

Abstract
An endoplasmic reticulum (ER)‐resident protein that regulates cytosolic and ER free‐Ca2+ concentration by induction of store‐operated calcium entry: that is the original definition of STIM2 and its function. While its activity strongly depends on the amount of calcium stored in the ER, its function goes further, to intracellular signalling and gene expression. Initially under‐studied owing to the prominent function of STIM1, STIM2 came to be regarded as vital in mice, gradually emerging as an important player in the nervous system, and cooperating with STIM1 in the immune system. STIM2 has also been proposed as a relevant player in pathological conditions related to ageing, Alzheimer's and Huntington's diseases, autoimmune disorders and cancer. The discovery of additional functions, together with new splicing forms with opposite roles, has clarified existing controversies about STIM2 function in SOCE. With STIM2 being essential for life, but apparently not for development, newly available data demonstrate a complex and still intriguing behaviour that this review summarizes, updating current knowledge of STIM2 function.

Keywords: calcium entry, STIM2, store‐operated calcium entry
Abbreviations
- AD
Alzheimer's disease
- [Ca2+]c
cytosolic free‐Ca2+ concentration
- [Ca2+]ER
ER free‐Ca2+ concentration
- CAD
CRAC activation domain
- CaM
calmodulin
- CaMKII
Ca2+/calmodulin‐dependent kinase II
- CRAC
Ca2+ release‐activated Ca2+
- CREB
cAMP response element‐binding protein
- EAE
experimental autoimmune encephalomyelitis
- ER
endoplasmic reticulum
- FasL
Fas ligand
- FcγR
Fc γ receptor
- GC
germinal centre
- HD
Huntington's disease
- IFN‐γ
interferon γ
- IP3
inositol 1,4,5‐trisphosphate
- IP3R
IP3 receptors
- LTD
long‐term depression
- LTP
long‐term potentiation
- MSN
medium spiny neurons
- PASMC
pulmonary arterial smooth muscle cell
- PIP2
phosphatidylinositol 4,5‐bisphosphate
- PIP3
phosphatidylinositol 1,4,5‐trisphosphate
- PM
plasma membrane
- SAM
sterile α‐motif
- SOAR
STIM1 Orai1 activating region
- SOC
store‐operated Ca2+
- SOCE
store‐operated Ca2+ entry
- TNF‐α
tumour necrosis factor α
Introduction
The coupling of an agonist to its receptor triggers a number of mechanisms that initiate a wide variety of cellular functions. The agonist–receptor association leads to the activation of phospholipase C isoforms, which catalyse the synthesis of inositol 1,4,5‐trisphosphate (IP3) and diacylglycerol from phosphatidylinositol 4,5‐bisphosphate (PIP2). IP3 activates receptors (IP3Rs) that are calcium (Ca2+) channels mainly located in the endoplasmic reticulum (ER), which mediate the release of Ca2+ from the ER to the cytosol. This intracellular Ca2+ mobilization is the starting point for cellular functions such as excitation, cell secretion, gene expression and proliferation (Patterson et al. 2004; Van Petegem, 2015). In order to avoid exhaustion, the cell must refill depleted ER Ca2+ stores by uptake of extracellular Ca2+. Here, store‐operated Ca2+ entry (SOCE) plays a key function facilitating the replenishment of depleted intracellular Ca2+ stores. The concept of SOCE as a mechanism operated by the degree of filling of the intracellular Ca2+ stores was proposed by Jim Putney in 1986. Mimicking an electronic capacitor, loaded Ca2+ stores abrogate SOCE, while depleted stores trigger it (Putney, 1986; Berridge, 1995). SOCE has been reported to occur through the Ca2+ release‐activated Ca2+ (CRAC) complex, involving the STromal Interaction Molecule (STIM)1 and Orai1, which generate a current (I CRAC) that is highly selective for Ca2+, and the store‐operated Ca2+ (SOC) complex, which involves interaction between STIM1, Orai1 and TRPC1 and mediates a current (I SOC) of monovalent, as well as divalent, cations (Desai et al. 2015).
The current model of SOCE proposes the STIM family as the sensors of the ER free‐Ca2+ concentration ([Ca2+]ER), which is integrated by two ER‐resident transmembrane proteins, STIM1 and STIM2 (Liou et al. 2005; Roos et al. 2005; Zhang et al. 2005; Brandman et al. 2007). Both proteins detect decreases in [Ca2+]ER and interact with SOCE channels to, mostly, activate them. A biphasic model was proposed to explain STIM activation after a decline in [Ca2+]ER (Brandman et al. 2007). The estimated [Ca2+]ER in resting conditions ranges between 400 and 600 μm, while the average of cytosolic free Ca2+ concentration ([Ca2+]c) is around 40–60 nm (Barrero et al. 1997; Yu & Hinkle, 2000; Demaurex & Frieden, 2003). The starting minimal depletion of [Ca2+]ER (below 400 μm approx.) is firstly detected by STIM2, which is more sensitive to [Ca2+]ER changes due to its lower affinity for Ca2+ (Brandman et al. 2007). Activated STIM2 triggers small and prolonged store‐operated currents in order to refill slightly depleted stores (Zhou et al. 2009), thus correcting small fluctuations in [Ca2+]ER. In fact, overexpression of STIM2 often leads to moderate increases in [Ca2+]c, [Ca2+]ER and SOCE (Brandman et al. 2007; Gruszczynska‐Biegala et al. 2011), while its knockdown has the opposite effects (Brandman et al. 2007; Berna‐Erro et al. 2009; Darbellay et al. 2010). If the extent of ER depletion goes further, reaching [Ca2+]c below 200 μm, STIM1 becomes activated and triggers larger and transient store‐operated currents to replenish moderately or highly depleted internal stores (Brandman et al. 2007; Liou et al. 2007; Zhou et al. 2009). Both STIM isoforms physically interact with each other in order to achieve this task (Williams et al. 2001). Thus, the consensus model of SOCE comprises two phases of STIM activation to face decreases in [Ca2+]ER (for review see Lopez et al. 2016). STIM1 forms mostly monomers, but also dimers, in resting conditions that once activated aggregate in clusters localized in ER–plasma membrane (PM) junctions where they recruit SOCE channels in the form of monomers, dimmers or octamers, activating them and eliciting SOCE (Liou et al. 2005; Zhang et al. 2005; Peinelt et al. 2006; Soboloff et al. 2006b; Stathopulos et al. 2006; Xu et al. 2006; Hoover & Lewis, 2011; Li et al. 2011; He et al. 2012; Balasuriya et al. 2014). The SOCE signalplex also includes a number of proteins involved in self‐regulation. Among them, SARAF is able to interact with STIMs (Palty et al. 2012; Jha et al. 2013) and Orais (Albarran et al. 2016a,b) to modulate SOCE and prevent Ca2+ overload. STIM2 also might cooperate with STIMATE in the early steps of puncta generation to remodel ER–PM junctions (Quintana et al. 2015). In this review, we will focus on the recent discoveries regarding STIM2 in cell function and physiology.
Protein and gene structure
Identified and characterized in 2001 (Williams et al. 2001), the Stim2 gene appeared in vertebrates approximately 500 million years ago, probably by duplication of an ancestral Stim gene in the Euteleostomi lineage (Williams et al. 2001; Cai, 2007; Collins & Meyer, 2011). The simplest organism identified bearing a STIM‐like gene was the single‐celled eukaryotic and colony‐forming choanoflagellate Monosiga brevicollis (Cai, 2008). Interestingly, a second round of duplication was undergone 350 million years ago only in fish, giving rise to four STIM fish genes (Stim1a, b and Stim2a, b) (Cai, 2007). In mammals, no additional STIM isoforms have been yet described. The human STIM2 gene is located at chromosome 4 region 15.2 (4p15.2) and comprises 14 exons, 12 previously characterized (Williams et al. 2001) plus an additional exon (exon 9) and an alternative exon (exon 13) recently described (Miederer et al. 2015) (Fig. 1). Alternative splicing gives rise to three mRNA isoforms, the already known variant (Stim2.2; also STIM2α), a new shorter variant containing an alternative exon 13 lacking 444 bp (Stim2.3), and a third larger splicing variant that includes the new exon 9 (Stim2.1; also STIM2β) (Miederer et al. 2015; Rana et al. 2015). On the other hand, bioinformatic tools predict the existence of four additional protein‐coding STIM2 mRNA isoforms in human (http://www.ensembl.org/). Protein translation gives rise to transmembrane pre‐proteins that contain an ER signal peptide (Williams et al. 2001, revised in Graham et al. 2011), a highly conserved Ca2+‐binding domain (‘EF’‐hand) in the N‐terminal region, followed by a ‘hidden’ EF‐hand motif and a sterile α‐motif (SAM), all located in the luminal region of the ER (Fig. 1) (Williams et al. 2001, 2002). The EF‐hand motif is the Ca2+ sensor while the hidden EF‐hand and the SAM domain are critical for maintaining the stability of the N‐terminal region and the oligomerization between STIM proteins, respectively (Liou et al. 2005; Zhang et al. 2005; Baba et al. 2006; Stathopulos et al. 2006; Zheng et al. 2008). The C‐terminal region is located in the cytosolic compartment and separated from the N‐terminal region by a single transmembrane domain. The C‐terminal region contains an ezrin/radixin/moesin domain, highly conserved among the STIM isoforms, which includes two coiled‐coil structures (Williams et al. 2001; Soboloff et al. 2006a). This region mediates the interaction between STIM1 and TRPC Ca2+ channels (Huang et al. 2006; Yuan et al. 2007), which by extrapolation might also happen with STIM2 (Shalygin et al. 2015). The CRAC activation domain (CAD, also known as STIM1 Orai1 activating region; SOAR) overlaps with this region (Wang et al. 2014). As the name suggests, this region is involved in direct binding to and activation of Orai (Wang et al. 2014; Rana et al. 2015), the interaction of STIM1 with TRPC Ca2+ channels (Lee et al. 2014), as well as the communication with calmodulin (CaM) (Miederer et al. 2015). The presence of overlapping and mutually exclusive binding sites for both CaM and Orai1 in CAD might have a Ca2+‐dependent impact on STIM2 function. The exon 9 of the STIM2.1 variant inserts an additional amino acid sequence (383‐VAASYLIQ‐392) within the CAD domain. This insertion abrogates the interaction with Orais and their subsequent activation (Rana et al. 2015). Adjacent to the CAD region, a proline‐ and histidine‐rich (P/H) region is located at a similar position to that of the serine‐ and proline‐rich (S/P) region observed in STIM1, whose function is still unclear (Williams et al. 2001; Ercan et al. 2012). At the end of the C‐terminal region there is a CaM‐binding region (Bauer et al. 2008) and a polybasic lysine (Lys)‐rich domain involved in the interaction with PIP2, phosphatidylinositol 1,4,5‐trisphosphate (PIP3) and CaM (Ercan et al. 2009; Bhardwaj et al. 2013). In contrast to STIM1, STIM2 also contains a consensus sequence for a di‐lysine ER‐retention signal (K(X)KXX) in this region, restricting its expression to the ER (Ercan et al. 2012). STIM2 suffers co‐ and post‐translational modifications that include inefficient signal peptide cleavage and not fully understood glycosylation or phosphorylation events (Williams et al. 2001; Graham et al. 2011).
Figure 1. STIM2 splicing variants.

The STIM2 gene generates three different splicing variants: STIM2.2 (top panel), the first isoform described and capable of interacting with Orai and TRP channels in the PM (Williams et al. 2001); STIM2.1 (middle panel) with an insert of additional amino acids in the CAD/SOAR region (383‐VAASYLIQ‐392), which impairs STIM2.1 association with Orai and TRP channels (Miederer et al. 2015; Rana et al. 2015); STIM2.3 (bottom panel), with an alternative C‐terminal region (587‐CIHLGLGACKSE) that lacks the CaM binding and the K‐rich domains present in the other two longer isoforms (Miederer et al. 2015).
Expression and binding partners
STIM proteins are widely found in many tissues (Oritani & Kincade, 1996; Sabbioni et al. 1997; Williams et al. 2001; Oh‐Hora et al. 2008; Berna‐Erro et al. 2009), and their ratio varies with cell type, suggesting a possible role of the expression ratio in fine‐tuning intracellular Ca2+ content (Abell et al. 2011) or SOCE (for a review see Lopez et al. 2012).
STIM2 is the predominant protein in murine dendritic cells, mammary epithelial cells during lactation, human melanocytes, mouse spinal cord dorsal horn neurons, forebrain and hippocampus, while STIM1 predominates in the rest of the studied tissues (Berna‐Erro et al. 2009; Bandyopadhyay et al. 2011; McAndrew et al. 2011; Stanisz et al. 2012, 2014; Xia et al. 2014). In hippocampal neurons, STIM2.2 is expressed in both soma and dendrites, especially in 40% of dendritic spines such as large and mushroom spines (Sun et al. 2014; Garcia‐Alvarez et al. 2015a). The new STIM2.1 variant is also widely expressed among tissues and species (Miederer et al. 2015; Rana et al. 2015). The ratio within STIM2 variants differs between naive and stimulated T cells, and during myoblast differentiation (Miederer et al. 2015; Rana et al. 2015), suggesting a dynamic interaction among them during development.
The most important binding partners are STIM1 and SOC channels, such as members of the Orai family, which greatly determine and define the function of STIM2 (for review see Lopez et al. 2012). In addition to the already known binding partners such as CaM, SERCA3, calnexin, exportin1 and transportin1, STIM2 binds to PIP2 and PIP3 through its Lys‐rich domain (Fig. 1), which is required for formation of ER–PM contacts and anchoring to the ER (Ercan et al. 2009, 2012; Bhardwaj et al. 2013). It was found also complexed with protein kinase A, A kinase anchor protein 150 (AKAP150) and glutamate receptor subunit A1 (GluA1) in cultured rat neuron lysates, an interaction that is involved in synaptic shaping and activity (Garcia‐Alvarez et al. 2015a). The co‐immunoprecipitation with type 1 IP3R receptor (IP3R1) and thrombospondin 1 in bovine aortic endothelial cells and Jurkat cells, respectively, also suggests a physical interactios among them (Beliveau et al. 2014; Duquette et al. 2014).
An updated consensus model of SOCE
Different studies have reported that STIM1 and STIM2 heteromerize upon store depletion, with diverse consequences such as SOCE stimulation, modulation or inhibition. In fact, there is a debate about how STIM2 might regulate SOCE and cellular Ca2+ homeostasis due to an apparent controversy among the experimental results reported. Thus, different groups have observed a modest (Liou et al. 2005; Oh‐Hora et al. 2008; Lu et al. 2009; Darbellay et al. 2010; Schuhmann et al. 2010) or no effect (Liou et al. 2005; Peel et al. 2006; Spassova et al. 2006; Brandman et al. 2007) on SOCE of STIM2 overexpression or downregulation depending on the cell line or the experimental conditions, suggestive of a minor or redundant function. On the other hand, either an important increase or an inhibition of SOCE was observed in diverse human cell lines when STIM2 was co‐expressed with Orai1 or expressed alone, respectively (Soboloff et al. 2006a,b). What could be the reason for such controversy? Analysis of their molecular evolution highlighted that, while few structural differences between the invertebrate Stim and mammalian STIM1 have been reported, mammalian STIM2 suffered a substantial divergence. Thus, STIM1 might have retained most properties of its ancestral homologue, while the appearance of STIM2 added new functional properties (Cai, 2007). For instance, it is clear that the interaction of Orai channels with STIM1 or STIM2 leads to different I CRAC amplitudes. STIM1 triggers a robust I CRAC, while STIM2 is a poorer I CRAC activator (Soboloff et al. 2006a,b; Zhou et al. 2009). Among other differences, this weak activation is due to the presence of a leucine (Leu485) instead of a phenylalanine (Phe394) in the CAD region, as well as differences in the amino acid sequence (Wang et al. 2014) or in other regions such as the SAM domain (Zheng et al. 2011). Both STIM isoforms can also bind each other, giving extra features to regulate I CRAC amplitude (for review see Lopez et al. 2012). In addition, the recent discovery of the inhibitory STIM2.1 variant might help to clarify further these apparently contradictory reports. The former STIM2.2 promotes SOCE, while the new isoform, STIM2.1, inhibits it. Moreover, the inhibitory effect of STIM2.1 is Ca2+‐dependent. In the absence of cytosolic Ca2+ (chelated with BAPTA), STIM2.1 is unable to inhibit STIM1‐ or STIM2.2‐mediated SOCE, while it successfully reduces SOCE in the presence of 150 nm [Ca2+]c (Miederer et al. 2015). Therefore, since STIM2.1 is able to heteromerize and inhibit STIM2.2 and STIM1, the expression ratio between these three proteins might influence SOCE amplitude, giving rise to variable consequences of STIM2 expression, such as a facilitative, redundant or inhibitory effect on SOCE (Miederer et al. 2015; Rana et al. 2015).
Furthermore, a new report in salivary acinar cells proposed that STIM2.2 might function as an adaptor protein, conferring additional abilities on STIM1 once heteromerized, such as sensing the otherwise undetectable low [Ca2+]ER decreases in cases of mild cell stimulation (Ong et al. 2015). According to this, the presence of STIM2 might promote STIM heteromerization and an eventual STIM1–Orai1 assembly upon mild stimulation, promoting STIM1‐dependent SOCE in conditions where the stimulus would not be strong enough to activate STIM1.
Additionally, inefficient signal peptide cleavage generates a cytosolic STIM2.2 pre‐protein subpopulation, not targeted to the ER. This cytosolic population is able to bind and activate Orai1 channels in a store‐independent mode, and it has been proposed to be responsible for the control of [Ca2+]c (Parvez et al. 2008; Graham et al. 2011). Cytosolic inhibitory STIM2.1 subpopulations generated by this mechanism have not been detected yet (Miederer et al. 2015; Rana et al. 2015).
In summary, several models describing how STIM2 might control intracellular Ca2+ homeostasis have been developed (Fig. 2), presenting a plastic and dynamic protein with different variants working in a multimodal way. In cells with high STIM1:STIM2.2 expression ratios, decreases in [Ca2+]ER activate STIMs in a biphasic way, small decreases and fluctuations in [Ca2+]c or [Ca2+]ER activate the cytosolic or ER‐resident STIM2.2 subpopulation, respectively, while stronger cell stimulation generates bigger [Ca2+]ER drops that need higher SOCE and activate STIM (for review see Lopez et al. 2012) (Fig. 2 A). In cells with a similar expression of STIM1 and STIM2.2, the latter might act as an adaptor protein conferring on STIM1 the ability to detect small drops in [Ca2+]ER and generating active STIM1–STIM2.2 oligomers that trigger STIM1‐dependent SOCE in response to mild stimuli (Ong et al. 2015) (Fig. 2 B). In cells with a low STIM1:STIM2.2 expression ratio, such as murine dendritic cells, melanocytes, mammary epithelial cells during lactation and neurons (Lopez et al. 2012; Stanisz et al. 2012, 2014), STIM2 might partially compensate STIM1 function generating a small SOCE (Liou et al. 2005; Soboloff et al. 2006b; Berna‐Erro et al. 2009; Stanisz et al. 2014) (Fig. 2 C). The level of expression of the inhibitory STIM2.1 variant might also determine the [Ca2+]c and [Ca2+]ER in resting conditions as well. Cells with lower STIM2.1 expression would have higher basal [Ca2+]c, [Ca2+]ER and higher SOCE. In contrast, cells with higher expression of STIM2.1 might have lower basal [Ca2+]c, [Ca2+]ER and smaller SOCE (Fig. 2 D).
Figure 2. Function of STIM2 isoforms depending on their expression.
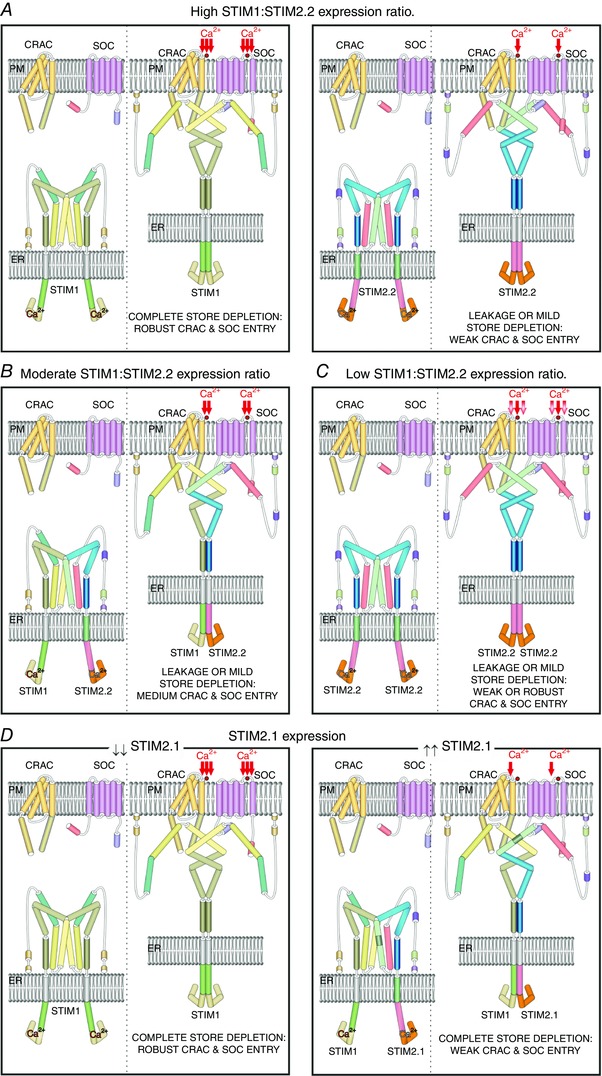
Scheme of the four proposed models of the role of STIM2 isoforms in SOCE. Each figure depicts the STIM proteins in resting (left) and activated state (right). A, in cells where STIM1 is highly expressed, STIM2.2 homodimers mediate weak SOCE to refill the ER from either leakage or mild store depletion (Brandman et al. 2007). B, cells that express similar levels of STIM1 and STIM2.2 present heterodimers of STIM1–STIM2.2, which allow earlier responses of STIM1 triggering increased SOCE in response to mild ER depletion (Soboloff et al. 2006b; Ong et al. 2015). C, in cells that either do not express STIM1 or express a high STIM2.2:STIM1 ratio, SOCE is triggered by STIM2.2 culminating in weak or robust Ca2+ entry, depending on the stimuli and always smaller than those triggered by STIM1 (Liou et al. 2005; Soboloff et al. 2006b; Berna‐Erro et al. 2009). D, cells expressing low levels of the inhibitory isoform STIM2.1 do not show impaired STIM1 function presenting robust SOCE, whereas those with high levels of STIM2.1 form heterodimers of STIM1–STIM2.1 or STIM2.2–STIM 2.1 activating weak SOCE (Wang et al. 2014; Rana et al. 2015).
Functional aspects of STIM2
Studies in mice lacking STIM2 demonstrated an essential function of this protein in sustaining life, since they progressively die starting at the age of 4–8 weeks (Table 1) (Oh‐Hora et al. 2008; Berna‐Erro et al. 2009). These mice display alterations in heart electrophysiology, but not severe enough to explain sudden death (Vetter, 2013). Besides this, the pathological mechanism underlying sudden death in the absence of STIM2 remains unidentified. Thus, a pending issue is to figure out how the cellular function of STIM2 is translated to the physiology of a living organism. The most relevant functional aspects of STIM2 are summarized below.
Table 1.
Phenotypes observed in STIM1‐ and STIM2‐deficient mice
| STIM1 | STIM2 | STIM1/STIM2 | References |
|---|---|---|---|
| Nervous system | |||
| Biochemistry | |||
| Reduced levels of synaptic pCaMKII, PSD95 and surface exposure of GluA1 in hippocampal neurons. | Enhanced phosphorylation of GluA1, CREB and Cav1.2 in hippocampus. | Sun et al. (2014); Garcia‐Alvarez et al. (2015b); Yap et al. (2017) | |
| Cell function | |||
| Abrogated mGluR1‐dependent TRPC3 activation and synaptic signalling in Purkinje neurons. |
|
Enhanced synaptic LTP in CA3–CA1 hippocampal neurons. | Hartmann et al. (2014); Sun et al. (2014); Garcia‐Alvarez et al. (2015b); Yap et al. (2017) |
| Mice | |||
| Purkinje neuron‐specific deletion alters cerebellar motor behaviour. | Resistance to transient hypoxic brain damage and delayed working memory. | Berna‐Erro et al. (2009); Hartmann et al. (2014) | |
| Adult neuron‐specific deletion generates a mild working memory delay. | Adult neuron‐specific deletion does not affect working memory. | Adult neuron‐specific deletion impairs working memory. | Garcia‐Alvarez et al. (2015b) |
| Immune system | |||
| Macrophages | |||
| Cell function | |||
| Defective FcγR‐mediated phagocytosis but normal chemotaxis and TLR4‐induced secretion of TNFα and IL‐6. | Reduced FcγR‐mediated phagocytosis, chemotaxis and TLR4‐induced secretion of TNFα and IL‐6. | Braun et al. (2009); Sogkas et al. (2015) | |
| Normal function of macrophages. | Normal function of macrophages. | Normal function of macrophages. | Vaeth et al. (2015) |
| Mice | |||
| Normal progression of Thg‐induced inflammation but resistance to experimental immune thrombocytopenia, anaphylaxis, IgG‐induced AIHA, and acute pneumonitis. | Normal IgG‐induced AIHA progression but reduced Thg‐peritonitis and severe LPS inflammation. | Braun et al. (2009); Sogkas et al. (2015) | |
| T cells | |||
| Biochemistry | |||
| Defective IFN‐γ, IL‐23R, IL‐2 and IL‐17 production, partial expression of CD40L by CD4+ T cells. | Decreased IFN‐γ, IL‐2 and IL‐17 production, partial expression of CD40L by CD4+ T cells. |
|
Oh‐Hora et al. (2008, 2013); Ma et al. (2010); Schuhmann et al. (2010); Shaw et al. (2014) |
| Cell function | |||
|
|
|
Oh‐Hora et al. (2008); Ma et al. (2010); Schuhmann et al. (2010); Shaw et al. (2014); Vaeth et al. (2016) |
| Mice | |||
| Presence of lymphoproliferative phenotype but protected against EAE. | Delayed EAE progression. |
|
Oh‐Hora et al. (2008, 2013); Beyersdorf et al. (2009); Ma et al. (2010); Schuhmann et al. (2010); Cheng et al. (2012); Shaw et al. (2014); Vaeth et al. (2016) |
| B cells | |||
|
|
|
Matsumoto et al. (2011) |
The nervous system
STIM2 demonstrated the most prominent function in brain (Table 1). The function of STIM2 in the nervous system was studied for the first time in 2009, using transgenic mice lacking the protein (Berna‐Erro et al. 2009). Cortical neurons isolated from E19 embryos showed a reduction in both SOCE and basal [Ca2+]c in agreement with previous observations (Brandman et al. 2007). Adult individuals displayed a deficit in spatial learning and memory when assessed by the Elevated Plus Maze test, similar to that observed after blockade of N‐methyl‐d‐aspartate (NMDA)‐type ionotropic glutamate receptors (Morris et al. 1986). Therefore, unexpected functions for STIM2 in synaptic transmission or plasticity were hypothesized and opened the question of its function in brain (Berna‐Erro et al. 2009). Indeed, an earlier report already suggested the function of SOCE in neuronal plasticity (Baba et al. 2003). Later, the involvement of neuronal SOCE in cellular network activity was suggested, supporting this idea (Steinbeck et al. 2011). However, a more recent study observed unaltered spatial memory in STIM‐deficient mice when only STIM2 was absent, but a clear memory defect when Stim1 or both Stim genes were genetically ablated (Garcia‐Alvarez et al. 2015b). The mice strain used loses STIM1 and STIM2 expression specifically in the hippocampus and neocortical tissues only from the third postnatal week, when most neural circuits are already formed, being probably completely removed at 3 months of age (Tsien et al. 1996), while the earlier study used constitutive knockout mice that lack STIM2 from the embryonic stage and die at 12 weeks of age. Whether the defective memory is due to an altered development of all the circuits that encode memory needs to be clarified.
Later reports further explored the function of STIM2 in neurons demonstrating a robust role in synaptic function and organization, as well as in formation or maintenance of dendritic spines. STIM2 was found to be present more in mature (mushroom) spines than in the soma (Sun et al. 2014; Garcia‐Alvarez et al. 2015a), colocalizing with Ca2+/calmodulin‐dependent kinase II (CaMKII), a molecule involved in postsynaptic plasticity and long‐term potentiation (LTP)‐induced formation of this type of spine (Sanhueza et al. 2011; Sun et al. 2014). Lack of STIM2 in murine hippocampal neurons abolishes synaptic SOCE and CaMKII auto‐phosphorylation, which reduces the number of mushroom spines, linked to memory, but proportionally increased the number of thin (immature) spines (Sanhueza et al. 2011; Sun et al. 2014; Garcia‐Alvarez et al. 2015a; Sweatt, 2016). Thus, the overall density of spines seems to be unaffected. Consistent with this, a recent report has demonstrated impaired early LTP and long‐term depression (LTD) at CA3–CA1 hippocampal synapses in adult mice bearing a specific STIM2 deletion in CaMKIIα positive neurons (Yap et al. 2017). The identity of the SOC channel activated by STIM2 in spines is not yet clear, but the similar impaired spatial memory phenotype found in mice lacking TRPC1 points to the Ca2+ channel as a good candidate (Xing et al. 2016). Another candidate might be its binding partner, Orai2 (Brandman et al. 2007; Parvez et al. 2008; Bandyopadhyay et al. 2011; Berna‐Erro et al. 2012), whose mRNA transcript is highly expressed in mouse hippocampal neurons (Berna‐Erro et al. 2009). Therefore, it is conceivable that loss of STIM2 function in dendritic regions might lead to learning and memory deficiency, which was initially identified in STIM2‐deficient mice (Berna‐Erro et al. 2009).
STIM2 participates in the turnover of cholesterol content in neuronal PM. Cholesterol is an important molecule for synaptic transmission (for review see Zhang & Liu, 2015). The content of synaptic cholesterol upon glutamate release after kainic acid injection changed in wild‐type mice, but not in mice lacking STIM2, suggesting that cholesterol loss from the PM in neurons subjected to glutamate excitotoxicity largely depends on high levels of intracellular Ca2+ and a functional STIM2 (Sodero et al. 2012).
Furthermore, a recent study has revealed in spinal cord astrocytes that STIM2 participates in lipopolysaccharide (LPS)‐induced cytokine production (Gao et al. 2016). STIM2 knockdown significantly reduces SOCE and LPS‐induced interleukin (IL)‐6 and tumour necrosis factor α (TNF‐α) production, cytokines that are involved and upregulated in pain hypersensitivity (Gao et al. 2016).
Finally, massive neuronal loss in the hippocampus has been also observed in STIM2‐deficient mice, suggesting an essential function in long‐term and synaptic‐dependent neuronal survival (Sun et al. 2014). Due to the remarkable function of STM2 in neurons, a relevant role in cognitive diseases has been postulated (see below).
The immune system
Studies in human patients lacking functional SOCE demonstrated its crucial role in the immune system (Table 1). Patients bearing a dysfunctional version of STIM1 or Orai1 develop life‐threatening severe combined immunodeficiency (SCID)‐like, or Stormorken syndrome (Feske et al. 2006; Picard et al. 2009; Misceo et al. 2014; Morin et al. 2014). Accumulating evidence shows that, despite the role of STIM1 in the immune system being more prominent than that of STIM2, there is a clear cooperative function between both STIMs. Thus, studies in STIM1‐deficient mice showed an uncompleted phenotype due to partial compensation by STIM2 (Oh‐Hora et al. 2008, 2013; Ma et al. 2010; Matsumoto et al. 2011; Shaw et al. 2014).
In T cells, the key cellular function of SOCE is to initiate the translocation of nuclear factor of activated T cells (NFAT) from the cytosol to the nucleus, a necessary step to initiate important T cell processes, via the CaM–calcineurin pathway (Jain et al. 1993). In this context, STIM2 partially contributes to SOCE in CD4+ T cells, regulating sustained SOCE after ‘STIM1‐driven’ T cell activation and promoting interferon γ (IFN‐γ), IL‐2 and IL‐17 production. Thus, murine T cells lacking STIM2 display reduced but not abrogated SOCE (Oh‐Hora et al. 2008; Ma et al. 2010; Schuhmann et al. 2010). The consequence is a decrease in the number of Th17 cells, which are involved in the regulation of autoimmune responses (Ma et al. 2010; Schuhmann et al. 2010). STIM2 is also required, together with STIM1, to differentiate naive CD8+ T cells into cytotoxic terminal effector (Teff) cells, and also for degranulation, expression of Fas ligand (FasL), and production of TNF‐α and IFN‐γ, and is therefore necessary for the cytotoxic function (Weidinger et al. 2013; Shaw et al. 2014). Memory and recall responses in CD8+ T cells also require the function of both isoforms in CD4+ T cells, since STIMs seem to control the expression of CD40 ligand in these cells (Duttagupta et al. 2009). Interestingly, a recent report demonstrated that STIM1‐ and STIM2‐mediated SOCE also has a dual role eliciting protective B cell responses against viral infections and preventing humoral autoimmunity (Vaeth et al. 2016). CD4+ T follicular helper (Tfh) cells promote the development of mature B cells into memory and plasma cells, and therefore the generation of specific high‐affinity antibodies and humoral immunity after viral infection. This process is carried out at particular places inside secondary lymph nodes called germinal centres (GCs), and comprises B cell clonal selection, affinity maturation and mutation of their antibody genes. In contrast, T follicular regulatory (Tfr) cells derived from regulatory T (Treg) cells limit the GC reaction (Vaeth et al. 2016). Hence, both cell types have opposing roles in regulating humoral immunity against viral infection. In this context, SOCE is critically necessary to promote Tfh and Tfr differentiation because it elicits NFAT‐mediated expression of essential transcription factors such as BATF, Bcl‐6 and IRF4. Thus, mice lacking both STIM1 and STIM2 specifically in CD4+ T cells fail to develop sufficient functional Tfh cells to generate proper humoral immunity responses and die after chronic viral infection. In addition, aged transgenic mice are unable to develop sufficient functional Tfr cells to limit spontaneous GC formation, GC reaction, autoantibody production and therefore protection against autoimmune disease (Vaeth et al. 2016). In summary, transgenic mice lacking both isoforms in T cells fail to trigger cell immunity after exposure to viral infection, are unable to prevent tumour engraftment (see below) and show fatal chronification of viral infections (Weidinger et al. 2013; Shaw et al. 2014; Vaeth et al. 2016).
In B cells, removal of single STIM1 or STIM2 has been found to have a minor impact on SOCE while removal of both isoforms abrogates SOCE upon B cell receptor stimulation, indicating a strong cooperative function in these cells (Matsumoto et al. 2011). B cells lacking both STIMs develop well and show unaltered antibody responses, but the absence of SOCE leads to deficient proliferation and absence of anti‐inflammatory IL‐10 production due to defective activation of NFAT (Matsumoto et al. 2011).
In macrophages, the function of STIM2 is not clear. A study has reported that STIM2 mediates cell migration and cytokine TNF‐α and IL‐6 production upon G protein‐coupled receptor and Toll‐like receptor 4 (TLR4) activation, in contrast to the modest contribution of SOCE in IgG Fc γ receptor (FcγR)III‐ and FcγRIV‐mediated responses (Sogkas et al. 2015). They also reported that STIM2‐deficient mice are resistant to inflammation mediated by macrophages due to deficient macrophage recruitment and to a reduced production of TNF‐α, IL‐6 and IL‐1β (Sogkas et al. 2015). Thus, these results suggest a remarkable function of STIM2 in innate immunity. By contrast, another group reported preserved macrophage functions in STIM‐deficient cells (Vaeth et al. 2015); therefore, further studies are necessary to solve this apparent discrepancy.
Finally, removal of STIM2 in murine microglial cells has been reported to reduce SOCE and impair nucleotide‐induced migration and phagocytosis (Heo et al. 2015; Michaelis et al. 2015). Since it has been suggested that microglia participates in remodelling of connectivity in the developing brain, these studies present STIM2 and SOCE as possible targets for the treatment of aberrant microglial functions that may lead to inflammatory, neuro‐developmental and psychiatric disorders.
Other cellular functions of STIM2
STIM2 has also been found to play an important functional role in other cells and tissues. Despite very little being known about the function of STIM2 in the skeletal muscle, as compared to its partner STIM1, in vitro studies have revealed that the interaction of STIM2 with STIM1 controls SOCE, which is required for both proper human myoblast differentiation and myotube excitation–contraction coupling (Darbellay et al. 2010). However, no apparent abnormality in skeletal muscle development in STIM2‐deficient mice was observed (Table 1) (Berna‐Erro et al. 2009).
Finally, it is noteworthy that the absence of STIM2 in salivary glands diminished agonist‐dependent saliva secretion in mice upon mild but not strong stimulus, indicating a potential role in salivary gland function (Ong et al. 2015).
STIM2 and disease
In contrast to STIM1 or Orai1, there is no evidence of diseases caused by single mutated STIM2 alleles in human. However, the literature describes pathological phenotypes in patients bearing alterations in the chromosome region 4p15.2, where STIM2 is located. For instance, deletions in chromosome 4 involving the region 4p16.1–p14 have been associated to mild or moderate cognitive disability accompanied with morphological face alterations, tall thin body figure, hyperextensible joints, etc. (Chitayat et al. 1995; Chen et al. 2013). The presence of other genes in this region, many of them involved in brain development such as PCDH7, ARAP2 and CCKAR, which are altered together with STIM2 in many cases, makes it impossible to guess the contribution of STIM2 to these phenotypes. Therefore, most of the information we have about STIM2 and disease has been obtained in vitro or from rat and transgenic murine models (Table 1). In this context, the proposed role of STIM2 as a contributor to pathological conditions ranges from neurodegenerative, autoimmune and lung disorders to cancer.
The nervous system
Most studies made in nervous system support the idea of a relevant role in brain pathologies such as ischaemic damage, Alzheimer's disease (AD) and Huntington's disease (HD) (Table 1). The first study on STIM2 function in murine brain revealed that in the presence of this protein brain damage under transient anoxic conditions is greater (Berna‐Erro et al. 2009). Thus STIM2‐deficient neurons displayed increased resistance to hypoxia, probably by preventing Ca2+ overload in the cell and the subsequent Ca2+‐dependent apoptosis that happens during hypoxic conditions. This initial analysis gave a starting point to study the link between STIM2, brain and pathologies associated with abnormal intracellular Ca2+ homeostasis. A similar reduced brain injury was later reported in STIM2‐knockdown mice subjected to traumatic brain injury, the most frequent neurological disease (Rao et al. 2015).
Recent studies also linked disturbances in STIM2 expression or function with memory loss in AD and with neurodegeneration in HD, giving further evidence of its important contribution in brain pathologies associated with altered intracellular Ca2+ homeostasis. For instance, a connection has been found between the presence of presenilin 1 and expression of STIMs in murine embryonic cells (Bojarski et al. 2009). The absence of native presenilin 1 or the presence of mutated presenilins associated with familiar AD clearly decreased STIM2 expression and SOCE. In fact, STIM2 but not STIM1 was found downregulated in cortical samples obtained from AD patients (Sun et al. 2014). In addition to the characteristic neurodegenerative disorder, AD is accompanied by a pronounced decrease of mushroom spine numbers and gradual memory loss. As commented above, STIM2 downregulation is involved in spine loss and memory decline. Consistent with this, STIM2 overexpression has been found to correct the phenotype in two murine models of AD, the presenilin1 M146V knock‐in (PS1‐M146V KI) and the amyloid precursor protein knock‐in (Sun et al. 2014; Zhang et al. 2015).
In human neuroblastoma cells, the expression of a mutated presenilin 1 (M146V) linked to AD decreases SOCE. Surprisingly, this phenotype could be rescued only by knocking down STIM2 in these cells, indicating a STIM2 gain‐of‐function in contrast to what was reported using murine models. In that case, STIM2 might act as an ‘inhibitor’ of STIM1‐dependent SOCE (Ryazantseva et al. 2013). More research has to be done in order to identify the predominant STIM2 variant in these cells.
Mutant huntingtin (mHtt) protein causes defective striatal neuron function, reduction of synaptic contacts, neurodegeneration and eventually HD (for review see Nithianantharajah & Hannan, 2013). Seeking determinants of synaptic loss in this disease, the elevated synaptic SOCE, as a consequence of increased STIM2 expression, was proposed as a pathological mechanism (Wu et al. 2016). Using medium spiny neurons (MSNs) isolated from a murine model of HD (FVB‐Tg[YAC128]53Hay/J) (Slow et al. 2003) that express an mHtt, researchers discovered that mHtt binds to IP3R1 and increases its sensitivity for IP3. The increased steady‐state activity of sensitized IP3R1 reduces [Ca2+]ER, which, in turn, overactivates STIM2 and promotes sustained and synaptotoxic SOCE in YAC128 MSN spines. Additionally, increased STIM2 expression was found in these cells and in YAC128 mouse striatum. The key finding was that knockout of STIM2 restored altered synaptic SOCE and rescued spine loss in YAC128 MSNs, pointing again to STIM2 as a necessary mediator of this pathological mechanism also in HD. An interesting question is why decreased STIM2 contributes to spine loss in AD, while in HD it is increased STIM2 that leads to decreased spine numbers. The most probable explanation is that neurons require SOCE amplitudes in stabilized spines to be set within a narrow range in order to maintain them over time (Wu et al. 2016).
The immune system
Concerning the role of STIM2 in immune disorders, it has been reported that the reduction of both STIM2 and STIM1 in human T cells is associated with the autoimmune response against salivary and other exocrine glands in Sjögren's syndrome (Cheng et al. 2012). This disease is characterized by progressive lymphocytic infiltration into the gland, leading to gland destruction and secretory defects. The diminished STIM1 expression in infiltrating T lymphocytes and, together with STIM2, in peripheral blood mononuclear cells seems to be a key feature, since double STIM1/STIM2 knockout mice develop Sjögren's‐like syndrome.
STIM2 has been reported to play a role in autoreactive T cell regulation during autoimmune central nervous system inflammation in mice (Ma et al. 2010; Schuhmann et al. 2010). Mice lacking STIM2 developed a delayed version of experimental autoimmune encephalomyelitis (EAE), associated with defective lymphocytic proliferation and a reduction of IFN‐γ/IL‐17 production by neuroantigen‐specific T cells. Thus, STIM2 function might be linked to IL‐17 secretion in these cells, which in turn depends especially on the Ca2+‐regulated transcription factor NFAT (Liu et al. 2004). Since Th17 cells have been linked to regulatory effects on autoreactive immune responses, the defective function of these cells in the absence of STIM2 might explain the protective phenotype.
Finally, a number of pathogens, such as Shigella, Salmonella and Yersinia, suppress macrophage responses via stimulation of Ca2+ influx and facilitation of the activation of cAMP response element‐binding protein (CREB). In this context, it has been reported that Mycobacterium tuberculosis increases the expression of L‐type Ca2+ channels in the PM via upregulation of CREB phosphorylation. STIM2, together with STIM1, contributes to Ca2+ voltage‐gated channel subunit α 1S (CACNA1S) upregulation, and knockdown of any of them decreases surface CACNA1S expression in macrophages infected with M. tuberculosis (Antony et al. 2015).
The vascular system
Enhanced pulmonary arterial smooth muscle cell (PASMC) proliferation plays an important role in vascular remodelling in patients with pulmonary arterial hypertension, a disease that ends in death if untreated. In this context, PASMCs switch from a contractile or quiescent phenotype to a proliferative phenotype. Upregulated expression of STIM2, together with TRPC6, and Orai2, and, therefore, SOCE, may play an important role in the transition of contractile PASMCs to the proliferative phenotype, contributing to pathological vascular remodelling (Fernandez et al. 2015).
STIM2 and cancer
Altered Ca2+ homeostasis is unlikely to be an aetiological factor of cancer, but growing evidence indicates remodelled SOCE as an important contributor to many hallmarks of cell cancer physiology (for review see Villalobos et al. 2016). In the case of STIM2, an altered STIM1/STIM2 expression ratio has been found in a study comprising 295 breast tumours, with the poorest prognosis for those tumours displaying high STIM1/STIM2 expression ratios (McAndrew et al. 2011). In accordance with this, an increase in STIM2 expression has been associated with a less invasive phenotype in colorectal cancer, pointing to a tumour cell growth suppressor in contrast to STIM1 (Aytes et al. 2012). Similar results were observed in human melanoma, where STIM2 is highly expressed and silencing of Orai1/STIM2 caused enhanced proliferation but reduced invasive and migratory ability (Stanisz et al. 2014). Increased STIM2 expression has been also found in glioblastoma multiforme tumours (Ruano et al. 2006). Therefore, the use of the STIM1/STIM2 ratio as a marker of tumour aggressiveness seems to be promising and might be further evaluated.
On the other hand, STIM2 expression is reduced in some human colon carcinoma cells, while it is present in normal human mucosa cells. STIM2 knockdown reduces SOCE in normal cells but promotes apoptosis resistance, suggesting that downregulation of STIM2 in tumour cells might be a mechanism to acquire apoptosis resistance (Sobradillo et al. 2014).
In breast cancer cells expressing oestrogen receptor (ER+), SOCE strongly depends on STIM2/STIM1 and Orai3, while SOCE is mostly promoted by STIM1 and Orai1 in breast cancer cells not expressing oestrogen receptor (ER–) (Motiani et al. 2010). The causes and consequences of this phenotypical differences still remain unclear although SOCE responses are clearly larger in ER− than in ER+ cells (Motiani et al. 2010).
Concluding remarks
As summarized in this review, STIM2 seems to play a complementary role in the control of intracellular Ca2+ homeostasis when STIM1 is prominently present. Far from contradicting the classical idea about STIM2 and its specialized control in regulating basal [Ca2+]c, new evidence expands STIM2's function and reveals its versatility, which seems to be behind some discrepant results observed in the past. The reason is unclear: maybe the complex behaviour of STIM proteins, the heterogeneity of the experimental models used, or a hypothetical high ability of SOCE or STIMs to change and rearrange themselves upon experimental manipulation. An explanation was given by Brandman, who observed decreased SOCE as the STIM2 overexpression period increases from 9 to 24 h, indicative of slow adaptive rearrangements, for instance against prolonged increased [Ca2+]c (Brandman et al. 2007). New studies suggested that those slow rearrangements might comprise the expression of inhibitory STIM2 splicing forms, which are sensitive to [Ca2+]c. STIM2 can also work as an adaptor protein and increase the sensitivity of STIM1 to small [Ca2+]ER changes. Together with the unexplored effect of phosphorylation and glycosylation on STIM2 function, these results may help to fully understand its heterogeneous behaviour at the cellular level in the near future.
Due to their different kinetics and binding partners, the expression ratio of both isoforms also will determine the contribution of STIM2 to SOCE and the final cellular response, since they represent differences in the interaction with other components of SOCE. But the most important progress has been made in the study of STIM2 function in different tissues. At this level, studies also reported contradictory results. For instance, transgenic murine models to study innate immunity revealed both irrelevant and important functions of STIM2 in general SOCE, in cell migration, cytokine production and macrophage function (Braun et al. 2009; Sogkas et al. 2015; Vaeth et al. 2015). Similarly, studies on the nervous system using transgenic mice lacking STIM2 showed opposite results for learning and working memory (Berna‐Erro et al. 2009; Garcia‐Alvarez et al. 2015b). In the nervous system, STIM2 seems to have the predominant function, and despite the presence of SOCE in this tissue, this function is not clear (for review see Lu & Fivaz, 2016), and the tendency is to accept the existence of a particular version of SOCE in neurons. Interestingly, groups that used constitutive knockout mice for these studies showed significant results, while those that used conditional knockout mice observed irrelevant results. Constitutive knockout mice lack STIM2 since egg fertilization, while conditional knockout technology uses a Cre‐Lox recombination system to remove STIM2 in a specific cell type in a chosen stage of development. The absence of phenotype in conditional knockout mice lacking STIM2 might reflect its importance in these tissues during development.
Most studies indicate that the function of STIMs cannot be understood separate from each other due to their strong interaction, and that there is a faint line between cooperative and specialized functions among them (for review see Lopez et al. 2012). For example, experiments on transgenic mice reported the highest physiological impact when both STIM genes were genetically ablated, and redundant functions when only one (mostly STIM2) was silenced, pointing to a synergistic function between them (Oh‐Hora et al. 2008; Ma et al. 2010; Matsumoto et al. 2011; Mancarella et al. 2013). Thus, the physiological role of STIMs might be difficult to understand if they are separately addressed.
Additional information
Competing interests
The authors declare no conflict of interests.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by MINECO (Grant BFU2013‐45564‐C2‐1‐P and BFU2016‐74932‐C2‐1‐P) and Gobierno de Extremadura‐FEDER (GR15029).
Acknowledgements
A.B.‐E. is supported by UPF ‘MINECO‐Plan Nacional de I+D+i(2008‐2012)‐SAF 2012‐38140’.
Biographies
Alejandro Berna Erro is a posdoctoral researcher specializing in Molecular Biology and currently working at the University Ponpeu Fabra. He received his PhD from Julius‐Maximilians University of Würzburg. His research focuses on intracellular Ca2+ signalling regulated by SOCE or TRP Ca2+ channels. Isaac Jardin is a “Juan de la Cierva‐Postdoctoral Researcher” currently working in the Department of Physiology at the University of Extremadura, where he obtained his PhD. His main research focuses on intracellular Ca2+ homeostasis, concretely the mechanisms that fine‐tune store‐operated Ca2+ entry mediated by STIM, Orai and TRPC proteins.

Gines M. Salido is Full Professor in Physiology at The University of Extremadura since 1991. He received his D. Sc. from University of Granada (Spain) in 1981. He is currently the coordinator of the Cell Physiology Research Group at the University of Extremadura and his research is focused on the intracellular mechanisms of Ca2+ homeostasis. Juan A Rosado is a University Lecturer in Physiology at the University of Extremadura. After obtaining his PhD he worked for 2 years in the Department of Physiology of the University of Cambridge (UK) and then returned to the University of Extremadura. His research focuses on intracellular Ca2+ homeostasis, especially the mechanism of store‐operated Ca2+ entry, STIM, Orai and TRPC proteins.
References
- Abell E, Ahrends R, Bandara S, Park BO & Teruel MN (2011). Parallel adaptive feedback enhances reliability of the Ca2+ signaling system. Proc Natl Acad Sci USA 108, 14485–14490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albarran L, Lopez JJ, Amor NB, Martin‐Cano FE, Berna‐Erro A, Smani T, Salido GM & Rosado JA (2016a). Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store‐operated calcium entry. Sci Rep 6, 24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albarran L, Lopez JJ, Woodard GE, Salido GM & Rosado JA (2016b). Store‐operated Ca2+ entry‐associated regulatory factor (SARAF) plays an important role in the regulation of arachidonate‐regulated Ca2+ (ARC) channels. J Biol Chem 291, 6982–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony C, Mehto S, Tiwari BK, Singh Y & Natarajan K (2015). Regulation of L‐type voltage gated calcium channel CACNA1S in macrophages upon mycobacterium tuberculosis infection. PLoS One 10, e0124263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytes A, Mollevi DG, Martinez‐Iniesta M, Nadal M, Vidal A, Morales A, Salazar R, Capella G & Villanueva A (2012). Stromal interaction molecule 2 (STIM2) is frequently overexpressed in colorectal tumors and confers a tumor cell growth suppressor phenotype. Mol Carcinog 51, 746–753. [DOI] [PubMed] [Google Scholar]
- Baba A, Yasui T, Fujisawa S, Yamada RX, Yamada MK, Nishiyama N, Matsuki N & Ikegaya Y (2003). Activity‐evoked capacitative Ca2+ entry: implications in synaptic plasticity. J Neurosci 23, 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, Numaga T, Mori Y, Iino M, Hikida M & Kurosaki T (2006). Coupling of STIM1 to store‐operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci USA 103, 16704–16709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasuriya D, Srivats S, Murrell‐Lagnado RD & Edwardson JM (2014). Atomic force microscopy (AFM) imaging suggests that stromal interaction molecule 1 (STIM1) binds to Orai1 with sixfold symmetry. FEBS Lett 588, 2874–2880. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay BC, Pingle SC & Ahern GP (2011). Store‐operated Ca2+ signaling in dendritic cells occurs independently of STIM1. J Leukoc Biol 89, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrero MJ, Montero M & Alvarez J (1997). Dynamics of [Ca2+] in the endoplasmic reticulum and cytoplasm of intact HeLa cells. A comparative study. J Biol Chem 272, 27694–27699. [DOI] [PubMed] [Google Scholar]
- Bauer MC, O'Connell D, Cahill DJ & Linse S (2008). Calmodulin binding to the polybasic C‐termini of STIM proteins involved in store‐operated calcium entry. Biochemistry 47, 6089–6091. [DOI] [PubMed] [Google Scholar]
- Beliveau E, Lessard V & Guillemette G (2014). STIM1 positively regulates the Ca2+ release activity of the inositol 1,4,5‐trisphosphate receptor in bovine aortic endothelial cells. PLoS One 9, e114718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berna‐Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G & Nieswandt B (2009). STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal 2, ra67. [DOI] [PubMed] [Google Scholar]
- Berna‐Erro A, Galan C, Dionisio N, Gomez LJ, Salido GM & Rosado JA (2012). Capacitative and non‐capacitative signaling complexes in human platelets. Biochim Biophys Acta 1823, 1242–1251. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1995). Capacitative calcium entry. Biochem J 312, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyersdorf N, Braun A, Vogtle T, Varga‐Szabo D, Galdos RR, Kissler S, Kerkau T & Nieswandt B (2009). STIM1‐independent T cell development and effector function in vivo. J Immunol 182, 3390–3397. [DOI] [PubMed] [Google Scholar]
- Bhardwaj R, Muller HM, Nickel W & Seedorf M (2013). Oligomerization and Ca2+/calmodulin control binding of the ER Ca2+‐sensors STIM1 and STIM2 to plasma membrane lipids. Biosci Rep 33, e00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarski L, Pomorski P, Szybinska A, Drab M, Skibinska‐Kijek A, Gruszczynska‐Biegala J & Kuznicki J (2009). Presenilin‐dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer's disease. Biochim Biophys Acta 1793, 1050–1057. [DOI] [PubMed] [Google Scholar]
- Brandman O, Liou J, Park WS & Meyer T (2007). STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131, 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A, Gessner JE, Varga‐Szabo D, Syed SN, Konrad S, Stegner D, Vogtle T, Schmidt RE & Nieswandt B (2009). STIM1 is essential for Fcgamma receptor activation and autoimmune inflammation. Blood 113, 1097–1104. [DOI] [PubMed] [Google Scholar]
- Cai X (2007). Molecular evolution and functional divergence of the Ca2+ sensor protein in store‐operated Ca2+ entry: stromal interaction molecule. PLoS One 2, e609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X (2008). Unicellular Ca2+ signaling ‘toolkit’ at the origin of metazoa. Mol Biol Evol 25, 1357–1361. [DOI] [PubMed] [Google Scholar]
- Collins SR & Meyer T (2011). Evolutionary origins of STIM1 and STIM2 within ancient Ca2+ signaling systems. Trends Cell Biol 21, 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Lee MJ, Chern SR, Wu PS, Su JW, Chen YT, Lee MS & Wang W (2013). Prenatal diagnosis and molecular cytogenetic characterization of a de novo proximal interstitial deletion of chromosome 4p (4p15.2–>p14). Gene 529, 351–356. [DOI] [PubMed] [Google Scholar]
- Cheng KT, Alevizos I, Liu X, Swaim WD, Yin H, Feske S, Oh‐hora M & Ambudkar IS (2012). STIM1 and STIM2 protein deficiency in T lymphocytes underlies development of the exocrine gland autoimmune disease, Sjogren's syndrome. Proc Natl Acad Sci USA 109, 14544–14549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitayat D, Ruvalcaba RH, Babul R, Teshima IE, Posnick JC, Vekemans MJ, Scarpelli H & Thuline H (1995). Syndrome of proximal interstitial deletion 4p15: report of three cases and review of the literature. Am J Med Genet 55, 147–154. [DOI] [PubMed] [Google Scholar]
- Darbellay B, Arnaudeau S, Ceroni D, Bader CR, Konig S & Bernheim L (2010). Human muscle economy myoblast differentiation and excitation‐contraction coupling use the same molecular partners, STIM1 and STIM2. J Biol Chem 285, 22437–22447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaurex N & Frieden M (2003). Measurements of the free luminal ER Ca2+ concentration with targeted “cameleonˮ fluorescent proteins. Cell Calcium 34, 109–119. [DOI] [PubMed] [Google Scholar]
- Desai PN, Zhang X, Wu S, Janoshazi A, Bolimuntha S, Putney JW & Trebak M (2015). Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci Signal 8, ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette M, Nadler M, Okuhara D, Thompson J, Shuttleworth T & Lawler J (2014). Members of the thrombospondin gene family bind stromal interaction molecule 1 and regulate calcium channel activity. Matrix Biol 37, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttagupta PA, Boesteanu AC & Katsikis PD (2009). Costimulation signals for memory CD8+ T cells during viral infections. Crit Rev Immunol 29, 469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan E, Chung SH, Bhardwaj R & Seedorf M (2012). Di‐arginine signals and the K‐rich domain retain the Ca2+ sensor STIM1 in the endoplasmic reticulum. Traffic 13, 992–1003. [DOI] [PubMed] [Google Scholar]
- Ercan E, Momburg F, Engel U, Temmerman K, Nickel W & Seedorf M (2009). A conserved, lipid‐mediated sorting mechanism of yeast Ist2 and mammalian STIM proteins to the peripheral ER. Traffic 10, 1802–1818. [DOI] [PubMed] [Google Scholar]
- Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D & Yuan JX (2015). Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol 308, C581–C593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M & Rao A (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. [DOI] [PubMed] [Google Scholar]
- Gao X, Xia J, Munoz FM, Manners MT, Pan R, Meucci O, Dai Y & Hu H (2016). STIMs and Orai1 regulate cytokine production in spinal astrocytes. J Neuroinflammation 13, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Alvarez G, Lu B, Yap KA, Wong LC, Thevathasan JV, Lim L, Ji F, Tan KW, Mancuso JJ, Tang W, Poon SY, Augustine GJ & Fivaz M (2015a). STIM2 regulates PKA‐dependent phosphorylation and trafficking of AMPARs. Mol Biol Cell 26, 1141–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Alvarez G, Shetty MS, Lu B, Yap KA, Oh‐Hora M, Sajikumar S, Bichler Z & Fivaz M (2015b). Impaired spatial memory and enhanced long‐term potentiation in mice with forebrain‐specific ablation of the Stim genes. Front Behav Neurosci 9, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham SJ, Dziadek MA & Johnstone LS (2011). A cytosolic STIM2 preprotein created by signal peptide inefficiency activates ORAI1 in a store‐independent manner. J Biol Chem 286, 16174–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszczynska‐Biegala J, Pomorski P, Wisniewska MB & Kuznicki J (2011). Differential roles for STIM1 and STIM2 in store‐operated calcium entry in rat neurons. PLoS One 6, e19285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Karl RM, Alexander RP, Adelsberger H, Brill MS, Ruhlmann C, Ansel A, Sakimura K, Baba Y, Kurosaki T, Misgeld T & Konnerth A (2014). STIM1 controls neuronal Ca2+ signaling, mGluR1‐dependent synaptic transmission, and cerebellar motor behavior. Neuron 82, 635–644. [DOI] [PubMed] [Google Scholar]
- He J, Yu T, Pan J & Li H (2012). Visualisation and identification of the interaction between STIM1s in resting cells. PLoS One 7, e33377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo DK, Lim HM, Nam JH, Lee MG & Kim JY (2015). Regulation of phagocytosis and cytokine secretion by store‐operated calcium entry in primary isolated murine microglia. Cell Signal 27, 177–186. [DOI] [PubMed] [Google Scholar]
- Hoover PJ & Lewis RS (2011). Stoichiometric requirements for trapping and gating of Ca2+ release‐activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1). Proc Natl Acad Sci USA 108, 13299–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S & Worley PF (2006). STIM1 carboxyl‐terminus activates native SOC, I crac and TRPC1 channels. Nat Cell Biol 8, 1003–1010. [DOI] [PubMed] [Google Scholar]
- Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T & Rao A (1993). The T‐cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature 365, 352–355. [DOI] [PubMed] [Google Scholar]
- Jha A, Ahuja M, Maleth J, Moreno CM, Yuan JP, Kim MS & Muallem S (2013). The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J Cell Biol 202, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, So I, Shin DM, Muallem S & Yuan JP (2014). Molecular determinants mediating gating of Transient Receptor Potential Canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J Biol Chem 289, 6372–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu L, Deng Y, Ji W, Du W, Xu P, Chen L & Xu T (2011). Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res 21, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T & Meyer T (2007). Live‐cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA 104, 9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr & Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+‐store‐depletion‐triggered Ca2+ influx. Curr Biol 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XK, Lin X & Gaffen SL (2004). Crucial role for nuclear factor of activated T cells in T cell receptor‐mediated regulation of human interleukin‐17. J Biol Chem 279, 52762–52771. [DOI] [PubMed] [Google Scholar]
- Lopez E, Salido GM, Rosado JA & Berna‐Erro A (2012). Unraveling STIM2 function. J Physiol Biochem 68, 619–633. [DOI] [PubMed] [Google Scholar]
- Lopez JJ, Albarran L, Gomez LJ, Smani T, Salido GM & Rosado JA (2016). Molecular modulators of store‐operated calcium entry. Biochim Biophys Acta 1863, 2037–2043. [DOI] [PubMed] [Google Scholar]
- Lu B & Fivaz M (2016). Neuronal SOCE: myth or reality? Trends Cell Biol 26, 890–893. [DOI] [PubMed] [Google Scholar]
- Lu W, Wang J, Peng G, Shimoda LA & Sylvester JT (2009). Knockdown of stromal interaction molecule 1 attenuates store‐operated Ca2+ entry and Ca2+ responses to acute hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 297, L17–L25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, McCarl CA, Khalil S, Luthy K & Feske S (2010). T‐cell‐specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur J Immunol 40, 3028–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancarella S, Potireddy S, Wang Y, Gao H, Gandhirajan RK, Autieri M, Scalia R, Cheng Z, Wang H, Madesh M, Houser SR & Gill DL (2013). Targeted STIM deletion impairs calcium homeostasis, NFAT activation, and growth of smooth muscle. FASEB J 27, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T & Baba Y (2011). The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin‐10 production. Immunity 34, 703–714. [DOI] [PubMed] [Google Scholar]
- McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, Smart CE, Brown MA, Kenny PA, Roberts‐Thomson SJ & Monteith GR (2011). ORAI1‐mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther 10, 448–460. [DOI] [PubMed] [Google Scholar]
- Michaelis M, Nieswandt B, Stegner D, Eilers J & Kraft R (2015). STIM1, STIM2, and Orai1 regulate store‐operated calcium entry and purinergic activation of microglia. Glia 63, 652–663. [DOI] [PubMed] [Google Scholar]
- Miederer AM, Alansary D, Schwar G, Lee PH, Jung M, Helms V & Niemeyer BA (2015). A STIM2 splice variant negatively regulates store‐operated calcium entry. Nat Commun 6, 6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misceo D, Holmgren A, Louch WE, Holme PA, Mizobuchi M, Morales RJ, De Paula AM, Stray‐Pedersen A, Lyle R, Dalhus B, Christensen G, Stormorken H, Tjonnfjord GE & Frengen E (2014). A dominant STIM1 mutation causes Stormorken syndrome. Hum Mutat 35, 556–564. [DOI] [PubMed] [Google Scholar]
- Morin G, Bruechle NO, Singh AR, Knopp C, Jedraszak G, Elbracht M, Bremond‐Gignac D, Hartmann K, Sevestre H, Deutz P, Herent D, Nurnberg P, Romeo B, Konrad K, Mathieu‐Dramard M, Oldenburg J, Bourges‐Petit E, Shen Y, Zerres K, Ouadid‐Ahidouch H & Rochette J (2014). Gain‐of‐function mutation in STIM1 (P.R304W) is associated with Stormorken syndrome. Hum Mutat 35, 1221–1232. [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS & Baudry M (1986). Selective impairment of learning and blockade of long‐term potentiation by an N‐methyl‐D‐aspartate receptor antagonist, AP5. Nature 319, 774–776. [DOI] [PubMed] [Google Scholar]
- Motiani RK, Abdullaev IF & Trebak M (2010). A novel native store‐operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor‐positive versus estrogen receptor‐negative breast cancer cells. J Biol Chem 285, 19173–19183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithianantharajah J & Hannan AJ (2013). Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience‐dependent mediators of cognitive and psychiatric symptoms in Huntington's disease. Neuroscience 251, 66–74. [DOI] [PubMed] [Google Scholar]
- Oh‐Hora M, Komatsu N, Pishyareh M, Feske S, Hori S, Taniguchi M, Rao A & Takayanagi H (2013). Agonist‐selected T cell development requires strong T cell receptor signaling and store‐operated calcium entry. Immunity 38, 881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh‐Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S & Rao A (2008). Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9, 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HL, de Souza LB, Zheng C, Cheng KT, Liu X, Goldsmith CM, Feske S & Ambudkar IS (2015). STIM2 enhances receptor‐stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum‐plasma membrane junctions. Sci Signal 8, ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oritani K & Kincade PW (1996). Identification of stromal cell products that interact with pre‐B cells. J Cell Biol 134, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Raveh A, Kaminsky I, Meller R & Reuveny E (2012). SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 149, 425–438. [DOI] [PubMed] [Google Scholar]
- Parvez S, Beck A, Peinelt C, Soboloff J, Lis A, Monteilh‐Zoller M, Gill DL, Fleig A & Penner R (2008). STIM2 protein mediates distinct store‐dependent and store‐independent modes of CRAC channel activation. FASEB J 22, 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson RL, Boehning D & Snyder SH (2004). Inositol 1,4,5‐trisphosphate receptors as signal integrators. Annu Rev Biochem 73, 437–465. [DOI] [PubMed] [Google Scholar]
- Peel SE, Liu B & Hall IP (2006). A key role for STIM1 in store operated calcium channel activation in airway smooth muscle. Respir Res 7, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan‐Huberson M, Lis A, Fleig A, Penner R & Kinet JP (2006). Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat Cell Biol 8, 771–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux‐Laucat F, Rechavi G, Rao A, Fischer A & Feske S (2009). STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med 360, 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW Jr (1986). A model for receptor‐regulated calcium entry. Cell Calcium 7, 1–12. [DOI] [PubMed] [Google Scholar]
- Quintana A, Rajanikanth V, Farber‐Katz S, Gudlur A, Zhang C, Jing J, Zhou Y, Rao A & Hogan PG (2015). TMEM110 regulates the maintenance and remodeling of mammalian ER‐plasma membrane junctions competent for STIM‐ORAI signaling. Proc Natl Acad Sci USA 112, E7083–E7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Yen M, Sadaghiani AM, Malmersjo S, Park CY, Dolmetsch RE & Lewis RS (2015). Alternative splicing converts STIM2 from an activator to an inhibitor of store‐operated calcium channels. J Cell Biol 209, 653–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao W, Zhang L, Peng C, Hui H, Wang K, Su N, Wang L, Dai SH, Yang YF, Chen T, Luo P & Fei Z (2015). Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim Biophys Acta 1852, 2402–2413. [DOI] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G & Stauderman KA (2005). STIM1, an essential and conserved component of store‐operated Ca2+ channel function. J Cell Biol 169, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano Y, Mollejo M, Ribalta T, Fiano C, Camacho FI, Gomez E, de Lope AR, Hernandez‐Moneo JL, Martinez P & Melendez B (2006). Identification of novel candidate target genes in amplicons of Glioblastoma multiforme tumors detected by expression and CGH microarray profiling. Mol Cancer 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazantseva M, Skobeleva K & Kaznacheyeva E (2013). Familial Alzheimer's disease‐linked presenilin‐1 mutation M146V affects store‐operated calcium entry: does gain look like loss? Biochimie 95, 1506–1509. [DOI] [PubMed] [Google Scholar]
- Sabbioni S, Barbanti‐Brodano G, Croce CM & Negrini M (1997). GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res 57, 4493–4497. [PubMed] [Google Scholar]
- Sanhueza M, Fernandez‐Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW & Lisman J (2011). Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci 31, 9170–9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmann MK, Stegner D, Berna‐Erro A, Bittner S, Braun A, Kleinschnitz C, Stoll G, Wiendl H, Meuth SG & Nieswandt B (2010). Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J Immunol 184, 1536–1542. [DOI] [PubMed] [Google Scholar]
- Shalygin A, Skopin A, Kalinina V, Zimina O, Glushankova L, Mozhayeva GN & Kaznacheyeva E (2015). STIM1 and STIM2 proteins differently regulate endogenous store‐operated channels in HEK293 cells. J Biol Chem 290, 4717–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Weidinger C, Vaeth M, Luethy K, Kaech SM & Feske S (2014). CD4+ and CD8+ T cell‐dependent antiviral immunity requires STIM1 and STIM2. J Clin Invest 124, 4549–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR & Hayden MR (2003). Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 12, 1555–1567. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA & Gill DL (2006a). STIM2 is an inhibitor of STIM1‐mediated store‐operated Ca2+ entry. Curr Biol 16, 1465–1470. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W & Gill DL (2006b). Orai1 and STIM reconstitute store‐operated calcium channel function. J Biol Chem 281, 20661–20665. [DOI] [PubMed] [Google Scholar]
- Sobradillo D, Hernandez‐Morales M, Ubierna D, Moyer MP, Nunez L & Villalobos C (2014). A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J Biol Chem 289, 28765–28782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodero AO, Vriens J, Ghosh D, Stegner D, Brachet A, Pallotto M, Sassoe‐Pognetto M, Brouwers JF, Helms JB, Nieswandt B, Voets T & Dotti CG (2012). Cholesterol loss during glutamate‐mediated excitotoxicity. EMBO J 31, 1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogkas G, Stegner D, Syed SN, Vogtle T, Rau E, Gewecke B, Schmidt RE, Nieswandt B & Gessner JE (2015). Cooperative and alternate functions for STIM1 and STIM2 in macrophage activation and in the context of inflammation. Immun Inflamm Dis 3, 154–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA & Gill DL (2006). STIM1 has a plasma membrane role in the activation of store‐operated Ca2+ channels. Proc Natl Acad Sci USA 103, 4040–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanisz H, Saul S, Muller CS, Kappl R, Niemeyer BA, Vogt T, Hoth M, Roesch A & Bogeski I (2014). Inverse regulation of melanoma growth and migration by Orai1/STIM2‐dependent calcium entry. Pigment Cell Melanoma Res 27, 442–453. [DOI] [PubMed] [Google Scholar]
- Stanisz H, Stark A, Kilch T, Schwarz EC, Muller CS, Peinelt C, Hoth M, Niemeyer BA, Vogt T & Bogeski I (2012). ORAI1 Ca2+ channels control endothelin‐1‐induced mitogenesis and melanogenesis in primary human melanocytes. J Invest Dermatol 132, 1443–1451. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Li GY, Plevin MJ, Ames JB & Ikura M (2006). Stored Ca2+ depletion‐induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF‐SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem 281, 35855–35862. [DOI] [PubMed] [Google Scholar]
- Steinbeck JA, Henke N, Opatz J, Gruszczynska‐Biegala J, Schneider L, Theiss S, Hamacher N, Steinfarz B, Golz S, Brustle O, Kuznicki J & Methner A (2011). Store‐operated calcium entry modulates neuronal network activity in a model of chronic epilepsy. Exp Neurol 232, 185–194. [DOI] [PubMed] [Google Scholar]
- Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL 3rd & Bezprozvanny I (2014). Reduced synaptic STIM2 expression and impaired store‐operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82, 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2016). Neural plasticity and behavior ‐ sixty years of conceptual advances. J Neurochem 139, 179–199. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER & Tonegawa S (1996). Subregion‐ and cell type‐restricted gene knockout in mouse brain. Cell 87, 1317–1326. [DOI] [PubMed] [Google Scholar]
- Vaeth M, Eckstein M, Shaw PJ, Kozhaya L, Yang J, Berberich‐Siebelt F, Clancy R, Unutmaz D & Feske S (2016). Store‐operated Ca2+ entry in follicular T cells controls humoral immune responses and autoimmunity. Immunity 44, 1350–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal‐Celhay C, Zahra A, Kozhaya L, Weidinger C, Philips J, Unutmaz D & Feske S (2015). Ca2+ signaling but not store‐operated ca2+ entry is required for the function of macrophages and dendritic cells. J Immunol 195, 1202–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F (2015). Ryanodine receptors: allosteric ion channel giants. J Mol Biol 427, 31–53. [DOI] [PubMed] [Google Scholar]
- Vetter S (2013). Electrophysiological characterization of STIM2 knockout mice. Thesis Dissertation, Universität Würzburg.
- Villalobos C, Sobradillo D, Hernandez‐Morales M & Nunez L (2016). Remodeling of calcium entry pathways in cancer. Adv Exp Med Biol 898, 449–466. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J & Gill DL (2014). Distinct Orai‐coupling domains in STIM1 and STIM2 define the Orai‐activating site. Nat Commun 5, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidinger C, Shaw PJ & Feske S (2013). STIM1 and STIM2‐mediated Ca2+ influx regulates antitumour immunity by CD8+ T cells. EMBO Mol Med 5, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ & Dziadek MA (2001). Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J 357, 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RT, Senior PV, Van Stekelenburg L, Layton JE, Smith PJ & Dziadek MA (2002). Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N‐linked glycosylation. Biochim Biophys Acta 1596, 131–137. [DOI] [PubMed] [Google Scholar]
- Wu J, Ryskamp DA, Liang X, Egorova P, Zakharova O, Hung G & Bezprozvanny I (2016). Enhanced store‐operated calcium entry leads to striatal synaptic loss in a Huntington's disease mouse model. J Neurosci 36, 125–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Pan R, Gao X, Meucci O & Hu H (2014). Native store‐operated calcium channels are functionally expressed in mouse spinal cord dorsal horn neurons and regulate resting calcium homeostasis. J Physiol 592, 3443–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing R, Zhang Y, Xu H, Luo X, Chang RC, Liu J & Yang X (2016). Spatial memory impairment by TRPC1 depletion is ameliorated by environmental enrichment. Oncotarget 7, 27855–27873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Lu J, Li Z, Yu X, Chen L & Xu T (2006). Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun 350, 969–976. [DOI] [PubMed] [Google Scholar]
- Yap KA, Shetty MS, Garcia‐Alvarez G, Lu B, Alagappan D, Oh‐Hora M, Sajikumar S & Fivaz M (2017). STIM2 regulates AMPA receptor trafficking and plasticity at hippocampal synapses. Neurobiol Learn Mem 138, 54–61. [DOI] [PubMed] [Google Scholar]
- Yu R & Hinkle PM (2000). Rapid turnover of calcium in the endoplasmic reticulum during signaling. Studies with cameleon calcium indicators. J Biol Chem 275, 23648–23653. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Huang GN, Worley PF & Muallem S (2007). STIM1 heteromultimerizes TRPC channels to determine their function as store‐operated channels. Nat Cell Biol 9, 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wu L, Pchitskaya E, Zakharova O, Saito T, Saido T & Bezprozvanny I (2015). Neuronal store‐operated calcium entry and mushroom spine loss in amyloid precursor protein knock‐in mouse model of Alzheimer's disease. J Neurosci 35, 13275–13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J & Liu Q (2015). Cholesterol metabolism and homeostasis in the brain. Protein Cell 6, 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA & Cahalan MD (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Stathopulos PB, Li GY & Ikura M (2008). Biophysical characterization of the EF‐hand and SAM domain containing Ca2+ sensory region of STIM1 and STIM2. Biochem Biophys Res Commun 369, 240–246. [DOI] [PubMed] [Google Scholar]
- Zheng L, Stathopulos PB, Schindl R, Li GY, Romanin C & Ikura M (2011). Auto‐inhibitory role of the EF‐SAM domain of STIM proteins in store‐operated calcium entry. Proc Natl Acad Sci USA 108, 1337–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Mancarella S, Wang Y, Yue C, Ritchie M, Gill DL & Soboloff J (2009). The short N‐terminal domains of STIM1 and STIM2 control the activation kinetics of Orai1 channels. J Biol Chem 284, 19164–19168. [DOI] [PMC free article] [PubMed] [Google Scholar]