

Abstract
Dental enamel is one of the most remarkable examples of matrix‐mediated biomineralization. Enamel crystals form de novo in a rich extracellular environment in a stage‐dependent manner producing complex microstructural patterns that are visually stunning. This process is orchestrated by specialized epithelial cells known as ameloblasts which themselves undergo striking morphological changes, switching function from a secretory role to a cell primarily engaged in ionic transport. Ameloblasts are supported by a host of cell types which combined represent the enamel organ. Fully mineralized enamel is the hardest tissue found in vertebrates owing its properties partly to the unique mixture of ionic species represented and their highly organized assembly in the crystal lattice. Among the main elements found in enamel, Ca2+ is the most abundant ion, yet how ameloblasts modulate Ca2+ dynamics remains poorly known. This review describes previously proposed models for passive and active Ca2+ transport, the intracellular Ca2+ buffering systems expressed in ameloblasts and provides an up‐dated view of current models concerning Ca2+ influx and extrusion mechanisms, where most of the recent advances have been made. We also advance a new model for Ca2+ transport by the enamel organ.
Keywords: calcium, calcium channel, calcium regulation, calcium signalling, calcium transport
Abbreviations
- AMBN
ameloblastin
- AMEL
amelogenin
- 2‐APB
2‐aminoethyldiphenyl borinate
- CAD
channel activation domain
- CaBP
Ca2+ binding protein
- CC
coil‐coil
- CCb9
CC fragment b9
- CNX
calnexin
- CRAC
Ca2+ release activated Ca2+
- EMPs
enamel matrix proteins
- ENAM
enamelin
- ER
endoplasmic reticulum
- GBHA
glyoxal bis(2‐hydroxyanil)
- HRP
horseradish peroxidase
- IP3
inositol‐1,4,5‐trisphosphate
- IP3Rs
inositol‐1,4,5‐trisphosphate receptors
- KLK4
kallikrein 4
- MMP20
matrix metalloprotease 20
- NCKX
K+‐dependent Na+/Ca2+ exchanger
- NCX
K+‐independent Na+/Ca2+ exchanger
- NFAT
nuclear factor of activated T cells
- PLC
phospholipase C
- PM
plasma membrane
- PMCA
plasma membrane Ca2+‐ATPase
- PtdIns(4,5)P2 or PIP2
phosphatidylinositol 4,5‐bisphosphate
- RA
ruffled‐ended ameloblast
- RyR
ryanodine receptor
- SA
smooth‐ended ameloblast
- SOAR
STIM‐ORAI activating region
- SOCE
store‐operated Ca2+ entry
- SAM
sterile alpha motif
- SERCA
sarco/endoplasmic reticulum ATPase
- SR
sarcoplasmic reticulum
- STIM
stromal interaction molecule
Introduction
It would be intuitive to think that the role of Ca2+ in mineralization would be best understood by examining the most highly mineralized tissue of vertebrates, dental enamel. Paradoxically, the potentially versatile role of Ca2+ and its impact in the formation and mineralization of enamel is vastly underappreciated, remaining one of the main challenges in the biology of this tissue. Part of the problem is the enamel forming cells themselves, the ameloblasts, as they are non‐proliferative and difficult to isolate and culture successfully. Ameloblasts also engage in a number of morphological changes across developmental stages concomitant with changes in their genetic profile adding to the complexity of the system. Yet they remain a particularly attractive model for potentially decoding relevant Ca2+ signatures in mineralizing systems specifically, and as a unique cell Ca2+ case study.
Enamel is the white outer cover of the tooth crown. In fully mineralized enamel, Ca2+ is the main ionic species represented in the crystal structure. Thus much attention has been centred on the extracellular role of Ca2+ as a mineralizing agent. Ca2+ research in enamel biology indeed offers a wealth of possibilities to assess the versatility of this cation. This review will define the signalling role of Ca2+ in enamel providing an overview of the different models proposed for the transport of Ca2+ and how these might be redefined in light of recently reported data.
Amelogenesis
Amelogenesis is a term used to describe both the formation (volume of tissue) and mineralization of enamel. Tooth enamel is a bioceramic of complex design and a prime example of cell mediated apatitic crystal formation via matrix–crystal interactions. Mineralized enamel is four times harder than bone.
Ameloblasts derive from ectodermal epithelium and tightly control amelogenesis, a process commonly divided into two main stages termed secretory and maturation stages (Fig. 1). Secretory ameloblasts are polarized tall (∼70 μm) post‐mitotic cells with a specialized process at the distal end (see below) known as the Tomes’’ process that plays a key role in enamel matrix secretion and the formation of enamel microstructure (Boyde, 1989). Secretory ameloblasts synthesize and secrete a protein‐rich scaffold formed by a core of enamel matrix proteins (EMPs) that is subsequently removed during maturation leaving behind a highly organized biomaterial. Morphologically, secretory ameloblasts undergo a number of transformations losing the Tomes’ process and reducing their height by about 50% to become maturation stage ameloblasts. The relevance of these changes can be more clearly appreciated when the histological transformations in cell anatomy and concomitant reorganization of organelle distribution occurring throughout amelogenesis are observed (see Fig. 1). This is even more significant when one considers the full spectrum of the functions associated with each cell stage.
Figure 1. Schematic diagram of histological changes in amelogenesis.
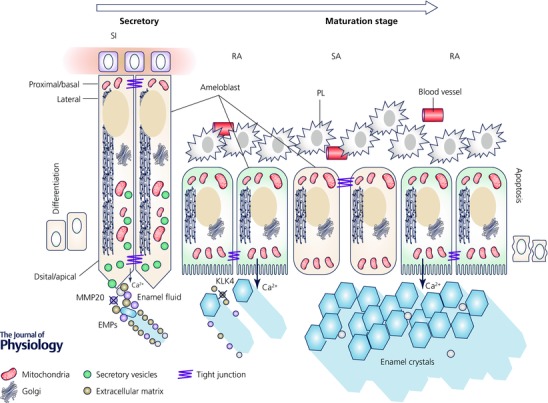
The histological development of enamel crystals goes hand in hand with changes in ameloblast morphology. Undifferentiated epithelial cells receive signals to transform into secretory ameloblast cells of some 75 μm tall and ∼5 μm in diameter with a specialized distal cell process (Tomes’ process) which plays an important role in matrix exocytosis. These same cells will retransform into shorter cells (∼35 μm tall) during maturation devoid of the Tomes’ process. In maturation stage, ameloblasts undergo cyclical changes from a cell with a distal ruffled border, the ruffled‐ameloblast (RA), to a cell with a smooth distal border, the smooth‐ameloblast (SA). Tight junctions are found at the basal and apical pole of secretory ameloblasts. The apical or distal pole is closest to the enamel crystals. In RA cells, tight junctions are found only at the apical pole but in SA cells they are located at the basal pole. Organellar distribution differs in cells at each stage (see text for details). SI = stratum intermedium, PL = papillary layer, EMPs = enamel matrix proteins. MMP20 and KLK4 are the main proteases in AMEL processing. See also organellar distribution at each stage.
During the maturation stage, the relatively long and thin crystals that had formed previously now increase to their full width and thickness, abutting each other almost entirely, a process facilitated by the increased activity of ameloblasts to transport ions and the removal of matrix (Robinson, 2014) (Fig. 1). Maturation stage ameloblasts are shorter (∼40 μm) with an enlarged nucleus more centrally placed, and intercellular spaces increase. The most characteristic change at this stage is the switch from a cell with an apical ruffled (also called striated) border (RA) that is reformed into a smooth‐ended border (SA) (Fig. 1) in a series of cycles impacting on ionic transport (Josephsen & Fejerskov, 1977; McKee et al. 1989). The reasons for this change are poorly understood.
Enamel: matrix proteins and crystal growth
Fully formed enamel is an acellular tissue containing tightly packed crystallites and is the hardest and most durable vertebrate tissue containing ∼95% mineral by weight (Smith, 1998).
Enamel matrix proteins
The composition of the proteinaceous scaffold provided by ameloblasts and its exact role in promoting crystal growth is still a subject of debate. The main protein secreted by ameloblasts (∼90%) is amelogenin (AMEL) (Termine et al. 1980), an unstable 25 kDa hydrophobic protein cleaved by the matrix metalloprotease 20 (MMP20) (Robinson et al. 1998). It is commonly considered that AMEL self‐assembles into nanospheres that adsorb to apatite inhibiting lateral crystal growth (Fincham et al. 1994; Brookes et al. 1995; Moradian‐Oldak, 2001). Alternatively, in the presence of both Ca2+ and phosphate in in vitro conditions under controlled pH, AMEL self‐assembles into structures called nanoribbons which have the capacity to align into structures some micrometres long resembling enamel crystals (Martinez‐Avila et al. 2011, 2012). Other proteins considered part of the unique ensemble of matrix proteins manufactured by ameloblasts include ameloblastin (AMBN) and enamelin (ENAM) which also show, albeit with considerable quantitative differences relative to AMEL, a pattern characterized by highest expression during the secretory stage (Smith & Nanci, 1996). Collectively, these proteins are regarded as structural proteins as they are involved at varying levels in determining the correct microstructure of enamel. In the maturation stage, the serine protease kallikrein 4 (KLK4) substitutes MMP20 for the proteolytic processing of peptides. Mutations to the genes encoding for AMELX, AMBN, ENAM, MMP20 or KLK4 result in amelogenesis imperfecta, a term used to clinically describe a broad range of abnormal enamel phenotypes (Hart et al. 2000; Gibson et al. 2001; Paine et al. 2003; Wright et al. 2003; Kim et al. 2005).
It should be highlighted that enamel formation is clearly not a process that solely depends on Ca2+ supply. Other important factors such as local extracellular pH, which is not stable throughout amelogenesis, has a major impact on crystal growth (Lacruz et al. 2010). Extracellular pH is heavily modulated by the activity of ameloblasts (Lacruz et al. 2010). Moreover, hydroxyapatite‐like crystals formed de novo in the extracellular environment require the presence of calcium's largest partner, phosphate, to be able to generate the intial stages of crystal growth. However, this review is intended to focus on aspects of Ca2+ transport and signalling in ameloblasts.
Enamel crystal formation
Thousands of individual crystals (∼50 nm in diameter) are bundled into larger structures known as prisms of some 5 μm in diameter. Enamel prisms, each formed by a single ameloblast, are considered the basic microstructural unit of enamel (Boyde, 1989) (Fig. 2). Enamel crystals are seeded within the enamel fluid during the secretory stage in close contact with the apical end of the cell (Fig. 1). Analysis of pig enamel fluid showed that it differed in its composition from serum showing lower total Ca2+ concentration ([Ca2+] was 10−3 m for serum and 10−4 m for enamel) supporting the notion that it represents a specialized micro‐compartment (Aoba & Moreno, 1987). In the secretory stage, the bulk of Ca2+ in the enamel fluid appears to be non‐ionic with as much as 85% of the total Ca2+ bound, possibly to AMEL‐derived products (Moreno & Aoba, 1987; Robinson et al. 1998). Crystal formation requires supersaturation of the enamel fluid by ions in secretory and maturation stages although the stoichiometry of the fluid and that of the minerals at each stage are different (Aoba, 1996). As discussed above, enamel is a highly mineralized biomaterial containing ‘hydroxyapatite‐like’ crystals that is best considered as a non‐stoichiometric carbonated Ca2+ hydroxyapatite also incorporating ions, such as Na+, Mg2+, Cl− and Fe3+, which compete for space in the crystal lattice and thus influence the properties of enamel (Young, 1974; Aoba, 1996). By weight, Ca2+ represents about 36% of the minerals contained in matured enamel as measured in dried samples, about twice as much as the next ion species represented (Aoba & Moreno, 1987). Much of this Ca2+ is incorporated during the maturation stage as the thin enamel crystals seeded during the preceding secretory stage now expand in width and thickness filling the spaces previously occupied by fluids and organics. Smith reports that about 86% of the Ca2+ found in enamel enters the tissue during maturation stage (Smith, 1998).
Figure 2. Electron micrograph showing enamel crystals and prisms.

Enamel crystals are needle‐like structures that elongate for hundreds of micrometres. These crystals are formed by accumulation of Ca2+ and phosphate. Thousands of these crystals are bundled forming a prism, which is the basic microstructural unit of mineralized enamel. The proper development of crystals and prisms are modulated by ameloblasts and their products. Field‐width is approximately 12 microns.
Ca2+ transport in enamel
Changes in systemic Ca2+ can directly impact on enamel. For example, enamel cells express the vitamin D receptor (VDR; Davideau et al. 1996) and in conditions of normal calcaemia, low vitamin D levels result in several abnormal dental phenotypes highlighting the role of systemic Ca2+ levels in the formation of dental enamel (Berdal et al. 1993). It also underscores its relevance for understanding Ca2+ transport by enamel epithelium.
The mechanisms involved in Ca2+ transport by enamel organ cells are far from clear (Bawden, 1989; Takano, 1995; Smith, 1998; Hubbard, 2000) and many of the methods used to report differences in Ca2+ dynamics are outdated (see below). The proposed models, however, agree that Ca2+ incorporation into enamel arises not from the underlying mesenchymal zone of the dentine, but from the enamel organ itself (Reith & Boyde, 1978). The main implication is that Ca2+ travels in the basolateral to apical direction and across the barrier formed by ameloblast cells (Fig. 1). Whether this transport occurs via an active transcellular route or passively across intercellular spaces (paracellular transit) has been the subject of a number of studies.
Because secretory and maturation stage ameloblasts are organized in cell cohorts with each cell bound to its neighbouring cell by tight junctions (Fig. 1), there are important constraints on the paracellular/intercellular or passive movement of ions to the forming enamel. Lanthanum tracer studies showed that it penetrated the proximal but not the distal intercellular junctions of secretory ameloblasts (Takano & Crenshaw, 1980), whereas injections of radiolabelled Ca2+ (45Ca) in 6‐day old rats suggested an intercellular route (Hanawa et al. 1990). Secretory stage ameloblasts stained with glyoxal bis(2‐hydroxyanil) (GBHA) did not find evidence of staining in the intercellular spaces but found strong reactions within the cells (Takano et al. 1989).
In maturation ameloblasts, tight junctions reorganize their localization. In the predominant ruffled‐ended phase, tight junctions are only found near the basolateral pole, this being reversed in the smooth‐ended phase with junctions found apically (Fig. 1) (Josephsen & Fejerskov, 1977). Regardless, neither lanthanum nor horseradish peroxidase (HRP) could penetrate across the distal junctions of ruffled ameloblasts or the proximal junctions of smooth‐ended cells (Takano & Crenshaw, 1980; Takano, 1995). This cellular reorganization consisting of ruffled‐to‐smooth waves is a system available only to ameloblast cells and seems to be advantageous to these cells. In the smooth‐ended phase, ameloblasts appear to rely on intracellular transport. Physiological limitations imposed in this system include the relatively long period of time between the two maturation stage cell morphologies, and that about 70% of all maturation ameloblasts are ruffled‐ended (Josephsen & Fejerskov, 1977; Smith, 1998). 45Ca studies suggest that Ca2+ is incorporated in bulk through the ruffled‐border stage (Takano & Crenshaw, 1980; Reith & Boyde, 1981; McKee et al. 1989). Although aptly pointed out by Smith that ameloblasts appear to optimally utilize both the gated (ruffled) and non‐gated (smooth) ionic transport system (Smith, 1998), reviewing the data discussed above and largely based on the lack of diffusion of lanthanum and HRP across intercellular junctions, Bawden, Takano and Hubbard strongly suggested that the dominant paradigm for the transport of Ca2+ was via a transcellular route (Bawden, 1989; Takano, 1995; Hubbard, 2000). Given the wider acceptance now of a dominant active Ca2+ transport system by the enamel organ, this in turn requires a number of molecular mechanisms common to most cells involving intracellular Ca2+ stores, release channels that enable moderate increases in [Ca2+]i and a Ca2+ influx system. Here we examine the available evidence.
Ca2+ release via stores
Cytoplasmic Ca2+ increase can occur either from the release of stored Ca2+ in the luminal compartments of a number of intracellular organelles, or from influx of Ca2+ from outside of the cell. While the majority of intracellular Ca2+ is stored in endoplasmic reticulum (ER; ∼1 mm; Prins & Michalak, 2011) or sarcoplasmic reticulum (SR) in muscle cells, intracellular Ca2+ is also found in mitochondria, endolysosomal compartments, Golgi apparatus and peroxisomes (Prins & Michalak, 2011).
Release of Ca2+ from the ER/SR can be controlled by Ca2+ itself, or by a group of intracellular messengers such as inositol‐1,4,5‐trisphosphate (IP3), cyclic ADP ribose (cADPR), nicotinic acid adenine dinucleotide phosphate (NAADP) and sphingosine‐1‐phosphate (S1P), which either stimulate or modulate Ca2+ release. The most common channels associated with the ER are inositol 1,4,5‐trisphosphate receptors (IP3Rs) and/or ryanodine receptors (RyRs) (Fill & Copello, 2002; Stathopulos et al. 2012).
Inositol receptors
Binding of the G‐coupled receptor on the cell membrane to its ligand induces tyrosine phosphorylation and activation of phospholipase C (PLC). PLC hydrolyses the phospholipid phosphatidylinositol 4,5‐bisphosphate (PtdIns(4,5)P 2 or PIP2), a component of cell membranes, to release soluble IP3 and diacylglycerol (DAG). Within seconds, IP3 binds to its receptor known as IP3R located on the surface of the ER increasing [Ca2+]i (Wagner & Yule, 2012). The IP3R channel consists of large proteins (300 kDa) (Hajnoczky et al. 2000; Szabadkai et al. 2006). Three predominant IP3R isoforms have been described (IP3R1, IP3R2 and IP3R3) in vertebrates with 60–80% homology in their amino acid sequences (Shah et al. 2015). IP3Rs are encoded by several genes with a number of isoforms formed through splicing (Foskett et al. 2007; Mikoshiba, 2007). A number of disease states have been described associated with the different IP3R subtypes (Mikoshiba, 2015). IP3R1 deficiency causes abnormal fertilization and severe neurological disorders (Miyazaki et al. 1992; Matsumoto et al. 1996; Foskett, 2010; Higo et al. 2010). IP3R2 overexpression results in hypertrophy of cardiac muscle (Nakayama et al. 2010) whereas IP3R2 mutations inhibit sweat secretion (Klar et al. 2014). IP3R3 is important in taste perception, hair growth and osteoclast formation.
Ryanodine receptors
RyRs are high‐conductance, cation selective, tetrameric ligand‐gated Ca2+ release channels that mediate Ca2+ release from the SR essential for muscle contraction. The three known mammalian isoforms (RyR1, RyR2 and RyR3) display a high degree of similarity in the peptide sequence and three‐dimensional structure (Baker et al. 2015). RyR1 is predominantly expressed in skeletal muscle and in cerebellar Purkinje neurons (Takeshima et al. 1989; Zorzato et al. 1990; Furuichi et al. 1994; Hertle & Yeckel, 2007). RyR2 is the most abundant isoform in the brain and is greatly expressed in cardiac muscle (Nakai et al. 1990; Otsu et al. 1990; Lai et al. 1992; Hertle & Yeckel, 2007). RyR3 was first identified in the brain and is mainly found in cortical and hippocampal regions involved in learning and memory (Hakamata et al. 1992; Futatsugi et al. 1999; Hertle & Yeckel, 2007). RyR3 is also expressed in the diaphragm (Marks et al. 1989). Several mutations in both RyR1 and RyR2 are associated with human disorders such as malignant hyperthermia (Denborough, 1998) and later central core disease, catecholaminergic polymorphic ventricular tachycardia, and arrhythmogenic right ventricular dysplasia (Lanner et al. 2010).
IP3Rs and RyRs in enamel cells
IP3Rs were first identified in dental enamel cells using radioligand binding (Hubbard, 1996). However, the cellular localization, isoforms and potential difference in expression level in secretory and maturation stage cells were not reported (Hubbard, 1996). Our group reported recently that the mouse‐derived ameloblast‐like cell line, LS8 cells (Chen et al. 1992), express IP3Rs and RyRs at the mRNA and protein level, IP3R3 and RyR2 being the predominant homologues in this cell line (Nurbaeva et al. 2015b). In primary cells, IP3R expression was detected in secretory and maturation stage rat ameloblasts at considerably higher levels than RyR expression. Immunofluorescence analysis by confocal microscopy revealed differences in cellular localization for IP3Rs with IP3R1 and IP3R3 found largely in the cytoplasm and IP3R2 was found only in the cell nuclei of both secretory and maturation enamel organ cells (Nurbaeva et al. 2015a). As RyRs are most commonly identified in excitable cells (Bennett et al. 1996), it is likely that ER Ca2+ release is mediated by IP3Rs in ameloblasts and LS8 cells but this has yet to be assessed in more detail.
Store operated Ca2+ entry
Depletion of ER Ca2+ stores activates store‐operated Ca2+ entry (SOCE) channels (Prakriya & Lewis, 2015). The best characterized SOCE channels that are functional in many cell types and organs are Ca2+ release‐activated Ca2+ (CRAC) channels, which are highly Ca2+ selective (Prakriya & Lewis, 2015). CRAC channels are composed of ORAI subunits, which belong to a small family of conserved integral plasma membrane (PM) proteins that form the pore of the CRAC channel. The best characterized family member is ORAI1, but ORAI2 and ORAI3 may also contribute to SOCE in certain tissues. CRAC channels are activated by two proteins located in the membrane of the ER, stromal interaction molecule (STIM) 1 and STIM2, which sense the ER Ca2+ concentration and bind to ORAI proteins upon Ca2+ depletion from the ER to enable sustained Ca2+ entry (Shaw et al. 2013).
ORAI and STIM proteins
STIM1 and its homologue STIM2 are single‐pass transmembrane proteins located in the ER with a luminal N‐terminus and a cytosolic C‐terminus (Shaw et al. 2013; Prakriya & Lewis, 2015). In non‐stimulated cells, the Ca2+ concentration in the ER is high (in the same range as that in the extracellular medium, ∼1 mm) and Ca2+ is bound to a canonical EF‐hand Ca2+ binding domain in the N‐terminus of STIM1 and STIM2 which is located proximal to a sterile alpha motif (SAM). The STIM C‐terminus contains three coil‐coil (CC) domains and a polybasic region (Prakriya & Lewis, 2015). The second and third CC domains form a domain variously known as CRAC channel activation domain (CAD) (Park et al. 2009), STIM‐ORAI activating region (SOAR) (Yuan et al. 2009) or CC fragment b9 (CCb9) (Kawasaki et al. 2009) that is necessary and sufficient for CRAC channel activation (Fig. 3 A). ORAI1 is a tetraspanning PM protein that assembles in a hexameric protein complex and forms the pore of the CRAC channel (Fig. 3 A) (Feske et al. 2006; Prakriya et al. 2006; Vig et al. 2006; Yeromin et al. 2006). ORAI1 contains intracellular N‐ and C‐termini through which it interacts with STIM1 and STIM2 as well as several other proteins that were shown to regulate CRAC channel function and SOCE (Prakriya & Lewis, 2015).
Figure 3. STIM1 and ORAI1 mutations affecting enamel and localization in ameloblasts.
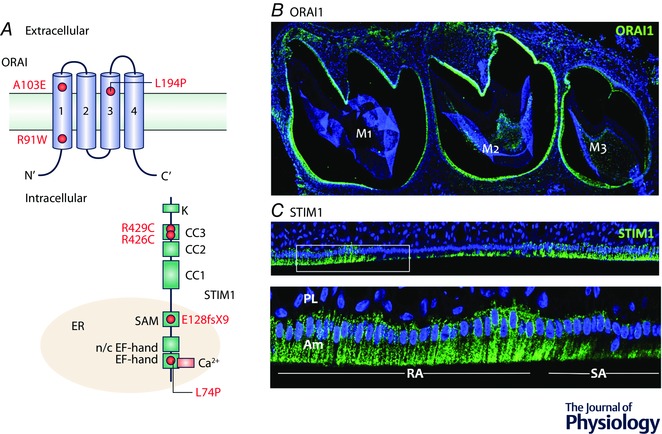
A, diagrammatic representation of STIM1 and ORAI1 protein structure. STIM1 is a single pass membrane in the endoplasmic reticulum (ER) with the N‐terminus found in the ER lumen. ORAI1 is a plasma membrane (PM) bound protein with four transmembrane domains. Both N‐ and C‐termini are cytosolic. The red dots in each protein mark the known mutations at each domain impacting enamel development. ER = endoplasmic reticulum, SAM = sterile alpha motif, CC = coil‐coil domain. B, ORAI1 localization by immunofluorescence microscopy in mouse molar (M) ameloblasts showing limited distribution of this protein. C, STIM1 localization in rat ameloblasts. Immunofluorescence staining shows differences in STIM1 localization between ruffled‐ameloblasts (RA) and smooth‐ameloblasts (SA). In RA cells STIM1 localizes throughout the cytosol whereas in SA cells, STIM1 signals markedly decrease suggesting a more important role for STIM1 during the RA stage. In B and C DAPI is shown in blue. Am = ameloblasts, PL = papillary layer.
Ca2+ entry in enamel cells via CRAC channels
In the past, Ca2+ influx into ameloblasts has been largely considered a passive event (Bawden, 1989; Takano, 1995; Hubbard, 2000). Our recent work elucidated new aspects of the Ca2+ entry process implicating CRAC channels as key modulators. (Nurbaeva et al. 2015a,b). Our data emanated from a genome wide study in which we compared rat enamel organ cells from the maturation and secretory stage (Lacruz et al. 2012a). These genomic data identified Stim1 and Stim2 transcripts as being up‐regulated in maturation, which was confirmed by Western blot analysis (Lacruz et al. 2012a). While STIM1 and STIM2 proteins were expressed in the enamel organ, little was known about their putative function in this system. A few years earlier, we had reported that patients with loss‐of‐function or null mutations in STIM1 and ORAI1 genes present with a hypocalcified form of amelogenesis imperfecta (McCarl et al. 2009; Picard et al. 2009; Fuchs et al. 2012), which strongly suggested that both proteins and thus CRAC channels are important in enamel formation. However, physiological data demonstrating CRAC channel activity in enamel cells was missing. To directly demonstrate a role of CRAC channels in dental enamel, we first used ameloblast‐like LS8 cells to develop and test protocols used in many other cells to investigate SOCE via the CRAC channel. Passive depletion of ER Ca2+ stores of LS8 cells with thapsigargin, an inhibitor of the sarco/endoplasmic reticulum ATPase (SERCA), followed by re‐addition of extracellular Ca2+ to the cells resulted in a marked increase in [Ca2+]i due to SOCE. By contrast, LS8 cells treated with a number of inhibitors (Synta 66, 2‐APB, BTP2) that have been used to suppress CRAC channel function lacked the thapsigargin‐induced [Ca2+]i increase, indicating that ameloblast‐derived cells indeed have functional CRAC channels and SOCE. This conclusion is supported by data showing that the major protein components of the CRAC channel, i.e. STIM1, STIM2, ORAI1, ORAI2 and ORAI3, as well as SERCA2, which mediates reuptake of Ca2+ into the ER, are expressed in these cells (Nurbaeva et al. 2015b).
To confirm that CRAC channel components are present in primary enamel cells and to investigate their expression throughout the development of the enamel organ, we studied dental tissues from mice and rats by RT‐PCR and immunofluorescence staining. ORAI1, ORAI2 and ORAI3 as well as STIM1 and STIM2 mRNAs are all expressed in rodent teeth at both the maturation and secretory stages of enamel development (Nurbaeva et al. 2015a). We noted that mRNA expression of all ORAI and STIM homologues increased in maturation stage consistent with an increased Ca2+ transport function at this stage. mRNA analysis showed that ORAI1 was the predominant ORAI isoform (see also Fig. 3 B for protein expression) and as expected, its expression in ameloblasts was enriched at the PM of RA cells consistent with its role as a pore‐subunit of the channel (Nurbaeva et al. 2015a). Uncharacteristically, another study reported a cytosolic expression of ORAI1 in ameloblasts (Zheng et al. 2015). We found that STIM1 was localized throughout the cytosol of maturation stage ameloblasts (Nurbaeva et al. 2015a) as also reported by Simmer and Hu's group (Wang et al. 2014). Further analysis showed that this was the case only in RA cells whereas SA cells showed significantly decreased immunoreactivity (Fig. 3 C) suggesting that Ca2+ uptake via CRAC channels may predominantly occur in RA cells. Although STIM2 mRNA expression was somewhat more abundant than that of STIM1 in ameloblasts isolated at the maturation stage, at the protein level we had previously reported that STIM1 protein levels are higher than those of STIM2 (Lacruz et al. 2012a).
The isolation of enamel organ cells from the secretory or maturation stages was originally reported by Smith and Nanci (Smith & Nanci, 1989). Using their method to manually separate each cell type enabled us to directly investigate SOCE in primary enamel organ cells after stimulation with thapsigargin to passively deplete ER Ca2+ stores and to induce SOCE. Cells from both stages of enamel development showed Ca2+ release from the ER when treated with thapsigargin, and a strong increase in [Ca2+]i following re‐addition of Ca2+ to the extracellular bath solution, indicating that both cell types have functional SOCE. Supporting this conclusion, Ca2+ entry was severely inhibited when secretory and maturation stage ameloblasts were pre‐treated with the CRAC channel inhibitor Synta 66 (Nurbaeva et al. 2015a) and 2‐aminoethyldiphenyl borinate (2‐APB) (author's unpublished observations) demonstrating that CRAC channels mediate SOCE in enamel cells. It should be pointed out that neither Synta 66 nor 2‐APB completely abrogated Ca2+ entry as there was a small increase in [Ca2+]i. It might be possible that other Ca2+ channels also contribute to Ca2+ influx in enamel cells. Some members of the transient receptor potential (TRP) channel family have been detected in pre‐secretory ameloblasts (Liu et al. 2015). However, direct functional evidence supporting a role of these channels in Ca2+ influx is missing and the potent inhibition of Ca2+ influx in ameloblasts using CRAC channel blockers suggests that SOCE is a dominant pathway for Ca2+ influx in enamel cells.
Mutations in STIM1 and ORAI1 cause amelogenesis imperfecta in patients
Homozygosity for autosomal recessive loss‐of‐function or null mutations in STIM1 or ORAI1 genes that abolish CRAC channel function and SOCE is associated with hypocalcified amelogenesis imperfecta (McCarl et al. 2009; Picard et al. 2009; Fuchs et al. 2012; Lacruz & Feske, 2015). For ORAI1, a number of mutations in different regions of the protein (R91W, Feske et al. 2006; A103E, L194P and A88SfsX25, McCarl et al. 2009; see also Fig. 3 A) have been recognized that cause severe, often lethal immune dysfunction. The dental phenotype of ORAI1‐deficient patients is similar in most patients being characterized by dysplastic enamel. The photographs of the patients’ teeth show a high degree of enamel attrition in both primary and secondary teeth with dentine exposure and enamel discolouration (McCarl et al. 2009).
The first reported null mutation in STIM1 associated with an enamel defect was identified in three siblings homozygous for a frameshift mutation in the N‐terminus of STIM1 (E128RfsX9 (or E136X)) (Fig. 3 A) (Picard et al. 2009). Another STIM1 mutation associated with an enamel defect was identified in a patient homozygous for a missense mutation located in the SOAR/CAD/CCb9 domain of STIM1 (R429C) (Fuchs et al. 2012). The black and white oral photographs of this patient (reported in Fuchs et al. 2012) suggest that his teeth may have preserved residual enamel but they show advanced wear in all teeth. More recently, another patient with a homozygous missense mutation in the SOAR/CAD/CCb9 domain of STIM1 (R426C) was reported (Wang et al. 2014). The dental phenotype of this patient has been described to include enamel attrition with cream‐coloured or brown teeth, although the patient's teeth were of similar size and shape as those of a control (Wang et al. 2014). Intriguingly, whereas the patient with R429C mutation suffered from the full clinical phenotype associated with CRAC channelopathy, including immunodeficiency, muscular and enamel dysplasia (Fuchs et al. 2012), the patient with R426C mutation only presented with an enamel defect (Wang et al. 2014), suggesting that his mutation may be a hypomorph and that some STIM1 function and SOCE are preserved. The most recently reported missense mutation in STIM1 results in the substitution of leucine 74 in the STIM1 EF‐hand domain with proline (L74P) (Parry et al. 2015) and was found in two teenaged cousins. The STIM1 p.L74P mutation resulted in hypomineralized amelogenesis imperfecta with discoloured enamel in both primary and secondary teeth (Parry et al. 2015).
Collectively, these data demonstrate that loss‐of‐function or null mutations in STIM1 or ORAI1 that impair SOCE result in rapid wear of enamel with discolouration of teeth and exposure of the underlying dentine, whereas the shape and size of teeth appeared to be normal. The advanced enamel wear reported in all patients strongly implies that a major effect of the mutations is hypomineralized enamel. However, all of these studies are based solely on oral photographs and in some cases dental X‐rays. Because of the limited availability of patient tissue, they do not allow an in‐depth analysis of the enamel tissue or possible changes in ameloblast function or morphology, and they have not investigated whether Ca2+ entry is altered in patient ameloblasts.
It is noteworthy that no mutations in STIM2, ORAI2 or ORAI3 genes have been reported to date in human patients, but this does not exclude a potential role of these genes in SOCE and enamel development.
Signalling role of Ca2+ in enamel
The bulk of data available on Ca2+ transport by enamel epithelium reflect the overwhelming interest in the role of secreted Ca2+ and its incorporation into mineral deposits. Yet many of the processes that temporally precede mineralization remain poorly understood. Among these, is the intriguing concept that Ca2+ may act as a secondary messenger which modulates a number of processes during amelogenesis including the regulation of enamel protein expression.
It is well known that a rise in [Ca2+]i has broad versatility in cell signalling (Berridge et al. 2003) and thus we investigated the potential effects of a CRAC channel mediated [Ca2+]i rise in the expression of enamel genes. We found that stimulating LS8 cells with thapsigargin for 30 min in the presence of extracellular Ca2+, the mRNA levels of the main enamel genes (Amelx, Ambn, Enam) significantly increased. This effect was reversed if LS8 cells were pre‐treated with 2‐APB, a CRAC channel inhibitor (Nurbaeva et al. 2015b), indicating the SOCE‐mediated Ca2+ entry impacted enamel gene expression in LS8 cells. The same positive effect in the expression of enamel genes was found in primary enamel cells dissected from mouse enamel organs stimulated with thapsigargin and although here we did not test these cells using inhibitors, the fact that we observed a positive response after passive depletion of ER Ca2+ stores using thapsigargin is indicative of SOCE involvement in mediating the expression of enamel genes (Nurbaeva et al. 2015b). Changes in gene expression were also assessed at other time points (1 h, 1.5 h) in LS8 cells but these showed only a moderate up‐regulation of mRNA for these genes. These data suggest that modulation of enamel genes by SOCE is very sensitive. Protein changes analysed by Western blot showed that AMBN expression increased in primary cells after 1 h of stimulation with thapsigargin, supporting the relatively fast action of SOCE on enamel protein expression. Using a small interfering (si)RNA knock‐down approach in HAT‐7 ameloblast‐like cells, others have also shown that in cells transfected with an siRNA against ORAI1, Amelx and Ambn expression declined supporting the role of CRAC channels in modulating enamel gene expression (Zheng et al. 2015).
An increase in [Ca2+]i can also occur in response to Ca2+ discharge from intracellular stores into the cytosol. Our preliminary unpublished data suggest that thapsigargin stimulation by itself in the absence of Ca2+ does not impact the mRNA levels of enamel genes indicating that the changes in gene expression described above are the result of SOCE activation and is not due to release from intracellular stores.
Ca2+ sensing molecules
Previous in vitro studies have shown that exposure of the immortalized ameloblast‐like cell line PABSo‐E cells to extracellular Ca2+ at various concentrations had a positive effect on the expression of AMELX and other enamel genes, and mediated ameloblast cell differentiation (Bronckers et al. 2006; Chen et al. 2009). The Ca2+ sensing receptor (CaSR) was identified in the PABSo‐E cell line suggesting that CaSR might be a mechanism used by these cells to interpret changes in extracellular Ca2+ to regulate cell function (Mathias et al. 2001). Exposure of PABSo‐E cells to varying concentrations of extracellular Ca2+ resulted in a concentration‐dependent rise of [Ca2+]i measured by Fura‐2 (Mathias et al. 2001). CaSR is indeed involved in the regulation of diverse cellular processes including ion channel activity via activation of PLC, which is a common pathway involved in ER Ca2+ release via IP3Rs and the activation of SOCE (Ye et al. 1996, 1997; Chattopadhyay et al. 1998). Thus, the extracellular Ca2+ concentration, sensed by CaSR, could plausibly regulate the uptake of Ca2+ into enamel cells via activation of SOCE. However, unpublished data from our lab detected only a minimal rise in [Ca2+]i when primary enamel cells loaded with Fura‐2 were subjected to a change in solution from 0 mm Ca2+ to 2 mm Ca2+.
Ca2+ binding proteins (CaBPs)
Cytoplasmic Ca2+ binding proteins
Hundreds of cellular proteins have been adapted to bind Ca2+, acting either as Ca2+ sensors or buffers. Ca2+ buffers generally do not undergo major conformational changes upon binding to Ca2+ and comprise only a small subset of cytosolic proteins of the EF‐hand family including parvalbumins, calbindin 9kDa, calbindin 28kDa and calretinin. The majority of EF‐hand proteins belong to the group of Ca2+ sensors. Binding of Ca2+ to Ca2+ binding sensor protein induces conformational changes enabling them to interact with specific targets. Prototypical examples of Ca2+ sensors are calmodulin (Chin & Means, 2000), calcineurin (a Ca2+/calmodulin‐dependent protein phosphatase) and some of the proteins from the S100 protein family. However, Ca2+ sensors may also function as Ca2+ buffers.
Parvalbumins
Parvalbumin is an EF‐hand type Ca2+‐binding albumin protein with molecular weight of 12 kDa (gene symbol: PVALB). This protein is divided into three domains each containing an EF‐hand (Swain et al. 1989). The global role of palvalbumin is to serve as a cytosolic Ca2+ buffer, while Ca2+‐dependent conformational changes can activate its Ca2+ sensor function (Cox et al. 1999).
Localization of palvalbumin during different stages of enamel formation has been investigated using immunogold cytochemistry (Davideau et al. 1993). In early secretory ameloblasts, palvalbumin distribution was limited, being found in association with the Tomes’ process. In older secretory cells this pattern changed, becoming distributed throughout the cell. During the maturation stage, parvalbumin was more abundant in the central area of the cell and in the distal pole of RA cells than in corresponding areas of smooth‐ended ameloblasts (Davideau et al. 1993).
Calbindin 9kDa and calbindin 28kDa
Calbindin 9kDa (gene symbol: S100G) is a protein with four α‐helical regions forming an EF‐hand pair consisting of a canonical (EF2) and a non‐canonical (EF1) domain (Kordel et al. 1993). Calbindin 9kDa undergoes Ca2+‐induced conformational changes but functions predominantly as a Ca2+ buffer rather than as a Ca2+ sensor (Skelton et al. 1994). Calbindin 28kDa (gene symbol: CALB1) has six EF‐hand domains (Cheung et al. 1993). Calbindin 28kDa's Ca2+‐dependent conformational changes indicate additional Ca2+ sensor functions (Berggard et al. 2002).
A number of studies reported the expression of calbindin 9kDa and calbindin 28kDa in ameloblasts of rat molars and incisors (Berdal et al. 1993, 1996; Hubbard, 1995, 1996; Turnbull et al. 2004; Kutuzova et al. 2006; Lee et al. 2007; Hubbard et al. 2011). For calbindin 9kDa, immunolocalization, radioimmunoassay and mRNA analysis showed higher levels during the maturation stage (Table 1) (Taylor et al. 1984; Berdal et al. 1991, 1993). In contrast, calbindin 28kDa expression level was higher in rat secretory ameloblasts (Table 1) (Hubbard, 1995). It has been suggested that fixation methods may influence the distribution of calbindin 9kDa and calbindin 28kDa seen by immunolabelling. Using this method, both calbindins localized in the nucleus and cytoplasm, but only calbindin 9kDa was observed in mitochondria in ameloblasts, as also described in other cells (Berdal et al. 1991). High expression levels of calbindin 9kDa during the maturation stage might suggest that calbindin 9kDa plays an important role during enamel maturation. However, calbindin 9kDa mutations do not result in any dental phenotype (Kutuzova et al. 2006; Lee et al. 2007). Moreover, calbindin 28kDa null mutant mice also lack a dental phenotype (Hubbard, 1995, 1996; Turnbull et al. 2004). Data from several sources identified that calbindin 9kDa and calbindin 28kDa gene expression appeared to be regulated by vitamin D in ameloblasts (Taylor, 1984; Taylor et al. 1984; Berdal et al. 1993), with these cells also expressing vitamin D receptors (Berdal et al. 1993).
Table 1.
Ca2+ signalling molecules in ameloblasts. Arrows indicate whether expression was high or low at each stage
| Ameloblasts | |||||
|---|---|---|---|---|---|
| Name of molecules | Secretory | Maturation | Methods | Mutation shows dental phenotype | Reference |
| Cytoplasmic Ca2+ binding molecules | |||||
| (i) Ca2+ buffers | |||||
| Parvalbumin | Same | Same | Protein expression | ? | (Davideau et al. 1993) |
| Calbindin‐D9k | ↓ | ↑ | mRNA, protein expression | No | (Hubbard et al. 2011), (Berdal et al. 1993), (Berdal et al. 1996), (Kutuzova et al. 2006) |
| Calbindin‐D28k | ↑ | ↓ | Protein expression | No | (Hubbard, 1995), (Berdal et al. 1993), (Berdal et al. 1996), (Turnbull et al. 2004) |
| Calretinin | Expressed during differentiation stage! | Protein expression | No | (Hubbard et al. 2011), (Schurmans et al. 1997) | |
| (ii) Ca2+ sensors | |||||
| Calmodulin | Same | Same | Protein expression | ? | (Hubbard, 1995) |
| Calcineurin | Same | Same | — | ? | (Hubbard, 1995) |
| ER Ca2+ binding proteins | |||||
| Calreticulin | ↓ | ↑ | Protein expression | ? | (Hubbard, 1996) |
| Endoplasmin | ↓ | ↑ | Protein expression | ? | (Hubbard, 1996) |
| ERp72 | ↑ | ↓ | Protein expression | ? | (Hubbard et al. 2000) |
| Calnexin | Same | Same | Immunofluorescence | ? | (Nurbaeva et al. 2015a) |
| ER Ca2+ signalosomes | |||||
| STIM1 | ↓ | ↑ | mRNA, protein expression | Yes | (Lacruz et al. 2012a), (Wang et al. 2014; Nurbaeva et al. 2015a), (Picard et al. 2009; Feske, 2011) |
| STIM2 | ↓ | ↑ | mRNA, protein expression | No | (Lacruz et al. 2012a), (Nurbaeva et al. 2015a) |
| IP3R | — | — | Radioligand binding | — | (Hubbard, 1996) |
| IP3R1, IP3R2, IP3R3 | Same | Same | Protein expression | ? | (Nurbaeva et al. 2015a) |
| RyR1,RyR2, RyR3 | Not expressed (?) | Not expressed (?) | mRNA, protein expression | ? | (Nurbaeva et al. 2015a) |
| Ca2+ entry | |||||
| ORAI1 | ↓ | ↑ | mRNA, protein, function | Yes | (Feske, 2011; Nurbaeva et al. 2015a), (McCarl et al. 2009) |
| ORAI2 | ↓ | ↑ | mRNA, function(?) | ? | (Nurbaeva et al. 2015a) |
| ORAI3 | ↓ | ↑ | mRNA, function(?) | ? | (Nurbaeva et al. 2015a) |
| Ca2+ extrusion molecules | |||||
| (i) Ca2+ exchangers | |||||
| NCKX1 | ↑ | ↓ | mRNA | ? | (Hu et al. 2012) |
| NCKX2 | Same | Same | mRNA | ? | (Hu et al. 2012) |
| NCKX3 | ↑ | ↓ | mRNA | ? | (Hu et al. 2012) |
| NCKX4 | ↓ | ↑ | mRNA, protein expression | Yes | (Hu et al. 2012), (Parry et al. 2013), (Wang et al. 2014) |
| NCKX5 | ↓ | ↑ | mRNA | ? | (Hu et al. 2012) |
| NCKX6 | Same | Same | mRNA | ? | (Hu et al. 2012) |
| NCX | — | — | Function | — | (Okumura et al. 2010) |
| NCX1 | Same | Same | mRNA, protein, | ? | (Okumura et al. 2010; Lacruz et al. 2012b) |
| NCX2 | Not expressed | Not expressed | mRNA, protein | N/A | (Okumura et al. 2010) |
| NCX3 | ↑ | ↓ | mRNA, protein | ? | (Okumura et al. 2010; Lacruz et al. 2012b) |
| (ii) Ca2+ pumps | |||||
| SERCA1 | Not expressed | Not expressed | mRNA | N/A | (Franklin et al. 2001), (Nurbaeva et al. 2015a) |
| SERCA2 | ↓ | ↑ | mRNA, protein, function | ? | (Franklin et al. 2001), (Nurbaeva et al. 2015a) |
| SERCA3 | Not expressed | Not expressed | mRNA | N/A | (Franklin et al. 2001), (Nurbaeva et al. 2015a) |
| PMCA | ? | ? | EM, protein | — | (Salama et al. 1987), (Borke et al. 1993) |
| PMCA1 | Expressed (?) | Expressed (?) | mRNA, protein | ? | (Borke et al. 1993) |
| PMCA2 | Not expressed | — | mRNA | N/A | (Borke et al. 1993) |
| PMCA3 | Not expressed | — | mRNA | N/A | (Borke et al. 1993) |
| PMCA4 | Expressed (?) | Expressed (?) | mRNA, protein | ? | (Borke et al. 1993) |
Calretinin
Human calretinin (31 kDa; gene symbol: CALB2) consists of 271 amino acids and has six EF‐hand domains, five of which are able to bind Ca2+ (Schwaller et al. 1997; Stevens & Rogers, 1997). Calretinin Ca2+‐dependent conformational changes suggest that calretinin may also have Ca2+‐sensor functions (Billing‐Marczak & Kuznicki, 1999).
In enamel cells, calretinin is expressed during the differentiation stage when generalized epithelial cells become pre‐ameloblasts but no data have been reported for secretory or maturation stages (Hubbard et al. 2011). Mutations to calretinin do not appear to have a major impact in enamel as no dental phenotypes have been reported.
Calmodulin
Calmodulin (16 kDa; gene symbol: CALM) is a ubiquitous, highly conserved EF‐hand containing a Ca2+ binding domain. This protein localizes to the cytosol and to the nucleus (Maier & Bers, 2002) and binds up to four Ca2+ via each EF‐hand motif. Calmodulin undergoes Ca2+‐dependent conformational changes that increase its affinity for target proteins (Maier & Bers, 2002). Calmodulin is involved in many processes including growth, proliferation and also in the immune system. As a Ca2+ sensor, calmodulin has the ability to detect and respond to a range of changes in [Ca2+]i (Chin & Means, 2000). Using immunoblot analysis it has been shown that in enamel cells, calmodulin expression was similar throughout the different stages of amelogenesis (Table 1) (Hubbard, 1995).
Calcineurin
Calcineurin is a Ca2+/calmodulin‐dependent protein phosphatase consisting of two subunits, the catalytic A (CnA) and the regulatory subunit B (CnB) of 60 kDa and 19 kDa, respectively (Ke & Huai, 2003). The latter contains four Ca2+ binding EF‐hand motifs and is bound to CnA in conditions of resting Ca2+ levels (Furman & Norris, 2014). In mammals, there are three calcineurin isoforms (CnAα, CnAβ, CnAγ) and two CnB isoforms (CnB1 and CnB2), being highly conserved. Calcineurin is involved in many biological processes of which probably the best studied is its association with signal transduction pathways mediating T‐cell activation thus making it a good candidate for drugs targeting immunosuppression (Crabtree, 2001). Calcineurin de‐phosphorylates nuclear factor of activated T cells (NFAT), a process required for NFAT nuclear translocation and signalling activity (Crabtree, 2001).
In ameloblasts, CnAα, CnAβ and CnB1 were identified by in situ hybridization during tooth differentiation at embryonic day 15 and by immunohistochemistry in secretory stage ameloblasts (Oshima & Watanabe, 2012). RA cells showed strong reactivity to CnAβ and CnB1 suggesting a role of calcineurin associated with the mineralization stage (Oshima & Watanabe, 2012).
Ca2+ binding proteins in ER lumen
The total concentration of Ca2+ within the ER lumen is approximately 1 mm, with free Ca2+ in the range of approximately 200 μm and the remainder buffered via Ca2+ binding proteins such as calreticulin, endoplasmin and ERp72 (Michalak & Opas, 2009). These proteins also play other roles within the cell being involved in protein folding and regulation of Ca2+ release (Michalak & Opas, 2009).
Calreticulin
Calreticulin is a 46 kDa protein responsible for buffering up to 50% of ER Ca2+ in non‐excitable cells (Nakamura et al. 2001a, b). Structurally, calreticulin consists of three distinct domains N, P and C. N and P domains are implicated in chaperone function and the C domain is enriched in negatively charged amino acid residues responsible for its Ca2+ buffering capabilities (Nakamura et al. 2001b). It binds Ca2+ with high capacity and low affinity (Nakamura et al. 2001b). In vivo and in vitro models of calreticulin deficiency and overexpression have shown how tight control of protein folding and Ca2+ homeostasis by calreticulin are necessary for proper function and development (Mesaeli et al. 1999; Nakamura et al. 2001a).
Endoplasmin
Endoplasmin is an ER resident protein of 94 kDa, with a Ca2+ buffering role (Macer & Koch, 1988; Van et al. 1989). It binds Ca2+ with high capacity (Macer & Koch, 1988; Van et al. 1989). Endoplasmin binds to ER luminal peptides and this binding is increased in the absence of Ca2+, consistent with its role as a response to ER stress (Ying & Flatmark, 2006; Biswas et al. 2007).
ERp72
The primary function of ERp72 appears to be to act as a molecular chaperone (Nigam et al. 1994), where it isomerizes disulfide bonds (Rupp et al. 1994). Though it does bind Ca2+, the chaperone activity of ERp72 is unaffected by Ca2+ concentrations (Rupp et al. 1994) and overexpression of ERp72 does not increase ER Ca2+ stores, suggesting that its protein folding activity is more important than its Ca2+ binding function (Lievremont et al. 1997).
Detection of calreticulin and endoplasmin by Western blot showed that both were up‐regulated in rat maturation ameloblasts (Table 1) (Hubbard, 1996), which might suggest that luminal ER [Ca2+] is higher in maturation relative to secretory and requires compensation by these Ca2+ buffering proteins. In contrast, ERp29 was up‐regulated in rat secretory stage ameloblasts (Table 1), which might indicate the involvement of this protein in enamel specific protein production as ERp29 is linked to protein folding and chaperone activities (Hubbard et al. 2000).
Calnexin
Calnexin (CNX) is a 90 kDa type‐I ER integral protein involved in protein folding following glucose removal of newly synthesized peptides (Ni & Lee, 2007). CNX contains luminal and cytosolic domains with the luminal domain (globular domain) containing a Ca2+ binding site (Dudek et al. 2015). Abnormal CNX function has been associated with Alzheimer's disease (Ni & Lee, 2007). In enamel cells, CNX has been found in LS8 cells and in secretory and maturation stage ameloblasts (Nurbaeva et al. 2015a,b). In LS8 cells, its localization differed from that of the ER pump SERCA2 (see Pumps and exchangers section) being more limited in its distribution (Nurbaeva et al. 2015b). In maturation stage ameloblasts, CNX showed a similar distribution to both SERCA2 and STIM1 (Nurbaeva et al. 2015a).
Pumps and exchangers
Tight regulation of [Ca2+]i is important and necessary for all cells in order to avoid Ca2+ toxicity. An important mechanism to maintain [Ca2+]i within a normal range is through the activity of various pumps and exchangers which remove Ca2+ from the cytoplasm such as ER/SR Ca2+‐ATPase (SERCA) or plasma membrane Ca2+‐ATPase (PMCA) pumps, or extruding Ca2+ out of the cell such as K+‐independent Na+/Ca2+ (NCX) and K+‐dependent Na+/Ca2+ exchangers (NCKX).
Pumps
Two main types of ATPase pumps mobilize Ca2+ with distinct roles. SERCAs move cytosolic Ca2+ into the ER lumen, whereas PMCAs pump cytosolic Ca2+ out of the cell. SERCA maintains low [Ca2+]i by exchanging protons for two Ca2+ per ATP hydrolysed, and PMCA transports one Ca2+ out of the cell per ATP hydrolysed. PMCA and SERCA pumps have lower transport rates but high Ca2+ affinity and thus they can respond only to modest elevations in [Ca2+]i.
SERCA
SERCA pumps are encoded by three different genes, ATP2A1–3, each encoding several protein isoforms. ATP2A1 gene encodes SERCA1a and b isoforms, ATP2A2 gene encodes SERCA2a, b and c, and ATP2A3 gene encodes six isoforms of SERCA3a–f, respectively (Hovnanian, 2007; Zarain‐Herzberg et al. 2014). SERCA expression shows distinct patterns of tissue distribution (Missiaen et al. 2000; Shull et al. 2003; Periasamy & Kalyanasundaram, 2007) so that mutations in each SERCA isoform differentially affect organ system function resulting in a number of diseases such as respiratory failure, Brody myopathy, dominant skin disease and Darier disease (Foggia & Hovnanian, 2004; Hovnanian, 2004, 2007).
SERCA in ameloblasts
SERCA was first identified in enamel organ cell extracts by RT‐PCR, cDNA cloning, Northern analysis and immunoblotting all of which suggested that SERCA2b was the main isoform expressed by the enamel organ (Franklin et al. 2001). No SERCA2a transcript has been detected in enamel cells (Franklin et al. 2001). It was also reported that SERCA2b expression increased at the maturation stage (Table 1) (Franklin et al. 2001). We have since identified the ATP2A1–3 genes in rat secretory and maturation ameloblasts as well as in LS8 cells by RT‐PCR (Nurbaeva et al. 2015a,b) and confirmed that SERCA2 was the predominant SERCA at both the transcript and protein levels (Nurbaeva et al. 2015a). SERCA2 localization in maturation stage ameloblasts showed wide cytosolic distribution overlapping with that of the ER protein CNX (Nurbaeva et al. 2015a). Thapsigargin is the most widely used SERCA inhibitor, which results in cytosolic Ca2+ increases as SERCA no longer pumps Ca2+ into the ER lumen leading to a passive depletion of Ca2+ stores through poorly understood Ca2+ leaks in the ER. Rat secretory and maturation ameloblasts as well as LS8 cells showed [Ca2+]i increase following stimulation with thapsigargin in the absence of extracellular Ca2+ due to Ca2+ release from the ER stores, providing direct evidence of a functional SERCA (Nurbaeva et al. 2015a,b).
PMCAs
PMCAs (∼140 kDa) include a subfamily of the P‐type ATPase superfamily characterized by the formation of a phosphorylated enzyme during ATP hydrolysis (Strehler, 2015). Four distinct PMCA isoforms (PMCA 1–4) are commonly identified in mammals encoded by distinct genes (ATP2B1, ATB2B2, ATP2B3 and ATP2B4 in humans) located on separate chromosomes (Strehler et al. 2007). PMCA1 and PMCA4 are ubiquitous, with the former having a housekeeping role being expressed during early development (Brini & Carafoli, 2011). PMCA2 is widely distributed in all areas of the brain and is also found in the organ of Corti in the middle ear (Furuta et al. 1998; Dumont et al. 2001). PMCA2 also regulates Ca2+ homeostasis in the retina. PMCA3 is highly expressed in skeletal muscles (Lopreiato et al. 2014; Strehler, 2015). PMCA2 mutations result in deafness and equilibrium disturbances as well as impaired retinal function (Duncan et al. 2006). Overexpression of PMCA2 increased the Ca2+ content in milk (Reinhardt et al. 2004). PMCA3 has never been successfully knocked out suggesting an essential function of this isoform. Mutations in ATP2B3 result in male sterility (Schuh et al. 2004).
PMCA in ameloblasts
Expression and plasma membrane association of PMCA was shown in both secretory and maturation stage ameloblasts in the rat incisors using enzyme cytochemistry, immunochemical techniques and mRNA expression (Inage & Weinstock, 1979; Crenshaw & Takano, 1982; Sasaki & Garant, 1986; Takano et al. 1986; Salama et al. 1987, 1989; Sasaki et al. 1987; Eisenmann et al. 1990; Borke et al. 1993, 1995; Takano, 1995; Zaki et al. 1996). mRNA transcript analysis showed that PMCA‐1 and PMCA‐4 are the predominant isoforms in human and rat ameloblasts (Table 1) (Borke et al. 1995; Zaki et al. 1996), as we have also identified in our lab (unpublished data). However, there seems to be a lack of consensus concerning expression pattern differences in stages as well as disagreement on its cellular localization. One study reported that PMCA expression in the secretory stage was associated with the entire PM (Salama et al. 1987). During early and late maturation, RA showed high PMCA intensity along the distal ruffled border (Salama et al. 1987). During early and late maturation, SA cells showed substantial PMCA expression along the lateral and proximal surfaces but not at the distal plasma membrane (Salama et al. 1987). A subsequent study by Borke and colleagues investigated the expression of PMCA epitopes in rat incisor formation and mineralization using a monoclonal antibody and found that PMCA epitopes are present in all stages of amelogenesis and the location and intensity of staining of PMCA epitopes also varies with the progress of tissue mineralization (Borke et al. 1993). Early maturation ameloblasts often exhibited greater staining intensity than secretory ameloblasts (Borke et al. 1993). More recently, detection and distribution of PMCA in ameloblasts used a specific monoclonal antibody and immunogold cytochemistry analysed by electron microscopy (Zaki et al. 1996). The highest concentration of gold particles were observed in the distal membranes of early‐maturation ameloblasts relative to late‐maturation and secretory stage cells (Zaki et al. 1996).
In addition, PMCA activity was sensitive to the calmodulin inhibitor trifluoperazine. Trifluoperazine yielded partial systemic inhibition of enamel mineralization during the secretory stage but had no effect during the maturation stage (Sasaki et al. 1987). It also induced morphological changes in secretory stage ameloblasts. Enamel crystallites of trifluoperazine‐injected rats appeared to be less electron‐dense than those from controls. In part, some of the discrepancies in the results described above have been attributed to a variety of chemicals and processing of tissues used by different laboratories (Takano, 1995). It should also be noted that Ca2+‐ATPases have also been described in mitochondria and Golgi complex in ameloblasts (Sasaki et al. 1997) and it has been considered that in Golgi saccules, Ca2+ might be condensed into secretory granules (Eisenmann et al. 1982).
Exchangers
Besides SERCA and PMCA, there are other Ca2+ clearance mechanisms in cells, such as NCX and NCKX. An important difference between these exchangers and the pumps is that the turnover rate of exchangers is orders of magnitude higher than that of ATP‐driven Ca2+ pumps (Herchuelz et al. 2007).
NCX and NCKX
Three NCX and six NCKX isoforms have been identified by molecular cloning in mammalian cells (Herchuelz et al. 2007; Lytton, 2007; Visser & Lytton, 2007; Visser et al. 2007). NCX isoforms remove a single Ca2+ in exchange for three Na+ (Yu & Choi, 1997), whereas NCKX isoforms co‐transport one K+ and one Ca2+ in exchange for four Na+ (Lytton, 2007). NCKX and NCX are bidirectional electronic transporters depending on the prevailing electrochemical driving forces, i.e. the Na+ and Ca2+ concentrations and the membrane potential, exchangers accomplishing Ca2+ entry or Ca2+ exit. NCKX1 is predominantly expressed on rod photo‐receptors and platelets, NCKX4 are found in the brain, and NCKX3 and 4 isoforms are predominant in aorta. NCKX2 is expressed in retinal cone photo‐receptors and retinal ganglion cells, NCKX3 is expressed in uterus and intestine, and NCKX4 is in lung thymus (Schnetkamp, 2004). NCX and NCKX isoforms are expressed in human lung macrophages, dendritic cells and blood monocytes (Staiano et al. 2009; Shumilina et al. 2010; Heise et al. 2011). NCX and NCKX are encoded by the SLC8A and SLC24A4 genes, respectively. NCX1, NCX3 and NCKX1–6 isoforms have been demonstrated to be expressed on rat ameloblasts albeit showing different expression levels (Okumura et al. 2010; Hu et al. 2012).
NCKX and NCX in ameloblasts
The expression of Na+/Ca2+ exchanger family NCX in enamel cells was first reported by Okumura and colleagues (Okumura et al. 2010). NCX1 and NCX3 but not NCX2 were found in these cells both showing an apical or apico‐lateral distribution. NCX1 also showed limited basal reactivity. NCX involvement in Ca2+ transport was further demonstrated by electrophysiological analysis of whole cell recordings as well as the use of pharmacological inhibitors of NCX. These data are unique in that they represent the only study to date, to the best of our knowledge, where ameloblasts have been used to perform electrophysiological experiments. However, a caveat in the experimental design is that the cells used derived from 7‐day‐old rats, which makes it unlikely that the data obtained fully represented the maturation stage. Indeed, despite the use of alkaline phosphatase as a maturation stage marker, the histological analysis reveals only moderate amounts of tissue thickness formed in enamel and dentine, and cells labelled as maturation stage ameloblasts show the presence of stratum intermedium cells (Okumura et al. 2010), all of which is inconsistent with maturation. Despite this, the data implicate NCX in Ca2+ transport in at least one type of ameloblast (secretory). This is an important advance in deciphering the molecular tools available to ameloblasts to export an excess of intracellular Ca2+ to the enamel compartment. Our assessment of NCX1 and NCX3 expression by RT‐PCR showed that neither of these genes increased expression during the maturation stage and hence we suggested the possibility of additional exchangers would be implicated in Ca2+ extrusion at this critical stage (Lacruz et al. 2012a). It might be the case that Ca2+‐ATPases combined with NCX activity provide secretory ameloblasts with the necessary mechanisms to enable sufficient Ca2+ to be extruded through the apical pole to ensure elongation of the thin enamel crystals.
Soon after reporting on NCX, our genome wide study comparing secretory vs. maturation stage enamel organs, identified the expression of NCKX4, a Na+/K+/Ca2+ exchanger, as being markedly up‐regulated in maturation (Lacruz et al. 2012a) and investigated the expression of the NCKX family in enamel cells. We found that all members of the NCKX family were expressed at varying levels in both secretory and maturation stages but that NCKX4 dominated the expression profile during the maturation stage, thus suggesting an important role in enamel formation (Hu et al. 2012). Moreover, on evidence from peroxidase staining, secretory stage ameloblasts did not show reactivity to NCKX4 (Hu et al. 2012). Maturation stage ameloblasts showed strong reactivity at the apical end (Hu et al. 2012). Immunofluorescence studies shown here confirmed the predominant expression of NCKX4 at the apical pole but only in RA cells. SA cells showed a different staining pattern being more diffusely distributed through the cell (Fig. 4) highlighting potential differences in Ca2+ extrusion between the two maturation stage cell types. The localization of NCKX4 to the apical end of the cell places it in the right side of the cells to be involved in Ca2+ extrusion (Fig. 4). This finding was supported by subsequent reports characterizing severe enamel defects in patients and in mouse models with mutations to SLC24A4 (the gene encoding for NCKX4) (Parry et al. 2013; Wang et al. 2014). Four mutations in SLC24A4 gene have been identified resulting in amelogenesis imperfecta such as Arg339Ter (C.1015C>T), Ser499Cys (C.1495A>T) (Parry et al. 2013), Ala146Val (Wang et al. 2014) and Leu436Arg (G.165151 T>G; C.1317 T>G) (Herzog et al. 2015) (Fig. 4 A). These reports described the enamel phenotype in human patients as displaying a yellow‐brown discolouration having increased opacity but with normal enamel volume and crown morphology, although affected teeth seemed susceptible to premature enamel loss (Parry et al. 2013). However, in mice, enamel was only present at the base of the tooth (cervical margin) and was missing from the remainder of the tooth (Parry et al. 2013). Thus, the role of NCKX4 in Ca2+ extrusion in ameloblasts has been strengthened although it would be desirable to obtain electrophysiological data to confirm this.
Figure 4. NCKX4 mutations and localization in ameloblasts.
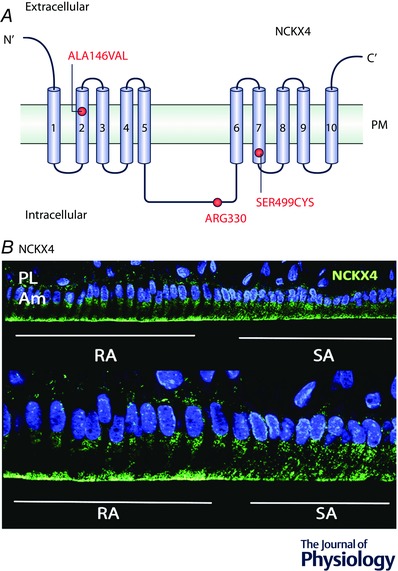
A, schematic representation of NCKX4 protein structure showing the 10 transmembrane domains and corresponding loops. The red dots indicate the mutations at each domain that have been associated with enamel deficiencies. PM = plasma membrane. B, NCKX4 localization in rat ameloblasts. Immunofluorescence microscopy analysis of NCKX4 localization in RA and SA ameloblasts. In RA cells, NCKX4 is largely localized at the apical (distal) pole of the cell. In SA cells, NCKX4 distribution becomes more diffused suggesting a principal role for this protein in Ca2+ extrusion during the RA stage. DAPI is shown in blue. RA = ruffled‐ameloblasts, SA = smooth‐ameloblasts, Am = ameloblasts, PL = papillary layer.
Voltage independent and voltage dependent Ca2+ channels
Ameloblast cells are considered non‐excitable cells and hence it is interesting to find that L‐type voltage‐gated (Cav1.2) channels are associated with abnormal enamel (Papineau & Wilson, 2014). Mutations in the gene encoding for Cav1.2 (CACNA1C) result in hypoplastic amelogenesis imperfecta (Papineau & Wilson, 2014). However, there is at present no available data on CACNA1C expression in enamel cells. In contrast, voltage‐independent K+/Ca2+ activated channel KCNN4, activated by intracellular Ca2+, was reported by us as being up‐regulated during the maturation stage in a genome wide study (Lacruz et al. 2012a), although at present we have no other expression data and no mutations to this gene have been linked with abnormal enamel.
Developing a model for Ca2+ signalling and transport in enamel
A summary of Ca2+ associated proteins thus far identified in enamel cells is shown in Fig. 5. It is now well accepted that the transport of ions to form and mature enamel crystals follows a basal to apical route across the ameloblast cell barrier. The principal mode of transport appears to be the transcellular route but the contribution of a paracellular passage of ions during the RA to SA cycles cannot be discounted. Ca2+ influx has been regarded as a passive event and was tightly coupled to a concentration gradient difference between the low cytosolic [Ca2+] and the much greater extracellular [Ca2+]. The rise in [Ca2+]i via passive influx occurred near the basolateral pole partly modulated by the hypothetical action of unknown exchangers located midway along the cell body that extruded an excess of Ca2+ (e.g. Bawden, 1989; Takano, 1995; Hubbard, 2000). A key point in these models was the consideration that CaBP enabled intracellular transport across the cell, which also benefited from a lower [Ca2+] at the apical pole so that CaBP acted as Ca2+ shuttles in the basal‐to‐apical direction without raising [Ca2+]i to toxic levels. As far as the extrusion of Ca2+ into the enamel was concerned, PMCAs were largely involved in this process (Bawden, 1989).
Figure 5. Ca2+ toolkit in ameloblasts.
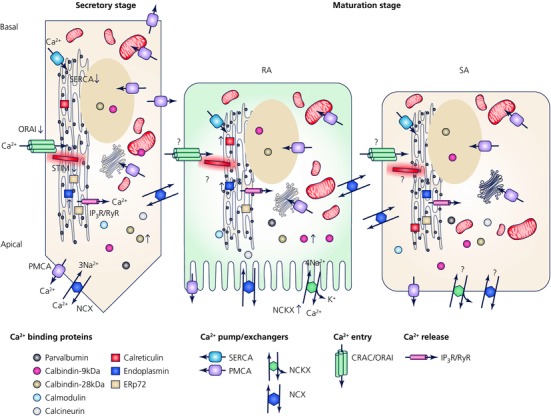
Schematic representation of Ca2+ handling proteins that have been reported to date in secretory and maturation stage ameloblasts. See text for details. RA = ruffled‐ameloblasts, SA = smooth‐ameloblasts.
However, Hubbard reworked this model invoking not a CaBP cytosolic transport route but an organellar‐based transfer of Ca2+ across the cell. In fact, Hubbard (Hubbard, 2000) considered the possibility that SOCE might be involved in Ca2+ transport although evidence for this was limited. More recently, Feske also considered the potential role of SOCE in Ca2+ transport in enamel epithelium (Feske, 2009). Indeed, our recent data strongly support this model for Ca2+entry (Nurbaeva et al. 2015a,b). Our functional studies showing markedly decreased Ca2+ entry in enamel organ cells pre‐treated with the CRAC inhibitor Synta 66 demonstrated that this path is important for Ca2+ influx into these cells. Strong supporting evidence is found in the severe enamel phenotypes in patients with mutations to either STIM1 or ORAI1. Clearly, CRAC channel function is tied to enamel development and mineralization. A caveat, however, is that to fully understand the effects of SOCE deficiency in enamel formation, the dentition of patients with STIM1/ORAI1 mutations and that of animal models should be analysed in detail. Unfortunately, Stim1‐deficient animals do not survive well after birth (Oh‐Hora et al. 2008) and to date no comprehensive analysis of the enamel of teeth derived from human patients has been performed. Despite this important caveat, Ca2+ entry in enamel cells can now be largely considered as being modulated by CRAC channels.
Intracellular Ca2+ transport across the cell remains the biggest unknown. Hubbard's group had discounted the importance of CaBP as no enamel defects had been identified in mice lacking a number of abundantly expressed CaBP in enamel cells (Hubbard et al. 2011). This might direct a focus towards an organellar route. The ER forms a continuous tunnel‐like network of structures transporting Ca2+ within this network in acinar and brain cells (Mogami et al. 1997; Park et al. 2000; Levine & Rabouille, 2005), but this is yet to be tested in ameloblasts, although it remains an attractive possibility. Within the ER lumen, enamel cells express calreticulin, endoplasmin, ERp72 and calnexin, which contribute to luminal Ca2+ buffering (Fig. 5). The ER membrane bound SERCA pumps are indeed expressed and have an active role in enamel cells as evidenced by inhibiting its function with thapsigargin. Thus, SERCA2 modulates cytosolic Ca2+ buffering in enamel cells although SERCA2 mutations do not seem to affect enamel despite its relative abundance. One could speculate that in such cases other SERCA types could compensate enabling some level of Ca2+ sequestration into the ER. Other cytosolic and ER luminal Ca2+ buffers are known to be expressed in enamel cells (e.g. parvalbumins, calbindins, calretinin, calmodulin, calcineurin) and can in principle play roles in modulating [Ca2+] but these roles might also be either limited or stimulate compensatory mechanisms as no mutations to any of these proteins result in enamel defects (Table 1).
Alternative models for the safe intracellular transport of Ca2+ have been suggested. One such possibility is the packing of Ca2+ into vesicles within the Golgi saccules which are then exocytosed at the apical pole (Eisenmann et al. 1982). In addition, mitochondrial mobility coupled with strong GBHA signals identified in mitochondria in secretory and maturation ameloblasts, suggested a potential role for these Ca2+ storage organelles in transporting Ca2+ towards the apical pole (Takano et al. 1989). Yet it is somewhat surprising that GBHA did not identify signals emanating from the principal intracellular stores, namely the ER. Regardless, the role of mitochondria in intracellular Ca2+ transport remains to be investigated.
The extrusion mechanisms appear also to have been decoded to some extent. PMCAs are expressed in enamel cells as reported by a number of studies so they probably play a Ca2+ extrusion role. A lower expression of PMCAs in the secretory stage relative to maturation is in keeping with the notion that Ca2+ requirements increase during the latter stage. Furthermore, PMCA expression in the distal ruffled border of RA cells has prompted some investigators to suggest that this cellular localization might enable proton removal from the enamel zone (Hubbard, 2000). Protons are released during enamel crystal formation which could impact local pH (Lacruz et al. 2012b) so PMCAs would thus contribute to this clearing process while pumping Ca2+ outside the cell. PMCAs require ATP for transport and thus have more limited clearing dynamics when compared with NCX and NCKX exchangers as these are ATP independent (Fig. 5).
Neither a genome wide analysis comparing secretory and maturation stage enamel organs nor mRNA screening suggests that NCXs are up‐regulated in maturation which might point to a more housekeeping role for this exchanger family. Importantly no mutations to NCX have been linked to disruptions of the enamel whereas a number of mutations in the NCKX4 encoding gene (SLC24A4) result in phenotypes resembling amelogenesis imperfecta (Parry et al. 2013; Wang et al. 2014). This points to a more substantial role for NCKX4 despite both having similar Ca2+ kinetics exchanging only one Ca2+.
Thus, a current conceptual model for gated Ca2+ transport has the following characteristics (see also Fig. 5): Ca2+ uptake is dominated by the function of CRAC channels, which are probably activated in response to PLC mediated IP3R Ca2+ release from the ER in response to a yet unknown agonist. The cytosolic rise of [Ca2+] via ORAI1 opening is compensated by common cytosolic buffers. SERCA2 replenishes luminal Ca2+, which is bound then to a number of luminal buffers. In secretory stage ameloblasts, Ca2+ entry might be mediated by STIM2 as its better known homologue STIM1 is nearly absent at this stage. ORAI1 is expressed at low levels in secretory ameloblasts but is clearly present. Efflux of Ca2+ in secretory cells might be mediated by NCX and PMCA extruding Ca2+ principally across the apical pole of the cell. The reported expression of PMCAs in the lateral cell membrane of secretory ameloblasts is intriguing as the intercellular space is extremely limited and the passage of ions to the enamel is hampered by the presence of tight junctions apically. Thus, lateral efflux of Ca2+ may be used as a mechanism to remove a small amount of Ca2+ which might slowly diffuse basally to be recycled or it can slowly leak across the tight junctions distally. In maturation stage ameloblasts we envision a NCKX4 dominated Ca2+ clearing function with PMCA playing a lesser role. More specifically, this pattern might be more closely associated with RA cells, whereas in SA cells this is less clear. In this regard, the important morphological changes in organellar distribution that are seen in the latter stage might have an impact in Ca2+ transport but this is far from clear. Regardless, these data combined enable us to construct a working model depicting the main molecular components enabling Ca2+ transport in enamel epithelium (Fig. 5).
Conclusion
Despite recent efforts in deciphering aspects of Ca2+ dynamics in enamel formation and mineralization, many unknowns still remain. Enamel cells are now recognized as a cell model expressing CRAC channels and show many similar components of the Ca2+ dynamic toolkit found in many other cell systems opening expansive future research avenues. For example we have previously suggested that increased levels of [Ca2+]i via CRAC channels have a direct effect in the expression of enamel genes although how this process is modulated remains unknown. This direct signalling mechanism can be a potent mediator of many cellular responses leading to protein synthesis and export in enamel cells as it is recognized in other cell systems. ER Ca2+ release in itself does not appear to affect the response of enamel genes suggesting that a more sustained or an elevated rise in [Ca2+]i is required. Considering that CRAC channel mediated Ca2+ influx is necessary for the activation of NFAT known to have a role in vertebrate development and bone homeostasis, its potential role in enamel development given that NFATs are expressed in enamel cells (our unpublished data) might be of interest. A better understanding of Ca2+ dynamics in the complex system represented by enamel cells should thus have an immediate appeal to those interested not only in enamel biology, but also to the larger community involved in Ca2+ signal transduction and Ca2+ regulation at large.
Additional information
Competing interests
S.F. is a cofounder of Calcimedica; the other coauthors declare no conflict of interest.
Funding
This work was funded by NIH/National Institute of Dental and Craniofacial Research (NIDCR) awards (DE022799 and DE025639) to R.S.L. and NIH grant AI097302 to S.F.
Acknowledgements
We would like to thank Mike Hubbard, Charles E. Smith and Michael Paine for discussions.
Biographies
Meerim Nurbaeva obtained her PhD at the University of Tübingen, Institute of Physiology, under Florian Lang and has been a postdoctoral researcher at the NYU College of Dentistry with Rodrigo S. Lacruz since 2014.

Miriam Eckstein has been a senior research technician at NYU College of Dentistry with Rodrigo S. Lacruz since 2013.
Stefan Feske earned his MD at the University of Freiburg in Germany, before conducting postdoctoral studies with Anjana Rao in Harvard Medical School. He is currently an Associate Professor of Pathology at NYU Medical School.
Rodrigo S. Lacruz earned his MSc and PhD at the University of the Witwatersrand in Johannesburg and conducted postdoctoral work at the University of Southern California, School of Dentistry. Since 2013 he has been an Assistant Professor in the Department of Basic Science and Craniofacial Biology at the NYU College of Dentistry.
This review was presented at “Advances and Breakthroughs in Calcium Signaling”, which took place in Honolulu, Hawaii, 7–9 April 2016.
References
- Aoba T (1996). Recent observations on enamel crystal formation during mammalian amelogenesis. Anat Rec 245, 208–218. [DOI] [PubMed] [Google Scholar]
- Aoba T & Moreno EC (1987). The enamel fluid in the early secretory stage of porcine amelogenesis: chemical composition and saturation with respect to enamel mineral. Calcif Tissue Int 41, 86–94. [DOI] [PubMed] [Google Scholar]
- Baker MR, Fan G & Serysheva II (2015). Single‐particle cryo‐EM of the ryanodine receptor channel in an aqueous environment. Eur J Transl Myol 25, 4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawden JW (1989). Calcium transport during mineralization. Anat Rec 224, 226–233. [DOI] [PubMed] [Google Scholar]
- Bennett DL, Cheek TR, Berridge MJ, De Smedt H, Parys JB, Missiaen L & Bootman MD (1996). Expression and function of ryanodine receptors in nonexcitable cells. J Biol Chem 271, 6356–6362. [DOI] [PubMed] [Google Scholar]
- Berdal A, Hotton D, Kamyab S, Cuisinier‐Gleizes P & Mathieu H (1991). Subcellular co‐localization and co‐variations of two vitamin D‐dependent proteins in rat ameloblasts. Arch Oral Biol 36, 715–725. [DOI] [PubMed] [Google Scholar]
- Berdal A, Hotton D, Pike JW, Mathieu H & Dupret JM (1993). Cell‐ and stage‐specific expression of vitamin D receptor and calbindin genes in rat incisor: regulation by 1,25‐dihydroxyvitamin D3. Dev Biol 155, 172–179. [DOI] [PubMed] [Google Scholar]
- Berdal A, Hotton D, Saffar JL, Thomasset M & Nanci A (1996). Calbindin‐D9k and calbindin‐D28k expression in rat mineralized tissues in vivo. J Bone Miner Res 11, 768–779. [DOI] [PubMed] [Google Scholar]
- Berggard T, Miron S, Onnerfjord P, Thulin E, Akerfeldt KS, Enghild JJ, Akke M & Linse S (2002). Calbindin D28k exhibits properties characteristic of a Ca2+ sensor. J Biol Chem 277, 16662–16672. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD & Roderick HL (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4, 517–529. [DOI] [PubMed] [Google Scholar]
- Billing‐Marczak K & Kuznicki J (1999). Calretinin – sensor or buffer – function still unclear. Pol J Pharmacol 51, 173–178. [PubMed] [Google Scholar]
- Biswas C, Ostrovsky O, Makarewich CA, Wanderling S, Gidalevitz T & Argon Y (2007). The peptide‐binding activity of GRP94 is regulated by calcium. Biochem J 405, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borke JL, Zaki AE, Eisenmann DR, Ashrafi SH, Ashrafi SS & Penniston JT (1993). Expression of plasma membrane Ca++ pump epitopes parallels the progression of enamel and dentin mineralization in rat incisor. J Histochem Cytochem 41, 175–181. [DOI] [PubMed] [Google Scholar]
- Borke JL, AE Zaki, Eisenmann DR & Mednieks MI (1995). Localization of plasma membrane Ca2+ pump mRNA and protein in human ameloblasts by in situ hybridization and immunohistochemistry. Connect Tissue Res 33, 139–144. [DOI] [PubMed] [Google Scholar]
- Boyde A (1989). Enamel In Teeth. Handbook of Microscopic Anatomy, vol. 5/6, ed. Berkovizt BKB, Boyde A, Frank RM, Höhling HJ, Moxham BJ, Nalbandian J. & Tonge CH, pp. 309–473. Springer, Berlin, Heidelberg. [Google Scholar]
- Brini M & Carafoli E (2011). The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb Perspect Biol 3 a004168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronckers AL, Bervoets TJ, Woltgens JH & Lyaruu DM (2006). Effect of calcium, given before or after a fluoride insult, on hamster secretory amelogenesis in vitro. Eur J Oral Sci 114, Suppl. 1, 116–122; discussion 127–119, 380. [DOI] [PubMed] [Google Scholar]
- Brookes SJ, Robinson C, Kirkham J & Bonass WA (1995). Biochemistry and molecular biology of amelogenin proteins of developing dental enamel. Arch Oral Biol 40, 1–14. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay N, Ye CP, Yamaguchi T, Kifor O, Vassilev PM, Nishimura R & Brown EM (1998). Extracellular calcium‐sensing receptor in rat oligodendrocytes: expression and potential role in regulation of cellular proliferation and an outward K+ channel. Glia 24, 449–458. [PubMed] [Google Scholar]
- Chen J, Zhang Y, Mendoza J & Denbesten P (2009). Calcium‐mediated differentiation of ameloblast lineage cells in vitro. J Exp Zool B Mol Dev Evol 312B, 458–464. [DOI] [PubMed] [Google Scholar]
- Chen LS, Couwenhoven RI, Hsu D, Luo W & Snead ML (1992). Maintenance of amelogenin gene expression by transformed epithelial cells of mouse enamel organ. Arch Oral Biol 37, 771–778. [DOI] [PubMed] [Google Scholar]
- Cheung WT, Richards DE & Rogers JH (1993). Calcium binding by chick calretinin and rat calbindin D28k synthesised in bacteria. Eur J Biochem 215, 401–410. [DOI] [PubMed] [Google Scholar]
- Chin D & Means AR (2000). Calmodulin: a prototypical calcium sensor. Trends Cell Biol 10, 322–328. [DOI] [PubMed] [Google Scholar]
- Cox JA, Durussel I, Scott DJ & Berchtold MW (1999). Remodelling of the AB site of rat parvalbumin and oncomodulin into a canonical EF‐hand. Eur J Biochem 264, 790–799. [DOI] [PubMed] [Google Scholar]
- Crabtree GR (2001). Calcium, calcineurin, and the control of transcription. J Biol Chem 276, 2313–2316. [DOI] [PubMed] [Google Scholar]
- Crenshaw MA & Takano Y (1982). Mechanisms by which the enamel organ controls calcium entry into developing enamel. J Dent Res Spec No, 1574–1579. [PubMed] [Google Scholar]
- Davideau JL, Celio MR, Hotton D & Berdal A (1993). Developmental pattern and subcellular localization of parvalbumin in the rat tooth germ. Arch Oral Biol 38, 707–715. [DOI] [PubMed] [Google Scholar]
- Davideau JL, Papagerakis P, Hotton D, Lezot F & Berdal A (1996). In situ investigation of vitamin D receptor, alkaline phosphatase, and osteocalcin gene expression in oro‐facial mineralized tissues. Endocrinology 137, 3577–3585. [DOI] [PubMed] [Google Scholar]
- Denborough M (1998). Malignant hyperthermia. Lancet 352, 1131–1136. [DOI] [PubMed] [Google Scholar]
- Dudek E, Millott R, Liu WX, Beauchamp E, Berthiaume LG & Michalak M (2015). N‐myristoyltransferase 1 interacts with calnexin at the endoplasmic reticulum. Biochem Biophys Res Commun 468, 889–893. [DOI] [PubMed] [Google Scholar]
- Dumont RA, Lins U, Filoteo AG, Penniston JT, Kachar B & Gillespie PG (2001). Plasma membrane Ca2+‐ATPase isoform 2a is the PMCA of hair bundles. J Neurosci 21, 5066–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JL, Yang H, Doan T, Silverstein RS, Murphy GJ, Nune G, Liu X, Copenhagen D, Tempel BL, Rieke F & Krizaj D (2006). Scotopic visual signaling in the mouse retina is modulated by high‐affinity plasma membrane calcium extrusion. J Neurosci 26, 7201–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann DR, Ashrafi S & Zaki AE (1982). Multi‐method analysis of calcium localization in the secretory ameloblast. J Dent Res Spec No, 1555–1562. [PubMed] [Google Scholar]
- Eisenmann DR, Salama AH, Zaki AM & Ashrafi SH (1990). Cytochemical localization of calcium and Ca2+,Mg2+‐adenosine triphosphatase in colchicine‐altered rat incisor ameloblasts. J Histochem Cytochem 38, 1469–1478. [DOI] [PubMed] [Google Scholar]
- Feske S (2009). ORAI1 and STIM1 deficiency in human and mice: roles of store‐operated Ca2+ entry in the immune system and beyond. Immunol Rev 231, 189–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S (2011). Immunodeficiency due to defects in store‐operated calcium entry. Ann NY Acad Sci 1238, 74–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M & Rao A (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. [DOI] [PubMed] [Google Scholar]
- Fill M & Copello JA (2002). Ryanodine receptor calcium release channels. Physiol Rev 82, 893–922. [DOI] [PubMed] [Google Scholar]
- Fincham AG, Moradian‐Oldak J, Simmer JP, Sarte P, Lau EC, Diekwisch T & Slavkin HC (1994). Self‐assembly of a recombinant amelogenin protein generates supramolecular structures. J Struct Biol 112, 103–109. [DOI] [PubMed] [Google Scholar]
- Foggia L & Hovnanian A (2004). Calcium pump disorders of the skin. Am J Med Genet C Semin Med Genet 131C, 20–31. [DOI] [PubMed] [Google Scholar]
- Foskett JK (2010). Inositol trisphosphate receptor Ca2+ release channels in neurological diseases. Pflugers Arch 460, 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH & Mak DO (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin IK, Winz RA & Hubbard MJ (2001). Endoplasmic reticulum Ca2+‐ATPase pump is up‐regulated in calcium‐transporting dental enamel cells: a non‐housekeeping role for SERCA2b. Biochem J 358, 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S, Rensing‐Ehl A, Speckmann C, Bengsch B, Schmitt‐Graeff A, Bondzio I, Maul‐Pavicic A, Bass T, Vraetz T, Strahm B, Ankermann T, Benson M, Caliebe A, Folster‐Holst R, Kaiser P, Thimme R, Schamel WW, Schwarz K, Feske S & Ehl S (2012). Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J Immunol 188, 1523–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL & Norris CM (2014). Calcineurin and glial signaling: neuroinflammation and beyond. J Neuroinflammation 11, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi T, Furutama D, Hakamata Y, Nakai J, Takeshima H & Mikoshiba K (1994). Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain. J Neurosci 14, 4794–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta H, Luo L, Hepler K & Ryan AF (1998). Evidence for differential regulation of calcium by outer versus inner hair cells: plasma membrane Ca‐ATPase gene expression. Hear Res 123, 10–26. [DOI] [PubMed] [Google Scholar]
- Futatsugi A, Kato K, Ogura H, Li ST, Nagata E, Kuwajima G, Tanaka K, Itohara S & Mikoshiba K (1999). Facilitation of NMDAR‐independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron 24, 701–713. [DOI] [PubMed] [Google Scholar]
- Gibson CW, Yuan ZA, Hall B, Longenecker G, Chen E, Thyagarajan T, Sreenath T, Wright JT, Decker S, Piddington R, Harrison G & Kulkarni AB (2001). Amelogenin‐deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem 276, 31871–31875. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Madesh M & Pacher P (2000). Control of apoptosis by IP3 and ryanodine receptor driven calcium signals. Cell Calcium 28, 349–363. [DOI] [PubMed] [Google Scholar]
- Hakamata Y, Nakai J, Takeshima H & Imoto K (1992). Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain. FEBS Lett 312, 229–235. [DOI] [PubMed] [Google Scholar]
- Hanawa M, Takano Y & Wakita M (1990). An autoradiographic study of calcium movement in the enamel organ of rat molar tooth germs. Arch Oral Biol 35, 899–906. [DOI] [PubMed] [Google Scholar]
- Hart S, Hart T, Gibson C & Wright JT (2000). Mutational analysis of X‐linked amelogenesis imperfecta in multiple families. Arch Oral Biol 45, 79–86. [DOI] [PubMed] [Google Scholar]
- Heise N, Shumilina E, Nurbaeva MK, Schmid E, Szteyn K, Yang W, Xuan NT, Wang K, Zemtsova IM, Duszenko M & Lang F (2011). Effect of dexamethasone on Na+/Ca2+ exchanger in dendritic cells. Am J Physiol Cell Physiol 300, C1306–C1313. [DOI] [PubMed] [Google Scholar]
- Herchuelz A, Kamagate A, Ximenes H & Van Eylen F (2007). Role of Na/Ca exchange and the plasma membrane Ca2+‐ATPase in β cell function and death. Ann NY Acad Sci 1099, 456–467. [DOI] [PubMed] [Google Scholar]
- Hertle DN & Yeckel MF (2007). Distribution of inositol‐1,4,5‐trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience 150, 625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog CR, Reid BM, Seymen F, Koruyucu M, Tuna EB, Simmer JP & Hu JC (2015). Hypomaturation amelogenesis imperfecta caused by a novel SLC24A4 mutation. Oral Surg Oral Med Oral Pathol Oral Radiol 119, e77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo T, Hamada K, Hisatsune C, Nukina N, Hashikawa T, Hattori M, Nakamura T & Mikoshiba K (2010). Mechanism of ER stress‐induced brain damage by IP3 receptor. Neuron 68, 865–878. [DOI] [PubMed] [Google Scholar]
- Hovnanian A (2004). Darier's disease: from dyskeratosis to endoplasmic reticulum calcium ATPase deficiency. Biochem Biophys Res Commun 322, 1237–1244. [DOI] [PubMed] [Google Scholar]
- Hovnanian A (2007). SERCA pumps and human diseases. Subcell Biochem 45, 337–363. [DOI] [PubMed] [Google Scholar]
- Hu P, Lacruz RS, Smith CE, Smith SM, Kurtz I & Paine ML (2012). Expression of the sodium/calcium/potassium exchanger, NCKX4, in ameloblasts. Cells Tissues Organs 196, 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard MJ (1995). Calbindin28kDa and calmodulin are hyperabundant in rat dental enamel cells. Identification of the protein phosphatase calcineurin as a principal calmodulin target and of a secretion‐related role for calbindin28kDa. Eur J Biochem 230, 68–79. [DOI] [PubMed] [Google Scholar]
- Hubbard MJ (1996). Abundant calcium homeostasis machinery in rat dental enamel cells. Up‐regulation of calcium store proteins during enamel mineralization implicates the endoplasmic reticulum in calcium transcytosis. Eur J Biochem 239, 611–623. [DOI] [PubMed] [Google Scholar]
- Hubbard MJ (2000). Calcium transport across the dental enamel epithelium. Crit Rev Oral Biol Med 11, 437–466. [DOI] [PubMed] [Google Scholar]
- Hubbard MJ, McHugh NJ & Carne DL (2000). Isolation of ERp29, a novel endoplasmic reticulum protein, from rat enamel cells. Evidence for a unique role in secretory‐protein synthesis. Eur J Biochem 267, 1945–1957. [DOI] [PubMed] [Google Scholar]
- Hubbard MJ, McHugh NJ & Mangum JE (2011). Exclusion of all three calbindins from a calcium‐ferry role in rat enamel cells. Eur J Oral Sci 119, Suppl. 1, 112–119. [DOI] [PubMed] [Google Scholar]
- Inage T & Weinstock A (1979). Localization of the enzyme ATPase in the rat secretory ameloblast by means of electron microscopy. J Dent Res 58, 1010–1011. [DOI] [PubMed] [Google Scholar]
- Josephsen K & Fejerskov O (1977). Ameloblast modulation in the maturation zone of the rat incisor enamel organ. A light and electron microscopic study. J Anat 124, 45–70. [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Lange I & Feske S (2009). A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun 385, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke H & Huai Q (2003). Structures of calcineurin and its complexes with immunophilins‐immunosuppressants. Biochem Biophys Res Commun 311, 1095–1102. [DOI] [PubMed] [Google Scholar]
- Kim JW, Seymen F, Lin BP, Kiziltan B, Gencay K, Simmer JP & Hu JC (2005). ENAM mutations in autosomal‐dominant amelogenesis imperfecta. J Dent Res 84, 278–282. [DOI] [PubMed] [Google Scholar]
- Klar J, Hisatsune C, Baig SM, Tariq M, Johansson AC, Rasool M, Malik NA, Ameur A, Sugiura K, Feuk L, Mikoshiba K & Dahl N (2014). Abolished InsP3R2 function inhibits sweat secretion in both humans and mice. J Clin Invest 124, 4773–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordel J, Skelton NJ, Akke M & Chazin WJ (1993). High‐resolution structure of calcium‐loaded calbindin D9k. J Mol Biol 231, 711–734. [DOI] [PubMed] [Google Scholar]
- Kutuzova GD, Akhter S, Christakos S, Vanhooke J, Kimmel‐Jehan C & Deluca HF (2006). Calbindin D(9k) knockout mice are indistinguishable from wild‐type mice in phenotype and serum calcium level. Proc Natl Acad Sci USA 103, 12377–12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS & Feske S (2015). Diseases caused by mutations in ORAI1 and STIM1. Ann NY Acad Sci 1356, 45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS, Nanci A, Kurtz I, Wright JT & Paine ML (2010). Regulation of pH during amelogenesis. Calcif Tissue Int 86, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS, Smith CE, Bringas P Jr, Chen YB, Smith SM, Snead ML, Kurtz I, Hacia JG, Hubbard MJ & Paine ML (2012a). Identification of novel candidate genes involved in mineralization of dental enamel by genome‐wide transcript profiling. J Cell Physiol 227, 2264–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS, Smith CE, Moffatt P, Chang EH, Bromage TG, Bringas P Jr, Nanci A, Baniwal SK, Zabner J, Welsh MJ, Kurtz I & Paine ML (2012b). Requirements for ion and solute transport, and pH regulation during enamel maturation. J Cell Physiol 227, 1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai FA, Dent M, Wickenden C, Xu L, Kumari G, Misra M, Lee HB, Sar M & Meissner G (1992). Expression of a cardiac Ca2+‐release channel isoform in mammalian brain. Biochem J 288, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD & Hamilton SL (2010). Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol 2, a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GS, Lee KY, Choi KC, Ryu YH, Paik SG, Oh GT & Jeung EB (2007). Phenotype of a calbindin‐D9k gene knockout is compensated for by the induction of other calcium transporter genes in a mouse model. J Bone Miner Res 22, 1968–1978. [DOI] [PubMed] [Google Scholar]
- Levine T & Rabouille C (2005). Endoplasmic reticulum: one continuous network compartmentalized by extrinsic cues. Curr Opin Cell Biol 17, 362–368. [DOI] [PubMed] [Google Scholar]
- Lievremont JP, Rizzuto R, Hendershot L & Meldolesi J (1997). BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+ . J Biol Chem 272, 30873–30879. [DOI] [PubMed] [Google Scholar]
- Liu C, Niu Y, Zhou X, Xu X, Yang Y, Zhang Y & Zheng L (2015). Cell cycle control, DNA damage repair, and apoptosis‐related pathways control pre‐ameloblasts differentiation during tooth development. BMC Genomics 16, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopreiato R, Giacomello M & Carafoli E (2014). The plasma membrane calcium pump: new ways to look at an old enzyme. J Biol Chem 289, 10261–10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J (2007). Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J 406, 365–382. [DOI] [PubMed] [Google Scholar]
- McCarl CA, Picard C, Khalil S, Kawasaki T, Rother J, Papolos A, Kutok J, Hivroz C, Ledeist F, Plogmann K, Ehl S, Notheis G, Albert MH, Belohradsky BH, Kirschner J, Rao A, Fischer A & Feske S (2009). ORAI1 deficiency and lack of store‐operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol 124, 1311–1318.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macer DR & Koch GL (1988). Identification of a set of calcium‐binding proteins in reticuloplasm, the luminal content of the endoplasmic reticulum. J Cell Sci 91, 61–70. [DOI] [PubMed] [Google Scholar]
- McKee MD, Warshawsky H & Nanci A (1989). Cyclical incorporation of 33P into rat incisor enamel in vivo as visualized by whole‐mount radioautography. Arch Oral Biol 34, 989–993. [DOI] [PubMed] [Google Scholar]
- Maier LS & Bers DM (2002). Calcium, calmodulin, and calcium‐calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol 34, 919–939. [DOI] [PubMed] [Google Scholar]
- Marks AR, Tempst P, Hwang KS, Taubman MB, Inui M, Chadwick C, Fleischer S & Nadal‐Ginard B (1989). Molecular cloning and characterization of the ryanodine receptor/junctional channel complex cDNA from skeletal muscle sarcoplasmic reticulum. Proc Natl Acad Sci USA 86, 8683–8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Avila O, Wu S, Kim SJ, Cheng Y, Khan F, Samudrala R, Sali A, Horst JA & Habelitz S (2012). Self‐assembly of filamentous amelogenin requires calcium and phosphate: from dimers via nanoribbons to fibrils. Biomacromolecules 13, 3494–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Avila OM, Wu S, Cheng Y, Lee R, Khan F & Habelitz S (2011). Self‐assembly of amelogenin proteins at the water‐oil interface. Eur J Oral Sci 119, Suppl. 1, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias RS, Mathews CH, Machule C, Gao D, Li W & Denbesten PK (2001). Identification of the calcium‐sensing receptor in the developing tooth organ. J Bone Miner Res 16, 2238–2244. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K & Noda T (1996). Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5‐trisphosphate receptor. Nature 379, 168–171. [DOI] [PubMed] [Google Scholar]
- Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, Opas M, MacLennan DH & Michalak M (1999). Calreticulin is essential for cardiac development. J Cell Biol 144, 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak M & Opas M (2009). Endoplasmic and sarcoplasmic reticulum in the heart. Trends Cell Biol 19, 253–259. [DOI] [PubMed] [Google Scholar]
- Mikoshiba K (2007). IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem 102, 1426–1446. [DOI] [PubMed] [Google Scholar]
- Mikoshiba K (2015). Role of IP3 receptor signaling in cell functions and diseases. Adv Biol Regul 57, 217–227. [DOI] [PubMed] [Google Scholar]
- Missiaen L, Robberecht W, van den Bosch L, Callewaert G, Parys JB, Wuytack F, Raeymaekers L, Nilius B, Eggermont J & De Smedt H (2000). Abnormal intracellular Ca2+ homeostasis and disease. Cell Calcium 28, 1–21. [DOI] [PubMed] [Google Scholar]
- Miyazaki S, Yuzaki M, Nakada K, Shirakawa H, Nakanishi S, Nakade S & Mikoshiba K (1992). Block of Ca2+ wave and Ca2+ oscillation by antibody to the inositol 1,4,5‐trisphosphate receptor in fertilized hamster eggs. Science 257, 251–255. [DOI] [PubMed] [Google Scholar]
- Mogami H, Nakano K, Tepikin AV & Petersen OH (1997). Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell 88, 49–55. [DOI] [PubMed] [Google Scholar]
- Moradian‐Oldak J (2001). Amelogenins: assembly, processing and control of crystal morphology. Matrix Biol 20, 293–305. [DOI] [PubMed] [Google Scholar]
- Moreno EC & Aoba T (1987). Calcium bonding in enamel fluid and driving force for enamel mineralization in the secretory stage of amelogenesis. Adv Dent Res 1, 245–251. [DOI] [PubMed] [Google Scholar]
- Nakai J, Imagawa T, Hakamat Y, Shigekawa M, Takeshima H & Numa S (1990). Primary structure and functional expression from cDNA of the cardiac ryanodine receptor/calcium release channel. FEBS Lett 271, 169–177. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Robertson M, Liu G, Dickie P, Nakamura K, Guo JQ, Duff HJ, Opas M, Kavanagh K & Michalak M (2001a). Complete heart block and sudden death in mice overexpressing calreticulin. J Clin Invest 107, 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Zuppini A, Arnaudeau S, Lynch J, Ahsan I, Krause R, Papp S, De Smedt H, Parys JB, Muller‐Esterl W, Lew DP, Krause KH, Demaurex N, Opas M & Michalak M (2001b). Functional specialization of calreticulin domains. J Cell Biol 154, 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K, Lorenz JN, Blatter LA, Bers DM & Molkentin JD (2010). The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res 107, 659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M & Lee AS (2007). ER chaperones in mammalian development and human diseases. FEBS Lett 581, 3641–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam SK, Goldberg AL, Ho S, Rohde MF, Bush KT & Sherman M (1994). A set of endoplasmic reticulum proteins possessing properties of molecular chaperones includes Ca2+‐binding proteins and members of the thioredoxin superfamily. J Biol Chem 269, 1744–1749. [PubMed] [Google Scholar]
- Nurbaeva MK, Eckstein M, Concepcion AR, Smith CE, Srikanth S, Paine ML, Gwack Y, Hubbard MJ, Feske S & Lacruz RS (2015a). Dental enamel cells express functional SOCE channels. Sci Rep 5, 15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurbaeva MK, Eckstein M, Snead ML, Feske S & Lacruz RS (2015b). Store‐operated Ca2+ entry modulates the expression of enamel genes. J Dent Res 94, 1471–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh‐Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S & Rao A (2008). Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9, 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura R, Shibukawa Y, Muramatsu T, Hashimoto S, Nakagawa K, Tazaki M & Shimono M (2010). Sodium‐calcium exchangers in rat ameloblasts. J Pharmacol Sci 112, 223–230. [DOI] [PubMed] [Google Scholar]
- Oshima S & Watanabe M (2012). Elevated expression of calcineurin subunits during active mineralization of developing mouse molar teeth. Eur J Oral Sci 120, 386–394. [DOI] [PubMed] [Google Scholar]
- Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM & MacLennan DH (1990). Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem 265, 13472–13483. [PubMed] [Google Scholar]
- Paine ML, Wang HJ, Luo W, Krebsbach PH & Snead ML (2003). A transgenic animal model resembling amelogenesis imperfecta related to ameloblastin overexpression. J Biol Chem 278, 19447–19452. [DOI] [PubMed] [Google Scholar]
- Papineau SD & Wilson S (2014). Dentition abnormalities in a Timothy syndrome patient with a novel genetic mutation: a case report. Pediatr Dent 36, 245–249. [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE & Lewis RS (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Petersen OH & Tepikin AV (2000). The endoplasmic reticulum as one continuous Ca2+ pool: visualization of rapid Ca2+ movements and equilibration. EMBO J 19, 5729–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, Holmes TD, Gamper N, El‐Sayed W, Hettiarachchi NT, Ahmed M, Cook GP, Logan CV, Johnson CA, Joss S, Peers C, Prescott K, Savic S, Inglehearn CF & Mighell AJ (2015). A homozygous STIM1 mutation impairs store‐operated calcium entry and natural killer cell effector function without clinical immunodeficiency. J Allergy Clin Immunol 137, 955–957.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, Poulter JA, Logan CV, Brookes SJ, Jafri H, Ferguson CH, Anwari BM, Rashid Y, Zhao H, Johnson CA, Inglehearn CF & Mighell AJ (2013). Identification of mutations in SLC24A4, encoding a potassium‐dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am J Hum Genet 92, 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy M & Kalyanasundaram A (2007). SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35, 430–442. [DOI] [PubMed] [Google Scholar]
- Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux‐Laucat F, Rechavi G, Rao A, Fischer A & Feske S (2009). STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med 360, 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A & Hogan PG (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. [DOI] [PubMed] [Google Scholar]
- Prakriya M & Lewis RS (2015). Store‐operated calcium channels. Physiol Rev 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins D & Michalak M (2011). Organellar calcium buffers. Cold Spring Harb Perspect Biol 3 a004069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt TA, Lippolis JD, Shull GE & Horst RL (2004). Null mutation in the gene encoding plasma membrane Ca2+‐ATPase isoform 2 impairs calcium transport into milk. J Biol Chem 279, 42369–42373. [DOI] [PubMed] [Google Scholar]
- Reith EJ & Boyde A (1978). Histochemical and electron probe analysis of secretory ameloblasts of developing rat molar teeth. Histochemistry 55, 17–26. [DOI] [PubMed] [Google Scholar]
- Reith EJ & Boyde A (1981). The arrangement of ameloblasts on the surface of maturing enamel of the rat incisor tooth. J Anat 133, 381–388. [PMC free article] [PubMed] [Google Scholar]
- Robinson C (2014). Enamel maturation: a brief background with implications for some enamel dysplasias. Front Physiol 5, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson C, Brookes SJ, Shore RC & Kirkham J (1998). The developing enamel matrix: nature and function. Eur J Oral Sci 106, Suppl. 1, 282–291. [DOI] [PubMed] [Google Scholar]
- Rupp K, Birnbach U, Lundström J, Van PN & Söling HD (1994). Effects of CaBP2, the rat analog of ERp72, and of CaBP1 on the refolding of denatured reduced proteins. Comparison with protein disulfide isomerase. J Biol Chem 269, 2501–2507. [PubMed] [Google Scholar]
- Salama AH, Eisenmann DR & Zaki AE (1989). Effect of cobalt on Ca2+‐Mg2+ ATPase in rat incisor maturation ameloblasts. Calcif Tissue Int 45, 298–304. [DOI] [PubMed] [Google Scholar]
- Salama AH, Zaki AE & Eisenmann DR (1987). Cytochemical localization of Ca2+‐Mg2+ adenosine triphosphatase in rat incisor ameloblasts during enamel secretion and maturation. J Histochem Cytochem 35, 471–482. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Colflesh DE & Garant PR (1987). Calcium transport by a calmodulin‐regulated Ca‐ATPase in the enamel organ. Adv Dent Res 1, 213–226. [DOI] [PubMed] [Google Scholar]
- Sasaki T & Garant PR (1986). Ultracytochemical demonstration of ATP‐dependent calcium pump in ameloblasts of rat incisor enamel organ. Calcif Tissue Int 39, 86–96. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Takagi M & Yanagisawa T (1997). Structure and function of secretory ameloblasts in enamel formation In Dental Enamel, ed. Chadwick D, Cardew G. & Ciba Foundation. Ciba Foundation Symposium, vol. 205 Wiley, Chichester, New York. [DOI] [PubMed] [Google Scholar]
- Schnetkamp PP (2004). The SLC24 Na+/Ca2+‐K+ exchanger family: vision and beyond. Pflugers Arch 447, 683–688. [DOI] [PubMed] [Google Scholar]
- Schuh K, Cartwright EJ, Jankevics E, Bundschu K, Liebermann J, Williams JC, Armesilla AL, Emerson M, Oceandy D, Knobeloch KP & Neyses L (2004). Plasma membrane Ca2+ ATPase 4 is required for sperm motility and male fertility. J Biol Chem 279, 28220–28226. [DOI] [PubMed] [Google Scholar]
- Schurmans S, Schiffmann SN, Gurden H, Lemaire M, Lipp HP, Schwam V, Pochet R, Imperato A, Bohme GA & Parmentier M (1997). Impaired long‐term potentiation induction in dentate gyrus of calretinin‐deficient mice. Proc Natl Acad Sci USA 94, 10415–10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B, Durussel I, Jermann D, Herrmann B & Cox JA (1997). Comparison of the Ca2+‐binding properties of human recombinant calretinin‐22k and calretinin. J Biol Chem 272, 29663–29671. [DOI] [PubMed] [Google Scholar]
- Shah SZ, Zhao D, Khan SH & Yang L (2015). Regulatory mechanisms of endoplasmic reticulum resident IP3 receptors. J Mol Neurosci 56, 938–948. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Qu B, Hoth M & Feske S (2013). Molecular regulation of CRAC channels and their role in lymphocyte function. Cell Mol Life Sci 70, 2637–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull GE, Okunade G, Liu LH, Kozel P, Periasamy M, Lorenz JN & Prasad V (2003). Physiological functions of plasma membrane and intracellular Ca2+ pumps revealed by analysis of null mutants. Ann NY Acad Sci 986, 453–460. [DOI] [PubMed] [Google Scholar]
- Shumilina E, Xuan NT, Matzner N, Bhandaru M, Zemtsova IM & Lang F (2010). Regulation of calcium signaling in dendritic cells by 1,25‐dihydroxyvitamin D3. FASEB J 24, 1989–1996. [DOI] [PubMed] [Google Scholar]
- Skelton NJ, Kordel J, Akke M, Forsen S & Chazin WJ (1994). Signal transduction versus buffering activity in Ca2+‐binding proteins. Nat Struct Biol 1, 239–245. [DOI] [PubMed] [Google Scholar]
- Smith CE (1998). Cellular and chemical events during enamel maturation. Crit Rev Oral Biol Med 9, 128–161. [DOI] [PubMed] [Google Scholar]
- Smith CE & Nanci A (1989). A method for sampling the stages of amelogenesis on mandibular rat incisors using the molars as a reference for dissection. Anat Rec 225, 257–266. [DOI] [PubMed] [Google Scholar]
- Smith CE & Nanci A (1996). Protein dynamics of amelogenesis. Anat Rec 245, 186–207. [DOI] [PubMed] [Google Scholar]
- Staiano RI, Granata F, Secondo A, Petraroli A, Loffredo S, Frattini A, Annunziato L, Marone G & Triggiani M (2009). Expression and function of Na+/Ca2+ exchangers 1 and 3 in human macrophages and monocytes. Eur J Immunol 39, 1405–1418. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Seo MD, Enomoto M, Amador FJ, Ishiyama N & Ikura M (2012). Themes and variations in ER/SR calcium release channels: structure and function. Physiology (Bethesda) 27, 331–342. [DOI] [PubMed] [Google Scholar]
- Stevens J & Rogers JH (1997). Chick calretinin: purification, composition, and metal binding activity of native and recombinant forms. Protein Expr Purif 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Strehler EE (2015). Plasma membrane calcium ATPases: From generic Ca2+ sump pumps to versatile systems for fine‐tuning cellular Ca2+ . Biochem Biophys Res Commun 460, 26–33. [DOI] [PubMed] [Google Scholar]
- Strehler EE, Caride AJ, Filoteo AG, Xiong Y, Penniston JT & Enyedi A (2007). Plasma membrane Ca2+ ATPases as dynamic regulators of cellular calcium handling. Ann NY Acad Sci 1099, 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain AL, Kretsinger RH & Amma EL (1989). Restrained least squares refinement of native (calcium) and cadmium‐substituted carp parvalbumin using X‐ray crystallographic data at 1.6‐Å resolution. J Biol Chem 264, 16620–16628. [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T & Rizzuto R (2006). Chaperone‐mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano Y (1995). Enamel mineralization and the role of ameloblasts in calcium transport. Connect Tissue Res 33, 127–137. [DOI] [PubMed] [Google Scholar]
- Takano Y & Crenshaw MA (1980). The penetration of intravascularly perfused lanthanum into the ameloblast layer of developing rat molar teeth. Arch Oral Biol 25, 505–511. [DOI] [PubMed] [Google Scholar]
- Takano Y, Matsuo S, Wakisaka S, Ichikawa H, Nishikawa S & Akai M (1989). Histochemical localization of calcium in the enamel organ of rat incisors in early‐stage amelogenesis. Acta Anat (Basel) 134, 305–311. [DOI] [PubMed] [Google Scholar]
- Takano Y, Ozawa H & Crenshaw MA (1986). Ca‐ATPase and ALPase activities at the initial calcification sites of dentin and enamel in the rat incisor. Cell Tissue Res 243, 91–99. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, Matsuo H, Ueda M, Hanaoka M, Hirose T & Numa S (1989). Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature 339, 439–445. [DOI] [PubMed] [Google Scholar]
- Taylor AN ( 1984). Tooth formation and the 28,000‐dalton vitamin D‐dependent calcium‐binding protein: an immunocytochemical study. J Histochem Cytochem 32, 159–164. [DOI] [PubMed] [Google Scholar]
- Taylor AN, Gleason WA Jr & Lankford GL (1984). Rat intestinal vitamin D‐dependent calcium‐binding protein: immunocytochemical localization in incisor ameloblasts. J Dent Res 63, 94–97. [DOI] [PubMed] [Google Scholar]
- Termine JD, Belcourt AB, Christner PJ, Conn KM & Nylen MU (1980). Properties of dissociatively extracted fetal tooth matrix proteins. I. Principal molecular species in developing bovine enamel. J Biol Chem 255, 9760–9768. [PubMed] [Google Scholar]
- Turnbull CI, Looi K, Mangum JE, Meyer M, Sayer RJ & Hubbard MJ (2004). Calbindin independence of calcium transport in developing teeth contradicts the calcium ferry dogma. J Biol Chem 279, 55850–55854. [DOI] [PubMed] [Google Scholar]
- Van PN, Peter F & Soling HD (1989). Four intracisternal calcium‐binding glycoproteins from rat liver microsomes with high affinity for calcium. No indication for calsequestrin‐like proteins in inositol 1,4,5‐trisphosphate‐sensitive calcium sequestering rat liver vesicles. J Biol Chem 264, 17494–17501. [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan‐Huberson M, Kraft S, Turner H, Fleig A, Penner R & Kinet JP (2006). CRACM1 is a plasma membrane protein essential for store‐operated Ca2+ entry. Science 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser F & Lytton J (2007). K+‐dependent Na+/Ca2+ exchangers: key contributors to Ca2+ signaling. Physiology (Bethesda) 22, 185–192. [DOI] [PubMed] [Google Scholar]
- Visser F, Valsecchi V, Annunziato L & Lytton J (2007). Exchangers NCKX2, NCKX3, and NCKX4: identification of Thr‐551 as a key residue in defining the apparent K+ affinity of NCKX2. J Biol Chem 282, 4453–4462. [DOI] [PubMed] [Google Scholar]
- Wagner LE 2nd & Yule DI (2012). Differential regulation of the InsP 3 receptor type‐1 and ‐2 single channel properties by InsP 3, Ca2+ and ATP. J Physiol 590, 3245–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Choi M, Richardson AS, Reid BM, Seymen F, Yildirim M, Tuna E, Gencay K, Simmer JP & Hu JC (2014). STIM1 and SLC24A4 are critical for enamel maturation. J Dent Res 93, 94S–100S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JT, Hart PS, Aldred MJ, Seow K, Crawford PJ, Hong SP, Gibson CW & Hart TC (2003). Relationship of phenotype and genotype in X‐linked amelogenesis imperfecta. Connect Tissue Res 44, Suppl. 1, 72–78. [PubMed] [Google Scholar]
- Ye C, Ho‐Pao CL, Kanazirska M, Quinn S, Rogers K, Seidman CE, Seidman JG, Brown EM & Vassilev PM (1997). Amyloid‐β proteins activate Ca2+‐permeable channels through calcium‐sensing receptors. J Neurosci Res 47, 547–554. [DOI] [PubMed] [Google Scholar]
- Ye C, Kanazirska M, Quinn S, Brown EM & Vassilev PM (1996). Modulation by polycationic Ca2+‐sensing receptor agonists of nonselective cation channels in rat hippocampal neurons. Biochem Biophys Res Commun 224, 271–280. [DOI] [PubMed] [Google Scholar]
- Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O & Cahalan MD (2006). Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying M & Flatmark T (2006). Binding of the viral immunogenic octapeptide VSV8 to native glucose‐regulated protein Grp94 (gp96) and its inhibition by the physiological ligands ATP and Ca2+ . FEBS J 273, 513–522. [DOI] [PubMed] [Google Scholar]
- Young RA (1974). Implications of atomic substitutions and other structural details in apatites. J Dent Res 53, 193–203. [DOI] [PubMed] [Google Scholar]
- Yu SP & Choi DW (1997). Na+‐Ca2+ exchange currents in cortical neurons: concomitant forward and reverse operation and effect of glutamate. Eur J Neurosci 9, 1273–1281. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF & Muallem S (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 11, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki AE, Hand AR, Mednieks MI, Eisenmann DR & Borke JL (1996). Quantitative immunocytochemistry of Ca2+‐Mg2+ ATPase in ameloblasts associated with enamel secretion and maturation in the rat incisor. Adv Dent Res 10, 245–251. [DOI] [PubMed] [Google Scholar]
- Zarain‐Herzberg A, Garcia‐Rivas G & Estrada‐Aviles R (2014). Regulation of SERCA pumps expression in diabetes. Cell Calcium 56, 302–310. [DOI] [PubMed] [Google Scholar]
- Zheng L, Zinn V, Lefkelidou A, Taqi N, Chatzistavrou X, Balam T, Nervina J, Papagerakis S & Papagerakis P (2015). Orai1 expression pattern in tooth and craniofacial ectodermal tissues and potential functions during ameloblast differentiation. Dev Dyn 244, 1249–1258. [DOI] [PubMed] [Google Scholar]
- Zorzato F, Fujii J, Otsu K, Phillips M, Green NM, Lai FA, Meissner G & MacLennan DH (1990). Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J Biol Chem 265, 2244–2256. [PubMed] [Google Scholar]