

Abstract
The AKT pathway is a fundamental signaling pathway that mediates multiple cellular processes, such as cell proliferation and survival, angiogenesis, and glucose metabolism. We recently reported that the immunophilin FKBP51 is a scaffolding protein that can enhance PHLPP‐AKT interaction and facilitate PHLPP‐mediated dephosphorylation of AKT at Ser473, negatively regulating AKT activation. However, the regulation of FKBP51‐PHLPP‐AKT pathway remains unclear. Here we report that a deubiquitinase, USP49, is a new regulator of the AKT pathway. Mechanistically, USP49 deubiquitinates and stabilizes FKBP51, which in turn enhances PHLPP's capability to dephosphorylate AKT. Furthermore, USP49 inhibited pancreatic cancer cell proliferation and enhanced cellular response to gemcitabine in a FKBP51‐AKT‐dependent manner. Clinically, decreased expression of USP49 in patients with pancreatic cancer was associated with decreased FKBP51 expression and increased AKT phosphorylation. Overall, our findings establish USP49 as a novel regulator of AKT pathway with a critical role in tumorigenesis and chemo‐response in pancreatic cancer.
Keywords: AKT, chemoresistance, deubiquitination, FKBP51
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
The serine/threonine kinase AKT, also known as protein kinase B (PKB), is a central node in cell signaling downstream of growth factors, cytokines, and other cellular stimuli. AKT functions as a central module to regulate multiple cellular functions, including cell proliferation and survival, angiogenesis, and glucose metabolism (Bellacosa et al, 2005; Engelman et al, 2006; Manning & Cantley, 2007). AKT hyper‐activation is associated with diverse pathophysiological states including tumorigenesis and chemoresistance (Vivanco & Sawyers, 2002; Luo et al, 2003). As a serine/threonine kinase, AKT controls these cellular functions by phosphorylating multiple substrates. For instance, AKT phosphorylates BAD on Ser136, which in turn creates a binding site for 14‐3‐3 proteins, releasing BAD from its target proteins and promoting cell survival (Datta et al, 1997, 2000; del Peso et al, 1997). AKT regulates cell growth and cell proliferation by phosphorylating TSC2 and PRAS40 (Manning et al, 2002; Kovacina et al, 2003). In addition, AKT phosphorylates and activates endothelial nitric oxide synthase (eNOS), which in turn stimulates vasodilation, vascular remodeling, and angiogenesis (Dimmeler et al, 1999; Fulton et al, 1999; Morbidelli et al, 2003). Furthermore, AKT regulates glycogen synthesis by phosphorylation and inhibition of GSK3 (Cross et al, 1995).
The phosphatidylinositol 3‐kinase (PI3K) is a critical upstream kinase of AKT signaling (Brazil & Hemmings, 2001). One of the main functions of PI3K is to synthesize the second messenger PtdIns(3,4,5)P3 (PIP3) from PtdIns(4,5)P2 (PIP2). PIP3 will further bind the pleckstrin homology (PH) domain of AKT and recruit AKT to the plasma membrane, where AKT is phosphorylated at Thr308 and Ser473 and activated (Alessi et al, 1997; Stephens et al, 1998; Sarbassov et al, 2005). Ubiquitination of AKT by the TRAF6 and Skp2‐SCF E3 ligases is also important for its recruitment to the plasma membrane (Yang et al, 2009; Chan et al, 2012). Full AKT activity requires phosphorylation of both Thr308 and Ser473. Thr308 is phosphorylated by the 3‐phosphoinositide‐dependent protein kinase PDK1 (Alessi et al, 1997), whereas Ser473 is phosphorylated by mammalian target of rapamycin (mTOR) complex 2 (mTORC2) (Sarbassov et al, 2005). A recent study showed AKT S477/T479 phosphorylation by CDK2 to be a critical layer of the AKT activation mechanism to regulate its physiological functions (Liu et al, 2014). On the other hand, AKT Thr308 and Ser473 phosphorylation can be dephosphorylated by protein phosphatase 2A (PP2A; Kuo et al, 2008; Padmanabhan et al, 2009; Rodgers et al, 2011) and PH domain leucine‐rich repeat protein phosphatase (PHLPP; Gao et al, 2005; Brognard et al, 2007), respectively. PHLPP‐AKT signaling is regulated in multiple layers, including transcription, posttranslational modification, and protein–protein interaction. For example, PHLPP was reported to be targeted by miR‐206 or miR‐190, which in turn activates AKT signaling (Beezhold et al, 2011; Cai et al, 2013); PHLPP‐AKT axis is regulated by ubiquitination–deubiquitination modification (Li et al, 2009, 2013; Zhiqiang et al, 2012; Gangula & Maddika, 2013). Previously, our laboratory showed that a scaffolding protein FKBP51 enhances dephosphorylation of AKT on Ser473 by connecting AKT to PHLPP (Pei et al, 2009). Previous studies also suggested the transcript of FKBP51 is regulated by androgen receptor (AR; Jaaskelainen et al, 2011; Magee et al, 2006) and FKBP51 balances the AR signaling and PI3K‐AKT pathway in prostate cancers (Carver et al, 2011; Mulholland et al, 2011). However, the regulation of this FKBP51‐PHLPP‐AKT signaling axis remains largely unknown.
Ubiquitin‐specific peptidase 49 (USP49) is a deubiquitinase containing ubiquitin‐specific protease domain and UBP‐type zinc finger domain. Except for a report suggesting USP49 regulates cotranscriptional pre‐mRNA splicing through deubiquitinating H2B (Zhang et al, 2013), the cellular function of USP49 remains largely unknown. Here, we report that USP49 regulates cancer cell proliferation and cancer cell response to therapeutic drugs through the AKT pathway. Mechanistically, USP49 deubiquitinated and stabilized FKBP51, which in turn enhanced dephosphorylation of AKT at Ser473. In addition, depletion of USP49 increased pancreatic cancer proliferation and chemoresistance in an AKT‐dependent manner. Furthermore, USP49 is downregulated in pancreatic cancers, which is correlated with the low expression of FKBP51 and increased phosphorylation of AKT at Ser473 suggesting USP49 may function as a potential prognostic marker in pancreatic cancer.
Results
USP49 is a FKBP51 binding protein
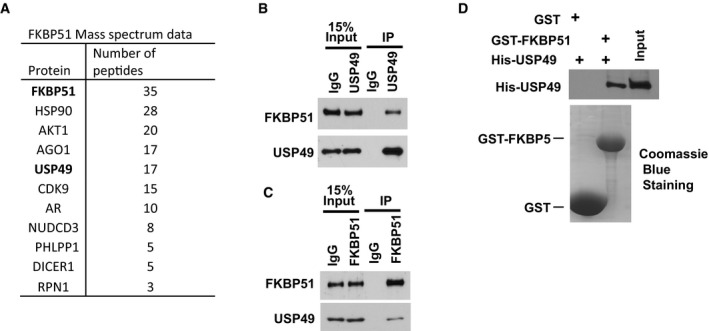
AKT is a serine/threonine‐specific protein kinase that plays a key role in multiple cellular processes such as glucose metabolism, apoptosis, cell proliferation, transcription, and cell migration (Bellacosa et al, 2005; Engelman et al, 2006; Manning & Cantley, 2007). Previous study in our laboratory suggested FKBP51 (FK506‐binding protein 51) acts as a scaffolding protein for AKT and PHLPP and promotes dephosphorylation of AKT, which in turn inhibits tumor cell proliferation (Pei et al, 2009). In addition, FKBP51 levels determine cellular sensitivity to chemotherapeutic drugs. However, how FKBP51 is regulated remains unclear. To further study the regulation of FKBP51‐PHLPP‐AKT axis in pancreatic cancer development, we used cells stably expressing FLAG‐FKBP51 to perform tandem affinity purification and mass spectrometry analysis. In addition to known FKBP51 interacting proteins such as AKT1 and HSP90 (Nair et al, 1997; Pei et al, 2009), we identified a deubiquitination enzyme, USP49, as a major FKBP51‐associated protein (Fig 1A). We confirmed the USP49‐FKBP51 interaction by coimmunoprecipitation. As shown in Fig 1B, USP49 coimmunoprecipitated with FKBP51. Reciprocal immunoprecipitation with FKBP51 antibodies also brought down USP49 (Fig 1C) suggesting an interaction between USP49 and FKBP51 in cells. We also confirmed that FKBP51 interacts with PHLPP1, PHLPP2, AKT1, and AKT2 in SU86 cells by co‐IP assay (Fig EV1A). To determine whether the USP49‐FKBP51 interaction is direct, we generated and purified recombinant USP49 and FKBP51. Purified His‐USP49 was able to interact with GST‐FKBP51 under cell‐free conditions, suggesting a direct interaction between USP49 and FKBP51 (Fig 1D).
Figure 1. USP49 is a FKBP51 binding protein.
-
AList of FKBP51‐associated proteins identified by mass spectrometric analysis. SU86 cells stably expressing FLAG‐SBP‐FKBP51 were generated and FKBP51 complexes were subjected to mass spectrometric analysis.
-
B, CSU86 cell lysates were subjected to immunoprecipitation with control IgG, (B) anti‐USP49, or (C) anti‐FKBP51 antibody. The immunoprecipitates were then blotted with the indicated antibodies.
-
DPurified recombinant GST, GST‐FKBP51, and His‐USP49 were incubated in cell‐free conditions. The interaction between FKBP51 and USP49 was then examined.
Figure EV1. USP49 interacts and stabilizes FKBP51.
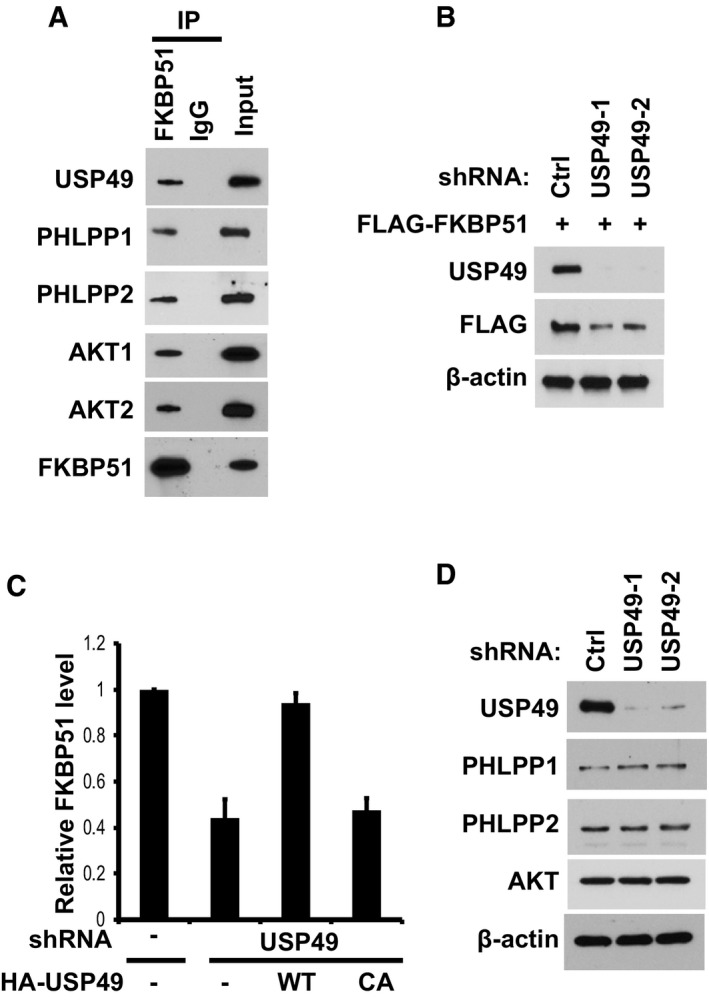
- SU86 cell lysates were subjected to immunoprecipitation with control IgG or anti‐FKBP51 antibody. The immunoprecipitates were then blotted with the indicated antibodies.
- SU86 cells stably expressing FLAG‐FKBP51 were infected with lentivirus encoding shRNAs against control or USP49, the cells were lysed, and Western blot was performed with the indicated antibodies.
- Quantification of the relative FKBP51 protein levels for Fig 2B. Data are represented as mean ± SEM of three independent experiments.
- SU86 cells stably expressing control or USP49 shRNAs were lysed and Western blot was performed with the indicated antibodies.
USP49 deubiquitinates and stabilizes FKBP51
Since USP49 is a deubiquitination enzyme, we hypothesized that USP49 could regulate the protein level of FKBP51. As shown in Fig 2A, when we depleted USP49 using two different USP49‐specific short hairpin RNAs (shRNA), we found that downregulation of USP49 decreased FKBP51 protein level with no effect on FKBP51 mRNA level (Figs 2A and EV1B). In addition, the decrease in FKBP51 level could be reversed by the addition of proteasome inhibitor MG132, suggesting that USP49 regulates FKBP51 levels in a proteasome‐dependent manner. Furthermore, reconstitution of wild‐type USP49 but not the catalytically inactive mutant (USP49CA) in USP49‐depleted cells restored FKBP51 protein levels (Figs 2B and EV1C). These results confirmed the specificity of our USP49 shRNAs and suggested USP49 deubiquitination activity is essential for regulation of FKBP51. However, depletion of USP49 did not affect PHLPPs or AKT protein levels (Fig EV1D). To further establish that USP49 regulates FKBP51 stability, we treated cells with cycloheximide (CHX) and determined the half‐life of FKBP51. As shown in Fig 2C, FKBP51 stability was dramatically decreased in USP49‐depleted cells. These results demonstrate that USP49 stabilizes FKBP51 in cells. We next examined whether USP49 regulates the level of FKBP51 ubiquitination in cells. As shown in Fig 2D, overexpressing wild‐type USP49 resulted in a significant decrease in polyubiquitination of FKBP51, while overexpressing USP49CA mutant failed to alter FKBP51 ubiquitination. Conversely, knocking‐down USP49 increased polyubiquitination of FKBP51 (Fig 2E). These results suggest that USP49 deubiquitinates FKBP51 in cells. To determine whether USP49 directly deubiquitinates FKBP51, we performed an in vitro deubiquitination assay. We purified wild‐type USP49 and USP49CA mutant from bacteria, and ubiquitinated FKBP51 from cells expressing FLAG‐FKBP51 and His‐Ub under denaturing conditions. We then incubated USP49 and ubiquitinated FKBP51 in a cell‐free system. We found WT USP49 but not the USP49CA mutant dramatically deubiquitinated FKBP51 in vitro (Fig 2F). Taken together, these results suggest that USP49 deubiquitinates FKBP51 both in vitro and in vivo.
Figure 2. USP49 deubiquitinates and stabilizes FKBP51.
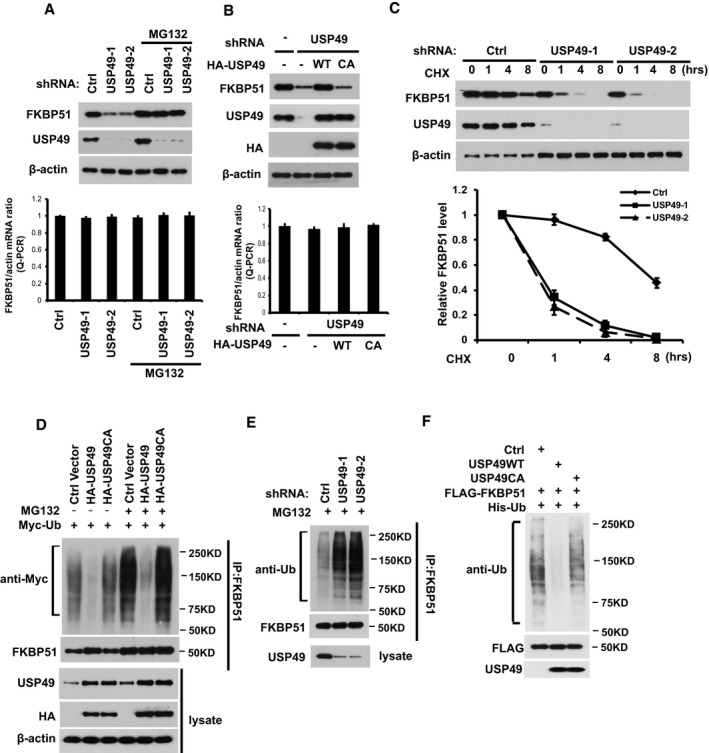
- SU86 cells stably expressing control or USP49 shRNAs were treated with vehicle or MG132. Half of the cells are lysed, and Western blot was performed with the indicated antibodies. Lower panel: The mRNAs were extracted from the rest of the cells and subjected to qRT–PCR. Error bars represent the SEM of three independent experiments.
- SU86 cells stably expressing control (Ctrl) or USP49 shRNAs were transfected with the indicated constructs. After 48 h, half of the cells were lysed and Western blot was performed with the indicated antibodies. Lower panel: The mRNAs were extracted from the rest of the cells and subjected to qRT–PCR. Error bars represent the SEM of three independent experiments.
- SU86 cells stably expressing control (Ctrl) or USP49 shRNAs were treated with CHX (0.1 mg/ml) and harvested at the indicated times. Cells were lysed and cell lysates were then blotted with the indicated antibodies. Lower panel: quantification of the FKBP51 protein levels relative to β‐actin. Error bars represent the SEM of three independent experiments.
- Cells transfected with the indicated constructs were treated with or without MG132 for 4 h before harvest. FKBP51 was immunoprecipitated and immunoblotted with the indicated antibodies.
- Cells stably expressing control (Ctrl) or USP49 shRNAs were treated with MG132 for 4 h before harvest. FKBP51 was immunoprecipitated and immunoblotted with the indicated antibodies.
- Deubiquitination of FKBP51 in vitro by USP49. Ubiquitinated FKBP51 was incubated with purified USP49 or USP49CA in vitro and then blotted with the indicated antibodies.
USP49 regulates cell proliferation and tumor growth through the AKT pathway
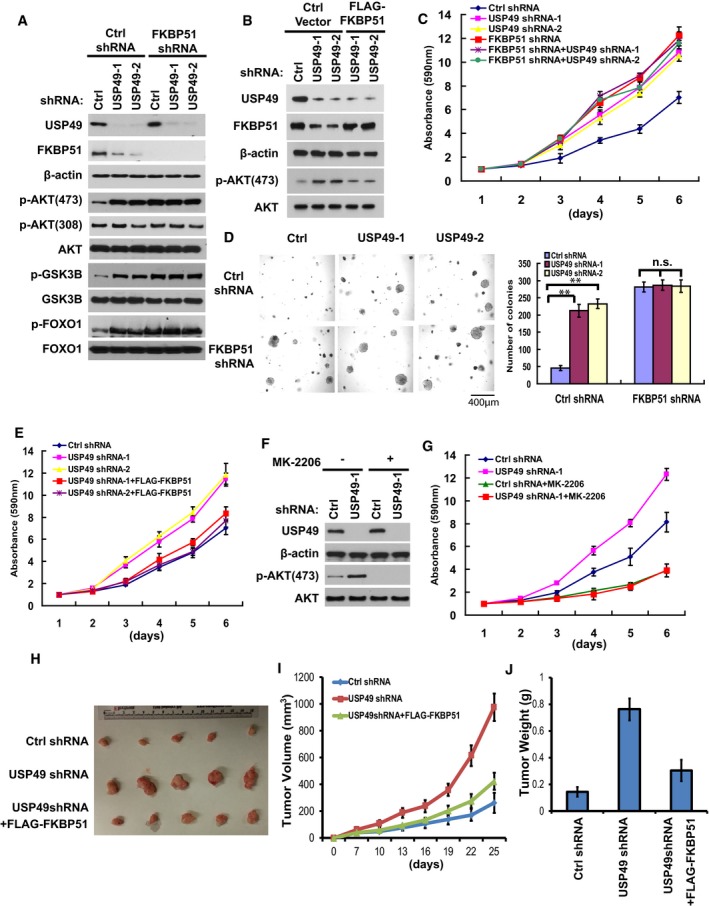
We have shown that USP49 regulates FKBP51 stability in cells. Our previous study clarified FKBP51 as a scaffolding protein for AKT and PHLPP and that it promotes dephosphorylation of AKT, which in turn inhibits tumor cell proliferation. We next examined whether USP49 regulates AKT signaling in a FKBP51‐dependent manner. As shown in Fig 3A, depletion of USP49 significantly increased the phosphorylation of AKT on serine 473 (S473), which is regulated by the FKBP51‐PHLPP axis. However, the phosphorylation of AKT on Threonine 308 (T308) did not change in USP49‐depleted cells (Fig 3A). Moreover, both AKT1 and AKT2 S473 phosphorylation were dramatically increased in USP49‐depleted cells (Fig EV2A and B). We also examined the phosphorylation of downstream substrates of AKT, such as GSK‐3β and FOXO1 (Cross et al, 1995; Nakae et al, 1999). We found depletion of USP49 also enhanced the phosphorylation of GSK‐3β (pSer9 on GSK‐3β) and FOXO1 (pSer256 on FoxO1) (Fig 3A). In addition, depletion of USP49 did not further increase the phosphorylation of AKT, GSK‐3β, and FOXO1 in FKBP51 knockdown cells (Fig 3A). On the other hand, overexpression of FKBP51 in USP49 knockdown cells completely reversed the effect of USP49 shRNA of upregulating the phosphorylation of AKT (Fig 3B). Taken together, these results suggest that USP49 regulates AKT signaling in a FKBP51‐dependent manner. Since AKT signaling is critical for pancreatic cancer cell proliferation, we next asked whether USP49 functions as a tumor‐suppressing protein by regulating FKBP51‐AKT pathway. Downregulation of USP49 by two different shRNAs both markedly increased pancreatic cancer cell proliferation, while USP49 knockdown had no apparent effect on proliferation of cells depleted of FKBP51 (Fig 3C). We also observed similar phenomenon in two other pancreatic cancer cell lines, CFPAC‐1 and Capan‐2 (Fig EV2C–F). In addition, a similar effect was observed when cancer cells were cultured in soft agar (Fig 3D). Notably, restoring FKBP51 expression fully reversed the tumor‐promoting effect of USP49 shRNAs (Fig 3E). We conclude from these data that USP49 inhibits pancreatic cancer cell growth by stabilizing FKBP51. To further confirm whether the tumor‐suppressing function of USP49 is dependent on FKBP51‐AKT axis, we examined cancer cell proliferation following treatment with the AKT inhibitor MK‐2206. As shown in Fig 3F, treatment with MK‐2206 totally abolished AKT phosphorylation suggesting it is efficient in blocking AKT signaling. Notably, depletion of USP49 dramatically increased pancreatic cancer cell proliferation in vehicle‐treated cells but not MK‐2206‐treated cells (Fig 3G), suggesting that USP49 has no additional effect on cell proliferation when AKT is blocked. Furthermore, we depleted USP49 and PHLPP1/2, both individually and in combination, in SU86 cells. As shown in Fig EV2G, knockdown of USP49 dramatically increased the phosphorylation of AKT on serine 473 in control cells. However, in PHLPP1/2 knockdown cells, further depletion of USP49 did not affect the phosphorylation of AKT. Furthermore, depletion of USP49 markedly increased pancreatic cancer cell proliferation, while USP49 knockdown had no further effect on proliferation of cells depleted of PHLPPs (Fig EV2H). Taken together, these results suggest that USP49 regulates AKT by regulating FKBP51‐PHLPP‐AKT axis.
Figure 3. USP49 regulates cell proliferation and tumor growth through the AKT pathway.
-
ASU86 cells stably expressing the indicated shRNAs were lysed and cell lysates were then blotted with the indicated antibodies.
-
BSU86 cells stably expressing USP49 shRNAs were transfected with the indicated constructs. After 48 h, cells were lysed and cell lysates were blotted with the indicated antibodies.
-
C, D(C) Cell proliferation and (D) anchorage‐independent growth in soft agar of the cells from (A) was assessed.
-
ECell proliferation of the cells from (B) was assessed.
-
FSU86 cells stably expressing USP49 shRNAs were treated with MK‐2206 (1 μM) for 4 h, cells were lysed and cell lysates were then blotted with the indicated antibodies.
-
GGrowth curves of USP49 shRNA‐transduced SU86 cells cultured in the presence or absence of the AKT inhibitor MK‐2206 (1 μM) were measured by MTS assay.
-
H–JSU86 cells stably expressing USP49 shRNAs were stably infected with retrovirus encoding FKBP51. 2 × 106 cells were subcutaneously injected into nude mice. Tumor volumes (I) were measured at indicated days. Mice were sacrificed after 4 weeks. Tumor images were acquired as shown in (H) and tumor weights were measured as shown in (J). n = 5; data points in (I) represent mean tumor volume ± SD. Data points in (J) represent mean tumor weight ± SD.
Figure EV2. USP49 regulates AKT phosphorylation through FKBP51‐PHLPP axis.
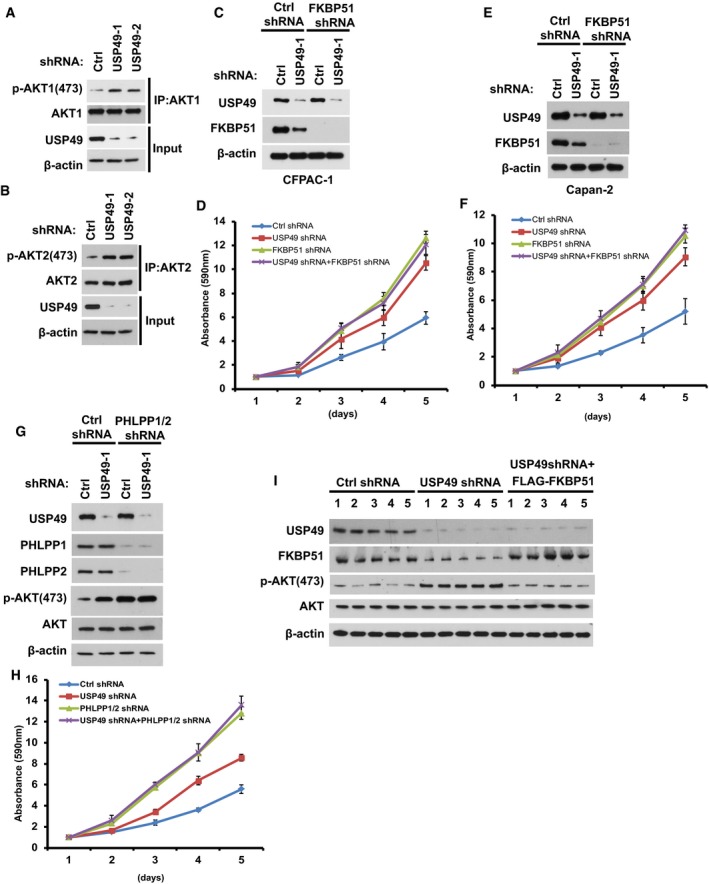
-
A, BSU86 cells stably expressing control or USP49 shRNAs were lysed and subjected to immunoprecipitation with (A) anti‐AKT1 or (B) anti‐AKT2 antibodies. The immunoprecipitates were then blotted with the indicated antibodies.
-
CCFPAC‐1 cells stably expressing USP49 shRNA were transfected with the indicated constructs. After 48 h, cells were lysed and cell lysates were blotted with the indicated antibodies.
-
DProliferation of the cells from (C) was assessed. Data are represented as mean ± SEM of three independent experiments.
-
ECapan‐2 cells stably expressing USP49 shRNA were transfected with the indicated constructs. After 48 h, cells were lysed and cell lysates were blotted with the indicated antibodies.
-
FProliferation of the cells from (E) was assessed. Data are represented as mean ± SEM of three independent experiments.
-
GSU86 cells stably expressing the indicated shRNAs were lysed and Western blot was performed with the indicated antibodies.
-
HProliferation of the cells from (G) was assessed. Data are represented as mean ± SEM of three independent experiments.
-
IThe tumors obtained from in vivo tumorigenesis experiments (Fig 3H) were lysed and Western blot was performed with the indicated antibodies.
To investigate the biological function of USP49 in pancreatic cancer cells in vivo, we subcutaneously implanted USP49‐depleted SU86 cells into nude mice and monitored tumor growth for 4 weeks. Mice bearing USP49 shRNA‐expressing SU86 cells showed increased tumor growth throughout the experiment compared to mice implanted with control shRNA‐infected cells (Fig 3H). Twenty‐five days after tumor cell implantation, we observed a fivefold increase in tumor volume (Fig 3I) and a fourfold increase in weight of tumors formed by USP49‐depleted SU86 cells (Fig 3J). Notably, restoring FKBP51 expression reversed the tumor‐promoting effect of USP49 knockdown (Fig 3H–J). We examined the expression of USP49, FKBP51, and phospho‐Akt levels in tumors obtained from the in vivo tumorigenesis experiments. As shown in Fig EV2I, FKBP51 level was dramatically decreased and p‐AKT was increased in USP49‐depleted samples. In addition, overexpression of FLAG‐FKBP51 rescued the effect of USP49 depletion on AKT phosphorylation. Furthermore, we stably overexpressed the phospho‐mimetic AKT (AKTS473D) in SU86 cells and found that knockdown of either USP49 or FKBP51 dramatically increased cell proliferation and tumorigenesis in control cells but not in cells stably overexpressing the phospho‐mimetic AKT (Fig EV3A–C). These results confirm that USP49 regulates cell proliferation and tumorigenesis in an AKT‐dependent manner. Taken together, these results suggest that loss of USP49 promotes tumorigenesis through downregulation of FKBP51.
Figure EV3. USP49 regulates tumor growth in the FKBP51‐AKT dependent manner.
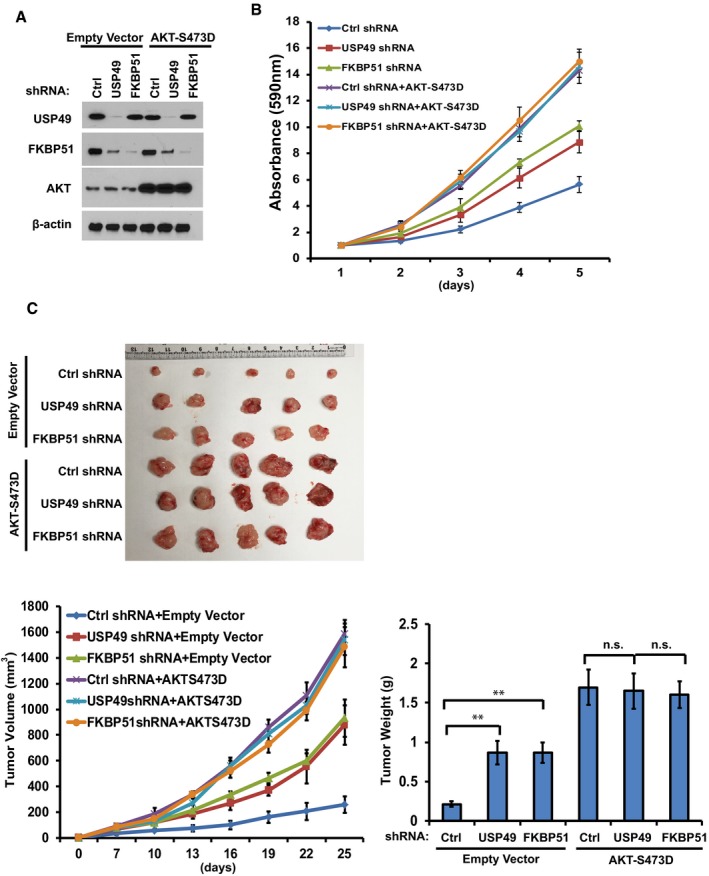
- Su86 cells stably expressing empty vector or AKTS473D were infected with lentivirus encoding shRNAs against control, USP49, or FKBP51. The cells were lysed and Western blot was performed with the indicated antibodies.
- Proliferation of the cells from (A) was assessed. Data are represented as mean ± SEM of three independent experiments.
- 2 × 106 cells from (A) were subcutaneously injected into nude mice. Tumor volumes were measured at indicated days. Mice were sacrificed after 4 weeks. Tumor images were acquired and tumor weights were measured as shown in the lower panels. n = 5; data points in the lower left graph represent mean tumor volume ± SD. Data points in the lower right graph represent mean tumor weight ± SD. **P < 0.01, statistical analyses were performed with ANOVA.
USP49 expression correlates with FKBP51 and pAKT‐S473 levels in clinical pancreatic cancer samples
We and others have reported that AKT is hyper‐phosphorylated and activated in pancreatic cancer (Pei et al, 2009; Liu et al, 2010, 2016). We also reported that FKBP51 is downregulated in pancreatic cancer (Pei et al, 2009). To determine the relevance of regulation of FKBP51‐AKT signaling by USP49 in patients, we performed immunohistochemical staining of p‐AKT, FKBP51, and USP49 on the pancreatic cancer tissue microarrays. As shown in Figs 4A–C and EV4A and B, downregulation of USP49 was detected in 74% (188 of 254) of pancreatic cancer samples, while 72% (125 of 174) of adjusted normal pancreatic tissues showed positive staining for USP49, suggesting loss of USP49 expression in human pancreatic tumors. We also observed downregulation of FKBP51 and upregulation of AKT S473 phosphorylation in pancreatic cancer (Fig 4A–C). Moreover, a significantly positive correlation between FKBP51 and USP49 protein levels (P < 0.0001 R = 0.455) was observed in these pancreatic carcinomas (Fig 4D). In addition, as shown in Fig 4E, we tested the correlation between USP49 and AKT phosphorylation (p‐Ser473) in the same pancreatic cancer samples and observed a strong negative correlation between USP49 and AKT phosphorylation (P < 0.0001 R = −0.315) because 76% (143 of 188) of USP49‐low samples had high AKT phosphorylation level. These results indicate that USP49 is downregulated in pancreatic cancers, which is positively correlated with FKBP51 expression and negatively correlated with AKT phosphorylation on Ser473. Furthermore, we examined the relationship between the expression of USP49, FKBP51, and pAKT473 and clinical outcome. As shown in Fig 4F–H, the pancreatic cancer patients with low USP49 and low FKBP51 expression had significantly decreased overall survival (OS). However, high pAKT473 expression in patients with pancreatic cancer was associated with decreased OS. Taken together, our results suggest that USP49 expression may function as an independent prognostic predictor for pancreatic ductal adenocarcinoma (PDAC) patients.
Figure 4. USP49 expression is correlated with FKBP51 and pAKT‐S473 in clinical pancreatic cancer samples.
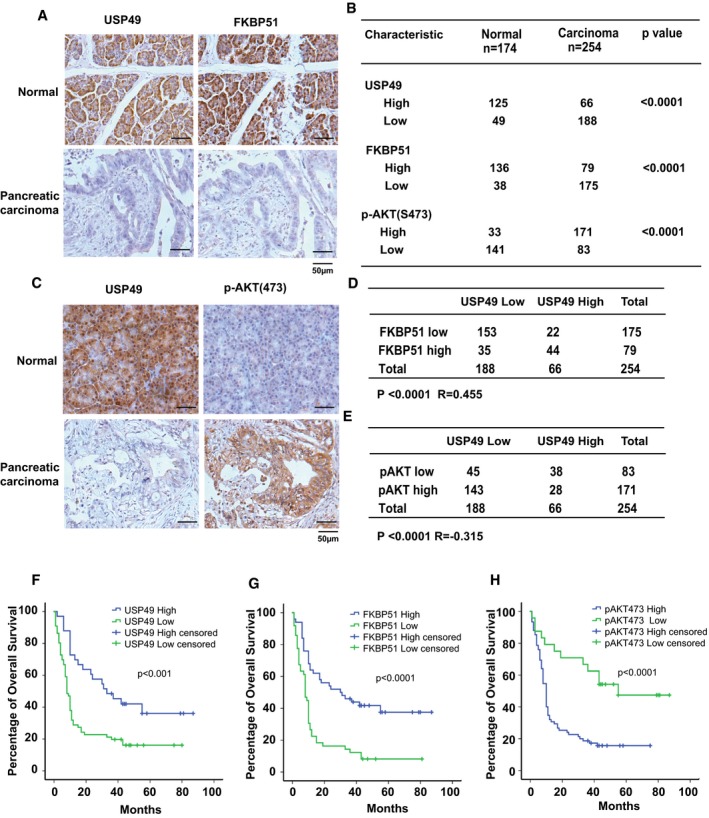
-
ARepresentative images of immunohistochemical staining of USP49 and FKBP51 in normal and pancreatic carcinoma.
-
BQuantification of USP49, FKBP51, and p‐AKT(S473) protein levels in normal and pancreatic carcinoma. Statistical analyses were performed with the χ2 test.
-
CRepresentative images of immunohistochemical staining of USP49 and p‐AKT(S473) in normal and pancreatic carcinoma.
-
DCorrelation study of USP49 and FKBP51 in pancreatic carcinoma. Statistical analyses were performed with the χ2 test. R, Pearson correlation coefficient.
-
ECorrelation study of USP49 and pAKT‐Ser473 in pancreatic carcinoma. Statistical analyses were performed with the χ2 test. R, Pearson correlation coefficient.
-
F–HSurvival analysis of PDAC patients by Kaplan–Meier plots and log‐rank tests. Patients were categorized into high and low expression of USP49, FKBP51, and pAKT‐Ser473 based on IHC staining scores. Censored groups entail non‐cancer deaths or those lost to follow‐up at last recorded follow‐up.
Figure EV4. USP49 is overexpressed in normal pancreatic tissues.
-
A, BRepresentative images of immunohistochemical staining of USP49, FKBP51, and p‐AKT(S473) in normal pancreatic tissues.
USP49 regulates response of pancreatic cancer cells to chemotherapy through AKT pathway
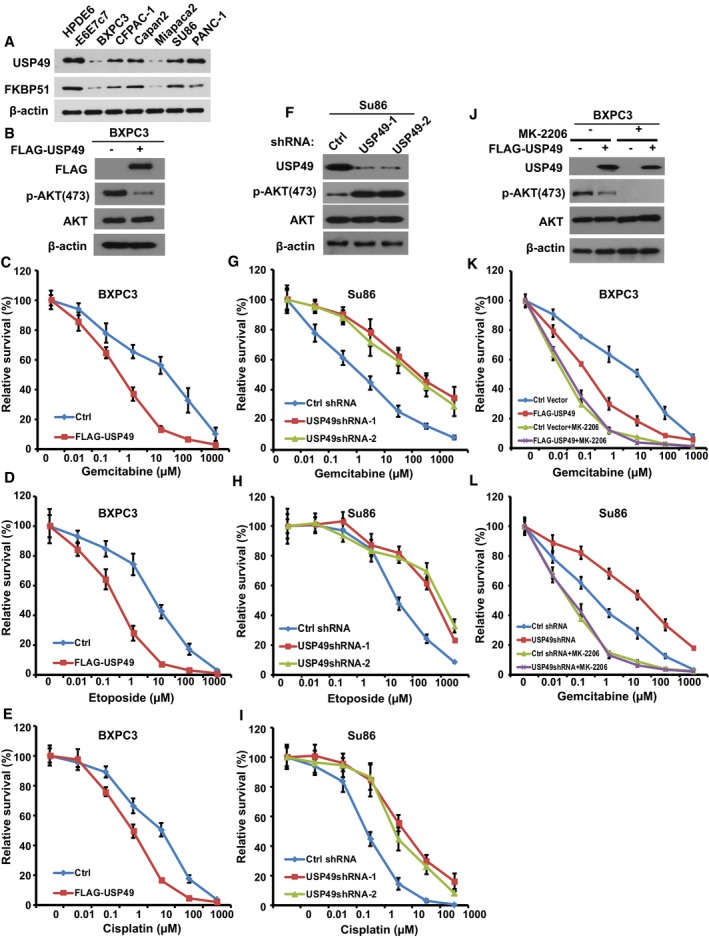
Because the FKBP51‐PHLPP‐AKT pathway also plays an important role in chemosensitivity, we next examined whether USP49 plays a role in pancreatic cancer response to chemotherapy. USP49 protein level was detected in multiple pancreatic cancer cell lines. As shown in Fig 5A, USP49 expression is significantly decreased in a high percentage of the pancreatic cancer cell lines; MiaPaCa2 and BxPC3 showed very low expression of USP49. SU86 and Capan‐2 showed 50% decrease in USP49 expression compared to normal pancreatic cell line (HPDE6). To test the role of USP49 in pancreatic cancer cell response to chemotherapy, we overexpressed USP49 in BxPC3 and MiaPaCa2 cells (Figs 5B and EV5A), which express USP49 at very low levels. The cells were subsequently treated with gemcitabine. As shown in Figs 5C and EV5B, overexpression of USP49 dramatically sensitized cells to gemcitabine. Next, multiple classes of chemotherapeutic drugs, including cisplatin and etoposide, were used to treat these cells. BXPC3 cells with overexpression of USP49 were significantly more sensitive to these drugs (Fig 5D and E). Conversely, we depleted USP49 with shRNA in SU86 and Capan‐2 cells and treated them with gemcitabine (Figs 5F and EV5C). Downregulation of USP49 resulted in increased resistance to gemcitabine (Figs 5G and EV5D), cisplatin, and etoposide (Fig 5H and I). However, overexpression or downregulation of USP49 did not further change response to gemcitabine in cells treated with AKT inhibitor MK‐2206 (Fig 5K and L). These results establish an important role of USP49 in regulating chemo‐response through AKT signaling in pancreatic cancers.
Figure 5. USP49 regulates response of pancreatic cancer cells to chemotherapy through the AKT pathway.
-
ACell lysates from pancreatic cancer cell lines were blotted with USP49 and FKBP51 antibodies. Lysates from normal pancreatic (HPDE6‐E6E7c7) epithelial cells were used as a control.
-
BBXPC3 cells were stably infected with retrovirus encoding control (Ctrl) or FLAG‐USP49. Cells were lysed and cell lysates were blotted with the indicated antibodies.
-
C–ECells from (B) were treated with (C) gemcitabine, (D) etoposide, or (E) cisplatin. Cell survival was determined.
-
FSU86 cells stably expressing USP49 shRNAs were lysed and cell lysates were blotted with indicated antibodies.
-
G–ICells from (F) were treated with (G) gemcitabine, (H) etoposide, or (I) cisplatin and cell survival was determined.
-
JBXPC3 cells stably infected with retrovirus encoding control (Ctrl) or FLAG‐USP49 were treated with MK‐2206 (1 μM) for 4 h, cells were lysed and cell lysates were then blotted with the indicated antibodies.
-
KCells from (J) were treated as indicated, and cell survival was determined.
-
LCells from Fig 3G were treated as indicated, and cell survival was determined.
Figure EV5. USP49 regulates response of pancreatic cancer cells to gemcitabine.
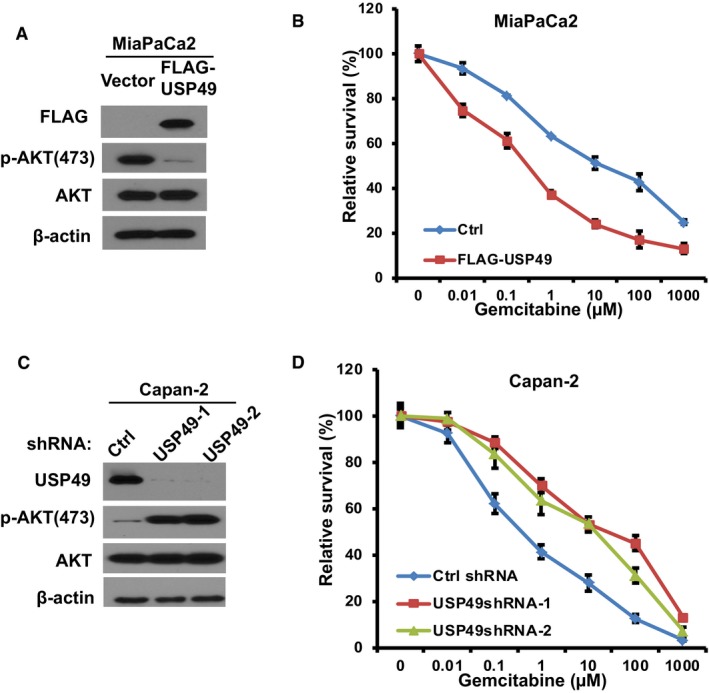
- MiaPaCa2 cells were stably infected with retrovirus encoding control (Ctrl) or FLAG‐USP49. Cells were lysed and cell lysates were blotted with indicated antibodies.
- Cells from (A) were treated with gemcitabine and cell survival was determined using MTS assay. The data presented are mean ± SD (n = 6).
- Capan‐2 cells stably expressing control (Ctrl) or USP49 shRNAs were lysed and cell lysates were blotted with indicated antibodies.
- Cells from (C) were treated with gemcitabine and cell survival was determined using MTS assay. The data presented are mean ± SD (n = 6).
Overall, our results suggest that USP49 negatively regulates AKT activation by stabilizing FKBP51 scaffolding protein and that hyper‐activation of AKT caused by the loss of USP49 might contribute to tumor proliferation and cancer cell resistance to chemotherapy.
Discussion
Pancreatic ductal adenocarcinoma is one of the most lethal cancers, with a 5‐year survival rate of 5% at diagnosis (Baer et al, 2015). Eighty percentage of patients develop local metastatic tumor at diagnosis. In these advanced stage patients, limited response to current therapies results in an extremely poor prognosis (Stathis & Moore, 2010). A better understanding of the molecular events involved in pancreatic cancer will help improve diagnosis and predictability of patient response to existing therapies. This will allow us to design new targeted therapeutic strategies with reduced adverse effects, allowing improved treatment response for this lethal pathology.
The AKT pathway is a fundamental signaling pathway that mediates multiple processes, such as cell proliferation, survival, and motility (Bellacosa et al, 2005; Engelman et al, 2006; Manning & Cantley, 2007). Increased activation of AKT pathway has been implicated in triggering tumorigenesis and conferring resistance to chemotherapy (Vivanco & Sawyers, 2002; Luo et al, 2003). AKT signaling pathway is highly relevant to pancreatic tumors (Baer et al, 2015; Sharma et al, 2015). More than 50% of pancreatic adenocarcinomas demonstrate activation of AKT. Recent reports also suggested that AKT is phosphorylated and thus activated under basal conditions in a variety of pancreatic cancer cell lines, possibly contributing to the anti‐apoptotic resistance (Parsons et al, 2010). Furthermore, significant positive correlation between activation of AKT and poor survival was reported in pancreatic cancer (Yamamoto et al, 2004). Thus, inhibiting AKT pathway may be a promising strategy to retard pancreatic cancer growth and overcome chemoresistance. Our previous study clarified the immunophilin FKBP51 as a scaffolding protein that can enhance PHLPP‐AKT interaction and facilitate PHLPP‐mediated dephosphorylation of AKT at Ser473 (Pei et al, 2009). This led to the hypothesis that FKBP51 could act as a tumor‐suppressing protein in the context of the AKT signaling pathway. Alteration in FKBP51 expression may result in chemoresistance in pancreatic cancers. Other groups have also confirmed the role of FKBP51‐PHLPP‐AKT cascade in tumorigenesis. For instance, downregulation of FKBP51 and preventing PHLPP‐mediated AKT inhibition contribute to castrate‐resistant prostate cancer (CRPC) proliferation. As a downstream target of androgen receptor, FKBP51 mediates the crosstalk between PI3K and AR pathway, which is one of the mechanisms for CRPC development (Carver et al, 2011; Mulholland et al, 2011). Another group showed that FKBP51 inhibits proliferation and stimulates apoptosis of glioma cells through AKT pathway (Yang et al, 2015). However, the role of FKBP51 in cancer is still controversial (Li et al, 2011; Staibano et al, 2011). FKBP51 was also reported to promote melanoma cancer cell proliferation through activation of the NF‐kappaB pathway (Romano et al, 2015).
Whether FKBP51‐PHLPP‐AKT pathway can be regulated in pancreatic cancer remains an unanswered question. Identifying new regulators of this pathway may contribute to improved understanding of the molecular events involved in pancreatic cancer and clarify new potential therapeutic targets in pancreatic cancer. In this study, by performing tandem affinity purification, we identified FKBP51 binding protein, USP49, functions as a deubiquitinates and stabilizes FKBP51, which in turn facilitates PHLPP‐mediated dephosphorylation of AKT‐Ser473. Thus, our study identified a new player in AKT signaling pathway. Furthermore, we propose that USP49 regulates tumorigenesis and chemoresistance in pancreatic cancer in an AKT‐dependent manner. The evidence is as follows: First, depletion of USP49 increased pancreatic cancer cell proliferation in vitro, tumorigenesis in vivo, and chemoresistance. On the other hand, overexpression of USP49 sensitized cancer cells to chemotherapy. However, the effects mediated by USP49 in cancer cell proliferation and chemoresistance were blunted by depletion of FKBP51 and inhibition of AKT. Overall, our study demonstrates USP49 as a new regulator of FKBP51‐PHLPP‐AKT pathway in pancreatic cancers. A previous study suggested USP49 deubiquitinates H2B‐ub and regulates RNA splicing (Zhang et al, 2013). H2B‐ub has also been shown to regulate FKBP51 transcription (Jaaskelainen et al, 2012). However, in our study, USP49 directly binds and stabilizes FKBP51 but does not affect its transcription, suggesting deubiquitination is the major regulation of FKBP51 by USP49. In addition, the FKBP51 gene is deleted in a small percentage of pancreatic cancer cases (1.8% TCGA), which cannot explain the large percentage of weak IHC staining of FKBP51 in pancreatic cancers. We propose that FKBP51 is downregulated in pancreatic cancer at the posttranscriptional level. Downregulation of USP49 maybe a potential mechanism. Indeed, low expression of USP49 in pancreatic cancer correlates with low FKBP51 expression in these samples. These evidences strongly support our hypothesis and clarify a physiological relationship between USP49 and FKBP51 in pancreatic cancer. Furthermore, the biological function of USP49 in sensitizing pancreatic cancer cell to gemcitabine is potentially important. Because gemcitabine represents the first line treatment for advanced and metastatic PDAC, therapeutic interventions targeting USP49 may improve outcome in combination with existing therapies. AKT is a central node in cell signaling downstream of growth factors, cytokines, and other cellular stimuli. AKT functions as a central module to regulate multiple cellular functions, including cell proliferation and survival, angiogenesis, and glucose metabolism. AKT is critical for cellular homeostasis; however, AKT hyper‐activation is associated with diverse pathophysiological states including tumorigenesis and chemoresistance. AKT activity should be tightly controlled, but total inhibition of AKT may not be a good strategy for chemotherapy since it kills not only cancer cells but also normal cells. AKT inhibitors are slowly advancing through clinical assessment. Among the inhibitors in clinical development are both allosteric inhibitors such as MK2206 and ATP competitive catalytic inhibitors such as GSK690693. In early‐phase clinical trials, both the allosteric and catalytic inhibitors of AKT have shown toxicities similar to those observed with the PI3K inhibitors, such as hyperglycemia, rash, stomatitis, and gastrointestinal side effects. USP49 regulates AKT Ser473 phosphorylation through FKBP51‐PHLPP axis. We speculate that activation of USP49 in cells may modestly adjust AKT overactivation but not totally abolish AKT activation, which may be less toxic and reduce the side effects of chemotherapy. In the future, we will screen and attempt to identify USP49 specific activators to elevate USP49 activity in cancer cells, which will in turn inhibit AKT activation and sensitize PDAC to chemotherapy.
Materials and Methods
Cell culture, plasmids, and antibodies
HEK293T and human pancreatic cancer cell lines SU86, BXPC3, CFPAC‐1, Capan‐2, MiaPaCa2, and PANC‐1 were purchased from ATCC and the identities of all cell lines were confirmed by the Medical Genome Facility at Mayo Clinic Center (Rochester, MN) using short tandem repeat profiling upon receipt. The cell lines were maintained in RPMI1640 with 10% FBS. HPDE6‐E6E7c7 cells were maintained in keratinocyte serum‐free medium supplemented with epidermal growth factor and bovine pituitary extract.
HA‐FLAG‐USP49 was purchased from Addgene (Plasmid #22586, provided by Dr. Wade Harper) and subcloned into pET28a vector (Clontech). USP49CA mutant was generated by site‐directed mutagenesis (Stratagene).
Anti‐USP49 and FKBP51 antibodies were purchased from Proteintech (18066‐1‐AP, 14155‐1‐AP). Antibodies against AKT (#9272), phospho‐AKT (Ser473) (#9271), phospho‐AKT (Thr308), GSK3β (#9315), and phospho‐GSK3β (#9316) were purchased from Cell Signaling Inc (Boston, MA). Antibodies against p‐Foxo1 (p‐Ser256) and Foxo1 were purchased from Santa Cruz (Santa Cruz Biotechnology, Santa Cruz, CA). Anti‐FLAG (m2) and anti‐HA antibodies were purchased from Sigma.
Tandem affinity purification
Cells stably expressing empty vector or FLAG‐tagged FKBP51 were lysed with high salt NETN buffer (20 mM Tris–HCl, pH 8.0, 300 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% Nonidet P‐40) containing 50 mM β‐glycerophosphate, 10 mM NaF, and 1 μg/ml each of pepstatin A and aprotinin on ice for 25 min. Cell lysates were diluted 1:1 with NET buffer (NETN buffer without NaCl) and incubated with anti‐FLAG beads (Sigma) overnight at 4°C. After washing with NETN buffer five times, the bound proteins were eluted with FLAG peptide for 1 h at 4°C. Protein samples were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and stained with Coomassie blue. The eluted proteins were detected by mass spectrometry performed by the Taplin Biological Mass Spectrometry Facility at Harvard.
Co‐immunoprecipitation (co‐IP) assay
SU86 cells were harvested and washed by PBS. Then, cells were lysed with NETN buffer (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P‐40) containing 50 mM β‐glycerophosphate, 10 mM NaF, and 1 mg/ml each of pepstatin A and aprotinin. Whole cell lysates obtained by centrifugation were incubated with 2 μg of indicated antibody and protein A or protein G Sepharose beads (Amersham Biosciences) for 4 h at 4°C. After washing with NETN buffer for three times, the immunocomplexes were separated by SDS–PAGE. Immunoblotting was performed following standard procedures.
Protein stability assay
To examine FKBP51 protein turnover, cycloheximide (0.1 mg/ml) was added to cell culture medium and cells were harvested at the indicated time points. Cells were then lysed in NETN buffer and cell lysates were subjected to Western blot with anti‐FKBP51, anti‐USP49, and anti‐β‐actin as indicated. Then, we quantified of the FKBP51 protein levels relative to β‐actin by ImageJ.
In vivo and in vitro deubiquitination assay
For the in vivo deubiquitination assay, HEK293T cells were transfected with HA‐USP49 wild‐type (WT) or mutant Cys 262 to Ala (CA mutant). After 2 days, cells were treated with MG132 (50 μM) for 4 h before being harvested. The cells were lysed in 120 μl 62.5 mM Tris–HCl (pH 6.8), 2% SDS, 10% glycerol, 20 mM NEM, and 1 mM iodoacetamide and directly boiled for 15 min. Then, the cell lysates were diluted 10 times with NETN buffer containing protease inhibitors, 20 mM NEM, and 1 mM iodoacetamide and centrifuged to remove cell debris. The cell extracts were subjected to immunoprecipitation with the indicated antibodies for 4 h at 4°C. After washing with NETN buffer (containing 20 mM NEM and 1 mM iodoacetamide) for four times, the immunocomplexes were separated by SDS–PAGE and blotted with anti‐Ub antibody.
For the deubiquitination assay in vitro, HEK293 cells were transfected together with the FLAG‐FKBP51 and His‐Ub expression vectors. After 2 days, the cells were treated with MG132 (50 μM) for additional 4 h, cells were lysed and ubiquitinated proteins were purified from the cell extracts with His beads under denatured conditions. Ubiquitinated proteins were eluted and diluted with FLAG‐lysis buffer (50 mM Tris–HCl pH 7.8, 137 mM NaCl, 10 mM NaF, 1 mM EDTA, 1% Triton X‐100, 0.2% Sarkosyl, 1 mM DTT, 10% glycerol, and fresh proteinase inhibitors). Ub‐FKBP51 was then further purified with FLAG beads and then eluted with FLAG‐peptides. The recombinant GST‐USP49 and USP49CA proteins were expressed in BL21 cells and purified following standard protocol. For the in vitro deubiquitination assay, ubiquitinated proteins were incubated with recombinant USP49 in deubiquitination buffer (50 mM Tris–HCl pH 8.0, 50 mM NaCl, 1 mM EDTA, 10 mM DTT, 5% glycerol) for 4 h at 37°C. After reaction, FKBP51 was immunoprecipitated with anti‐FKBP51. The immunocomplexes were separated by SDS–PAGE and blotted with indicated antibody.
Cell survival assay
Three thousand cells per well were plated in 96‐well plates. After 24 h, cells were treated with indicated drugs. Etoposide and cisplatin were purchased from Sigma‐Aldrich (St. Louis, MO) and gemcitabine was provided by Eli Lilly (Indianapolis, IN). After incubation with drugs for 72 h, the plates were read in a Safire2 microplate reader (Tecan AG, 14 Switzerland). Assays were performed in triplicate with the CellTiter 96 AQueous Non‐Radioactive Cell Proliferation Assay (Promega Corporation, Madison, WI).
Cell proliferation assay
To measure the cell proliferation, equal numbers of cells were plated in 24‐well plates in 10% serum containing medium. After 1 day, cells were washed with PBS and then fixed with 10% methanol and stained with 0.1% crystal violet on the indicated days. After staining for 30 min in room temperature, wells were washed three times with PBS, then air dried. The dye was extracted with 10% acetic acid. The absorbance of the crystal violet solution was measured at 590 nm.
Soft agar colony‐formation assays
SU86 cells were infected with lentivirus encoding the indicated shRNAs. After selected with puromycin, the indicated stable knockdown cells (5,000 cells per well in 6‐well plates) were plated in 0.3% (w/v) low‐melting temperature agarose on a 0.6% (w/v) agarose base layer, both of which contained complete medium. After 2 weeks, colonies were counted at room temperature under a light microscope (ECLIPSE 80i; Nikon) using a 4× NA 0.10 objective lens (Nikon).
Athymic nude mice tumor formation assay
SU86 cells infected with lentivirus encoding the indicated shRNAs together with FLAG‐tagged FKBP51 were injected subcutaneously and bilaterally into the flank of 5‐week‐old male athymic nude NCr nu/nu (NCI/NIH) mice using 19‐gauge needles. Each mouse received two injections of a 200 μl mixture of 2 × 106 cells in 100 μl of 1 × PBS and 100 μl of growth factor reduced MATRIGEL (BD Biosciences). Tumor growth was monitored for 4 weeks and tumor volume was calculated using the formula: 0.5 × L × H × W. At the end of the study, the tumors were surgically removed, weighed, and processed. Data were analyzed using ANOVA test. Mice were subjected to euthanasia if they display pain or distress, such as lethargy, lying down, not eating or drinking, weight loss greater than 10% body weight, or difficulty in breathing. According to the blinding procedures, two persons as a group performed all the mice experiment. One person performed the experiments and another one totally blinded to the experiment group measured the tumor volume and weight and analyzed the data.
Tissue microarray
The tissue arrays of pancreatic cancer samples were purchased from US Biomax (PA1001a, HPan‐Ade150CS‐01, PA483c) and Shanghai Outdo Biotech Company (HPan‐Ade180 Sur‐02). Immunohistochemical staining of USP49 (dilution 1:500), FKBP51 (dilution 1:500) and p‐AKT‐Ser473 (dilution 1:200) were carried out using IHC Select® HRP/DAB kit (Cat. DAB50, Millipore). The immunostaining was blindly scored by pathologists. The IHC score was calculated by combining the quantity score (percentage of positive stained cells) with the staining intensity score. The quantity score ranges from 0 to 4, that is 0, no immunostaining; 1, 1–10% of cells are stained; 2, 11–50% are positive; 3, 51–80% are positive; and 4, ≥81% of cells are positive. The staining intensity was scored as 0 (negative), 1 (weak), 2 (moderate), and 3 (strong). The score for each tissue was calculated by multiplying the intensity with the quantity score (the range of this calculation was therefore 0–12). An IHC score of 9–12 was considered a strong immunoreactivity; 5–8, moderate; 1–4, weak; and 0, negative. Samples with IHC score of more than 4 were considered to be high, and less than 4 were considered to be low. The score of tumor tissue was determined by comparing to the staining intensity of normal tubules on the same slide. χ2 test and the Pearson correlation coefficient were used for statistical analysis of the correlation between USP49, FKBP51, and p‐AKT‐Ser473.
Patients' specimens and follow‐up
Tissue microarray (TMA) chips that contain 99 cases of paired PDAC tumor and peri‐tumor specimens were purchased (HPan‐Ade180 Sur‐02; Shanghai Outdo Biotech Company). All specimens spotted on TMA chips included complete postoperative follow‐up for a period of 3–7 years. These tissue samples had been obtained before any anticancer treatment and with prior written consent from patients. The overall survival (OS) for the corresponding patients was calculated from the day of surgery to the day of death or to the last follow‐up (censored patients).
Statistics
For cell proliferation and soft agar assays, data are represented as the mean ± SEM of three independent experiments. For cell survival assay, data are represented as the mean ± SD (n = 6). For the animal studies, data are represented as the mean ± SD of 5 mice. Statistical analyses were performed with Student's t‐test, ANOVA, or χ2 test. Statistical significance is represented in figures by: *P < 0.05; **P < 0.01.
Author contributions
JY designed the experiments, analyzed results, and wrote the manuscript. KL, YL, YY, and JY carried out the experiments. LZ, YY and LL assisted with the analysis of IHC data. YY, CW, and YC assisted in the animal experiment. QH, SN and ZL assisted with the analysis of data. JY and ZL supervised the research. All authors discussed the results and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by National Basic Research Program of China (973 Program, Grant No. 2013CB530700), National Natural Science Foundation of China (81402286, 81572770, 31371367), National Institutes of Health (grant CA203971), NCI Pancreatic SPORE grant (CA102701), Mayo Clinic Ovarian Cancer SPORE (P50CA136393) and the Mayo Clinic Eagles 5th District Cancer Telethon Cancer Research Fund.
The EMBO Journal (2017) 36: 1434–1446
References
- Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M (1997) 3‐Phosphoinositide‐dependent protein kinase‐1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 7: 776–789 [DOI] [PubMed] [Google Scholar]
- Baer R, Cintas C, Therville N, Guillermet‐Guibert J (2015) Implication of PI3K/Akt pathway in pancreatic cancer: when PI3K isoforms matter? Adv Biol Regul 59: 19–35 [DOI] [PubMed] [Google Scholar]
- Beezhold K, Liu J, Kan H, Meighan T, Castranova V, Shi X, Chen F (2011) miR‐190‐mediated downregulation of PHLPP contributes to arsenic‐induced Akt activation and carcinogenesis. Toxicol Sci 123: 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Kumar CC, Di Cristofano A, Testa JR (2005) Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res 94: 29–86 [DOI] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA (2001) Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci 26: 657–664 [DOI] [PubMed] [Google Scholar]
- Brognard J, Sierecki E, Gao T, Newton AC (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell 25: 917–931 [DOI] [PubMed] [Google Scholar]
- Cai J, Fang L, Huang Y, Li R, Yuan J, Yang Y, Zhu X, Chen B, Wu J, Li M (2013) miR‐205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non‐small cell lung cancer. Cancer Res 73: 5402–5415 [DOI] [PubMed] [Google Scholar]
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL (2011) Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN‐deficient prostate cancer. Cancer Cell 19: 575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, Hazle JD, Yu D, Wei W, Sarbassov D, Hung MC, Nakayama KI, Lin HK (2012) The Skp2‐SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 149: 1098–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase‐3 by insulin mediated by protein kinase B. Nature 378: 785–789 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell 91: 231–241 [DOI] [PubMed] [Google Scholar]
- Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME (2000) 14‐3‐3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell 6: 41–51 [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999) Activation of nitric oxide synthase in endothelial cells by Akt‐dependent phosphorylation. Nature 399: 601–605 [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3‐kinases as regulators of growth and metabolism. Nat Rev Genet 7: 606–619 [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC (1999) Regulation of endothelium‐derived nitric oxide production by the protein kinase Akt. Nature 399: 597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangula NR, Maddika S (2013) WD repeat protein WDR48 in complex with deubiquitinase USP12 suppresses Akt‐dependent cell survival signaling by stabilizing PH domain leucine‐rich repeat protein phosphatase 1 (PHLPP1). J Biol Chem 288: 34545–34554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 18: 13–24 [DOI] [PubMed] [Google Scholar]
- Jaaskelainen T, Makkonen H, Palvimo JJ (2011) Steroid up‐regulation of FKBP51 and its role in hormone signaling. Curr Opin Pharmacol 11: 326–331 [DOI] [PubMed] [Google Scholar]
- Jaaskelainen T, Makkonen H, Visakorpi T, Kim J, Roeder RG, Palvimo JJ (2012) Histone H2B ubiquitin ligases RNF20 and RNF40 in androgen signaling and prostate cancer cell growth. Mol Cell Endocrinol 350: 87–98 [DOI] [PubMed] [Google Scholar]
- Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA (2003) Identification of a proline‐rich Akt substrate as a 14‐3‐3 binding partner. J Biol Chem 278: 10189–10194 [DOI] [PubMed] [Google Scholar]
- Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW (2008) Regulation of phosphorylation of Thr‐308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem 283: 1882–1892 [DOI] [PubMed] [Google Scholar]
- Li X, Liu J, Gao T (2009) beta‐TrCP‐mediated ubiquitination and degradation of PHLPP1 are negatively regulated by Akt. Mol Cell Biol 29: 6192–6205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lou Z, Wang L (2011) The role of FKBP5 in cancer aetiology and chemoresistance. Br J Cancer 104: 19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Stevens PD, Yang H, Gulhati P, Wang W, Evers BM, Gao T (2013) The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP‐dependent attenuation of Akt signaling in colon cancer. Oncogene 32: 471–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cheng Sun SH, Sun SJ, Huang C, Hu HH, Jin YB, Qiu ZJ (2010) Phosph‐Akt1 expression is associated with a favourable prognosis in pancreatic cancer. Ann Acad Med Singapore 39: 548–554 [PubMed] [Google Scholar]
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, Wan L, Singh A, Zhai B, Yuan M, Wang Z, Gygi SP, Lee TH, Lu KP, Toker A, Pandolfi PP et al (2014) Cell‐cycle‐regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508: 541–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Fang Y, Zhang H, Deng M, Gao B, Niu N, Yu J, Lee S, Kim J, Qin B, Xie F, Evans D, Wang L, Lou W, Lou Z (2016) HEATR1 Negatively Regulates Akt to Help Sensitize Pancreatic Cancer Cells to Chemotherapy. Cancer Res 76: 572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Manning BD, Cantley LC (2003) Targeting the PI3K‐Akt pathway in human cancer: rationale and promise. Cancer Cell 4: 257–262 [DOI] [PubMed] [Google Scholar]
- Magee JA, Chang LW, Stormo GD, Milbrandt J (2006) Direct, androgen receptor‐mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology 147: 590–598 [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex‐2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3‐kinase/akt pathway. Mol Cell 10: 151–162 [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morbidelli L, Donnini S, Ziche M (2003) Role of nitric oxide in the modulation of angiogenesis. Curr Pharm Des 9: 521–530 [DOI] [PubMed] [Google Scholar]
- Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, Wu H (2011) Cell autonomous role of PTEN in regulating castration‐resistant prostate cancer growth. Cancer Cell 19: 792–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SC, Rimerman RA, Toran EJ, Chen S, Prapapanich V, Butts RN, Smith DF (1997) Molecular cloning of human FKBP51 and comparisons of immunophilin interactions with Hsp90 and progesterone receptor. Mol Cell Biol 17: 594–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J, Park BC, Accili D (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin‐sensitive pathway. J Biol Chem 274: 15982–15985 [DOI] [PubMed] [Google Scholar]
- Padmanabhan S, Mukhopadhyay A, Narasimhan SD, Tesz G, Czech MP, Tissenbaum HA (2009) A PP2A regulatory subunit regulates C. elegans insulin/IGF‐1 signaling by modulating AKT‐1 phosphorylation. Cell 136: 939–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CM, Muilenburg D, Bowles TL, Virudachalam S, Bold RJ (2010) The role of Akt activation in the response to chemotherapy in pancreatic cancer. Anticancer Res 30: 3279–3289 [PMC free article] [PubMed] [Google Scholar]
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L (2009) FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16: 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Peso L, Gonzalez‐Garcia M, Page C, Herrera R, Nunez G (1997) Interleukin‐3‐induced phosphorylation of BAD through the protein kinase Akt. Science 278: 687–689 [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Vogel RO, Puigserver P (2011) Clk2 and B56beta mediate insulin‐regulated assembly of the PP2A phosphatase holoenzyme complex on Akt. Mol Cell 41: 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano S, Xiao Y, Nakaya M, D'Angelillo A, Chang M, Jin J, Hausch F, Masullo M, Feng X, Romano MF, Sun SC (2015) FKBP51 employs both scaffold and isomerase functions to promote NF‐kappaB activation in melanoma. Nucleic Acids Res 43: 6983–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 307: 1098–1101 [DOI] [PubMed] [Google Scholar]
- Sharma N, Nanta R, Sharma J, Gunewardena S, Singh KP, Shankar S, Srivastava RK (2015) PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 6: 32039–32060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staibano S, Mascolo M, Ilardi G, Siano M, De Rosa G (2011) Immunohistochemical analysis of FKBP51 in human cancers. Curr Opin Pharmacol 11: 338–347 [DOI] [PubMed] [Google Scholar]
- Stathis A, Moore MJ (2010) Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol 7: 163–172 [DOI] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument‐Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT (1998) Protein kinase B kinases that mediate phosphatidylinositol 3,4,5‐trisphosphate‐dependent activation of protein kinase B. Science 279: 710–714 [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3‐Kinase AKT pathway in human cancer. Nat Rev Cancer 2: 489–501 [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Tomita Y, Hoshida Y, Morooka T, Nagano H, Dono K, Umeshita K, Sakon M, Ishikawa O, Ohigashi H, Nakamori S, Monden M, Aozasa K (2004) Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res 10: 2846–2850 [DOI] [PubMed] [Google Scholar]
- Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, Lin HK (2009) The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325: 1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhang QX, Pei DS, Xu F, Li Y, Yu RT (2015) FK506‐binding protein 5 inhibits proliferation and stimulates apoptosis of glioma cells. Arch Med Sci 11: 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jones A, Joo HY, Zhou D, Cao Y, Chen S, Erdjument‐Bromage H, Renfrow M, He H, Tempst P, Townes TM, Giles KE, Ma L, Wang H (2013) USP49 deubiquitinates histone H2B and regulates cotranscriptional pre‐mRNA splicing. Genes Dev 27: 1581–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhiqiang Z, Qinghui Y, Yongqiang Z, Jian Z, Xin Z, Haiying M, Yuepeng G (2012) USP1 regulates AKT phosphorylation by modulating the stability of PHLPP1 in lung cancer cells. J Cancer Res Clin Oncol 138: 1231–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File