

Abstract
Key points
β‐Adrenergic stimulation enhances Ca2+ entry via L‐type CaV1.2 channels, causing stronger contraction of cardiac muscle cells.
The signalling pathway involves activation of protein kinase A (PKA), but the molecular details of PKA regulation of CaV1.2 remain controversial despite extensive research.
We show that PKA regulation of CaV1.2 can be reconstituted in Xenopus oocytes when the distal C‐terminus (dCT) of the main subunit, α1C, is truncated.
The PKA upregulation of CaV1.2 does not require key factors previously implicated in this mechanism: the clipped dCT, the A kinase‐anchoring protein 15 (AKAP15), the phosphorylation sites S1700, T1704 and S1928, or the β subunit of CaV1.2. The gating element within the initial segment of the N‐terminus of the cardiac isoform of α1C is essential for the PKA effect.
We propose that the regulation described here is one of two or several mechanisms that jointly mediate the PKA regulation of CaV1.2 in the heart.
Abstract
β‐Adrenergic stimulation enhances Ca2+ currents via L‐type, voltage‐gated CaV1.2 channels, strengthening cardiac contraction. The signalling via β‐adrenergic receptors (β‐ARs) involves elevation of cyclic AMP (cAMP) levels and activation of protein kinase A (PKA). However, how PKA affects the channel remains controversial. Recent studies in heterologous systems and genetically engineered mice stress the importance of the post‐translational proteolytic truncation of the distal C‐terminus (dCT) of the main (α1C) subunit. Here, we successfully reconstituted the cAMP/PKA regulation of the dCT‐truncated CaV1.2 in Xenopus oocytes, which previously failed with the non‐truncated α1C. cAMP and the purified catalytic subunit of PKA, PKA‐CS, injected into intact oocytes, enhanced CaV1.2 currents by ∼40% (rabbit α1C) to ∼130% (mouse α1C). PKA blockers were used to confirm specificity and the need for dissociation of the PKA holoenzyme. The regulation persisted in the absence of the clipped dCT (as a separate protein), the A kinase‐anchoring protein AKAP15, and the phosphorylation sites S1700 and T1704, previously proposed as essential for the PKA effect. The CaVβ2b subunit was not involved, as suggested by extensive mutagenesis. Using deletion/chimeric mutagenesis, we have identified the initial segment of the cardiac long‐N‐terminal isoform of α1C as a previously unrecognized essential element involved in PKA regulation. We propose that the observed regulation, that exclusively involves the α1C subunit, is one of several mechanisms underlying the overall PKA action on CaV1.2 in the heart. We hypothesize that PKA is acting on CaV1.2, in part, by affecting a structural ‘scaffold’ comprising the interacting cytosolic N‐ and C‐termini of α1C.
Keywords: calcium channel, heterologous expression, protein kinase A, regulation
Key points
β‐Adrenergic stimulation enhances Ca2+ entry via L‐type CaV1.2 channels, causing stronger contraction of cardiac muscle cells.
The signalling pathway involves activation of protein kinase A (PKA), but the molecular details of PKA regulation of CaV1.2 remain controversial despite extensive research.
We show that PKA regulation of CaV1.2 can be reconstituted in Xenopus oocytes when the distal C‐terminus (dCT) of the main subunit, α1C, is truncated.
The PKA upregulation of CaV1.2 does not require key factors previously implicated in this mechanism: the clipped dCT, the A kinase‐anchoring protein 15 (AKAP15), the phosphorylation sites S1700, T1704 and S1928, or the β subunit of CaV1.2. The gating element within the initial segment of the N‐terminus of the cardiac isoform of α1C is essential for the PKA effect.
We propose that the regulation described here is one of two or several mechanisms that jointly mediate the PKA regulation of CaV1.2 in the heart.
Abbreviations
- AKAP
A‐kinase‐anchoring protein
- AR
adrenergic receptor
- cAMP
cyclic AMP
- CFTR
cystic fibrosis transmembrane conductance regulator
- CS
catalytic subunit
- CT
C‐terminus
- dCT
distal CT
- IBa
Ba2+ current
- LNT
long‐N‐terminus
- NT
N‐terminus
- PKA
cAMP‐dependent protein kinase
- PKI
protein kinase inhibitor protein
- RS
regulatory subunit
- SNT
short‐N‐terminus
Introduction
The sympathetic system regulates cardiac function, largely through the β‐adrenergic receptors (β‐ARs), causing multiple changes in Ca2+ handling and consequently in force, rate and duration of cardiac contraction (Bers, 2008). Increased β‐adrenergic stimulation is harmful in chronic heart failure, and β‐AR‐blocking drugs are used to treat a wide range of cardiovascular disorders (Florea & Cohn, 2014). In the classical signalling scheme, adrenaline (epinephrine) or noradrenaline (norepinephrine) binding to β‐AR activates the G protein Gαs, stimulating adenylyl cyclase, which increases intracellular levels of cyclic AMP (cAMP). cAMP activates protein kinase A (cAMP‐dependent protein kinase, PKA) by dissociating the regulatory subunits (PKA‐RS) from the catalytic subunits (PKA‐CS). PKA‐CS phosphorylates key proteins involved in myocyte contraction, such as the L‐type voltage gated Ca2+ channel CaV1.2 in the cardiac sarcolemmal T‐tubules, major Ca2+‐handling proteins of the sarcoplasmic reticulum, certain proteins of the myofilaments, etc. (Wehrens et al. 2005; Bers, 2008; Vinogradova & Lakatta, 2009).
The CaV1.2 channel mediates the fundamental mechanism that initiates the cardiac cell contraction: depolarization‐induced Ca2+ entry via CaV1.2 triggers the Ca2+‐induced Ca2+ release from the sarcoplasmic reticulum, which underlies the excitation–contraction coupling (Bers, 2000). β‐Adrenergic stimulation, through the activation of PKA, increases Ca2+ entry via CaV1.2 by enhancing the channel's gating by voltage and increasing its open probability (Osterrieder et al. 1982; Reuter et al. 1982; Trautwein et al. 1982; Cachelin et al. 1983; Yue et al. 1990). The increased Ca2+ influx leads to strengthening of cardiac contraction. This β‐adrenergic action is an important mechanism for regulation of heartbeat by positive inotropic, positive chronotropic and positive lusitropic effects, taking place under a variety of physiological conditions. Nevertheless, despite extensive research, the mechanism linking β‐AR and PKA activation to CaV1.2 enhancement has not been fully elucidated (reviewed in: Weiss et al. 2013; Hofmann et al. 2014; Catterall, 2015; Morrow & Marx, 2015).
CaV1.2 in the heart comprises three subunits: the pore‐forming α1C, the extracellular α2/δ, and the cytosolic CaVβ subunit (Catterall, 2000; Dolphin, 2012; Campiglio & Flucher, 2015). Both α1C and CaVβ are phosphorylated by PKA in vitro and in vivo, and the leading concept is that PKA regulation is mediated by phosphorylation of specific sites in the channel, mainly in the C‐terminus (CT) of α1C (reviewed by Catterall, 2015).
The majority of α1C molecules in cardiomyocytes are proteolytically cleaved around amino acid (a.a.) 1800 (Hulme et al. 2005, 2006); the exact cleavage site is uncertain (Yang et al. 2013). The clipped distal CT (dCT) remains attached non‐covalently to the truncated α1C, and reduces current density and voltage sensitivity (Wei et al. 1994; Klockner et al. 1997; Gerhardstein et al. 2000; Gao et al. 2001; Crump et al. 2013). The dCT and its cleavage have been postulated to play an important role in β‐AR modulation of CaV1.2 in cardiomyocytes (Ganesan et al. 2006; Fuller et al. 2010; Domes et al. 2011; Fu et al. 2011, 2013, 2014).
Several mechanisms which assigned crucial roles to various PKA phosphorylation sites in α1C and CaVβ subunits, based on biochemical and heterologous expression data, have been proposed but remained controversial. Inconsistencies between studies prevail, even with the same mammalian cell line such as HEK (reviewed in Weiss et al. 2013), presumably due to variability of research methodologies or model systems. The importance of the cardiac cellular environment is not fully understood. Recent studies with animal models carrying targeted mutations and in transfected cardiac cells have challenged most of the proposed mechanisms (Ganesan et al. 2006; Lemke et al. 2008; Brandmayr et al. 2012; Jones et al. 2012; Yang et al. 2013).
An important proposed mechanism emerged from the reconstitution of CaV1.2 upregulation by PKA in the tsA‐201 mammalian cell line (Fuller et al. 2010). Essential requirements for reconstituting the PKA regulation were the truncation of the dCT of α1C, the presence of the clipped part of the CT (amino acids (a.a.) 1800–2171) as a separate protein, and the presence of the cardiac A‐kinase‐anchoring protein AKAP15 (also termed AKAP18α; Fraser et al. 1998). Mutagenesis suggested that serine 1700 (S1700) and threonine 1704 (T1704) were necessary for the PKA effect in tsA‐201 cells, and the proposed model attributed the CaV1.2 enhancement to a relief of dCT's inhibitory effect upon PKA phosphorylation of S1700 (Fuller et al. 2010). Follow‐up studies in a mouse model showed that replacement of serine 1700 or of threonine 1704 by an alanine residue reduced both ‘basal’ Ca2+ currents and sensitivity to β‐AR agonists (Fu et al. 2013, 2014). Nevertheless, the two mutations did not fully eliminate the β‐adrenergic regulation (Fu et al. 2013). A prominent β‐adrenergic stimulation of CaV1.2, similar to that seen in wild‐type cardiomyocytes, also persisted in a knock‐in genetically engineered mouse model with the alanine mutations S1700A and T1704A (Yang et al. 2013). The exclusive requirement for AKAP15, or another PKA‐anchoring protein, AKAP79, could not be confirmed either, since β‐adrenergic regulation endured in AKAP knock‐out mice (Nichols et al. 2010; Jones et al. 2012). These complex, controversial results may indicate redundancy, i.e. the existence of multiple pathways, of PKA control of CaV1.2 gating in native cardiomyocytes (Weiss et al. 2013). According to this view, some of the pathways suggested by heterologous studies may, in fact, mediate part of the PKA action (such as the phosphorylation of S1700), but their lack in genetically engineered animal models may be compensated by other pathways. Therefore, we posit that, whereas validation in genetically engineered animal models remains essential, further heterologous studies are needed to identify the putative novel or undetected components of the intricate mechanism of PKA regulation of CaV1.2.
We sought to utilize the experimental approach developed in tsA‐201 cells (Fuller et al. 2010) to reconstitute and better understand the PKA regulation of CaV1.2 in Xenopus oocytes. Oocytes, like other heterologous systems, lack the specific cellular architecture of cardiac cells which is undoubtedly important for reconstitution of the complete β‐AR cascade. However, they have been successfully used for identification of phosphorylation end‐points and additional necessary structural elements in PKA regulation of ion channels such as voltage‐gated Na+ channels and the cystic fibrosis transmembrane conductance regulator (CFTR) (e.g. Frohnwieser et al. 1997; Wilkinson et al. 1997; Csanady et al. 2005).
We found that, like in tsA‐201, cleavage of dCT was necessary for upregulation of the PKA‐dependent CaV1.2 current. However, PKA‐induced enhancement of CaV1.2 currents did not require the presence of either dCT as a separate protein, AKAP15, CaVβ, or the phosphorylation sites S1700 and T1704. The gating element located in the beginning of the N‐terminus (NT) of the cardiac isoform of α1C was necessary for cAMP‐dependent upregulation of CaV1.2 currents in Xenopus oocytes. These findings suggest that both cytosolic N‐ and C‐termini of α1C participate in the PKA regulation of the channel, pointing to a new direction in the search for pathway(s) of PKA regulation of cardiac CaV1.2.
Methods
Ethical approval
Experiments were approved by Tel Aviv University Institutional Animal Care and Use Committee (permits M‐08‐081 and M‐13‐002). Adult female Xenopus laevis frogs were purchased from Xenopus‐1 (Dexter, MI, USA), transported to Israel and maintained according to the Guidance on the housing and care of the African clawed frog Xenopus laevis (Research Animals Department, RSPCA, UK). The frogs were housed in Tel Aviv University Medical School Animal Facility and handled essentially as described (Kahanovitch et al. 2014). Female frogs were maintained at 20 ± 2°C on a 10 h light/14 h dark cycle. Frogs were anaesthetized in a 0.17% solution of procainemethanesulphonate (MS222), and portions of ovary were removed through an incision on the abdomen. The incision was sutured and the animal was held in a separate tank until it had fully recovered from the anaesthesia. Afterwards it was returned to a separate tank for post‐operational animals. The animals did not show any signs of post‐operational distress and were allowed to recover for at least 3 months until the next surgery. Following the final collection of oocytes, anaesthetized frogs were killed by decapitation and double pithing.
DNA constructs and RNA
The DNA constructs used were: the cardiac long‐N‐terminus (LNT) isoform of α1C from rabbit (Mikami et al. 1989; GenBank: X15539) and the corresponding mouse α1C isoform (NP_001242928; Link et al. 2009), CaVβ2b (GenBank: X64297.1; CaVβ2N4 according to the comprehensive nomenclature, Hofmann et al. 2014), α2δ1 (GenBank: M21948), AKAP15 (GenBank: AF047716) and enhanced green fluorescent protein (EGFP)‐fused Ht31 (Carr et al. 1992; Lynch et al. 2005). The short‐NT (SNT) α1C used here was the NTSL construct (Kanevsky & Dascal, 2006) in which the initial segment of the long‐NT (LNT) α1C (encoding the first 46 a.a.) was replaced with that encoded by exon 1 (16 a.a. long). DNAs of rabbit and mouse α1CΔ1821, in which the dCT is truncated, were prepared by the removal of the cDNA segment encoding the dCT from the plasmid and introducing a stop codon after valine 1821, by PCR. NT constructs of rabbit α1C were constructed by subcloning into a LNT α1CΔ1821 template (Shistik et al. 1998; Kanevsky & Dascal, 2006). Mouse α1CΔ5Δ1821 DNA (deletion of a.a. 2–5 in the mouse α1CΔ1821 protein) was constructed by PCR procedures and inserted into the pMXT vector. All mouse α1C constructs additionally contained the double mutation T1066Y, Q1070M, which renders the channel dihydropyridine insensitive but does not affect the regulation by PKA (Yang et al. 2013). The following mutations were made in CaVβ2b. To construct CaVβ‐4A, serines 296, 479/80 and 576 were replaced by alanines. To construct CaVβ2b‐CTtrunc, a stop codon was introduced after the nucleotide position corresponding to amino acid 471, together with the point mutation S296A (see also Pankonien et al. 2012). Mutations were introduced by PCR using the standard site‐directed mutagenesis. All constructs were confirmed by DNA sequencing. CFTR (GenBank: M28668) was in a pSP64 vector. All other cDNA constructs used for RNA synthesis were inserted into the pGEM‐GSB (Shistik et al. 1998) or pGEM‐HJ vectors, which are derivatives of pGEM‐HE (Liman et al. 1992) and contain 5′ and 3′ UTR from Xenopus β‐globin. The RNAs were prepared using a standard procedure described previously (Dascal & Lotan, 1992). The amount of injected RNA, per oocyte, for the full‐length α1C was 5 ng. For the α1CΔ1821 constructs, the injected RNA amounts were: 0.5–1 ng for LNT, LNTΔ20–46, LNTΔ5 and LNTΔ5Δ20–46, and 0.3–0.5 ng RNA for LNTΔ20, LNTΔ139, SNT, SNT+a.a.2–5 and SNT+a.a.20–46. When CaVβ was not expressed, we injected 7 ng RNA of α1CΔ1821 and α2δ1.
The following DNA constructs were used for protein purification in Escherichia coli: His‐tagged Cα‐subunit of PKA (His‐PKA‐CS, GenBank: NM_008854.5), glutathion‐S‐transferase (GST)‐fused PKA‐RIIβ (GST‐PKA‐RS; GenBank: NM_001030020) and His‐tagged human protein kinase inhibitor protein (PKI) (Olsen & Uhler, 1991) (GenBank: S76965.1).
Protein purification and pull‐down assays
Proteins were expressed in E. coli (BL21‐DE3). For PKA‐CS and PKI, respectively, cultures were grown in YT medium at 37°C to an A 600 of 0.6–0.8 and induced with 0.5 or 1 mm isopropyl β‐d‐thiogalactopyranoside for 6.5 h at 24°C (PKA‐CS) or for 3 h at 37°C (PKI), before being collected by centrifugation and stored frozen. Pellets were resuspended in buffer I for PKA‐CS (in mm: 50 KH2PO4, 20 Tris‐HCl, 100 NaCl, 5 β‐mercaptoethanol, pH 8.0), or buffer II for PKI (in mm: 20 Tris‐HCl, 300 NaCl, 0.1% Triton X‐100, pH 8.0) with the addition of 15 U ml−1 DNase‐I, 10 mg lysozyme, 1 mm phenylmethanesulfonyl fluoride (PMSF) and Protease Inhibitor Cocktail (Roche, Mannheim, Germany). Cells were lysed using a microfluidizer and clarified by centrifugation. Clarified lysate was loaded onto a Ni‐NTA column followed by washing (20 mm) and elution (80 mm) with imidazole. Then the protein was subjected to size‐exclusion chromatography on Superdex‐75 in buffer III–PKA‐CS (in mm: 20 KH2PO4, 20 KCl, 2 DTT, pH 7.5), or buffer IV–PKI (in mm: 20 Tris‐HCl, 300 NaCl, 2 DTT, pH 8). PKI was concentrated in a Vivaspin concentrator with 5000 Da cutoff (Sartorius, Göttingen, Germany). Buffer IV–PKI was used for PKI injection into oocytes (Figs 2 and 3).
Figure 2. Purified recombinant PKI inhibits the interaction between PKA‐CS and PKA‐RS and reduces basal I Ba .
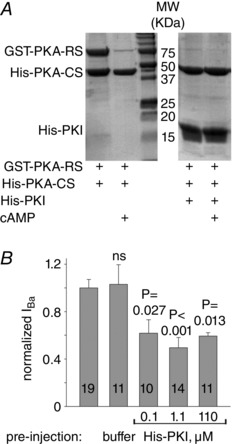
A, interaction of purified His‐PKA‐CS (10 μg, 0.8 μm) with GST‐PKA‐RS (RIIβ, 25 μg, 1.1 μm) was tested in the absence or presence of His‐PKI (25 μg, 7.6 μm), with and without 40 μm cAMP. His‐tagged proteins were pulled down with nickel beads, eluates were run on SDS‐PAGE gel and stained with Coomassie Blue. A representative experiment out of 4 is shown. B, PKI reduces basal I Ba. Oocytes expressing CaV1.2Δ1821 were pre‐injected with Tris‐NaCl buffer, His‐PKI, or not pre‐injected. The estimated final concentrations of PKI within the oocyte, assuming a 1 μl cell volume, are shown in μm. Maximal currents in each cell, obtained using an I–V protocol of 10 mV depolarization steps from −60 to 50 mV, were averaged and normalized to control group of the same day. Summary from 2 experiments is shown. Statistical significance was determined using one‐way ANOVA followed by Bonferroni t test.
Figure 3. Rp‐cAMPS and PKI inhibit cAMP‐dependent upregulation of I Ba .
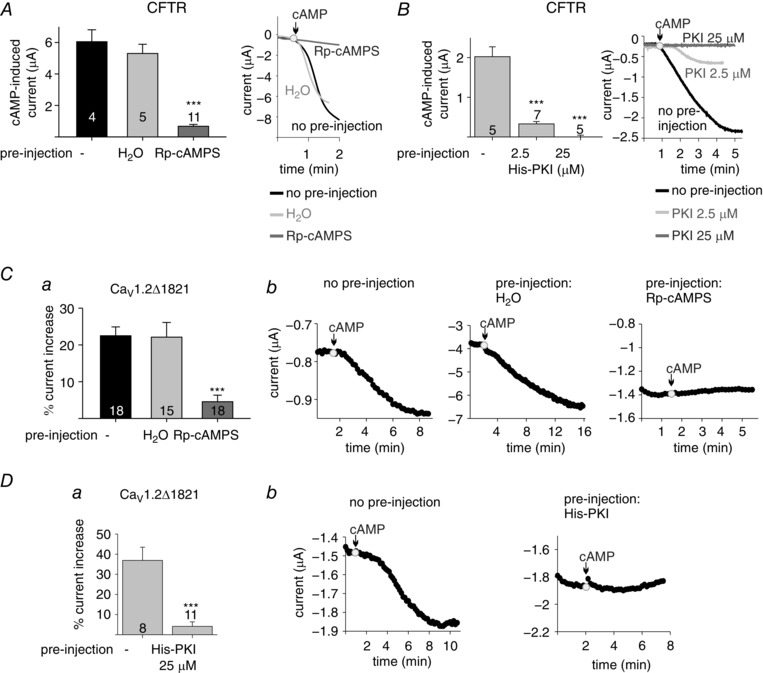
A and B, effects of PKA blockers on CFTR. Oocytes expressing the CFTR channel were used without any pretreatment or pre‐injected with 50 nl of H2O, Rp‐cAMPS (2 mm) (A) or PKI (B). Oocytes were held at −80 mV and currents were evoked in ND96 solution by injection of cAMP (200 μm). Left, averaged evoked CFTR currents following cAMP injection. Right, representative time course of current changes after cAMP injection. The experiments are from a representative batch of oocytes, out of 3. C and D, effects of PKA blockers on CaV1.2Δ1821. Oocytes expressing CaV1.2Δ1821 were used without pretreatment or pre‐injected with H2O, Rp‐cAMPS (2 mm) (C) or PKI (D). Following pre‐injection, cAMP (200 μm)‐dependent changes in I Ba were recorded as in Fig. 1. Averaged % increase in I Ba per oocyte, following cAMP injection (from 3 experiments) is shown in a; representative time courses of currents following cAMP injection are shown in b. In the set of experiments with Rp‐cAMPS, the cAMP‐induced increase in CaV1.2Δ1821 I Ba was relatively low (22.5% in the control group, less than the average 35.7% summarized from all experiments in Fig. 1 F), which could reflect low endogenous PKA levels or a high basal phosphorylation of the channel. Statistical significance was determined using one‐way ANOVA followed by Bonferroni test, except Da where a t test was applied. *** P < 0.001.
For GST‐PKA‐RS, the pellet was resuspended in PBS supplemented with 0.1% Triton X‐100, 15 U ml−1 DNase‐I, 10 mg of lysozyme, and 1 mm PMSF. The cells were homogenized and lysed with a microfluidizer, and centrifuged for 1 h. The supernatant was loaded onto a glutathione column pre‐equilibrated with PBS. Cell extract was loaded on the column and washed with PBS. Protein was eluted with buffer containing (in mm): 50 Tris pH 8.0, 100 NaCl, and 10 glutathione.
Pull‐down assays were performed with Ni‐NTA resin (Thermo Scientific, Waltham, MA, USA) and washed with buffer containing (in mm): 150 KCl, 50 Tris, 0.6 MgCl2, 1 CaCl2, 0.5% CHAPS, and 10 imidazole, pH 7.4. Elution was done with 100 mm imidazole. Eluates were run on SDS‐PAGE gel and stained with Coomassie Blue.
Electrophysiology
Oocytes were defolliculated by collagenase, injected with RNA and incubated for 2–3 days before recording at 20–22°C in NDE solution (in mm: 96 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2, 5 Hepes, 2.5 pyruvic acid; plus 50 mg l−1 gentamycin).
Whole‐cell Ba2+ currents (I Ba) in oocytes were measured using the two‐electrode voltage clamp technique (see Fig. 1 A) with a GeneClamp 500 amplifier (Molecular Devices, Sunnyvale, CA, USA). I Ba was recorded in most cases by 20 ms depolarizing pulses from a resting potential of −80 mV to 20 mV, with a 10 s interval between sweeps, in 40 mm Ba2+ solution (in mm: 40 Ba(OH)2, 50 NaOH, 2 KOH, and 5 Hepes, titrated to pH 7.5 with methanesulfonic acid). In some experiments (e.g. Fig. 7), for some α1C‐derived chimeras, I Ba amplitudes in 40 mm Ba2+ solution exceeded 5 μA. In such cases, to avoid artifacts resulting from oocyte series resistance and poor space clamp, recordings were made in 2 mm Ba2+ solution (in mm: 2 Ba(OH)2, 96 NaOH, 2 KOH, and 5 Hepes, titrated to pH 7.5 with methanesulfonic acid). These measurements were used to assess cAMP‐induced changes in I Ba amplitude only, and have not been included in amplitude summaries and activation curve fits. In the current–voltage (I–V) protocols, currents were elicited by 20 ms pulses from the holding potential −80 mV to voltages from −50 to +60 mV with 10 mV intervals and 10 s between sweeps. Currents measured in the presence of 200 μm Cd2+ were subtracted from total I Ba (Fig. 4 D and E) to yield the net I Ba. CFTR currents were measured at −80 mV in ND96 solution (in mm: 96 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2, 5 Hepes, pH 7.6).
Figure 1. CaV1.2Δ1821 is upregulated by PKA.
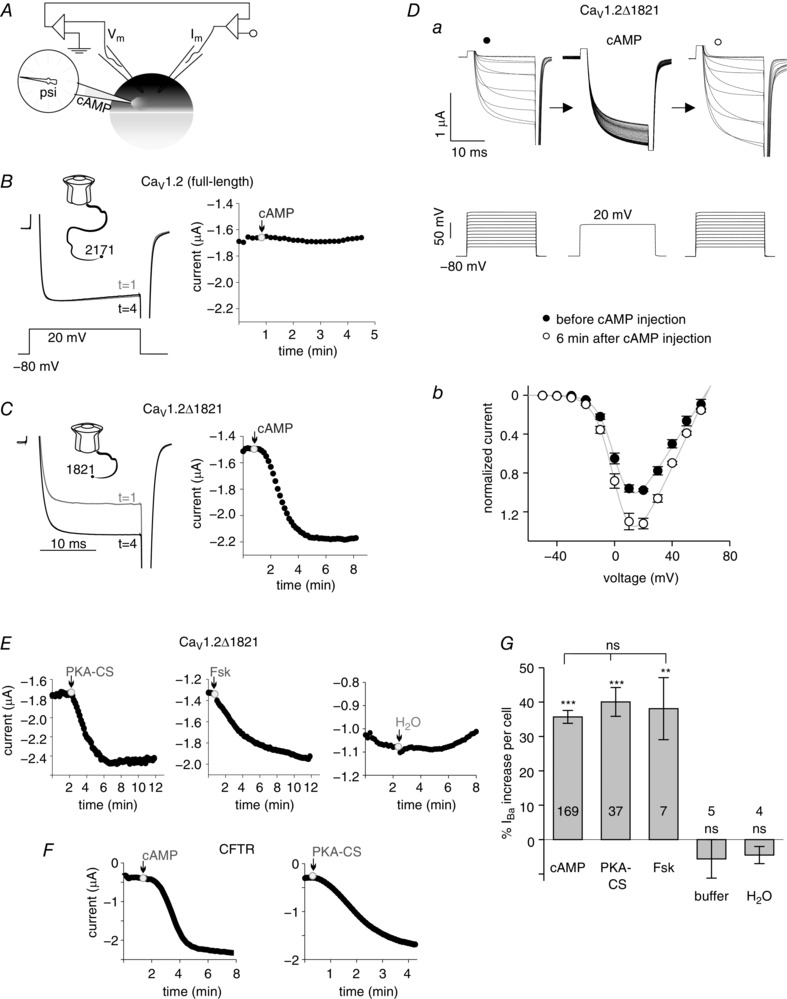
A, illustration of the setup. The oocyte was impaled with current and voltage electrodes and the injection micropipette. B and C, cAMP effects on CaV1.2. Left panels show I Ba of full‐length CaV1.2 (B) and CaV1.2Δ1821 (C) before cAMP injection (at time 1 min) and 4 min after cAMP injection, in representative cells. Insets above current traces show schematic drawings of the full‐length and truncated α1C, respectively. Lower inset in B shows the voltage step protocol. Right panels summarize the time course of cAMP effect in the same oocytes. cAMP injection is indicated by arrows. Final concentration in the oocyte was 200 μm. D, the I–V relations of CaV1.2Δ1821 in a representative cell. a, the stimulation protocol (lower panel) and the recorded currents (upper panel) before (left) and 6 min after cAMP (right) injection. The middle panel represents the sequence of currents recorded during the 6 min period following cAMP injection; the standard 20 ms voltage pulse was repeated every 10 s. b, normalized I–V curves (each point shows mean ± SEM), averaged from 5 oocytes. E, representative time course charts of I Ba, following injection of recombinant PKA‐CS, forskolin (Fsk) or water. F, representative time course charts of CFTR currents elicited by the injection of cAMP (left, 200 μm) or recombinant His‐PKA‐CS (right, 100 nm) into the oocyte. Currents were recorded at −80 mV in ND96 solution. G, summary of changes in I Ba in oocytes expressing CaV1.2Δ1821 following injection of the indicated compounds. Percentage change was calculated in each cell; number of cells tested is shown within bars. cAMP (200 μm; summary of 40 experiments); PKA‐CS (400 pm to 1 μm, 11 experiments); forskolin (100 μm, 1 experiment); Tris buffer (1 experiment); H2O (1 experiment). Statistical significance (indicated above the bars) of changes in I Ba was calculated for each treatment using paired t test. The comparison among all treatments using one‐way ANOVA showed no significant difference between groups injected with cAMP, PKA‐CS and forskolin. *** P < 0.001; ** P = 0.007; ns, not significant.
Figure 7. The initial segment of the long‐NT α1C is essential for the I Ba enhancement by cAMP.
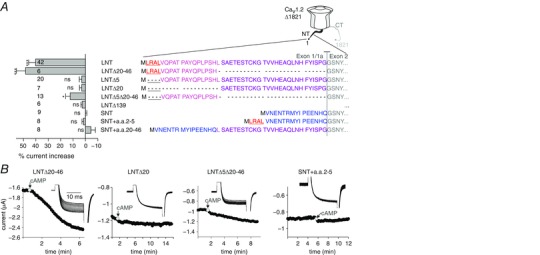
In this figure, LNT denotes the long‐NT initial segment of α1C encoded by exon 1a, and SNT denotes the short‐NT encoded by exon 1. A, regulation of chimeras and N‐terminal deletion mutants of rabbit α1CΔ1821 by cAMP (200 μm). Right panel, the N‐terminal amino acid sequences in α1CΔ1821 constructs used. Δ139 is a deletion of most of the NT, including exon 1a and most of exon 2. Colour codes: blue, SNT initial segment (encoded by exon 1); the other colours correspond to segments of the initial segment of LNT; first methionine is in black. Left panel shows the summary of cAMP effect (11 experiments). Statistical significance was determined by paired t test. *** P < 0.001; * P = 0.012; ns, not significant. B, representative time course charts of changes in I Ba following cAMP injection in some of the constructs. Insets show current traces in the same cells.
Figure 4. Overexpression of dCT reduces basal I Ba amplitude and depolarizes the activation curve.
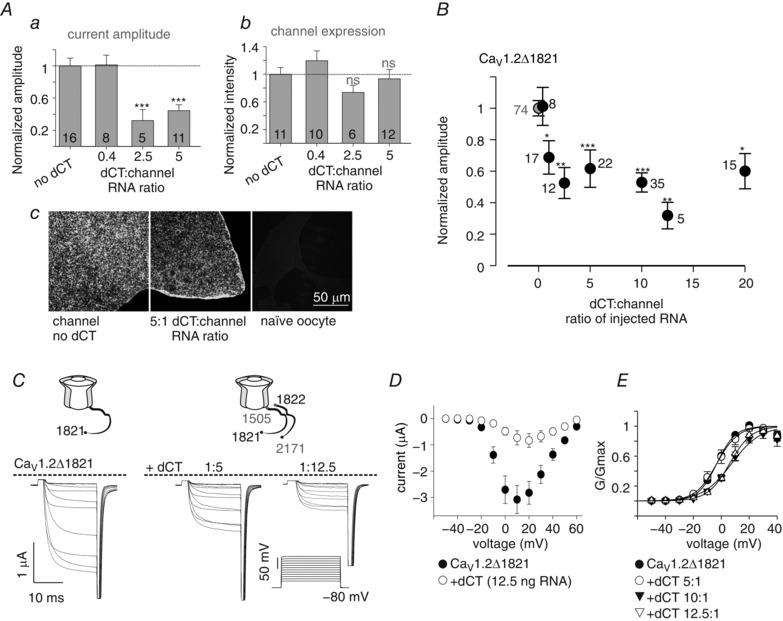
A, I Ba amplitudes at +20 mV (a) and plasma membrane expression (b) were measured in oocytes expressing CaV1.2Δ1821 together with increasing levels of dCT. In each experiment both current amplitude and surface labelling intensity were measured and normalized to the group expressing the channel alone. Statistical significance was determined by one‐way ANOVA followed by Bonferroni test. *** P < 0.001; ns, not significant. N = 3 experiments. c, examples of confocal images of giant membrane patches, where the channel levels were quantified with CaV1.2 antibody directed to an intracellular loop. B, dose‐dependent reduction of I Ba by coexpressed dCT. Oocytes were injected with 1 ng RNA CaV1.2Δ1821 together with increasing amounts of dCT RNA. The leftmost grey point represents channels expressed without dCT. In each experiment, I Ba amplitude was normalized to average I Ba in the control group expressing the channel alone. N = 11 experiments. Statistical significance was determined by one‐way ANOVA followed by Bonferroni test. * P < 0.05; ** P < 0.01; *** P < 0.001. C and D, current–voltage (I–V) relationships in oocytes expressing 1 ng CaV1.2Δ1821 and no dCT or 5 or 12.5 ng dCT RNA, from a representative experiment (n = 5–7 oocytes). Representative records of net I Ba (C) and averaged I–V curves (D). E, averaged conductance–voltage relationships from 3 experiments (n = 25 without dCT, and n = 5–9 with the different doses of dCT RNA). The Boltzmann equation fit parameters are shown in Table 2.
I–V curves were fitted to the Boltzmann equation in the form
where G max is the maximal Ba2+ conductance, V m is the membrane voltage, V rev is the reversal potential of the current, K a is the slope factor and V a is half‐maximum activation voltage. The parameters obtained for G max and V rev were then used to calculate fractional conductance at each V m and to construct the conductance–voltage (G–V) curve using the equation.
Injection of cAMP and other substances
In experiments that used pre‐injection of the compounds (Figs 2 B and 3), oocytes were injected with 50 nl of the substance solution 5–30 min prior to recording. Injection during recording was done with sharp capillary glass micropipettes filled with the desired substances (Fig. 1 A). The compound was pressure‐injected into the oocyte as explained in the Results section. The micropipette tip was trimmed so that application of pressure during 1–2 s extruded about 10 nl (1% of oocyte volume). Current stabilized during 1–5 min following insertion of the injection micropipette. Artifacts (sharp shifts in current, e.g. the right panel in Fig. 3 D) following the insertion of the micropipette or injection of the compound were usually minor. Records with insertion or injection artifacts that exceeded 10% of I Ba amplitude were discarded. The final concentrations of injected substances in the oocytes is indicated in the legends. cAMP (Sigma, A6885) was diluted in H2O and kept in 1, 20 and 40 mm stock solutions. Injection of 10 nl of cAMP from these stocks gave final concentrations of 10, 200 and 400 μm in the oocyte, respectively. Purified bovine heart PKA‐CS (Sigma, P2645) was diluted in Tris‐Mg buffer (50 mm Tris‐HCl, 10 mm MgCl2, pH 7.5) to 16 units μl−1. Forskolin (Alomone Labs, Jerusalem, Israel) was diluted in DMSO. Rp‐cAMPS (Sigma, A165) was diluted in H2O.
Giant membrane patches
Giant excised patches of oocyte membrane were prepared as described (Singer‐Lahat et al. 2000). Oocytes were mechanically devitellinized using tweezers in a hypertonic solution (in mm: 6 NaCl, 150 KCl, 4 MgCl2, 10 Hepes, pH 7.6) and transferred onto a coverslip in EGTA‐containing ND96 solution (in mm: 96 NaCl, 2 KCl, 1 MgCl2, 5 Hepes, 5 EGTA, pH 7.6), with their animal pole facing the coverslip, for 30–45 min. The oocytes were then removed with a jet of solution using a Pasteur pipette, leaving a giant membrane patch attached to the coverslip, with the cytosolic part facing the medium, and the extracellular surface facing the coverslip. The coverslip was washed thoroughly with fresh ND96 solution, and fixated using 4% formaldehyde in EGTA‐containing ND96 solution for 30 min. Coverslips were mounted on a glass slide. Giant membrane patches were stained with primary antibody for α1C (1:200; ACC‐003, Alomone Labs), followed by incubation with Cy3 fused secondary antibody. The fluorescent labelling was examined by a confocal laser scanning microscope (Zeiss 510 META), using a ×63 oil‐immersion objective. Cy3 was excited by a 488 nm laser and the intensities were measured in a 560–569 nm window in the spectral mode. In each experiment, all oocytes from the different groups were studied using a constant set of imaging parameters. Net fluorescence intensity per unit area was obtained by subtracting an averaged background signal measured in the same way on the coverslip outside the oocytes.
Data presentation and statistics
Imaging data on protein expression (Fig. 4 A) have been normalized as described previously (Kanevsky & Dascal, 2006). Fluorescence intensity in each giant membrane patch was calculated relative to the average signal in the oocytes of the control group of the same experiment. This procedure yields average normalized intensity ± SEM in all treatment groups as well as in the control group.
For experiments that involved the injection of substances during current recording, percentage change was relative to the current before injection (which was taken as 100%), in the same cell. The statistical significance of amplitude changes was analysed using a paired t test on raw data if normally distributed (using the Kolmogorov–Smirnov test), otherwise a Wilcoxon test was performed. Net CFTR current was defined as (I after injection − I before injection).
Statistical analysis of data obtained from different test samples (e.g. comparison of currents in oocytes pre‐injected with different substances) was performed as follows. Two‐group comparisons were performed using Student's t test. Multiple group comparison was done with one‐way ANOVA if the data were normally distributed. ANOVA on ranks was performed whenever the data did not distribute normally. A Bonferroni post hoc test was performed for normally distributed data and Dunn's post hoc test otherwise. Unless specified otherwise, the data in the graphs are presented as mean ± SEM and numbers within or near the bars or symbols indicate number of cells tested (n). Statistical analysis was performed with SigmaPlot 11 (Systat Software Inc., San Jose, CA, USA).
Results
C‐terminal truncation of α1C is necessary for PKA upregulation of CaV1.2 in Xenopus oocytes
The majority of previous studies showed that full‐length α1C is not significantly upregulated by cAMP or PKA in Xenopus oocytes (Singer‐Lahat et al. 1994; Charnet et al. 1995), as well as in mammalian CHO, HEK or tsA‐201 cell lines (Perez Reyes et al. 1994; Zong et al. 1995; Fuller et al. 2010). In view of the emerging importance of the C‐terminal cleavage of α1C for PKA regulation (Fuller et al. 2010; Domes et al. 2011; Fu et al. 2011), we decided to re‐evaluate the effect of PKA on CaV1.2 in Xenopus oocytes using a dCT‐truncated α1C. We found that this channel is prominently upregulated by PKA (Fig. 1).
We injected cAMP (to activate the endogenous PKA), purified PKA‐CS, or other compounds as shown in Fig. 1 A, and monitored the ensuing changes in CaV1.2 currents. First, we inserted the voltage and the current electrodes and measured the Ba2+ current (I Ba) using the two‐electrode voltage clamp technique. I Ba was elicited with test pulses to +20 mV every 10 s and continuously monitored during all stages of the experiment. When the current amplitude stabilized, a third micropipette was inserted for pressure injection. The stability of the current amplitude was again verified before the injection of the substances.
We used two distinct α1C constructs: the full‐length rabbit cardiac α1C (Mikami et al. 1989) (2171 a.a., illustration in Fig. 1 B), and the C‐terminally truncated α1C lacking the dCT after valine 1821, α1CΔ1821 (1821 a.a., Fig. 1 C). α1C or α1CΔ1821 were expressed, along with the auxiliary subunits CaVβ2b and α2δ1, by injecting corresponding RNAs at a 1:1:1 ratio. The resulting channels are termed here CaV1.2 or CaV1.2Δ1821, respectively. Consistent with previous reports (Wei et al. 1994; Ivanina et al. 2000; Gao et al. 2001; Hulme et al. 2006), I Ba amplitudes in CaV1.2Δ1821 were higher compared with the full‐length CaV1.2. In order to maintain similar macroscopic currents, we injected different amounts of the channel RNA: 0.5–1 ng RNA for CaV1.2Δ1821 and 5 ng RNA for CaV1.2.
The modulation of I Ba following the injection of cAMP is illustrated in Fig. 1 B for CaV1.2 and in Fig. 1 C for CaV1.2Δ1821. The left panels in Fig. 1 B and C show representative current traces at two time points: t = 1 min, just before cAMP injection, and t = 4 min, 3 min after cAMP injection. The time course of changes in I Ba in the same cells is shown in the right panels in Fig. 1 B and C. As expected, the full length CaV1.2 I Ba was not modulated by cAMP. In contrast, I Ba of the C‐terminally truncated CaV1.2Δ1821 increased following cAMP injection. Figure 1 D shows the effect of cAMP on current–voltage (I–V) relations in a representative oocyte, illustrating the increase in whole‐cell I Ba (Fig. 1 Da) and the ensuing change in the I–V curve (Fig. 1 Db). An increase in CaV1.2Δ1821 I Ba was also recorded following the injection of purified PKA‐CS protein or the adenylyl cyclase activator, forskolin (Fig. 1 E).
We used the cystic fibrosis transmembrane conductance regulator (CFTR) as the positive control for endogenous PKA activation by cAMP, and for PKA‐dependent current modulation. CFTR is a chloride channel gated by PKA phosphorylation along with ATP binding (Welsh et al. 1992; Gadsby & Nairn, 1999). CFTR's Cl− current (I Cl) is robustly activated by PKA in Xenopus oocytes (Bear et al. 1991; Uezono et al. 1993). As expected, both injected cAMP and PKA‐CS activated CFTR and elicited large I Cl, usually between 1 and 4 μA (Fig. 1 F).
The upregulation in CaV1.2Δ1821 currents is summarized in Fig. 1 G as percentage increase from basal current to current at the time of maximal increase in I Ba. Time to maximal increase varied between 5 and 15 min among oocytes. An average 35.7 ± 1.9% increase in I Ba (n = 164; range: 6–100%) was observed following the injection of cAMP to a final concentration of 200 μm in the oocyte. After injections of purified bovine heart PKA‐CS (final concentration in the oocyte 0.16 units μl−1), I Ba increased by 43 ± 5.5% (n = 20). With recombinant His‐tagged PKA‐CS (see Fig. 2; final concentration 100 nm to 1 μm), the increase was 36 ± 4.7% (n = 17). Figure 1 G shows pooled data (40 ± 4.2% increase in I Ba, n = 37; range: 9–114%) for all PKA‐CS experiments.
Contrary to cAMP, injection of water or Tris‐Mg buffer did not increase I Ba of CaV1.2Δ1821 (Fig. 1 E–G). This indicates that the current enhancement of I Ba seen in Fig. 1 C–E is not an injection artifact. The small and statistically non‐significant negative change in I Ba reflects the occasional small rundown of I Ba observed following buffer or water injection. This could be happening because of a deterioration of the cell's condition, possibly caused by the insertion of micropipettes.
Our data suggest that the concentrations of cAMP and PKA‐CS that we used have produced maximal upregulation of rabbit CaV1.2Δ1821 and of CFTR. All three compounds used (cAMP, PKA‐CS or forskolin) produced a similar increase in CaV1.2Δ1821 current (Fig. 1 F). Furthermore, in a separate experiment we verified that a 20‐fold dilution of cAMP yielded a similar increase in I Ba: 33.8 ± 5.5% (n = 5) with 10 μm cAMP vs. 33.75 ± 5.1% (n = 4) with 200 μm cAMP in the oocyte. Similarly, 10–400 μm cAMP induced CFTR currents of comparable sizes (Table 1).
Table 1.
cAMP at 10 μm causes maximal activation of CFTR
| cAMP concentration (μm) | cAMP‐evoked CFTR current (nA) | n |
|---|---|---|
| 400 | −1484 ± 132 | 11 |
| 200 | −1776 ± 282 | 8 |
| 10 | −1610 ± 207 | 10 |
cAMP‐evoked CFTR current: summary of two experiments; the difference in effect between the three cAMP doses was not significant (P = 0.611, one‐way ANOVA).
We purified and used recombinant proteins: His‐tagged PKA‐CS; His‐tagged, highly specific heat‐stable protein kinase inhibitor protein (PKI) (Walsh et al. 1971; Olsen & Uhler, 1991); and GST‐tagged PKA‐RIIβ regulatory subunit (PKA‐RS). To test their functionality, we performed a pull‐down assay in which His‐tagged PKA‐CS, immobilized on nickel agarose beads, bound and pulled down PKA‐RS (Fig. 2 A). In the presence of cAMP, the PKA holoenzyme complex dissociates to a large degree (Beavo et al. 1974; Kim et al. 2007). In concordance, our pull‐down assay showed that in the presence of 40 μm cAMP, PKA‐CS did not pull‐down PKA‐RS. PKI is a PKA‐CS pseudo‐substrate that competes with the PKA‐RS on binding to the active site cleft in PKA‐CS and also blocks the catalytic subunit directly (Scott et al. 1985; Knighton et al. 1991; Kim et al. 2005). When added, His‐PKI completely eliminated the interaction between PKA‐CS and PKA‐RS (Fig. 2 A). These experiments attest to the quality and functionality of the recombinant proteins used in our experiments.
The basal I Ba may be composed of current from non‐phosphorylated and endogenously phosphorylated channels. To measure the contribution of phosphorylated CaV1.2Δ1821 to basal I Ba, we injected 156 μm His‐PKI, 5–20 min before measuring I Ba. This caused a 40% reduction in I Ba compared to oocytes pre‐injected with buffer alone (Fig. 2 B). Thus, about 40% of the amplitude may be due to endogenous PKA activity. To calculate the full span of the enhancing effect of PKA on CaV1.2 in our experiments, we took into account the PKA‐dependent part of the basal current. A 40% increase in current due to cAMP or PKA‐CS, on top of only 60% of the basal current, would yield a 1.4/0.6 = 2.3‐fold increase of rabbit α1CΔ1821 current due to PKA‐CS activity.
To further examine the specificity of the cAMP and PKA‐CS effects, we used the PKI protein (a direct inhibitor of PKA‐CS) and an indirect PKA inhibitor, the cAMP diastereoisomer Rp‐cAMPS, which competes with cAMP on the PKA‐RS binding sites and inhibits the dissociation of PKA‐CS from PKA‐RS (Anand et al. 2010). First, we validated the experimental protocol with CFTR as a PKA activation reporter. We pre‐injected the inhibitors, and 5–30 min later measured the cAMP‐induced CFTR Cl− currents. Rp‐cAMPS was injected to obtain a final concentration of 2 mm, which is comparable to 1–5 mm used by others to inhibit the β‐adrenergic regulation of CaV1.2 in whole‐cell experiments in cardiomyocytes (Hartzell et al. 1991; Hirayama & Hartzell, 1997). Figure 3 A shows that Rp‐cAMPS inhibited the cAMP effect on CFTR by 89%, while pre‐injection of H2O did not significantly alter the cAMP effect. Pre‐injection of purified His‐PKI to 2.5 μm (final concentration in the oocyte) yielded an 84% inhibition of cAMP‐induced CFTR current, and a complete inhibition was achieved at 25 μm His‐PKI (Fig. 3 B). This concentration range is similar to that employed by others who used PKI or PKI peptides to inhibit β‐adrenergic regulation of CaV1.2 in cardiomyocytes (Kameyama et al. 1986; Parsons et al. 1991; Hartzell et al. 1995; Skeberdis et al. 1997).
Next, we examined the effects of PKA inhibitors on cAMP regulation of CaV1.2Δ1821 using the same experimental protocol as for CFTR. Rp‐cAMPS inhibited 80% of the cAMP‐induced effect. Pre‐injection of water did not alter the cAMP effect (Fig. 3 C). Similarly, the cAMP‐induced increase in I Ba of CaV1.2Δ1821 was reduced by 89% when oocytes were pre‐injected with His‐PKI to a final concentration of 25 μm (Fig. 3 D). Taken together, the results so far suggest that cAMP‐induced upregulation of both CFTR and CaV1.2Δ1821 channel currents in oocytes is a PKA‐specific modulation that requires dissociation of PKA‐CS from PKA‐RS.
dCT and AKAP15 are not essential for cAMP upregulation of CaV1.2Δ1821 in Xenopus oocytes
The results of Fig. 1 demonstrate that truncation of the dCT of α1C is essential for the PKA effect on CaV1.2. However, the presence of the ‘clipped’ dCT seems non‐obligatory. To better understand the role of dCT in cAMP modulation of the truncated CaV1.2Δ1821 in Xenopus oocytes, we coexpressed CaV1.2Δ1821 with the dCT as a separate protein. We first examined whether the coexpression of dCT produces the two characteristic effects on the truncated CaV1.2: a reduction in maximal inward I Ba amplitude and a depolarizing shift of the activation (conductance–voltage, G–V) curve (Hulme et al. 2006; Fuller et al. 2010; Crump et al. 2013).
Coexpression of dCT, at variable ratios of RNA concentrations of dCT and CaV1.2Δ1821 (‘dCT:channel RNA ratio’), caused a large reduction of 60–70% in I Ba amplitude when using 2.5:1 to 5:1 dCT:channel RNA ratios (Fig. 4 Aa). The expression of dCT in the same batch of oocytes did not alter the channel density in the plasma membrane, as demonstrated using immunocytochemistry in giant excised plasma membrane patches (Fig. 4 Ab and c). Thus, the reduction in I Ba following coexpression of dCT resulted from a modification of the gating properties, and was not due to a change in channel expression. dCT titration using a wider range of RNA doses, while keeping the level of CaV1.2Δ1821 constant, revealed that current inhibition by dCT was dose dependent, reaching a maximal 50–60% reduction at 2.5:1 dCT:channel RNA ratio (Fig. 4 B and C).
The coexpressed dCT also produced the second expected effect, the depolarizing shift of the G–V curve, increasing the half‐maximal activation voltage (V a). However, this effect took place only at dCT:channel RNA ratios of ≥10:1 (Fig. 4 D and E, Table 2), much higher than required for the maximal effect of dCT on I Ba amplitude. Evidently, the expression of the dCT protein further increased at RNA ratios higher than 5:1, as it produced an effect not seen at lower RNA doses. Thus, the saturation of dCT action on I Ba amplitude at a 2.5:1 dCT:channel ratio reflects a genuine maximal reduction in I Ba rather than a limitation of the oocyte's expression machinery (Oz et al. 2013). A similar distinction between the actions of dCT on I Ba amplitude and G–V curve was previously reported in mammalian tsA‐201 cells (Hulme et al. 2006; Fuller et al. 2010); it may imply distinct molecular mechanisms for the two effects.
Table 2.
Boltzmann I–V fit parameters
| Mean | n | SEM | t test | |
|---|---|---|---|---|
| CaV1.2Δ1821 (1 ng) | ||||
| G max | 45.9 | 25 | 4.2 | |
| V rev | 61.7 | 25 | 0.6 | |
| V a | −3.0 | 25 | 0.8 | |
| K a | 6.4 | 25 | 0.3 | |
| CaV1.2Δ1821 (1 ng) + dCT (5 ng) | ||||
| G max | 40.1 | 8 | 5.4 | |
| V rev | 62.9 | 8 | 1.0 | |
| V a | −2.3 | 8 | 2.9 | ns |
| K a | 6.7 | 8 | 0.6 | |
| CaV1.2Δ1821 (1 ng) + dCT (10 ng) | ||||
| G max | 24.0 | 9 | 2.0 | |
| V rev | 60.8 | 9 | 0.9 | |
| V a | 5.3 | 9 | 1.5 | *** |
| K a | 7.3 | 9 | 0.3 | |
| CaV1.2Δ1821 (1 ng) + dCT (12.5 ng) | ||||
| G max | 25.3 | 5 | 7.1 | |
| V rev | 70.4 | 5 | 5.7 | |
| V a | 6.8 | 5 | 2.5 | *** |
| K a | 7.5 | 5 | 1.0 | |
Statistical significance of the shift in half‐maximal voltage (V a) was determined using a t test, comparing to control group (no dCT expression). *** P < 0.001; ns, not significant.
It has been proposed that the cleaved dCT interacts with the proximal part of the CT in CaV1.2Δ1821, and this interaction is essential for the proper regulation by PKA. By binding both α1C (at the dCT) and PKA‐RS, A‐kinase‐anchoring proteins (AKAPs) anchor PKA to CaV1.2. AKAP15 is an integral part of the CaV1.2 regulatory complex in cardiomyocytes (Catterall, 2015), and it was also found indispensable for PKA modulation of CaV1.2 reconstituted in HEK cells (Fuller et al. 2010, 2014). In contrast, our data suggest that in Xenopus oocytes, neither dCT nor AKAP15 are essential for the cAMP effect on I Ba. We tested a range of dCT:channel and AKAP15:channel RNA ratios, but none have yielded a significant increase in cAMP effect on I Ba amplitude, compared with the channel alone (Fig. 5 A).
Figure 5. dCT and AKAP15 do not alter the cAMP‐dependent increase in I Ba .
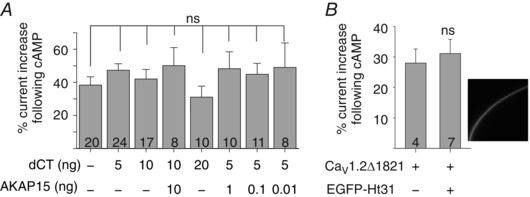
A, coexpression of dCT and AKAP15 does not alter the cAMP effect on CaV1.2Δ1821. Oocytes were injected with 1 ng RNA of CaV1.2Δ1821 and the indicated amounts of dCT and AKAP15 RNAs (summary of 5 experiments). B, coexpression of EGFP‐labelled Ht31 (5 ng RNA) does not affect the regulation of CaV1.2Δ1821 (1 ng RNA). Expression of EGFP‐Ht31 was confirmed by measuring fluorescence in intact oocytes. Summary from 2 experiments is shown.
We used the AKAP blocker Ht31 (Tröger et al. 2012; Dema et al. 2015) to test the possibility that endogenous AKAPs in the oocytes optimize the channel complex, obviating the need for exogenously expressed AKAP15. However, EGFP‐fused Ht31 peptide (Lynch et al. 2005), which was well expressed in the oocytes as verified by confocal imaging, did not affect the cAMP‐induced modulation of CaV1.2Δ1821 (Fig. 5 B). We conclude that, in Xenopus oocytes, reconstitution of PKA‐CS regulation of CaV1.2Δ1821 does not necessitate the presence of dCT or AKAP.
cAMP modulation of CaV1.2 in Xenopus oocytes does not require the α1C phosphorylation sites S1700 and T1704, or the CaVβ subunit
Serine‐1700 and threonine‐1704 were proposed to be crucial for PKA‐induced CaV1.2 upregulation reconstituted in HEK cells (Fuller et al. 2010). We mutated these amino acids, separately or together, to alanines. These mutations did not attenuate the cAMP‐dependent increase in CaV1.2Δ1821 currents in oocytes (Fig. 6 A).
Figure 6. cAMP‐dependent I Ba upregulation is not affected by alanine mutations of S1700 and T1704 in α1C, or by the CaVβ subunit.
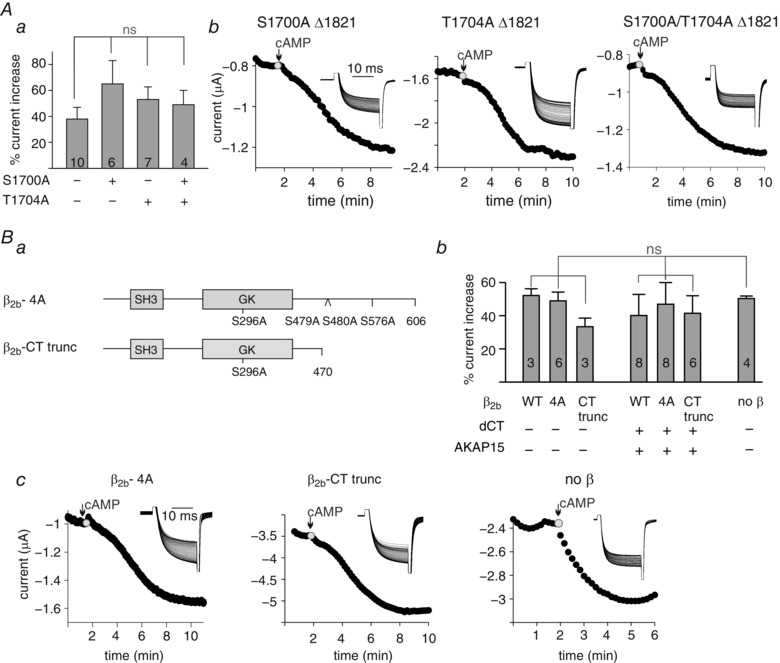
A, mutation S1700A, T1704A, or both to alanine do not alter the extent of cAMP upregulation of CaVΔ1821. a, summary of cAMP effect. N = 2 experiments. No statistical significance was found using one‐way ANOVA test. b, representative time course charts of changes in I Ba following cAMP injection (200 μm). Insets in b show I Ba current traces in the same cell. B, the presence of CaVβ2b subunit, or mutations in CaVβ2b phosphorylation sites, do not alter cAMP regulation. a, an illustration of the constructs used. SH3 and GK are structural modules within the β subunit. Numbers indicate amino acids. b, summary of % increase in I Ba per cell, following cAMP injection. Oocytes were injected with the following RNAs: 1 ng α1CΔ1821, α2δ1 and either CaVβ2b‐wild‐type (WT), CaVβ2b‐4A or CaVβ2b‐CTtrunc, with or without 2.5 ng AKAP15 and 5 ng dCT. α1CΔ1821 RNA (7 ng) was injected when CaVβ2b subunit was not present (α1CΔ1821 and α2δ1 only). No statistical significance was found using one‐way ANOVA test. c, representative time course charts of I Ba following cAMP injection (200 μm). Insets show current traces in the same cells.
The cardiac CaVβ subunit is phosphorylated in vivo following β‐adrenergic stimulation (Haase et al. 1996). However, the role of phosphorylation of CaVβ in PKA enhancement of CaV1.2 is controversial (compare Bunemann et al. 1999 vs. Miriyala et al. 2008; Brandmayr et al. 2012; Minobe et al. 2014, and see Discussion). To test the involvement of CaVβ in our system, we prepared a construct, termed β2b‐4A, with four alanine mutations in PKA phosphorylation sites: serine 296 (Pankonien et al. 2012), serines 479 and 480 (Bunemann et al. 1999) and serine 576 (Viard et al. 2004). To exclude the involvement of any unrecognized phosphorylation sites in the CT of CaVβ, another construct, truncated after amino acid 470, was prepared (β2b‐CTtrunc, Fig. 6 Ba). The two constructs, β2b‐4A and β2b‐CTtrunc, were expressed in the oocyte along with the α1CΔ1821 and α2δ subunits. The mutated CaVβ constructs did not attenuate the upregulation of I Ba following cAMP injection (Fig. 6 Bb and c). Coexpression of AKAP15 and dCT constructs together with α1CΔ1821 and α2δ and either β2b‐4A or β2b‐CTtrunc did not significantly alter the cAMP effect on I Ba (Fig. 6 Bb). Furthermore, α1CΔ1821 coexpressed with the α2δ subunit only, without CaVβ, showed cAMP modulation comparable to full subunit composition (Fig. 6 Bb and c), indicating the redundancy of CaVβ in the cAMP modulation observed in Xenopus oocytes.
N‐terminus of α1C is involved in cAMP‐dependent PKA upregulation of I Ba
The N‐terminus (NT) of the cardiac α1C encompasses an inhibitory module that regulates the channel's gating and is involved in CaV1.2 regulation by protein kinase C (PKC) (Shistik et al. 1998; Blumenstein et al. 2002; Weiss et al. 2012). We hypothesized that the NT may also be involved in PKA‐dependent regulation of CaV1.2.
The cytosolic NT domain of the cardiac ‘long‐NT’ (LNT) α1C isoform is 154 amino acids long. The first 46 amino acids are encoded by the variable exon 1a and the rest by the constant exon 2 (Blumenstein et al. 2002; Dai et al. 2002; Pang et al. 2003). The inhibitory module comprises the first 20 a.a. of the initial 46 a.a. segment (Kanevsky & Dascal, 2006). In the smooth muscle ‘short‐NT’ (SNT) isoform, also abundant in the brain (Biel et al. 1990; Koch et al. 1990; Snutch et al. 1991), the initial NT segment is 16 a.a. long and is encoded by exon 1 instead of 1a (Abernethy & Soldatov, 2002). The SNT α1C does not contain an inhibitory module, and is not upregulated by PKC in Xenopus oocytes (Kanevsky & Dascal, 2006).
We compared the cAMP regulation of LNT and SNT α1C isoforms, as well as a series of additional NT deletions and chimeric constructs, created on the template of the CT‐truncated α1CΔ1821 (numbering by LNT isoform) (Fig. 7 A). cAMP injection into oocytes expressing the SNT α1CΔ1821 isoform failed to enhance I Ba, in contrast with the cardiac LNT α1CΔ1821. The removal of most of the NT (LNTΔ139) or of the inhibitory gating element alone (the first 20 a.a. of the LNT; LNTΔ20) abolished the cAMP‐dependent increase in I Ba (Fig. 7 A and B). These results show that the initial 20 a.a. are essential for PKA regulation.
Importantly, addition of the inhibitory module (the first 20 a.a.) directly to the rest of NT encoded by exon 2 (by removing the last 26 amino acids of the exon 1a‐encoded segment; LNTΔ20–46) fully preserved the cAMP‐induced increase in I Ba. Since this construct is the exact counterpart of the SNT α1C except for the exchanged first short (20 or 16 a.a.) segment, we conclude that the first 20 a.a. of LNT are sufficient to support the PKA regulation in the context of α1CΔ1821.
Deletion of amino acids 2–5 in the inhibitory gating element of the LNT α1CΔ1821 eliminated the cAMP regulation (the LNTΔ5 construct), and greatly reduced it by ∼70% when both amino acids 2–5 and amino acids 20–46 of LNT α1CΔ1821 were deleted (LNTΔ5Δ20–46). Yet, adding amino acids 2–5 or amino acids 20–46 of the long NT to the SNT α1CΔ1821 did not render the SNT channel cAMP sensitive (chimeras ‘SNT+a.a.2–5’ and ‘SNT+a.a.20–46’). Thus, amino acids 2–5 are essential but not sufficient for cAMP‐dependent current upregulation.
The initial five amino acids of the LNT isoform are not fully conserved in different species. The a.a. sequence is MLRAL in rabbit, MLRAF in human, and MIRAF in mouse LNT α1C. We have tested for potential species variability in PKA regulation using mouse α1C. Figure 8 A–C shows that the dCT‐truncated mouse α1CΔ1821 was strongly regulated by the injection of purified PKA‐CS, with an average increase of 126 ± 23% (n = 12), which is almost 3‐fold greater than the average ∼40% increase observed in rabbit CaV1.2Δ1821 (compare to Fig. 1 G). Deletion of amino acids 2–5 (CaV1.2Δ5Δ1821) strongly reduced, but did not eliminate, the PKA‐CS effect: a residual 29 ± 12% increase was still present (Fig. 8 B and C). Full‐length α1C with the deletion of amino acids 2–5 (CaV1.2Δ5‐full) was not upregulated by PKA‐CS.
Figure 8. PKA‐CS enhances I Ba of dCT‐truncated mouse α1C .
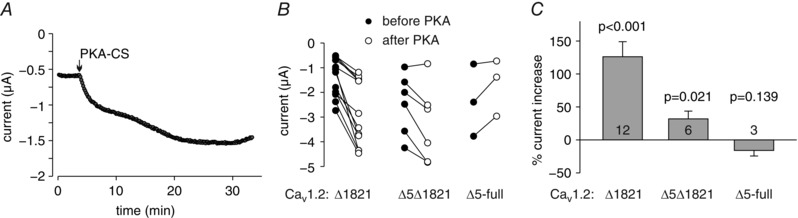
A, representative time course of I Ba changes in an oocyte expressing mouse CaV1.2Δ1821. The experimental protocols were as with rabbit CaV1.2. PKA‐CS was injected to a final concentration of about 4 μm. B, changes in I Ba in the three α1C constructs used, caused by PKA‐CS injection in individual oocytes. Filled circles show I Ba before PKA‐CS injection; open circles show I Ba after full development of the effect, 10–25 min after injection. C, summary of two experiments. Statistical significance was calculated using paired t test.
In summary, the experiments with the N‐terminal mutants of rabbit and mouse α1C suggest that the initial 20 amino acid segment of α1C uniquely present in the cardiac LNT isoform is essential for the cAMP and PKA regulation of CaV1.2Δ1821 in Xenopus oocytes. This segment is also sufficient to render the channel sensitive to PKA when the rest of the channel (starting from the exon 2‐encoded part) is present. The few partly conserved amino acids at the very beginning of this segment play an important role, and their removal eliminates >70% of the PKA effect.
Discussion
The intensive effort to understand the mechanism of PKA regulation of CaV1.2 has not yet yielded a unifying mechanistic insight. Studies in genetically modified cardiomyocytes and mice refuted the proposed crucial roles of putative target phosphorylation sites in α1C and β subunits suggested by in vitro and heterologous studies. Disappointingly, so far, studies with genetically engineered animals also did not positively resolve the puzzle (Weiss et al. 2013; Hofmann et al. 2014; Catterall, 2015; Morrow & Marx, 2015). Nevertheless, one major mechanistic insight is shared by most published works: the involvement of the distal C‐terminus (dCT) of α1C, and the importance of its naturally occurring post‐translational truncation (Fuller et al. 2010; Domes et al. 2011; Fu et al. 2011). With this in mind, in this work we have re‐examined the possibility of utilizing the Xenopus oocyte to search for additional unknown components of the PKA regulatory mechanism.
We succeeded to reconstitute the cAMP/PKA regulation of the dCT‐truncated CaV1.2 in Xenopus oocytes, a task which previously failed with the full‐length, non‐truncated α1C. A reproducible whole‐cell current increase of CaV1.2 from two species, rabbit and mouse, has been observed in single oocytes following intracellular application (injection) of cAMP or PKA‐CS. Although truncation of the dCT was essential, the presence of the clipped dCT or AKAP15 was not. The cAMP/PKA effect did not involve the phosphorylation sites S1700 and T1704 in α1C, or the β subunit of the channel, in line with recent animal model studies (Brandmayr et al. 2012; Yang et al. 2013). These findings pave the way for the further use of this versatile and easily manipulated heterologous system to pinpoint the molecular determinants of PKA modulation within CaV1.2. To this end, using a standard mutagenesis approach, we identified the initial segment of the cardiac LNT isoform of α1C as a previously unrecognized requisite structural element involved in PKA regulation. This finding raises the possibility that PKA is acting, in part, by affecting the hypothesized synergistic regulation of CaV1.2 by a scaffold formed by cytosolic N‐ and C‐termini of α1C (Ivanina et al. 2000; Dick et al. 2008; Benmocha Guggenheimer et al. 2016). We further propose that the new mechanism may be one of two or more complementary pathways of PKA regulation of CaV1.2 in the heart.
cAMP regulation of CaV1.2Δ1821 is PKA specific and requires dissociation of PKA subunits
Our results unequivocally demonstrate that CaV1.2, with its main α1C subunit truncated approximately at the same site as in the cardiomyocytes, is specifically regulated by PKA‐CS in Xenopus oocytes. The use of Xenopus oocytes allowed several advantages for the study of L‐type Ca2+ channels. First, we used two‐electrode voltage clamp with sharp electrodes and injected cAMP, PKA‐CS and other substances directly into the cell. This method avoids the dilution of the cytosol and the accompanying rundown of the channel's current inherent in whole‐cell patch clamp (Weiss et al. 2013) used in mammalian cell models. Further, the injection technique allowed Ba2+ currents to be measured via CaV1.2 before and after cAMP/PKA addition in the same cell, thus allowing sensitive detection of even small changes in whole‐cell currents. Finally, oocytes are highly suitable for titrated expression of proteins, due to precise control of the amount of injected RNA (e.g. Yakubovich et al. 2015).
As positive control for the functional effects of cAMP and PKA‐CS in Xenopus oocytes, we monitored PKA‐dependent changes in Cl− currents of the CFTR channel, a well‐characterized PKA effector (Gadsby & Nairn, 1999). CFTR's Cl− currents were greatly enhanced by the injection of cAMP or PKA‐CS, and specificity of PKA regulation was confirmed through inhibition by the inactive cAMP analogue Rp‐cAMPS and the highly specific PKA inhibitory protein PKI (Fig. 3). By using the same concentrations of Rp‐cAMPS and PKI, we confirmed the inhibition of cAMP‐dependent CaV1.2Δ1821 upregulation (Fig. 3 C and D). Thus, the regulation of CaV1.2 is PKA specific.
The results also strongly suggest that the observed cAMP regulation involved the classical mechanism of dissociation of PKA‐CS from the PKA‐RS which, in the absence of cAMP, is strongly associated with, and inhibits the activity of, PKA‐CS (Taylor et al. 2012). This is supported by the enhancing action of purified PKA‐CS and the strong blocking effect of Rp‐cAMPS, which acts mainly by preventing the dissociation of the PKA holoenzyme (Anand et al. 2010).
The discrepancies with previous reports
The open questions regarding the mechanism of PKA regulation of CaV1.2 have been thoroughly reviewed in the past (Weiss et al. 2013; Hofmann et al. 2014). There were disagreements on experimental findings even in the same expression system such as HEK cells or their derivative, the tsA cell line (for example, Gao et al. 1997 vs. Fuller et al. 2010 vs. Zong et al. 1995). At the same time, there was a general consensus on the absence of PKA upregulation of full‐length (untruncated) CaV1.2 across different heterologous systems (Perez Reyes et al. 1994; Singer‐Lahat et al. 1994; Zong et al. 1995), as confirmed here (Fig. 1). In the following we discuss how some of the discrepancies can be reconciled considering potential differences between cell types and the importance of colocalization of CaV1.2 and PKA in cardiomyocytes. Our emphasis is on the recent extensive studies of Fuller et al. done in tsA cells (Fuller et al. 2010, 2014). The two major points of disagreement with the latter work are the requirement for a simultaneous presence of dCT and AKAP, and the importance of phosphorylation of S1700 and T1704.
Truncation of dCT is crucial for PKA regulation of CaV1.2, but the dCT and AKAP15 are not essential
Our study agrees with that of Fuller et al. (2010) in one central aspect: as in tsA cells, in the oocytes the truncation of dCT of α1C is essential for PKA‐induced regulation of CaV1.2. In the oocytes, current via the dCT‐truncated channel, CaV1.2Δ1821, was increased by ∼40% (rabbit) or ∼130% (mouse α1C) by the injected cAMP or purified PKA‐CS, whereas the full‐length channel was not regulated (Figs 1 and 8).
In our hands, the PKA‐induced increase in current amplitude of CaV1.2Δ1821 in the oocytes did not require the clipped dCT, and cAMP‐induced increase in channel current was not altered in the very wide dCT titration range used here. The dCT was synthesized and expressed as a separate protein in the oocytes, and interacted with α1C, because it produced the two well‐documented effects on the truncated CaV1.2: a reduction in whole‐cell current (without any change in surface expression of α1C) and, at higher expression levels of the dCT, a depolarized shift in channel activation (Fig. 4) (Hulme et al. 2006; Fuller et al. 2010).
Further, titration of AKAP15 had no additional effects on cAMP‐dependent upregulation of CaV1.2Δ1821 (Fig. 5). We cannot completely rule out a role for an endogenous oocyte AKAP. However, two lines of evidence argue against this possibility. First, Ht31, a peptide that inhibits interaction of AKAPs with the PKA regulatory subunit and disrupts PKA anchoring by AKAPs, did not influence the effect of cAMP. Second, the dCT, which anchors both AKAP15 and AKAP79 to α1C (Catterall, 2015), was not needed. The expendability of AKAP does not contradict in vivo studies, because PKA regulation of cardiac CaV1.2 is preserved after knockout of AKAP15 and AKAP79/150 in mice (Nichols et al. 2010; Jones et al. 2012).
The above results are at odds with the reported necessity for the simultaneous presence of dCT and AKAP in the tsA‐201 cell line, where cAMP levels were elevated with the adenylyl cyclase activator, forskolin (Fuller et al. 2010, 2014). Nevertheless, a comparative analysis of oocyte and tsA‐201 data offers a plausible way to reconcile among them, and to explain why forskolin cannot regulate CaV1.2Δ1821 in the absence of dCT and AKAP in tsA‐201 cells. The logic is as follows:
-
(1)
It is widely agreed that AKAP is needed only for anchoring the PKA close to the channel; it is not implicated in the end‐point of regulation itself (Catterall, 2015).
-
(2)
AKAP15 is anchored to CaV1.2 via the dCT of α1C. The dCT is essential to allow close apposition of PKA and CaV1.2.
-
(3)
In tsA cells, no regulation of α1CΔ1800 by forskolin is seen without AKAP15, even when dCT is present. It follows that forskolin‐induced activation of endogenous PKA in tsA cells is insufficient for CaV1.2 regulation without the juxtaposition of PKA and α1C via AKAP.
-
(4)
In summary, we contend that the Fuller et al. (2010) data show the necessity of the dCT for anchoring of PKA to α1C via AKAP, but do not necessarily implicate the dCT in the end‐point regulation mechanism itself.
-
(5)
In the oocytes, we are providing the injected cell with ample cAMP or purified PKA‐CS, which obviously bypasses the need for PKA anchoring near the channel, since we see a bold regulation without adding AKAPs.
In partial support of this hypothesis, a previous study in HEK cells reported upregulation of CaV1.2 by infused PKA‐CS in the absence of dCT or AKAP, when α1C was truncated at amino acid 1905 (Bunemann et al. 1999).
We conclude that, on the factual level, the dispensability of dCT and AKAP15 in oocytes does not necessarily contradict the phenomena observed in HEK/tsA cells. However, on the mechanistic level, our findings do not support the elegant mechanism proposed by Catterall and colleagues (Fuller et al. 2010), where phosphorylation of S1700 removes a dCT‐imposed channel inhibition. This is because in the oocytes PKA regulates CaV1.2Δ1821 in the absence of dCT. Notably, Beam et al. arrived at a similar conclusion for the skeletal muscle CaV1.1, where dCT is truncated but does not associate with or modulate the channel, yet the channel is regulated by PKA, though rather modestly (Ohrtman et al. 2015). The expendability of dCT does not undermine the importance of the rest of the CT for PKA regulation. For instance, the fact that in the context of full‐length α1C the dCT impairs the effect of PKA hints upon a folding effect of the dCT, possibly by its binding to proximal CT.
Prominent phosphorylation sites in α1C and β subunits are not involved in PKA regulation of the truncated CaV1.2 in Xenopus oocytes
On the basis of experiments in HEK cells, phosphorylation of C‐terminal serines in the CaVβ subunit has been proposed to play a major role in PKA regulation of CaV1.2 (Bunemann et al. 1999). In our oocyte system, cAMP similarly regulated CaV1.2Δ1821 with or without the coexpression of the CaVβ subunit (Fig. 6). Involvement of the oocytes’ endogenous CaVβ3 subunit (Tareilus et al. 1997) is unlikely, since CaVβ subunit overexpression greatly changes the amplitude and activation of the expressed Ca2+ channels, suggesting that most of them are not pre‐associated with the endogenous CaVβ (Canti et al. 2001). By mutagenesis, we rule out the involvement of CaVβ phosphorylation sites 296, 479, 480 and 576. Our results corroborate the previous expression studies in the mammalian BHK cell line (Minobe et al. 2014) and in cardiomyocytes (Miriyala et al. 2008), and in genetically engineered mice, where truncation of CaVβ at position 450 (a.a. count according to rabbit CaVβ2b isoform used here) did not affect β‐adrenergic I Ca upregulation (Brandmayr et al. 2012). Taken together, our data suggest that phosphorylation of the CaVβ subunit is not involved in the PKA effect on CaV1.2Δ1821 reconstituted in Xenopus oocytes.
The role of PKA phosphorylation sites in the α1C subunit has been highly controversial. The involvement of S1928, a major PKA phosphorylation site previously thought to mediate the PKA effect (reviewed in Weiss et al. 2013), was ruled out by functional studies in genetically engineered cardiomyocytes and mice (Ganesan et al. 2006; Lemke et al. 2008). Phosphorylation of this serine has recently been found crucial for uncoupling of the direct interaction between β2AR and CaV1.2 (Patriarchi et al. 2016). In our system, S1928 is irrelevant because it is located in the dCT which is not needed for the PKA regulation of CaV1.2Δ1821.
Phosphorylation of two additional sites located in the proximal CT, S1700 and T1704 (Norman & Leach, 1994; Fuller et al. 2010), has been proposed to mediate the effect of PKA in CaV1.2 by weakening the interaction of dCT with a proximal CT‐acceptor site, PCRD (Fuller et al. 2010; Catterall, 2015). However, more recently it has been proposed that T1704 (which is not a PKA but rather a casein kinase site; Fuller et al. 2010) is probably not involved in PKA regulation but may contribute to the basal activity of the channel (Fu et al. 2014). In our case, alanine mutations of S1700 and T1704 in CaV1.2Δ1821 expressed in oocytes did not reduce the sensitivity to cAMP (Fig. 6 A). This result correlates with the reported lack of effect of mutating these residues in a mouse model with inducible expression of the mutated α1C (Yang et al. 2013). Nevertheless, studies of a different mouse model show that mutation of S1700 alters the regulation of CaV1.2 function, and PKA and Ca2+ handling in mouse heart (Fu et al. 2014; Yang et al. 2016), although the exact role of phosphorylation of S1700 in overall PKA effect on CaV1.2 itself may need further clarification. In heterologous systems, diverse cellular properties certainly may play a role, such that the fractional contribution of phosphorylation of S1700 (possibly followed by separation of the dCT) is prominent in tsA cells but not in the oocytes. Resolving this conundrum will require further study.
A novel mechanism involving the N‐terminus of α1C
We have utilized the newly developed experimental approach to look for additional molecular determinants of PKA regulation in α1C. The NT is an important regulatory domain that controls the channel's open probability, trafficking and inactivation (Shistik et al. 1998; Ivanina et al. 2000; Kanevsky & Dascal, 2006; Dick et al. 2008; Thomsen et al. 2009; Simms et al. 2015). Previously we have shown that PKC regulation of CaV1.2 expressed in Xenopus oocytes is controlled by the NT inhibitory gating element through a mechanism that does not involve phosphorylation of the gating element itself, and does not require the C‐terminal truncation of α1C (Shistik et al. 1998, 1999; Blumenstein et al. 2002). This gating element comprises the first 20 amino acids, out of the 46 amino acids encoded by exon 1a, of the ‘cardiac’ LNT isoform of α1C (Kanevsky & Dascal, 2006). Here we show that this 20 a.a. segment is essential for PKA regulation of cardiac CaV1.2 reconstituted in Xenopus oocytes (Fig. 7). This 20 a.a. segment is also sufficient for rendering the CaV1.2Δ1821 sensitive to PKA when added to the rest of α1C starting from the part of NT encoded by the obligatory exon 2. The partially homologous 16 a.a. segment of the short NT isoform, encoded by exon 1, does not carry this property, implying the presence and importance of a highly specific element or structure within the 20 amino acids of the cardiac LNT isoform of α1C. The first five amino acids may be part of this element, since the removal of this segment reduced the PKA effect by >70% (Figs 7 and 8). Nevertheless, the addition of five initial amino acids did not confer PKA regulation upon the SNT CaV1.2Δ1831. Thus, the first five amino acids are important, but not sufficient for the PKA effect. We have previously proposed that NT and CT may act together as one functional unit (an ‘NT–CT scaffold’) to regulate the channel's gating (Ivanina et al. 2000), and demonstrated a direct interaction (binding) between the NT and CT of α1C (Benmocha Guggenheimer et al. 2016). We hypothesize that the ‘NT–CT scaffold’ participates in PKA regulation of CaV1.2 gating.
One or more mechanisms of CaV1.2 regulation by PKA?
The regulation of α1C truncated at 1821 is at odds with two reports in genetically engineered cardiomyocytes and mice, where truncation of α1C at a.a. 1905 or 1796 produced channels with low or no sensitivity to β‐AR and PKA activators (Ganesan et al. 2006; Fu et al. 2011). Notably, however, in genetically engineered mice the expression of C‐terminally truncated channels is severely reduced, and localized expression of AKAP15 is also reduced (Domes et al. 2011; Fu et al. 2011). In cardiomyocytes, the truncated channels were expressed ectopically (Ganesan et al. 2006) and might have not been localized in the correct cellular compartment. The β2AR is also expected to uncouple from CaV1.2 in the absence of dCT of α1C (Patriarchi et al. 2016). Localization is crucial in cardiomyocytes (Balijepalli et al. 2006; McConnachie et al. 2006). Therefore, one reason for the observed lack of regulation could be insufficient anchoring of components of the β‐AR–PKA pathway in the vicinity of the channel, or insufficient increase in cAMP concentration at the channel's location. Under our conditions, we saturated the channel with an excess of injected cAMP or PKA‐CS to achieve the maximal possible response. Another (or an additional) possibility is that the mechanism described here is one of two or several redundant PKA regulation pathways (Weiss et al. 2013) that, together, produce the strong regulation of the channel observed in cardiomyocytes.
In this respect, it is interesting to compare the extent of increase in Ca2+ channel currents in cardiac cells of the relevant species with that seen in our system. In isolated rabbit cardiomyocytes, the increase in the current above basal level varies from 50 to 130% with isoproterenol (isoprenaline) or forskolin (Han et al. 1994; Shannon et al. 1995; Matsuda et al. 1996; Ginsburg & Bers, 2004). In the mouse, the current is increased by β‐AR activation anywhere from 45 to 200% above the basal level (Sako et al. 1997; Heubach et al. 1999; Lemke et al. 2008), 50–100% by forskolin, and up to 200% above baseline by purified PKA‐CS (Maltsev et al. 1999). The 40% increase caused by cAMP of PKA‐CS with rabbit CaV1.2Δ1821 and the ∼130% increase caused by PKA‐CS with mouse CaV1.2Δ1821 are comparable to the lower or medium values from this (admittedly incomplete) list. It is therefore plausible that the observed effect might account for only a part of the total PKA regulation. As discussed above, phosphorylation of S1700 may be the other molecular event contributing to the total regulation.
In summary, we propose that several PKA regulation pathways may exist under physiological conditions. The regulation of the truncated CaV1.2 observed in the Xenopus oocyte expression system appears to be a very basic one, inherent to α1C. It does not require the β subunit, AKAPs or the dCT (the latter actually hinders the regulation in the context of a full‐length, non‐truncated α1C). This expression system proved appropriate to observe the specific PKA regulation of the C‐terminally truncated channel and to uncover the potential role of the NT. In native heart cells, more layers of complexity are added by localization and compartmentalization, anchoring, and regulatory proteins and other factors. The basic regulation of the truncated α1C may coexist with AKAP15 and dCT‐dependent regulation (Catterall, 2015), or some as yet unknown regulation of the full‐length (non‐truncated) α1C, which may differentially contribute to the total PKA effect.
Additional information
Competing interests
None declared.
Author contributions
The experiments were performed in the Department of Physiology and Pharmacology, Sackler School of Medicine, Tel Aviv University; Max Delbrück Center for Molecular Medicine (MDC), Berlin, Germany; and Experimentelle und Klinische Pharmakologie und Toxikologie, Universität des Saarlandes, Homburg, Germany. S.O., I.P., H.H., V.F., E.K. and N.D. participated in the conception and design of the study, interpretation of the results and in editing the draft of the paper. A.B., I.P. and S.O. prepared the DNA constructs. S.O. and I.P. performed the experiments and analysed the data. S.O. and N.D. wrote the paper. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the German‐Israel Foundation for Research and Development (GIF; N.D., H.H. and E.K., grant no. I‐1210‐286.13/2012), the Fields Fund for Molecular Cardiology (N.D.), and Sonderforschungsbereich (SFB) 894 by the German Research Foundation (V.F.).
Authors’ present addresses
S. Oz: Leviev Heart Center, Sheba Medical Center, Tel Hashomer, Ramat Gan, Israel.
I. Pankonien: University of Lisbon, Faculty of Sciences, BioISI–Biosystems and Integrative Sciences Institute, Campo Grande, 1749‐016, Lisbon, Portugal.
Acknowledgements
We thank Dr Ronit S. Cherki for performing some of the two‐electrode voltage clamp experiments, Dr Sharon Weiss for critical reading of the manuscript, and the colleagues who kindly provided the cDNA constructs: W. A. Catterall (AKAP15), H. A. Lester (CFTR) and M. D. Uhler (PKI). This work was performed in partial fulfilment of the requirements for the PhD degree of S. Oz.
References
- Abernethy DR & Soldatov NM (2002). Structure‐functional diversity of human L‐type Ca2+ channel: perspectives for new pharmacological targets. J Pharmacol Exp Ther 300, 724–728. [DOI] [PubMed] [Google Scholar]
- Anand GS, Krishnamurthy S, Bishnoi T, Kornev A, Taylor SS & Johnson DA (2010). Cyclic AMP‐ and (R p)‐cAMPS‐induced conformational changes in a complex of the catalytic and regulatory (RIα) subunits of cyclic AMP‐dependent protein kinase. Mol Cell Proteomics 9, 2225–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balijepalli RC, Foell JD, Hall DD, Hell JW & Kamp TJ (2006). Localization of cardiac L‐type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2‐adrenergic regulation. Proc Natl Acad Sci USA 103, 7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear CE, Duguay F, Naismith AL, Kartner N, Hanrahan JW & Riordan JR (1991). Cl− channel activity in Xenopus oocytes expressing the cystic fibrosis gene. J Biol Chem 266, 19142–19145. [PubMed] [Google Scholar]
- Beavo JA, Bechtel PJ & Krebs EG (1974). Activation of protein kinase by physiological concentrations of cyclic AMP. Proc Natl Acad Sci USA 71, 3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmocha Guggenheimer A, Almagor L, Tsemakhovich V, Tripathy DR, Hirsch JA & Dascal N (2016). Interactions between N and C termini of α1C subunit regulate inactivation of CaV1.2 L‐type Ca2+ channel. Channels 10, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2000). Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res 87, 275–281. [DOI] [PubMed] [Google Scholar]
- Bers DM (2008). Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70, 23–49. [DOI] [PubMed] [Google Scholar]
- Biel M, Ruth P, Bosse E, Hullin R, Stuhmer W, Flockerzi V & Hofmann F (1990). Primary structure and functional expression of a high voltage activated calcium channel from rabbit lung. FEBS Lett 269, 409–412. [DOI] [PubMed] [Google Scholar]
- Blumenstein Y, Kanevsky N, Sahar G, Barzilai R, Ivanina T & Dascal N (2002). A novel long‐N‐terminus isoform of human L‐type Ca2+ channel is up‐regulated by protein kinase C. J Biol Chem 277, 3419–3423. [DOI] [PubMed] [Google Scholar]
- Brandmayr J, Poomvanicha M, Domes K, Ding J, Blaich A, Wegener JW, Moosmang S & Hofmann F (2012). Deletion of the C‐terminal phosphorylation sites in the cardiac β‐subunit does not affect the basic β‐adrenergic response of the heart and the Cav1.2 channel. J Biol Chem 287, 22584–22592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunemann M, Gerhardstein BL, Gao T & Hosey MM (1999). Functional regulation of L‐type calcium channels via protein kinase A‐ mediated phosphorylation of the β2 subunit. J Biol Chem 274, 33851–33854. [DOI] [PubMed] [Google Scholar]
- Cachelin AB, de Peyer JE, Kokubun S & Reuter H (1983). Ca2+ channel modulation by 8‐bromocyclic AMP in cultured heart cells. Nature 304, 462–464. [DOI] [PubMed] [Google Scholar]
- Campiglio M & Flucher BE (2015). The role of auxiliary subunits for the functional diversity of voltage‐gated calcium channels. J Cell Physiol 230, 2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti C, Davies A, Berrow NS, Butcher AJ, Page KM & Dolphin AC (2001). Evidence for two concentration‐dependent processes for β‐subunit effects on α1B calcium channels. Biophys J 81, 1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Hausken ZE, Fraser ID, Stofko‐Hahn RE & Scott JD (1992). Association of the type II cAMP‐dependent protein kinase with a human thyroid RII‐anchoring protein. Cloning and characterization of the RII‐binding domain. J Biol Chem 267, 13376–13382. [PubMed] [Google Scholar]
- Catterall WA (2000). Structure and regulation of voltage‐gated Ca2+ channels. Annu Rev Cell Dev Biol 16, 521–555. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2015). Regulation of cardiac calcium channels in the fight‐or‐flight response. Curr Mol Pharmacol 8, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnet P, Lory P, Bourinet E, Collin T & Nargeot J (1995). cAMP‐dependent phosphorylation of the cardiac L‐type Ca channel: a missing link? Biochimie 77, 957–962. [DOI] [PubMed] [Google Scholar]
- Crump SM, Andres DA, Sievert G & Satin J (2013). The cardiac L‐type calcium channel distal carboxy terminus autoinhibition is regulated by calcium. Am J Physiol Heart Circ Physiol 304, H455–H464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanady L, Seto‐Young D, Chan KW, Cenciarelli C, Angel BB, Qin J, McLachlin DT, Krutchinsky AN, Chait BT, Nairn AC & Gadsby DC (2005). Preferential phosphorylation of R‐domain serine 768 dampens activation of CFTR channels by PKA. J Gen Physiol 125, 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai B, Saada N, Echetebu C, Dettbarn C & Palade P (2002). A new promoter for α1C subunit of human L‐type cardiac calcium channel Cav1.2. Biochem Biophys Res Commun 296, 429–433. [DOI] [PubMed] [Google Scholar]
- Dascal N & Lotan I (1992). Expression of exogenous ion channels and neurotransmitter receptors in RNA‐injected Xenopus oocytes In Protocols in Molecular Neurobiology, vol. 13, ed. Longstaff A. & Revest P, pp. 205–225. Humana Press, Totowa, NJ, USA. [Google Scholar]
- Dema A, Perets E, Schulz MS, Deak VA & Klussmann E (2015). Pharmacological targeting of AKAP‐directed compartmentalized cAMP signalling. Cell Signal 27, 2474–2487. [DOI] [PubMed] [Google Scholar]
- Dick IE, Tadross MR, Liang H, Tay LH, Yang W & Yue DT (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of Cav channels. Nature 451, 830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2012). Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci 13, 542–555. [DOI] [PubMed] [Google Scholar]
- Domes K, Ding J, Lemke T, Blaich A, Wegener JW, Brandmayr J, Moosmang S & Hofmann F (2011). Truncation of murine Cav1.2 at Asp‐1904 results in heart failure after birth. J Biol Chem 286, 33863–33871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florea VG & Cohn JN (2014). The autonomic nervous system and heart failure. Circ Res 114, 1815–1826. [DOI] [PubMed] [Google Scholar]
- Fraser ID, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV & Scott JD (1998). A novel lipid‐anchored A‐kinase anchoring protein facilitates cAMP‐responsive membrane events. EMBO J 17, 2261–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohnwieser B, Chen LQ, Schreibmayer W & Kallen RG (1997). Modulation of the human cardiac sodium channel alpha‐subunit by cAMP‐dependent protein kinase and the responsible sequence domain. J Physiol 498, 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Westenbroek RE, Scheuer T & Catterall WA (2013). Phosphorylation sites required for regulation of cardiac calcium channels in the fight‐or‐flight response. Proc Natl Acad Sci USA 110, 19621–19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Westenbroek RE, Scheuer T & Catterall WA (2014). Basal and β‐adrenergic regulation of the cardiac calcium channel CaV1.2 requires phosphorylation of serine 1700. Proc Natl Acad Sci USA 111, 16598–16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Westenbroek RE, Yu FH, Clark JP, Marshall MR, Scheuer T & Catterall WA (2011). Deletion of the distal C terminus of CaV1.2 channels leads to loss of β‐adrenergic regulation and heart failure in vivo. J Biol Chem 286, 12617–12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MD, Emrick MA, Sadilek M, Scheuer T & Catterall WA (2010). Molecular mechanism of calcium channel regulation in the fight‐or‐flight response. Sci Signal 3, ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MD, Fu Y, Scheuer T & Catterall WA (2014). Differential regulation of CaV1.2 channels by cAMP‐dependent protein kinase bound to A‐kinase anchoring proteins 15 and 79/150. J Gen Physiol 143, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC & Nairn AC (1999). Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev 79, S77–S107. [DOI] [PubMed] [Google Scholar]
- Ganesan AN, Maack C, Johns DC, Sidor A & O'Rourke B (2006). β‐Adrenergic stimulation of L‐type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of α1C but not serine 1928. Circ Res 98, e11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Ten Eick R & Hosey MM (2001). C‐terminal fragments of the α1C (Cav1.2) subunit associate with and regulate L‐type calcium channels containing C‐terminally truncated α1C subunits. J Biol Chem 276, 21089–21097. [DOI] [PubMed] [Google Scholar]
- Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD & Hosey MM (1997). cAMP‐dependent regulation of cardiac L‐type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19, 185–196. [DOI] [PubMed] [Google Scholar]
- Gerhardstein BL, Gao T, Bünemann M, Puri TS, Adair A, Ma H & Hosey MM (2000). Proteolytic processing of the C terminus of the α1C subunit of L‐type calcium channels and the role of a proline‐rich domain in membrane tethering of proteolytic fragments. J Biol Chem 275, 8556–8563. [DOI] [PubMed] [Google Scholar]
- Ginsburg KS & Bers DM (2004). Modulation of excitation‐contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol 556, 463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase H, Bartel S, Karczewski P, Morano I & Krause EG (1996). In‐vivo phosphorylation of the cardiac L‐type calcium channel beta‐subunit in response to catecholamines. Mol Cell Biochem 163–164, 99–106. [DOI] [PubMed] [Google Scholar]
- Han J, Leem C, So I, Kim E, Hong S, Ho W, Sung H & Earm YE (1994). Effects of thyroid hormone on the calcium current and isoprenaline‐induced background current in rabbit ventricular myocytes. J Mol Cell Cardiol 26, 925–935. [DOI] [PubMed] [Google Scholar]
- Hartzell HC, Hirayama Y & Petit‐Jacques J (1995). Effects of protein phosphatase and kinase inhibitors on the cardiac L‐type Ca current suggest two sites are phosphorylated by protein kinase A and another protein kinase. J Gen Physiol 106, 393–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC, Mery PF, Fischmeister R & Szabo G (1991). Sympathetic regulation of cardiac calcium current is due exclusively to cAMP‐dependent phosphorylation. Nature 351, 573–576. [DOI] [PubMed] [Google Scholar]
- Heubach JF, Trebeß I, Wettwer E, Himmel HM, Michel MC, Kaumann AJ, Koch WJ, Harding SE & Ravens U (1999). L‐type calcium current and contractility in ventricular myocytes from mice overexpressing the cardiac β2‐adrenoceptor. Cardiovasc Res 42, 173–182. [DOI] [PubMed] [Google Scholar]
- Hirayama Y & Hartzell HC (1997). Effects of protein phosphatase and kinase inhibitors on Ca2+ and Cl− currents in guinea pig ventricular myocytes. Mol Pharmacol 52, 725–734. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Flockerzi V, Kahl S & Wegener JW (2014). L‐type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev 94, 303–326. [DOI] [PubMed] [Google Scholar]
- Hulme JT, Konoki K, Lin TW‐C, Gritsenko MA, Camp DG, Bigelow DJ & Catterall WA (2005). Sites of proteolytic processing and noncovalent association of the distal C‐terminal domain of CaV1.1 channels in skeletal muscle. Proc Natl Acad Sci USA 102, 5274–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Yarov‐Yarovoy V, Lin TW‐C, Scheuer T & Catterall WA (2006). Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C‐terminal domain. J Physiol 576, 87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanina T, Blumenstein Y, Shistik E, Barzilai R & Dascal N (2000). Modulation of L‐type Ca2+ channels by Gβγ and calmodulin via interactions with N‐ and C‐termini of α1C . J Biol Chem 275, 39846–39854. [DOI] [PubMed] [Google Scholar]
- Jones BW, Brunet S, Gilbert ML, Nichols CB, Su T, Westenbroek RE, Scott JD, Catterall WA & McKnight GS (2012). Cardiomyocytes from AKAP7 knockout mice respond normally to adrenergic stimulation. Proc Natl Acad Sci USA 109, 17099–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahanovitch U, Tsemakhovich V, Berlin S, Rubinstein M, Styr B, Castel R, Peleg S, Tabak G, Dessauer CW, Ivanina T & Dascal N (2014). Recruitment of Gβγ controls the basal activity of GIRK channels: crucial role of distal C‐terminus of GIRK1. J Physiol 592, 5373–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama M, Hescheler J, Hofmann F & Trautwein W (1986). Modulation of Ca current during the phosphorylation cycle in the guinea pig heart. Pflugers Arch 407, 123–128. [DOI] [PubMed] [Google Scholar]
- Kanevsky N & Dascal N (2006). Regulation of maximal open probability is a separable function of Cavβ subunit in L‐type Ca2+ channel, dependent on NH2 terminus of α1C (Cav1.2α). J Gen Physiol 128, 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Cheng CY, Saldanha SA & Taylor SS (2007). PKA‐I holoenzyme structure reveals a mechanism for cAMP‐dependent activation. Cell 130, 1032–1043. [DOI] [PubMed] [Google Scholar]
- Kim C, Xuong NH & Taylor SS (2005). Crystal structure of a complex between the catalytic and regulatory (RIα) subunits of PKA. Science 307, 690–696. [DOI] [PubMed] [Google Scholar]
- Klockner U, Mikala G, Eisfeld J, Iles DE, Strobeck M, Mershon JL, Schwartz A & Varadi G (1997). Properties of three COOH‐terminal splice variants of a human cardiac L‐type Ca2+‐channel α1‐subunit. Am J Physiol 272, H1372–H1381. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS & Sowadski JM (1991). Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate‐dependent protein kinase. Science 253, 414–420. [DOI] [PubMed] [Google Scholar]
- Koch WJ, Ellinor PT & Schwartz A (1990). cDNA cloning of a dihydropyridine‐sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced forms. J Biol Chem 265, 17786–17791. [PubMed] [Google Scholar]
- Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, Hofmann F & Moosmang S (2008). Unchanged β‐adrenergic stimulation of cardiac L‐type calcium channels in CaV1.2 phosphorylation site S1928A mutant mice. J Biol Chem 283, 34738–34744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Tytgat J & Hess P (1992). Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9, 861–871. [DOI] [PubMed] [Google Scholar]
- Link S, Meissner M, Held B, Beck A, Weissgerber P, Freichel M & Flockerzi V (2009). Diversity and developmental expression of L‐type calcium channel β2 proteins and their influence on calcium current in murine heart. J Biol Chem 284, 30129–30137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MJ, Baillie GS, Mohamed A, Li X, Maisonneuve C, Klussmann E, van Heeke G & Houslay MD (2005). RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with β‐arrestin to control the protein kinase A/AKAP79‐mediated switching of the β2‐adrenergic receptor to activation of ERK in HEK293B2 cells. J Biol Chem 280, 33178–33189. [DOI] [PubMed] [Google Scholar]
- McConnachie G, Langeberg LK & Scott JD (2006). AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med 12, 317–323. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Ji GJ, Wobus AM, Fleischmann BK & Hescheler J (1999). Establishment of β‐adrenergic modulation of L‐type Ca2+ current in the early stages of cardiomyocyte development. Circ Res 84, 136–145. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Hagiwara N, Shoda M, Kasanuki H & Hosoda S (1996). Enhancement of the L‐type Ca2+ current by mechanical stimulation in single rabbit cardiac myocytes. Circ Res 78, 650–659. [DOI] [PubMed] [Google Scholar]
- Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S & Numa S (1989). Primary structure and functional expression of the cardiac dihydropyridine‐sensitive calcium channel. Nature 340, 230–233. [DOI] [PubMed] [Google Scholar]
- Minobe E, Maeda S, Xu JJ, Hao LY, Kameyama A & Kameyama M (2014). A new phosphorylation site in cardiac L‐type Ca2+ channels (Cav1.2) responsible for its cAMP‐mediated modulation. Am J Physiol Cell Physiol 307, C999–C1009. [DOI] [PubMed] [Google Scholar]
- Miriyala J, Nguyen T, Yue DT & Colecraft HM (2008). Role of CaVβ subunits, and lack of functional reserve, in protein kinase A modulation of cardiac CaV1.2 channels. Circ Res 102, e54–e64. [DOI] [PubMed] [Google Scholar]
- Morrow JP & Marx SO (2015). Novel approaches to examine the regulation of voltage‐gated calcium channels in the heart. Curr Mol Pharmacol 8, 61–68. [DOI] [PubMed] [Google Scholar]
- Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF & McKnight GS (2010). Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L‐type calcium channels. Circ Res 107, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman RI & Leach RN (1994). Subunit structure and phosphorylation of the cardiac L‐type calcium channel. Biochem Soc Trans 22, 492–496. [DOI] [PubMed] [Google Scholar]
- Ohrtman JD, Romberg CF, Moua O, Bannister RA, Levinson SR & Beam KG (2015). Apparent lack of physical or functional interaction between CaV1.1 and its distal C terminus. J Gen Physiol 145, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SR & Uhler MD (1991). Isolation and characterization of cDNA clones for an inhibitor protein of cAMP‐dependent protein kinase. J Biol Chem 266, 11158–11162. [PubMed] [Google Scholar]
- Osterrieder W, Brum G, Hescheler J, Trautwein W, Flockerzi V & Hofmann F (1982). Injection of subunits of cyclic AMP‐dependent protein kinase into cardiac myocytes modulates Ca2+ current. Nature 298, 576–578. [DOI] [PubMed] [Google Scholar]
- Oz S, Benmocha A, Sasson Y, Sachyani D, Almagor L, Lee A, Hirsch JA & Dascal N (2013). Competitive and non‐competitive regulation of calcium‐dependent inactivation in CaV1.2 L‐type Ca2+ channels by calmodulin and Ca2+‐binding protein 1. J Biol Chem 288 12680–12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang L, Koren G, Wang Z & Nattel S (2003). Tissue‐specific expression of two human Cav1.2 isoforms under the control of distinct 5′ flanking regulatory elements. FEBS Lett 546, 349–354. [DOI] [PubMed] [Google Scholar]
- Pankonien I, Otto A, Dascal N, Morano I & Haase H (2012). Ahnak1 interaction is affected by phosphorylation of Ser‐296 on CaVβ2. Biochem Biophys Res Commun 421, 184–189. [DOI] [PubMed] [Google Scholar]
- Parsons TD, Lagrutta A, White RE & Hartzell HC (1991). Regulation of Ca2+ current in frog ventricular cardiomyocytes by 5′‐guanylylimidodiphosphate and acetylcholine. J Physiol 432, 593–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Qian H, Di Biase V, Malik ZA, Chowdhury D, Price JL, Hammes EA, Buonarati OR, Westenbroek RE, Catterall WA, Hofmann F, Xiang YK, Murphy GG, Chen CY, Navedo MF & Hell JW (2016). Phosphorylation of Cav1.2 on S1928 uncouples the L‐type Ca2+ channel from the β2 adrenergic receptor. EMBO J 35, 1330–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Reyes E, Yuan W, Wei X & Bers DM (1994). Regulation of the cloned L‐type cardiac calcium channel by cyclic‐AMP‐dependent protein kinase. FEBS Lett 342, 119–123. [DOI] [PubMed] [Google Scholar]
- Reuter H, Stevens CF, Tsien RW & Yellen G (1982). Properties of single calcium channels in cardiac cell culture. Nature 297, 501–504. [DOI] [PubMed] [Google Scholar]
- Sako H, Green SA, Kranias EG & Yatani A (1997). Modulation of cardiac Ca2+ channels by isoproterenol studied in transgenic mice with altered SR Ca2+ content. Am J Physiol 273, C1666–C1672. [DOI] [PubMed] [Google Scholar]
- Scott JD, Fischer EH, Takio K, Demaille JG & Krebs EG (1985). Amino acid sequence of the heat‐stable inhibitor of the cAMP‐dependent protein kinase from rabbit skeletal muscle. Proc Natl Acad Sci USA 82, 5732–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon KM, Klitzner TS, Chen F & Van Dop C (1995). Effects of triiodothyronine on retention of β‐adrenergic responsiveness of voltage‐gated transmembrane calcium current during culture of ventricular myocytes from neonatal rabbits. Pediatr Res 37, 277–282. [DOI] [PubMed] [Google Scholar]
- Shistik E, Ivanina T, Blumenstein Y & Dascal N (1998). Crucial role of N terminus in function of cardiac L‐type Ca2+ channel and its modulation by protein kinase C. J Biol Chem 273, 17901–17909. [DOI] [PubMed] [Google Scholar]
- Shistik E, Keren‐Raifman T, Idelson GH, Dascal N & Ivanina T (1999). The N‐terminus of the cardiac L‐type Ca2+ channel α1C subunit: The initial segment is ubiquitous and crucial for protein kinase C modulation, but it is not directly phosphorylated. J Biol Chem 274, 31145–31149. [DOI] [PubMed] [Google Scholar]
- Simms BA, Souza IA, Rehak R & Zamponi GW (2015). The CaV1.2 N terminus contains a CaM kinase site that modulates channel trafficking and function. Pflugers Arch 467, 677–686. [DOI] [PubMed] [Google Scholar]
- Singer‐Lahat D, Dascal N, Mittelman L, Peleg S & Lotan I (2000). Imaging plasma membrane proteins in large membrane patches of Xenopus oocytes. Pflugers Arch 440, 627–633. [DOI] [PubMed] [Google Scholar]
- Singer‐Lahat D, Lotan I, Biel M, Flockerzi V, Hofmann F & Dascal N (1994). Cardiac calcium channels expressed in Xenopus oocytes are modulated by dephosphorylation but not by cAMP‐dependent phosphorylation. Receptors Channels 2, 215–226. [PubMed] [Google Scholar]
- Skeberdis VA, Jurevičius J & Fischmeister R (1997). β2 Adrenergic activation of L‐type Ca++ current in cardiac myocytes. J Pharmacol Exp Ther 283, 452–461. [PubMed] [Google Scholar]
- Snutch TP, Tomlinson WJ, Leonard JP & Gilbert MM (1991). Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron 7, 45–57. [DOI] [PubMed] [Google Scholar]
- Tareilus E, Roux M, Qin N, Olcese R, Zhou J, Stefani E & Birnbaumer L (1997). A Xenopus oocyte β subunit: evidence for a role in the assembly/expression of voltage‐gated calcium channels that is separate from its role as a regulatory subunit. Proc Natl Acad Sci USA 94, 1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Ilouz R, Zhang P & Kornev AP (2012). Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol 13, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MB, Foster E, Nguyen KH & Sosunov EA (2009). Transcriptional and electrophysiological consequences of KChIP2‐mediated regulation of CaV1.2. Channels (Austin) 3, 308–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein W, Taniguchi J & Noma A (1982). The effect of intracellular cyclic nucleotides and calcium on the action potential and acetylcholine response of isolated cardiac cells. Pflugers Arch 392, 307–314. [DOI] [PubMed] [Google Scholar]
- Tröger J, Moutty MC, Skroblin P & Klussmann E (2012). A‐kinase anchoring proteins as potential drug targets. Br J Pharmacol 166, 420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezono Y, Bradley J, Min C, McCarty NA, Quick M, Riordan JR, Chavkin C, Zinn K, Lester HA & Davidson N (1993). Receptors that couple to 2 classes of G proteins increase cAMP and activate CFTR expressed in Xenopus oocytes. Receptors Channels 1, 233–241. [PubMed] [Google Scholar]
- Viard P, Butcher AJ, Halet G, Davies A, Nurnberg B, Heblich F & Dolphin AC (2004). PI3K promotes voltage‐dependent calcium channel trafficking to the plasma membrane. Nat Neurosci 7, 939–946. [DOI] [PubMed] [Google Scholar]
- Vinogradova TM & Lakatta EG (2009). Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP‐mediated PKA‐dependent Ca2+ cycling with surface membrane channels. J Mol Cell Cardiol 47, 456–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DA, Ashby CD, Gonzalez C, Calkins D, Fischer EH & Krebs EG (1971). Purification and characterization of a protein inhibitor of adenosine 3′,5′‐monophosphate‐dependent protein kinases. J Biol Chem 246, 1977–1985. [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE & Marks AR (2005). Intracellular calcium release and cardiac disease. Annu Rev Physiol 67, 69–98. [DOI] [PubMed] [Google Scholar]
- Wei X, Neely A, Lacerda AE, Olcese R, Stefani E, Perez Reyes E & Birnbaumer L (1994). Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac α1 subunit. J Biol Chem 269, 1635–1640. [PubMed] [Google Scholar]
- Weiss S, Keren‐Raifman T, Oz S, Ben Mocha A, Haase H & Dascal N (2012). Modulation of distinct isoforms of L‐type calcium channels by Gq‐coupled receptors in Xenopus oocytes: Antagonistic effects of Gβγ and protein kinase C. Channels 6, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Oz S, Benmocha A & Dascal N (2013). Regulation of cardiac L‐type Ca2+ channel CaV1.2 via the β‐adrenergic‐cAMP‐protein kinase A pathway: Old dogmas, advances, and new uncertainties. Circ Res 113, 617–631. [DOI] [PubMed] [Google Scholar]
- Welsh MJ, Anderson MP, Rich DP, Berger HA, Denning GM, Ostedgaard LS, Sheppard DN, Cheng SH, Gregory RJ & Smith AE (1992). Cystic fibrosis transmembrane conductance regulator: A chloride channel with novel regulation. Neuron 8, 821–829. [DOI] [PubMed] [Google Scholar]
- Wilkinson DJ, Strong TV, Mansoura MK, Wood DL, Smith SS, Collins FS & Dawson DC (1997). CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am J Physiol 273, L127–L133. [DOI] [PubMed] [Google Scholar]
- Yakubovich D, Berlin S, Kahanovitch U, Rubinstein M, Farhy Tselnicker I, Styr B, Keren‐Raifman T, Dessauer CW & Dascal N (2015). A quantitative model of the GIRK1/2 channel reveals that its basal and evoked activities are controlled by unequal stoichiometry of Gα and Gβγ. PLoS Comput Biol 11, e1004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Dai D‐F, Yuan C, Westenbroek RE, Yu H, West N, de la Iglesia HO & Catterall WA (2016). Loss of β‐adrenergic‐stimulated phosphorylation of CaV1.2 channels on Ser1700 leads to heart failure. Proc Natl Acad Sci USA 113, E7976–E7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Katchman A, Samad T, Morrow JP, Weinberg RL & Marx SO (2013). β‐Adrenergic regulation of the L‐type Ca2+ channel does not require phosphorylation of α1C Ser1700. Circ Res 113, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue DT, Herzig S & Marban E (1990). β‐Adrenergic stimulation of calcium channels occurs by potentiation of high‐activity gating modes. Proc Natl Acad Sci USA 87, 753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong X, Schreieck J, Mehrke G, Welling A, Schuster A, Bosse E, Flockerzi V & Hofmann F (1995). On the regulation of the expressed L‐type calcium channel by cAMP‐dependent phosphorylation. Pflugers Arch 430, 340–347. [DOI] [PubMed] [Google Scholar]