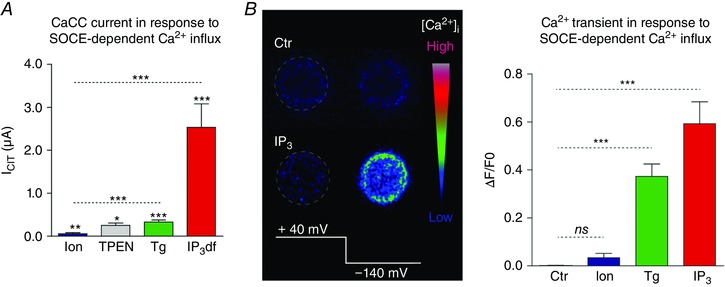
A, amplitudes of current through CaCC following store depletion with different agents: ionomycin, TPEN, thapsigargin (Tg), non‐hydrolyzable IP3 (IP3df). B, left, intracellular Ca2+ transient monitored by confocal microscopy in a voltage‐clamped oocyte using Oregon green BAPTA‐1 under control conditions (Ctr) or following IP3 injection. Ca2+ influx through SOCE was stimulated by hyperpolarizing the cell to −140 mV. Right, summary of the intracellular Ca2+ rise induced by the hyperpolarizing pulse to −140 mV when intracellular stores were depleted with ionomycin (Ion), thapsigargin (Tg) or IP3. Adapted from Courjaret and Machaca (2014).
Figure 5. Model of Ca2+ tunnelling.
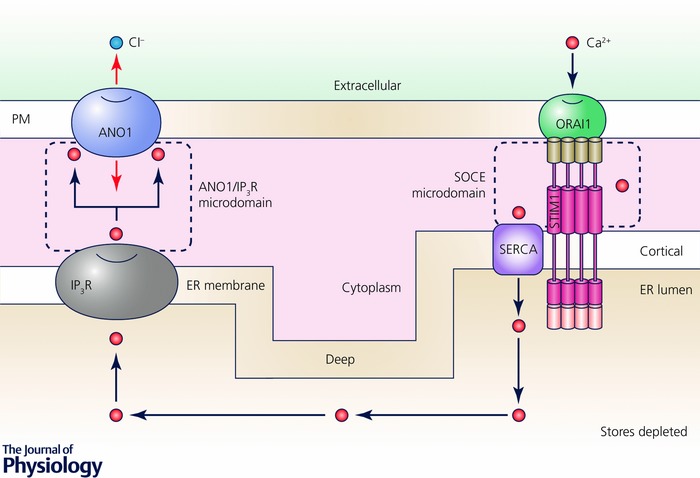
Cartoon depicting the mechanism of Ca2+ tunnelling downstream of Ca2+ store depletion and SOCE activation. Two distinct separate microdomains are at play. The SOCE microdomain, defined by STIM1‐Orai1 interactions, mediates Ca2+ flow from the extracellular space into the narrow ER–PM junction where it is taken up into the ER through the action of SERCA. The second domain is defined by IP3R release sites that localize close to CaCCs to selectively activate them. Ca2+ flows from the extracellular space through Orai1 resulting in a localized Ca2+ microdomain that is spatially limited due to rapid uptake of Ca2+ by SERCA into the ER lumen. With open IP3Rs, Ca2+ flowing into the ER leaks out through IP3R to activate CaCC.
Careful co‐localization experiments in the oocyte confirm this model and show that Orai1, STIM1 and SERCA localize to the SOCE clusters at ER–PM junctions, thereby creating a specialized Ca2+ handling domain that favours Ca2+ influx into the cytoplasm through Orai1 and uptake into the ER through SERCA (Courjaret & Machaca, 2014). Importantly, in the case of CaCC as a downstream Ca2+ effector, store depletion is associated with a dramatic remodelling of the Ca2+ signalling machinery. STIM1, Orai1 and SERCA localize to SOCE clusters at ER–PM junctions. In contrast, Ano1 is excluded from these junctions and localizes to other areas of the plasma membrane (Fig. 6). At rest, Ano1 is evenly distributed throughout the cell membrane of the oocyte, including the dense brush of microvilli where it serves an additional scaffolding function and regulates microvilli length (Courjaret et al. 2016b). Store depletion, while concentrating STIM1, Orai1 and to a lesser extend SERCA into the SOCE clusters, excludes Ano1 resulting in the patchy separation illustrated in Fig. 6. In other words, there is little to no co‐localization of the Ca2+ entry source (SOCE) and of the Ca2+ effector (CaCC), and the distance to be covered by Ca2+ ions in the cytoplasm from the mouth of the Orai channel to the CaCC is incompatible with a diffusion mechanism given the speed of activation of CaCCs and the measured size and distribution of SOCE puncta as compared to CaCC‐rich membrane domains (Fig. 6). To overcome the diffusion barrier and reach the CaCCs, Ca2+ ions transit through the ER and are released at the target spot by IP3R. This spatial reorganization of the Ca2+ signalling machinery mediating SOCE and Ca2+ release results in the delivery of Ca2+ flowing into the cell through SOCE to a distal effector, CaCC, without inducing a global Ca2+ rise or having to contend with the limiting cytoplasmic Ca2+ diffusion. This signalling module allows for the transport of information carried by Ca2+ influx across distances that exceed the SOCE microdomain and are in the micrometre range or ‘mid‐range’ between elementary and global signals.
Figure 6. Spatial reorganization of the Ca2+ signalling machinery and effectors in response to Ca2+ store depletion.
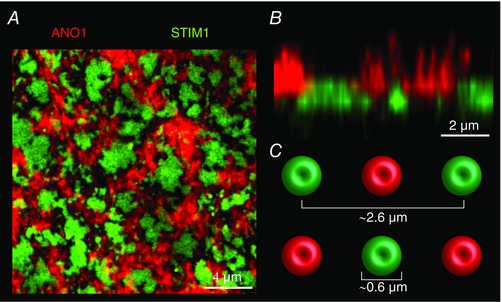
A, confocal imaging of Xenopus Ano1 tagged with mCherry (red) and of the ER Ca2+ sensor STIM1 tagged with green fluorescent protein (green) after store depletion induced by IP3 injection. The example is an extreme situation where STIM1 forms large fused clusters that exclude the CaCC Ano1. B, orthogonal reconstruction of a section of the oocyte in A. The microvilli covered by Ano1 are clearly visible as well as the separation between the SOCE domains and the domains painted by Ano1. C, dimensions and spatial spread of the SOCE clusters as measured in Courjaret and Machaca (2014).
Therefore, with SERCA active, the cytoplasmic Ca2+ transient within SOCE puncta at ER–PM junctions is transient and very localized, with the majority of the Ca2+ flux going into the ER and then leaking out at distal sites through IP3Rs to activate effectors with high specificity and efficiency. This is consistent with the small flux through Orai1 channels. Localization of SERCA to the STIM/Orai cluster is not restricted to Xenopus oocytes and has been reported in other cell types (Jousset et al. 2007; Sampieri et al. 2009; Alonso et al. 2012; Hogan, 2015). Therefore, following store depletion and the activation of SOCE, a pump–leak balance develops at the ER membrane with a point source pump pathway mediated by Orai1–STIM1–SERCA that is physically localized at SOCE puncta, and a leak pathway through open IP3Rs at distal sites to activate effectors such as CaCC (Fig. 5). We have previously proposed the term ‘Ca2+ teleporting’ to borrow an analogy from science fiction (Fort, 1931), to suggest rapid transport of Ca2+ through the ER lumen given the fact that ER stores are never fully depleted of Ca2+ (Courjaret et al. 2016a). Although a single Ca2+ ion is obviously not instantaneously traversing that distance through the ER lumen, the term teleporting nicely reflects the transfer of Ca2+ from the SOCE entry sites to CaCCs in a directed fashion to modulate CaCC current. In fact a direct physical interaction has been reported between the IP3receptor and Ano1 in neurons (Jin et al. 2013; Jin et al. 2014); whether a similar interaction exists in the oocyte remains unknown.
In addition to modulating the spatial aspects of Ca2+ signals and effector activation, Ca2+ tunnelling also modulates the temporal aspects of Ca2+ signals by favouring tonic over oscillatory Ca2+ signalling (Courjaret et al. 2016a). When Ca2+ stores are relatively full, IP3 production favours Ca2+ oscillations resulting in repetitive transient Ca2+ signals. In contrast, when SOCE is fully activated with depleted Ca2+ store, Ca2+ tunnelling mediates pump–leak balance at the ER membrane that favours tonic sustained Ca2+ signalling but inhibiting Ca2+ oscillations (Courjaret et al. 2016a). In this case Ca2+ tunnelling through the ER lumen targets the IP3 receptor itself and modulates its properties to favour tonic rather than oscillatory signalling. Therefore, Ca2+ tunnelling not only modulates the spatial aspects of Ca2+ signalling, but also affects the temporal features of Ca2+ signals in the same cell. This has significant implications for encoding specific cellular responses downstream of SOCE using Ca2+ tunnelling.
Is Ca2+ tunnelling active in other cells?
The Ca2+ tunnelling system is clearly functional in pancreatic acinar cells and in the frog oocyte. Both of these cell types are highly specialized for a specific function, secretion in the acinar cell and fertilization and support of early development in the oocyte. Therefore, the question arises as to whether this Ca2+ signalling mode is ubiquitous or unique to some highly specialized cell types. We currently do not know the answer to this question, but several arguments support the conclusion that this Ca2+ signalling modality is widespread. First, SOCE is ubiquitous in non‐excitable cells and present in excitable cells as well. SOCE is physiologically linked to IP3 receptor activation downstream of agonist stimulation. Therefore, the entire machinery supporting Ca2+ tunnelling is present. The spatial remodelling of the Ca2+ signalling machinery in response to store depletion outlined in the oocyte system translates and has been described in other cells as well. Furthermore, the IP3R has been shown to directly link to CaCC, a defined effector for Ca2+ tunnelling. Conceptually, the Ca2+ tunnelling mechanism is quite attractive as it allows for specific signalling to effectors through the SOCE pathway without inducing a global Ca2+ rise in the cytosol and without the need to localize multiple effectors into the physically limited space defined by ER–PM junctions where the SOCE machinery localizes.
In principle, Ca2+ tunnelling through the ER should be present in all cell types as it would seem unlikely that SERCA pumps and Ca2+ release channels should be exactly co‐localized. Therefore Ca2+ would always tunnel a bit between Ca2+ uptake and release sites. The length of the effective tunnel would vary between cell types depending on their function. The effectiveness and speed of Ca2+ tunnelling would depend critically on the concentrations of Ca2+ buffers in the ER lumen, their mobility as well as their binding and dissociation rate constants. In addition, it would depend on the degree of depletion of ER Ca2+ stores. In the pancreatic acinar cells, it has been shown directly that there are only minor reductions in [Ca2+]ER during physiological stimulation (Park et al. 2000). In general, it is unlikely that there would be a need for complete depletion of ER Ca2+ stores before SOCE is activated. This is indeed the case in the frog oocyte, where IP3‐dependent release of Ca2+ from the ER fully activates SOCE without emptying the stores completely (Courjaret et al. 2016a).
In the ER lumen of the pancreatic acinar cells, the movement of Ca2+ immediately after localized uncaging of caged Ca2+ (following maximal ACh‐induced Ca2+ release) has been directly monitored. The rate of rise of [Ca2+]ER decreases, as expected for a diffusional process, with increasing distance from the uncaging site (Park et al. 2000). At a distance of 10 μm from the uncaging site, the peak [Ca2+]ER occurs ∼2.5 s later than at the uncaging site itself. Complete re‐equilibration of [Ca2+] in the whole of the ER is attained 6–8 s after the uncaging event (Park et al. 2000). These data underestimate the speed of Ca2+ movement in the ER lumen under physiological conditions, because of the necessity of first having to evoke maximal release of Ca2+ from the ER in order to obtain a clear local increase in [Ca2+]ER upon Ca2+ uncaging. The free buffer concentration in these experiments (Park et al. 2000) would therefore have been higher than under more physiological conditions, where many of the buffers would already have been saturated with Ca2+.
One can readily postulate a long list of potential effectors that could be targeted by Ca2+ tunnelling with the most obvious being Ca2+‐regulated ion channels located at the plasma membrane such as CaCCs, Ca2+‐activated K+ channels (Liu et al. 1998), other integral membrane proteins such as adenylate cyclases (Halls & Cooper, 2011), and Ca2+ sensitive enzymes anchored at the plasma membrane through A‐kinase anchor proteins such as protein kinase C and phosphatase 2B (Esseltine & Scott, 2013). Ca2+ tunnelling effectors are likely to localize in the immediate vicinity of the release site, the IP3R, and this can include virtually all the downstream effectors of the IP3R that have been recently reviewed (Prole & Taylor, 2016). In the cytosol, organelles can also be a target for Ca2+ tunnelling, including lysosomes, nuclei, vesicles and mitochondria that can all localize next to IP3Rs. Mitochondria are of particular interest given their intimate interaction with SOCE and the localization of IP3R to ER‐mitochondria junctions (Parekh, 2003).
Currently there are few validated targets of Ca2+ tunnelling including CaCCs, Ca2+‐activated K+ channels, secretion in acinar cells, and the IP3R itself where we have shown that Ca2+ tunnelling can modulate IP3R activity switching it from a mode that favours Ca2+ oscillations to one that favours tonic Ca2+ signals (Courjaret et al. 2016a). There are also hints in the literature of potential additional effectors of Ca2+ tunnelling. In a human salivary gland cell line, the direct activation of the Ca2+‐activated K+ channel by SOCE is limited by the fast buffering of Ca2+ below the plasma membrane and can be restored when the ER Ca2+ pump is inhibited by thapsigargin. When SOCE and IP3 receptors are simultaneously activated (by stimulating muscarinic receptors with carbachol), Ca2+‐sensitive K+ channels are strongly activated, supporting the idea that SOCE ‘fuels’ the IP3 receptors when the stores are empty to provide an efficient activation of the K+ channel (Liu et al. 1998).
Conclusion
Herein we focus on findings from two distinctive specialized cell types, the pancreatic acinar cell and the frog oocyte, that led to proposing a novel model of Ca2+ signalling that we refer to as Ca2+ tunnelling. In pancreatic acinar cells, Ca2+ tunnelling allows the transport of Ca2+ flowing from the basolateral membrane to support transepithelial fluid transport and secretion of digestive enzymes. The tunnelling of Ca2+ through the ER lumen circumvents the slow diffusion of Ca2+ through the highly buffered cytosol and importantly delivers Ca2+ to effectors in the apical membrane without inducing a global [Ca2+]i rise, which would undoubtedly activate multiple other Ca2+‐dependent processes. In oocytes, Ca2+ tunnelling specifically and efficiently activates CaCCs downstream of SOCE without inducing a global Ca2+ rise. This activation occurs spatially in the mid‐range broader than the Ca2+ microdomain but more contained than a global [Ca2+]i rise. This again eludes the need for Ca2+ to diffuse long distances in the highly buffered cytosol and avoids a global [Ca2+]i rise while allowing the activation of a specific effector, CaCC, downstream of SOCE. In addition, Ca2+ tunnelling in the oocyte modulates the spatial features of Ca2+ signals favouring a tonic signal while inhibiting Ca2+ oscillations by acting on the IP3R itself, in this case as a downstream effector.
Of note is the mechanism underlying Ca2+ tunnelling with SOCE forming the Ca2+ entry pathway that fuels the whole process. Ca2+ entering the cell within the SOCE microdomain is unlikely to diffuse beyond the microdomain due both to the cytoplasmic Ca2+ buffering and also to the rapid uptake into the ER lumen through the action of SERCA. This is somewhat reminiscent of the capacitative Ca2+ entry model originally proposed by Jim Putney (Putney, 1986), where it was postulated that Ca2+ enters the cell directly from the extracellular space into the ER lumen. Although it is now clear that this is not the case, the limited diffusion of Ca2+ beyond the SOCE microdomain and the rapid uptake of Ca2+ flowing through SOCE into the ER lumen argue that a significant proportion of the signalling downstream of SOCE occurs through Ca2+ tunnelling.
Interestingly, the molecular mechanisms underlying Ca2+ tunnelling in acinar cells and oocytes are analogous. The machinery mediating Ca2+ tunnelling encompasses STIM1 and Orai1 (SOCE), the SERCA pump and the IP3R. Store depletion stabilizes the STIM1–Orai1 puncta at ER–PM junctions thus providing the source for Ca2+ entry from the extracellular space. Ca2+ flowing through SOCE channels is taken up by the SERCA pump into the ER lumen preventing its diffusion out of the SOCE microdomain. In turn ER Ca2+ is released through IP3Rs thus delivering it to the appropriate effectors (secretion, CaCC, IP3R) with high efficiency and specificity. The Ca2+ tunnelling machinery has been adapted to very different cell physiological needs in the oocyte as compared to the pancreatic acinar cell. In the oocyte it modulates resting membrane potential and the temporal features of Ca2+ signals, whereas in the acinar cell it drives enzyme secretion and fluid flow. Given that the molecular machinery underlying Ca2+ tunnelling is ubiquitous, it is likely that this pathway is involved in Ca2+ signalling in a plethora of other physiological functions. The remarkable functional link between SOCE, SERCA and IP3R conscripted to allow Ca2+ tunnelling results in the delivery of Ca2+ to effectors that could easily be missed experimentally and interpreted as signalling downstream of SOCE directly. It is therefore likely that Ca2+ tunnelling activates additional cell physiological events that remain to be defined.
Additional information
Competing interests
None declared.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
O.H.P. is a Medical Research Council Professor (G19/22/2).
Biographies
Ole Petersen CBE FRS is Medical Research Council Professor in the School of Biosciences at Cardiff University. He pioneered patch clamp single‐channel and whole‐cell recordings in epithelial cells and discovered messenger‐mediated calcium release from the nuclear envelope as well as calcium tunnelling through the endoplasmic reticulum. More recently, he has shown how inhibition of store‐operated calcium channels could be used to treat acute pancreatitis.

Khaled Machaca is Professor of Physiology and Biophysics at Weill Cornell Medicine and the Associate Dean for Research for the Qatar campus. He is interested in calcium signalling under physiological and pathological conditions. He has comprehensively documented the remodelling of the Ca2+ signalling machinery during oocyte maturation and made significant contributions to the regulation of store‐operated Ca2+ entry during the cell cycle, Orai1 trafficking and to the mechanisms governing oocyte meiotic arrest.
Contributor Information
Ole H Petersen, Email: PetersenOH@cardiff.ac.uk.
Khaled Machaca, Email: khm2002@qatar-med.cornell.edu.
References
- Allbritton NL, Meyer T & Stryer L (1992). Range of messenger action of calcium ion and inositol 1,4,5‐trisphosphate. Science 258, 1812–1815. [DOI] [PubMed] [Google Scholar]
- Alonso MT, Manjarres IM & Garcia‐Sancho J (2012). Privileged coupling between Ca2+ entry through plasma membrane store‐operated Ca2+ channels and the endoplasmic reticulum Ca2+ pump. Mol Cell Endocrinol 353, 37–44. [DOI] [PubMed] [Google Scholar]
- Ashby MC, Camello‐Almaraz C, Gerasimenko OV, Petersen OH & Tepikin AV (2003). Long distance communication between muscarinic receptors and Ca2+ release channels revealed by carbachol uncaging in cell‐attached patch pipette. J Biol Chem 278, 20860–20864. [DOI] [PubMed] [Google Scholar]
- Baker PF & Crawford AC (1972). Mobility and transport of magnesium in squid giant axons. J Physiol 227, 855–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belan PV, Gerasimenko OV, Tepikin AV & Petersen OH (1996). Localization of Ca2+ extrusion sites in pancreatic acinar cells. J Biol Chem 271, 7615–7619. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1997). Elementary and global aspects of calcium signalling. J Physiol 499, 291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (2016). The inositol trisphosphate/calcium signalling pathway in health and disease. Physiol Rev 96, 1261–1296. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD & Roderick HL (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4, 517–529. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P & Bootman MD (2000). The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ & Roderick HL (2002). Calcium signalling: more messengers, more channels, more complexity. Curr Biol 12, R563–R565. [DOI] [PubMed] [Google Scholar]
- Bugrim A, Fontanilla R, Eutenier BB, Keizer J & Nuccitelli R (2003). Sperm initiate a Ca2+ wave in frog eggs that is more similar to Ca2+ waves initiated by IP3 than by Ca2+ . Biophys J 84, 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callamaras N & Parker I (2000). Ca2+‐dependent activation of Cl– currents in Xenopus oocytes is modulated by voltage. Am J Physiol Cell Physiol 278, C667–C675. [DOI] [PubMed] [Google Scholar]
- Cancela JM, Gerasimenko OV, Gerasimenko JV, Tepikin AV & Petersen OH (2000). Two different but converging messenger pathways to intracellular Ca2+ release: the roles of nicotinic acid adenine dinucleotide phosphate, cyclic ADP‐ribose and inositol trisphosphate. EMBO J 19, 2549–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YM, Kim SH, Chung S, Uhm DY & Park MK (2006). Regional interaction of endoplasmic reticulum Ca2+ signals between soma and dendrites through rapid luminal Ca2+ diffusion. J Neurosci 26, 12127–12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courjaret R, Dib M & Machaca K (2016a). Store‐operated Ca2+ entry in oocytes modulate the dynamics of IP3‐dependent Ca2+ release from oscillatory to tonic. J Cell Physiol 232, 1095–1103. [DOI] [PubMed] [Google Scholar]
- Courjaret R, Hodeify R, Hubrack S, Ibrahim A, Dib M, Daas S & Machaca K (2016b). The Ca2+‐activated Cl– channel Ano1 controls microvilli length and membrane surface area in the oocyte. J Cell Sci 129, 2548–2558. [DOI] [PubMed] [Google Scholar]
- Courjaret R, Hubrack S, Daalis A, Dib M & Machaca K (2013). The Xenopus TRPV6 homolog encodes a Mg2+ ‐permeant channel that is inhibited by interaction with TRPC1. J Cell Physiol 228, 2386–2398. [DOI] [PubMed] [Google Scholar]
- Courjaret R & Machaca K (2014). Mid‐range Ca2+ signalling mediated by functional coupling between store‐operated Ca2+ entry and IP3‐dependent Ca2+ release. Nat Commun 5, 3916. [DOI] [PubMed] [Google Scholar]
- Courjaret R & Machaca K (2016). Xenopus oocyte as a model system to study store‐operated Ca2+ entry (SOCE). Front Cell Dev Biol 4, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I & Rizzuto R (2011). A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Rizzuto R & Pozzan T (2016). Enjoy the trip: calcium in the mitochondria back and forth. Annu Rev Biochem 85, 161–192. [DOI] [PubMed] [Google Scholar]
- Demuro A & Parker I (2006). Imaging single‐channel calcium microdomains. Cell Calcium 40, 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC & Healy JI (1997). Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386, 855–858. [DOI] [PubMed] [Google Scholar]
- Esseltine JL & Scott JD (2013). AKAP signalling complexes: pointing towards the next generation of therapeutic targets? Trends Pharmacol Sci 34, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S ( 2007). Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 7, 690–702. [DOI] [PubMed] [Google Scholar]
- Fort CH (1931). Lo! Cosimo Books, New York. [Google Scholar]
- Foskett JK, White C, Cheung KH & Mak DO (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Charlesworth RM, Sherwood MW, Ferdek PE, Mikoshiba K, Parrington J, Petersen OH & Gerasimenko OV (2015). Both RyRs and TPCs are required for NAADP‐induced intracellular Ca2+ release. Cell Calcium 58, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TO, Bychkova S, Peng S, Begg M, Gerasimenko OV & Petersen OH (2013). Ca2+ release‐activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci USA 110, 13186–13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko OV, Gerasimenko JV, Rizzuto RR, Treiman M, Tepikin AV & Petersen OH (2002). The distribution of the endoplasmic reticulum in living pancreatic acinar cells. Cell Calcium 32, 261–268. [DOI] [PubMed] [Google Scholar]
- Gerasimenko OV, Gerasimenko JV, Tepikin AV & Petersen OH (1995). ATP‐dependent accumulation and inositol trisphosphate‐ or cyclic ADP‐ribose‐mediated release of Ca2+ from the nuclear envelope. Cell 80, 439–444. [DOI] [PubMed] [Google Scholar]
- Goul‐Somero M & Jaffe LA (1984). Control of cell fusion at fertilization by membrane potential In Cell Fusion: Gene Transfer and Transformation, ed. Beers RF. & Bassett EG, pp. 27–38. Raven Press, New York. [Google Scholar]
- Halls ML & Cooper DM (2011). Regulation by Ca2+‐signaling pathways of adenylyl cyclases. Cold Spring Harb Perspect Biol 3, a004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer AM, Fasolato C & Pozzan T (1998). Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+]. J Cell Biol 140, 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG (2015). The STIM1‐ORAI1 microdomain. Cell Calcium 58, 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M & Penner R (1992). Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356. [DOI] [PubMed] [Google Scholar]
- Hou X, Pedi L, Diver MM & Long SB (2012). Crystal structure of the calcium release‐activated calcium channel Orai. Science 338, 1308–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe LA, Cross NL & Picheral B (1983). Studies of the voltage‐dependent polyspermy block using cross‐species fertilization of amphibians. Dev Biol 98, 319–326. [DOI] [PubMed] [Google Scholar]
- Jin X, Shah S, Du X, Zhang H & Gamper N (2014). Activation of Ca2+‐activated Cl– channel ANO1 by localized Ca2+ signals. J Physiol 594, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Shah S, Liu Y, Zhang H, Lees M, Fu Z, Lippiat JD, Beech DJ, Sivaprasadarao A, Baldwin SA & Gamper N (2013). Activation of the Cl– channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci Signal 6, ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jousset H, Frieden M & Demaurex N (2007). STIM1 knockdown reveals that store‐operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J Biol Chem 282, 11456–11464. [DOI] [PubMed] [Google Scholar]
- Kar P, Mirams GR, Christian HC & Parekh AB (2016). Control of NFAT isoform activation and NFAT‐dependent gene expression through two coincident and spatially segregated intracellular Ca2+ signals. Mol Cell 64, 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H & Augustine GJ (1990). Cytosolic Ca2+ gradients triggering unidirectional fluid secretion from exocrine pancreas. Nature 348, 735–738. [DOI] [PubMed] [Google Scholar]
- Kasai H, Li YX & Miyashita Y (1993). Subcellular distribution of Ca2+ release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell 74, 669–677. [DOI] [PubMed] [Google Scholar]
- Kasai H & Petersen OH (1994). Spatial dynamics of second messengers: IP3 and cAMP as long-range and associative messengers. Trends Neurosci 17, 95–101. [DOI] [PubMed] [Google Scholar]
- Kuruma A & Hartzell HC (1998). Dynamics of calcium regulation of Cl currents in Xenopus oocytes. Am J Physiol Cell Physiol 276, C161–C175. [DOI] [PubMed] [Google Scholar]
- Kuruma A & Hartzell HC (2000). Bimodal control of a Ca2+‐activated Cl– channel by different Ca2+ signals. J Gen Physiol 115, 59–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechleiter JD & Clapham DE (1992). Spiral waves and intracellular calcium signalling. J Physiol Paris 86, 123–128. [DOI] [PubMed] [Google Scholar]
- Lee MG, Ohana E, Park HW, Yang D & Muallem S (2012). Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol Rev 92, 39–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K‐H, Taschenberger H & Neher E (2017). Dynamics of volume‐averaged intracellular Ca2+ in a rat CNS nerve terminal during single and repetitive voltage‐clamp depolarizations. J Physiol 595, 3219–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T & Meyer T (2007). Live‐cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA 104, 9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr & Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+‐store‐depletion‐triggered Ca2+ influx. Curr Biol 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Rojas E & Ambudkar IS (1998). Regulation of KCa current by store‐operated Ca2+ influx depends on internal Ca2+ release in HSG cells. Am J Physiol Cell Physiol 275, C571–C580. [DOI] [PubMed] [Google Scholar]
- Luik RM, Wu MM, Buchanan J & Lewis RS (2006). The elementary unit of store‐operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER‐plasma membrane junctions. J Cell Biol 174, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, Burgoyne RD & Tepikin AV (2009). Ribosome‐free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP3 receptors. Curr Biol 19, 1648–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaca K (2007). Ca2+ signaling differentiation during oocyte maturation. J Cell Physiol 213 331–340. [DOI] [PubMed] [Google Scholar]
- Machaca K & Hartzell HC (1998). Asymmetrical distribution of Ca‐activated Cl channels in Xenopus oocytes. Biophys J 74, 1286–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaca K & Hartzell HC (1999). Reversible Ca gradients between the sub‐plasmalemma and cytosol differentially activate Ca‐dependent Cl currents. J Gen Physiol 113, 249–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaca K & Haun S (2002). Induction of maturation‐promoting factor during Xenopus oocyte maturation uncouples Ca2+ store depletion from store‐operated Ca2+ entry. J Cell Biol 156, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaca K, Qu Z, Kuruma A, Hartzell HC & McCarty N (2001). The endogenous calcium‐activated Cl channel in Xenopus oocytes: a physiologically and biophysically rich model system In Calcium Activates Chloride Channels, ed. Fuller CM, pp. 3–39. Academic Press, San Diego. [Google Scholar]
- Maruyama Y & Petersen OH (1994). Delay in granular fusion evoked by repetitive cytosolic Ca2+ spikes in mouse pancreatic acinar cells. Cell Calcium 16, 419–430. [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Petersen OH, Flanagan P & Pearson GT (1983). Quantification of Ca2+‐activated K+ channels under hormonal control in pig pancreas acinar cells. Nature 305, 228–232. [DOI] [PubMed] [Google Scholar]
- Matthews EK, Petersen OH & Williams JA (1973). Pancreatic acinar cells: acetylcholine‐induced membrane depolarization, calcium efflux and amylase release. J Physiol 234, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami H, Gardner J, Gerasimenko OV, Camello P, Petersen OH & Tepikin AV (1999). Calcium binding capacity of the cytosol and endoplasmic reticulum of mouse pancreatic acinar cells. J Physiol 518, 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami H, Nakano K, Tepikin AV & Petersen OH (1997). Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell 88, 49–55. [DOI] [PubMed] [Google Scholar]
- Morgan AJ & Jacob R (1994). Ionomycin enhances Ca influx by stimulating store‐regulated cation entry and not by direct action at the plasma membrane. Biochem J 300, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (1998). Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20, 389–399. [DOI] [PubMed] [Google Scholar]
- Nielsen SP & Petersen OH (1972). Transport of calcium in the perfused submandibular gland of the cat. J Physiol 223, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipchuk YV, Wakui M, Yule DI, Gallacher DV & Petersen OH (1990). Cytoplasmic Ca2+ oscillations evoked by receptor stimulation, G‐protein activation, internal application of inositol trisphosphate or Ca2+: simultaneous microfluorimetry and Ca2+ dependent Cl– current recording in single pancreatic acinar cells. EMBO J 9, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB (2003). Store‐operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J Physiol 547, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB (2008). Ca2+ microdomains near plasma membrane Ca2+ channels: impact on cell function. J Physiol 586, 3043–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB (2010). Store‐operated CRAC channels: function in health and disease. Nat Rev Drug Discov 9, 399–410. [DOI] [PubMed] [Google Scholar]
- Park MK, Ashby MC, Erdemli G, Petersen OH & Tepikin AV (2001a). Perinuclear, perigranular and sub‐plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J 20, 1863–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Lomax RB, Tepikin AV & Petersen OH (2001b). Local uncaging of caged Ca2+ reveals distribution of Ca2+‐activated Cl– channels in pancreatic acinar cells. Proc Natl Acad Sci USA 98, 10948–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Petersen OH & Tepikin AV (2000). The endoplasmic reticulum as one continuous Ca2+ pool: visualization of rapid Ca2+ movements and equilibration. EMBO J 19, 5729–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CCH, Toescu EC & Petersen OH (1991). Different patterns of receptor‐activated cytoplasmic Ca2+ oscillations in single pancreatic acinar cells: dependence on receptor type, agonist concentration and intracellular Ca2+ buffering. EMBO J 10, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH (1992). Stimulus‐secretion coupling: cytoplasmic calcium signals and the control of ion channels in exocrine acinar cells. J Physio l 448, 1–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH, Findlay I, Iwatsuki N, Singh J, Gallacher DV, Fuller CM, Pearson GT, Dunne MJ & Morris AP (1985). Human pancreatic acinar cells: studies of stimulus‐secretion coupling. Gastroenterology 89, 109–117. [DOI] [PubMed] [Google Scholar]
- Petersen OH & Maruyama Y (1984). Calcium‐activated potassium channels and their role in secretion. Nature 307, 693–696. [DOI] [PubMed] [Google Scholar]
- Petersen OH, Petersen CC & Kasai H (1994). Calcium and hormone action. Annu Rev Physiol 56, 297–319. [DOI] [PubMed] [Google Scholar]
- Petersen OH & Tepikin AV (2008). Polarized calcium signalling in exocrine gland cells. Annu Rev Physiol 70, 273–299. [DOI] [PubMed] [Google Scholar]
- Petersen OH, Tepikin A & Park MK (2001). The endoplasmic reticulum: one continuous or several separate Ca2+ stores? Trends Neurosci 24, 271–276. [DOI] [PubMed] [Google Scholar]
- Petersen OH & Ueda N (1976). Pancreatic acinar cells: the role of calcium in stimulus‐secretion coupling. J Physiol 254, 583–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH & Verkhratsky A (2016). Calcium and ATP control multiple vital functions. Philos Trans R Soc Lond B Biol Sci 371, 20150418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A & Hogan PG (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. [DOI] [PubMed] [Google Scholar]
- Prakriya M & Lewis RS (2015). Store‐operated calcium channels. Physiol Rev 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prole DL & Taylor CW (2016). Inositol 1,4,5‐trisphosphate receptors and their protein partners as signalling hubs. J Physiol 594, 2849–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW (1986). A model for receptor‐activated calcium entry. Cell Calcium 7, 1–12 [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C & Hogan PG (1997). Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15, 707–747. [DOI] [PubMed] [Google Scholar]
- Rizzuto R & Pozzan T (2006). Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86, 369–408. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G & Stauderman KA (2005). STIM1, an essential and conserved component of store‐operated Ca2+ channel function. J Cell Biol 169, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampieri A, Zepeda A, Asanov A & Vaca L (2009). Visualizing the store‐operated channel complex assembly in real time: identification of SERCA2 as a new member. Cell Calcium 45, 439–446. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Cheng T, Jan YN & Jan LY (2008). Expression cloning of TMEM16A as a calcium‐activated chloride channel subunit. Cell 134, 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scratcherd T, Hutson D & Case RM (1981). Ionic transport mechanisms underlying fluid secretion by the pancreas. Philos Trans R Soc Lond B Biol Sci 296, 167–178. [DOI] [PubMed] [Google Scholar]
- Shuai J & Parker I (2005). Optical single‐channel recording by imaging Ca2+ flux through individual ion channels: theoretical considerations and limits to resolution. Cell Calcium 37, 283–299. [DOI] [PubMed] [Google Scholar]
- Simon SM & Llinas RR (1985). Compartmentalization of the submembrane calcium activity during calcium influx and its significance in transmitter release. Biophys J 48, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Li GY, Plevin MJ, Ames JB & Ikura M (2006). Stored Ca2+ depletion‐induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF‐SAM region: An initiation mechanism for capacitive Ca2+ entry. J Biol Chem 281, 35855–35862. [DOI] [PubMed] [Google Scholar]
- Südhof TC ( 2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XP, Callamaras N, Marchant JS & Parker I (1998). A continuum of InsP3‐mediated elementary Ca2+ signalling events in Xenopus oocytes. J Physiol 509, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepikin AV, Voronina SG, Gallacher DV & Petersen OH (1992). Pulsatile Ca2+ extrusion from single pancreatic acinar cells during receptor‐activated cytosolic Ca2+ spiking. J Biol Chem 267, 14073–14076. [PubMed] [Google Scholar]
- Thastrup O, Dawson AP, Scharff O, Foder B, Cullen PJ, Drobak BK, Bjerrum PJ, Christensen SB & Hanley MR (1989). Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions 27, 17–23. [DOI] [PubMed] [Google Scholar]
- Thorn P, Lawrie AM, Smith PM, Gallacher DV & Petersen OH (1993). Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell 74, 661–668. [DOI] [PubMed] [Google Scholar]
- Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV & Petersen OH (1999). Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate‐evoked local cytosolic Ca2+ signals. EMBO J 18, 4999–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toescu EC & Petersen OH (1995). Region‐specific activity of the plasma membrane Ca2+ pump and delayed activation of Ca2+ entry characterize the polarized, agonist‐evoked Ca2+ signals in exocrine cells. J Biol Chem 270, 8528–8535. [DOI] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan‐Huberson M, Kraft S, Turner H, Fleig A, Penner R & Kinet JP (2006). CRACM1 is a plasma membrane protein essential for store‐operated Ca2+ entry. Science 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakui M, Osipchuk YV & Petersen OH (1990). Receptor‐activated cytoplasmic Ca2+ spiking mediated by inositol trisphosphate is due to Ca2+‐induced Ca2+ release. Cell 63, 1025–1032. [DOI] [PubMed] [Google Scholar]
- Wu MM, Buchanan J, Luik RM & Lewis RS (2006). Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 174, 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Thomas JM, Churchill GC, Garnham C, Lewis AM, Cancela JM, Patel S & Galione A (2005). Role of NAADP and cADPR in the induction and maintenance of agonist‐evoked Ca2+ spiking in mouse pancreatic acinar cells. Curr Biol 15, 874–878. [DOI] [PubMed] [Google Scholar]
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK & Oh U (2008). TMEM16A confers receptor‐activated calcium‐dependent chloride conductance. Nature 455, 1210–1215. [DOI] [PubMed] [Google Scholar]
- Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O & Cahalan MD (2006). Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Chung I, Liu Z, Goldin AL & Dong K (2004). A voltage‐gated calcium‐selective channel encoded by a sodium channel‐like gene. Neuron 42, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]