

Abstract
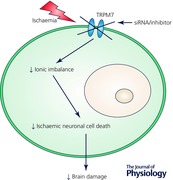
Transient receptor potential melastatin 7 (TRPM7) channel, a calcium‐permeable non‐selective divalent cation channel, is broadly expressed in various cells and tissues, including the brain. TRPM7 is thought to be coupled to the metabolic state and regulate calcium homeostasis in the cell. TRPM7 takes part in a wide range of cell biology processes that affect cell growth and proliferation, as well as in embryonic development and skeleton formation. TRPM7 plays a significant role in ischaemic and hypoxic brain injury and neuronal cell death. TRPM7, as a key non‐glutamate mechanism of cerebral ischaemia, also triggers an intracellular ionic imbalance and neuronal cell death in ischaemia and hypoxia. We have reported that TRPM7 is expressed in neurons of the hippocampus and cortex and activation of TRPM7 induced ischaemic neuronal cell death; suppression of TRPM7 with virally mediated gene silencing using siRNA reduced ischaemic neuronal cell death and improved neurobehavioural outcomes in vivo. Recently, we also demonstrated that inhibition of TRPM7 using pharmacological means promoted neuronal outgrowth in vitro and provided neuroprotection against brain injury to hypoxia in vivo. Thus, we have shown the contributions of TRPM7 in many physiological and pathophysiological processes, including hypoxia and ischaemia.
Keywords: cerebral ischaemia, hypoxia, ion channels, neuroprotection, TRPM7
Abbreviations
- AAV
adeno‐associated virus
- AET
anti‐excitotoxic therapies
- AMPA
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid
- ASIC
acid‐sensing ion channel
- HIE
hypoxic–ischaemic encephalopathy
- KATP
ATP‐sensitive potassium channel
- MCAO
middle cerebral artery occlusion
- NCX
sodium–calcium exchanger
- NMDA
N‐methyl‐d‐aspartate
- OGD
oxygen‐glucose deprivation
- STAIR
Stroke Therapy Academic Industry Roundtable
- tPA
tissue plasminogen activator
- TRP
transient receptor potential
- TRPM7
transient receptor potential melastatin 7
- VRAC
volume‐regulated anion channel
Introduction
Cerebral ischaemia and stroke (Dirnagl et al. 1999; Lipton, 1999) is a leading cause of mortality and a major cause of long‐term immobility in the world based on statistics from the World Health Organization (WHO) (WHO, 2014; Mozaffarian et al. 2016). Stroke has a high mortality rate, and stroke prevalence is projected to increase. There is no effective treatment for stroke, except for the tissue plasminogen activator (tPA), which has a limited therapeutic window (Zivin, 2009). Stroke has already shown significant social and economic impacts worldwide (American Stroke Association, 2016). In addition, hypoxia could cause neonatal hypoxic–ischaemic brain injury and subsequent early‐onset brain and behavioural disorders in children, termed hypoxic–ischaemic encephalopathy (HIE) (Vannucci, 2000; Nelson & Lynch, 2004). HIE is characterized by neurodevelopmental delay, motor and cognitive impairments, and epilepsy. Neonatal hypoxic–ischaemic brain injury and its related HIE have also caused noticeable burdens worldwide.
Stroke triggers intracellular calcium overload and ionic imbalance, eventually leading to neuronal cell death (Dirnagl et al. 1999; Lipton, 1999). The time course and sequence of events occurring in cerebral ischaemia includes (1) anoxic depolarization in seconds to minutes, (2) peri‐infarct depolarization in minutes, (3) excitotoxicity in minutes, to later cause (4) apoptosis and (5) inflammation in days. During cerebral ischaemia, the excitatory neurotransmitter glutamate is released from the brain, acts on the glutamate receptor channels, and triggers a calcium overload and neuronal cell death (Dirnagl et al. 1999; Lipton, 1999). Excitotoxicity, mediated through NMDA and AMPA receptor channels (Besancon et al. 2008; Tymianski, 2011), has been the central focus of stroke research for decades. Preventing the calcium overload is theoretically considered to be neuroprotective. Blocking calcium‐mediated glutamate receptor channels in vitro and in vivo inhibits intracellular calcium overload and prevents ischaemic brain damage. Even experimental studies have shown hopeful data; however, the subsequent clinical trials of anti‐excitotoxic therapies (AET) could not support AET as a further therapeutic development (Davis et al. 2000). As a result, stroke researchers began searching for unconventional mechanism(s) outside the traditional glutamate mechanism. In addition to the traditional glutamate excitotoxicity mediated through NMDA and AMPA receptor channels (Besancon et al. 2008; Tymianski, 2011), new data indicates that a non‐glutamate mechanism in cerebral ischaemia also causes intracellular ionic imbalance and neuronal cell death (Besancon et al. 2008; Tymianski, 2011). Ischaemic neuronal death is now accepted as a result of both glutamate‐mediated excitotoxicity and the newly discovered non‐glutamate mechanisms (Besancon et al. 2008; Tymianski, 2011). The newly accepted non‐glutamate mechanism undeniably contributes to the disappointing results of the AET clinical trials. Thus, we may need to consider both glutamate and non‐glutamate mechanisms in new drug development for stroke, as well as using multiple in vivo animal models of human disease based on the Stroke Therapy Academic Industry Roundtable (STAIR) Protocol (Stroke Therapy Academic Industry Roundtable (STAIR), 1999), which emphasizes the necessity for testing potential stroke drugs using multiple animal stroke models in multiple species. The non‐glutamate mechanism includes transient receptor potential (TRP) channels (Sun et al. 2009; Alim et al. 2013; Chen et al. 2015a), ATP‐sensitive potassium (KATP) channels (Sun et al. 2006, 2007, 2015; Liu et al. 2016), acid‐sensing ion channels (ASICs) (Xiong et al. 2004), hemichannels (Thompson et al. 2006), volume‐regulated anion channels (VRACs) (Alibrahim et al. 2013), sodium–calcium exchangers (NCXs) (Pignataro et al. 2004), and other non‐selective cation channels (Simard et al. 2006). Activation of these ion channels could be at the initial stages of cerebral ischaemia and/or later in the ischaemic events. For example, KATP channels are activated at the initiation of the ischaemic event during the anoxic depolarization; TRPM7 may be activated at the same time or shortly after the excitotoxicity; and TRPM2 may be activated in the later stages as it is involved in inflammation. Ion channels are the third largest target in drug development (Dabrowski et al. 2008). Thus, the non‐glutamate mechanism is a target for neuroprotection.
Transient receptor potential channels
Ion channels play many fundamental roles in physiological and pathophysiological functions in the brain. We have been working on the non‐glutamate mechanism in cerebral ischaemia and hypoxia, including TRP channels, KATP channels and VRAC channels. Here, the focus is on the role of TRP channels in cerebral ischaemia and hypoxia.
Transient receptor potential channels, also termed TRP channels, are a group of non‐selective cation channels located on the cell membrane of various cell types (Clapham, 2003; Pedersen et al. 2005; Wu et al. 2010). TRP channels were originally discovered in fruit fly (Drosophila) (Minke et al. 1975) photoreceptors where they participated in phototransduction (Monteilh‐Zoller et al. 2003). The channel received its name, transient receptor potential, because the Drosophila photoreceptors in the fly carrying the mutant trp gene initiated a transient response to light (Minke et al. 1975). The TRP channel superfamily now has approximately 30 mammalian TRP channels (Wu et al. 2010). The TRP channels, classified based on their homologous sequences, are divided into six sub‐families: (1) TRPC (canonical), (2) TRPV (vanilloid), (3) TRPM (melastatin), (4) TRPA (ankyrin), (5) TRPML (mucolipin), and (6) TRPP (polycystin). TRP channels can be activated by different physical and chemical stimuli and play many physiological and pathological functions in various cells (Clapham, 2003; Pedersen et al. 2005; Wu et al. 2010).
The TRPM family has eight members, TRPM 1 to 8. TRPM7 (Clapham, 2003), the seventh member of the TRPM channel sub‐family, is widely expressed in many tissues and cells including the brain and neurons. TRPM7 is a calcium‐permeable non‐selective divalent cation channel and is also permeable to other trace metal ions, i.e. Zn2+ ≈ Ni2+ >> Ba2+ > Co2+ > Mg2+ ≥ Mn2+ ≥ Sr2+ ≥ Cd2+ ≥ Ca2+ ions (Monteilh‐Zoller et al. 2003).
TRPM7 plays an important role in a wide range of physiological and pathophysiological functions, and its channel activities can be modulated by diverse intracellular and extracellular factors (Penner & Fleig, 2007), such as Mg2+ and Mg2+‐complexed nucleotides (such as MgATP and MgGTP) (Takezawa et al. 2004; Demeuse et al. 2006), extracellular pH (Jiang et al. 2005; Li et al. 2007), shear stress (Oancea et al. 2006), etc. TRPM7 participates in a wide scope of cell biology processes ranging from cell proliferation, cell growth and cell adhesion (Nadler et al. 2001; Inoue & Xiong, 2009); TRPM7 overexpression reduces cell viability (Nadler et al. 2001; Su et al. 2006; Chen et al. 2010). TRPM7 also regulates embryonic development (Jin et al. 2008) and skeleton formation (Elizondo et al. 2005). Globally knocking out TRPM7 has been shown to be embryonically lethal in mice (Jin et al. 2008). Thus, TRPM7 is essential for development. TRPM7 channels are thought to be activated during ischaemia based on the metabolic state of the cell. Therefore, the favourable condition for TRPM7 activation during ischaemia would be low concentrations of Mg2+‐nucleotides (Demeuse et al. 2006) and acidic conditions (Rehncrona, 1985; Li et al. 2007).
In the brain, TRPM7 plays key roles both under physiological conditions, e.g. cell growth (Nadler et al. 2001; Inoue & Xiong, 2009; Turlova et al. 2016), and under pathophysiological conditions, e.g. hypoxia‐ and ischaemia‐induced neuronal cell death (Aarts et al. 2003; Sun et al. 2009; Chen et al. 2015a) and survival of brain tumour cells (Chen et al. 2015b,c). In addition to showing the role of TRPM7 in ischaemia (Sun et al. 2009), we have recently also demonstrated the following: (1) inhibition of TRPM7 in vitro enhances neurite outgrowth and maturation in mouse culture hippocampal cells (Turlova et al. 2016); (2) TRPM7 plays a role in neonatal hypoxic–ischaemic brain injury in mice in vivo (Chen et al. 2015a); and (3) TRPM7 also plays an important role in cell survival in glioma cell lines in vitro (Chen et al. 2015b,c). Here, the role of TRPM7 in neuronal cell death and brain damage during ischaemia and hypoxia in vivo (Sun et al. 2009; Chen et al. 2015a), its pharmacology and its potential in drug development for stroke will be further discussed.
Molecular and pharmacological reagents are available for TRPM7; these include channel activators and blockers (Zierler et al. 2011; Chubanov et al. 2014; Chen et al. 2015a; Turlova et al. 2016), antibodies (Sun et al. 2009; Chen et al. 2015a,; Turlova et al. 2016), and siRNA (Sun et al. 2009; Turlova et al. 2016). These will be beneficial for studying TRPM7, and many studies have used various TRPM7 reagents.
TRPM7 plays a key role in anoxic cell death in cultured neurons (Aarts et al. 2003). We have demonstrated that TRPM7 plays a significant role in ischaemic brain damage and neuronal cell death in vivo (Sun et al. 2009). The study showed that virally mediated gene silencing of TRPM7 in vivo with siRNA increased neuronal cell survival and improved neurobehavioural outcomes after cerebral ischaemia (Sun et al. 2009). Later, we also showed that TRPM7 plays many important roles both in vitro and in vivo: inhibiting TRPM7 in vitro enhances hippocampal neuronal cell outgrowth (Turlova et al. 2016); blocking TRPM7 in vivo reduces brain damage in hypoxia (Chen et al. 2015a); and suppressing TRPM7 in vitro decreases glioma cell survival in vitro (Chen et al. 2015b,c).
TRPM7 in cerebral ischaemia and hypoxia
In the event of cerebral ischaemia and/or hypoxia, calcium overload and ionic imbalance inside the neuronal cells have been the accepted cellular and molecular mechanisms for ischaemic and/or hypoxic neuronal cell death and brain damage (Besancon et al. 2008; Tymianski, 2011). In addition to the traditional glutamate mechanism, which is mainly focused on glutamate receptor channel‐mediated excitotoxicity, the non‐glutamate mechanism also triggers intracellular ionic imbalance and initiates ischaemic and/or hypoxic neuronal cell death and brain damage in stroke (Besancon et al. 2008; Tymianski, 2011). The non‐glutamate mechanism contains many other ion channels and newly described calcium‐mediated non‐selective cation channels, e.g. ATP‐sensitive potassium channels (KATP) (Sun et al. 2006, 2007, 2015; Liu et al. 2016), transient receptor potential (TRP) channels (Sun et al. 2009; Alim et al. 2013; Chen et al. 2015a), volume‐regulated anion channels (VRACs) (Alibrahim et al. 2013), hemichannels (Thompson et al. 2006), acid‐sensing ion channels (ASICs) (Xiong et al. 2004), ion exchangers (Pignataro et al. 2004) and other non‐selective cation channels (Simard et al. 2006). In this report, the focus is on the neuronal cell death and brain damage mediated by TRPM7 in stroke and hypoxia in vivo.
An early in vitro study reported that TRPM7 plays a key role in anoxic‐induced neuronal cell death in culture (Aarts et al. 2003). This study originally described the so‐called I OGD currents in response to prolonged in vitro oxygen–glucose deprivation (OGD) challenge in primary culture cortical neurons, which eventually triggered a secondary neuronal cell death through a secondary increase in Ca2+ influx through the later identified TRPM7 (Aarts et al. 2003). The effects of the prolonged anoxic cell death were eventually unmasked with treatment of a cocktail of blockers aimed at blocking the glutamate NMDA and AMPA receptors and L‐type calcium channels using MK‐801, CNQX and nimodipine in the in vitro OGD experiment (Aarts et al. 2003). The study was facilitated by calcium imaging and electrophysiological approaches, as well as additional molecular biology verification. The in vitro study further used the TRPM7 siRNA directly against the TRPM7 in the mouse primary culture cortical neurons and showed that both the expression level of TRPM7 mRNA and the subsequent prolonged anoxia‐mediated neuronal cell death were reduced. Thus, this was the first study that revealed the key role of TRPM7 in mediating Ca2+ influx and subsequent anoxic neuronal cell death in vitro during prolonged OGD. As OGD is a simplified in vitro anoxia model for studying neuronal cell death, it is not a good representation of ‘ischaemia’ because it lacks other factors in vivo, such as blood, neuronal circuitry, network connectivity and the involvement of other brain cells. An unrelated in vivo study using the middle cerebral artery occlusion (MCAO) model reported that TRPM7 expression levels in mRNA and protein were increased after MCAO (Jiang et al. 2008), indicating that TRPM7 may be implicated in cerebral ischaemia. As a result, we still need to confirm the study in vivo and use animal models to further investigate the pathophysiological role of TRPM7 in ischaemia and/or hypoxia.
We have later confirmed that TRPM7 also plays an important role in cerebral ischaemia in vivo (Sun et al. 2009). In the report, we showed that virally mediated gene silencing of TRPM7 in hippocampal CA1 neurons in vivo suppressed mRNA and protein TRPM7 expression levels, reduced CA1 neuronal death and preserved behavioural outcomes after cerebral ischaemia. TRPM7 pharmacology was not clear, and selective TRPM7 blockers were not available at that time; the study utilized a virally mediated gene silencing shRNA approach to suppress TRPM7 in adult rat hippocampal CA1 neurons. The adeno‐associated viral vector (AAV) was used to package the TRPM7 shRNA, and a stereotaxic microinjection was used to deliver the AAV to the hippocampal CA1 area in vivo. We confirmed that the AAV infected the adult hippocampal CA1 neurons in vivo. We also showed suppression of TRPM7 in the infected hippocampal CA1 neurons at the mRNA level with RT‐PCR, at the protein level with Western Blot and immunohistochemistry, and at the functional level with electrophysiology. We verified that transient suppression of TRPM7 in the adult hippocampal CA1 neurons showed no noticeable effects on neuronal cell survival, fine structures and electrophysiological properties. We then revealed that TRPM7 suppression significantly reduced hippocampal CA1 cell death in vivo and preserved behavioural outcomes using a global ischaemia model. We demonstrated that the surviving hippocampal CA1 neuronal cells were also healthy with intact morphology and fine cell structures. We also proved that the surviving hippocampal CA1 neurons had well maintained electrophysiological properties. Lastly, we confirmed that suppressed TRPM7 in vivo not only reduced hippocampal cell death but also preserved the hippocampal‐associated behavioural tasks, e.g. fear‐associated and spatial‐navigation memory (fear conditioning (Cheng et al. 2006; Sun et al. 2009) and Morris water maze (Morris et al. 1982; Sun et al. 2009)). This study was the first detailed in vivo study showing the important role of TRPM7 in cerebral ischaemia.
We also recently showed that TRPM7 plays a significant role in brain damage in hypoxic–ischaemic brain injury in vivo (Chen et al. 2015a). We reported that the non‐selective TRPM7 inhibitor carvacrol significantly reduced the brain damage in a mouse hypoxic–ischaemic brain injury model (Chen et al. 2015a). The inhibition of TRPM7 also preserved behavioural outcomes after hypoxic–ischaemic brain injury (Chen et al. 2015a), and these included the geotaxic reflex (Ten et al. 2003; Sun et al. 2015; Chen et al. 2015a), which tests the vestibular and/or proprioceptive functions; the cliff avoidance reaction (Ten et al. 2003; Sun et al. 2015; Chen et al. 2015a), which assesses maladaptive impulsive behaviour; and the grip test (Liu et al. 2013; Sun et al. 2015; Chen et al. 2015a), which evaluates force and fatigability. Neuroprotective effects in reducing brain damage by TRPM7 inhibition in the hypoxic–ischaemic brain injury were arbitrated partly by promoting pro‐survival signalling (e.g. Akt signalling) and inhibiting pro‐apoptotic signalling (e.g. caspase‐3 and Bcl/Bax signalling) (Chen et al. 2015a).
Conclusions and future direction
TRPM7, a calcium‐mediated non‐selective divalent cation channel, is one of the newly described non‐glutamate mechanisms of neuronal cell death and brain damage in cerebral ischaemia and hypoxia. Both in vitro and in vivo studies have shown that TRPM7 plays a critical role in ischaemic and hypoxic neuronal cell death and brain damage. Thus, TRPM7 is a potential therapeutic target for drug development for stroke and hypoxic–ischaemic brain injury. With the new development of selective TRPM7 inhibitors (Zierler et al. 2011; Turlova et al. 2016), we could further study TRPM7 drug development in stroke and hypoxia following the Stroke STAIR protocol (Stroke Therapy Academic Industry Roundtable (STAIR), 1999).
Additional information
Competing interests
The author declares no conflicts of interest.
Funding
Supported by grants to H.‐S.S from the Heart and Stroke Foundation of Canada (G‐13‐0003069), the Canadian Institutes of Health Research (CIHR) China‐Canada Joint Health Research Initiative (CIHR, FRN no. 132571) and Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grants (RGPIN‐2016‐04574).
Biography
Hong‐Shuo Sun is a neuroscientist with expertise in ion channels, neuroprotection and drug development for stroke and other brain diseases. He first received his MD degree from Zhongshan Medical College, Sun Yat‐Sen University, Guangzhou, China. He then received his MSc degree in Pharmaceutical Sciences (Pharmacology) from the University of Alberta, Canada, and his PhD degree in Neuroscience from the University of Calgary, Canada. He later did his postdoctoral training at the University Health Network, Toronto, Canada. Currently, he is a tenured Associate Professor and PhD Supervisor in the Departments of Surgery, Physiology, and Pharmacology and the Institute of Medical Science, Faculty of Medicine at the University of Toronto, Canada.

This review was presented at “Advances and Breakthroughs in Calcium Signaling”, which took place in Honolulu, Hawaii, 7‐9 April 2016
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, Macdonald JF & Tymianski M (2003). A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877. [DOI] [PubMed] [Google Scholar]
- Alibrahim A, Zhao LY, Bae CY, Barszczyk A, Lf SC, Wang GL & Sun HS (2013). Neuroprotective effects of volume‐regulated anion channel blocker DCPIB on neonatal hypoxic–ischemic injury. Acta Pharmacol Sin 34, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alim I, Teves L, Li R, Mori Y & Tymianski M (2013). Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J Neurosci 33, 17264–17277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Stroke Association (2016). About stroke. Retrieved from http://www.strokeassociation.org/STROKEORG/AboutStroke/About-Stroke_UCM_308529_SubHomePage.jsp.
- Besancon E, Guo S, Lok J, Tymianski M & Lo EH (2008). Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci 29, 268–275. [DOI] [PubMed] [Google Scholar]
- Chen HC, Xie J, Zhang Z, Su LT, Yue L & Runnels LW (2010). Blockade of TRPM7 channel activity and cell death by inhibitors of 5‐lipoxygenase. PLoS One 5, e11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Xu B, Xiao A, Liu L, Fang X, Liu R, Turlova E, Barszczyk A, Zhong X, Sun CL, Britto LR, Feng ZP & Sun HS (2015a). TRPM7 inhibitor carvacrol protects brain from neonatal hypoxic–ischemic injury. Mol Brain 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WL, Barszczyk A, Turlova E, Deurloo M, Liu B, Yang BB, Rutka JT, Feng ZP & Sun HS (2015b). Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget 6, 16321–16340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WL, Turlova E, Sun CL, Kim JS, Huang S, Zhong X, Guan YY, Wang GL, Rutka JT, Feng ZP & Sun HS (2015c). Xyloketal B suppresses glioblastoma cell proliferation and migration in vitro through inhibiting TRPM7‐regulated PI3K/Akt and MEK/ERK signaling pathways. Mar Drugs 13, 2505–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng VY, Martin LJ, Elliott EM, Kim JH, Mount HT, Taverna FA, Roder JC, Macdonald JF, Bhambri A, Collinson N, Wafford KA & Orser BA (2006). α5GABAA receptors mediate the amnestic but not sedative‐hypnotic effects of the general anesthetic etomidate. J Neurosci 26, 3713–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubanov V, Schafer S, Ferioli S & Gudermann T (2014). Natural and synthetic modulators of the TRPM7 channel. Cells 3, 1089–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE (2003). TRP channels as cellular sensors. Nature 426, 517–524. [DOI] [PubMed] [Google Scholar]
- Dabrowski MA, Dekermendjian K, Lund PE, Krupp JJ, Sinclair J & Larsson O (2008). Ion channel screening technology. CNS Neurol Disord Drug Targets 7, 122–128. [DOI] [PubMed] [Google Scholar]
- Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G & Norris J (2000). Selfotel in acute ischemic stroke: possible neurotoxic effects of an NMDA antagonist. Stroke 31, 347–354. [DOI] [PubMed] [Google Scholar]
- Demeuse P, Penner R & Fleig A (2006). TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol 127, 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C & Moskowitz MA (1999). Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22, 391–397. [DOI] [PubMed] [Google Scholar]
- Elizondo MR, Arduini BL, Paulsen J, MacDonald EL, Sabel JL, Henion PD, Cornell RA & Parichy DM (2005). Defective skeletogenesis with kidney stone formation in dwarf zebrafish mutant for trpm7 . Curr Biol 15, 667–671. [DOI] [PubMed] [Google Scholar]
- Inoue K & Xiong ZG (2009). Silencing TRPM7 promotes growth/proliferation and nitric oxide production of vascular endothelial cells via the ERK pathway. Cardiovasc Res 83, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Tian SL, Zeng Y, Li LL & Shi J (2008). TrkA pathway(s) is involved in regulation of TRPM7 expression in hippocampal neurons subjected to ischemic‐reperfusion and oxygen‐glucose deprivation. Brain Res Bull 76, 124–130. [DOI] [PubMed] [Google Scholar]
- Jiang J, Li M & Yue L (2005). Potentiation of TRPM7 inward currents by protons. J Gen Physiol 126, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Desai BN, Navarro B, Donovan A, Andrews NC & Clapham DE (2008). Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322, 756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Du J, Jiang J, Ratzan W, Su LT, Runnels LW & Yue L (2007). Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J Biol Chem 282, 25817–25830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P (1999). Ischemic cell death in brain neurons. Physiol Rev 79, 1431–1568. [DOI] [PubMed] [Google Scholar]
- Liu R, Wang H, Xu B, Chen W, Turlova E, Dong N, Sun CL, Lu Y, Fu H, Shi R, Barszczyk A, Yang D, Jin T, Mannucci E, Feng ZP & Sun HS (2016). Cerebrovascular safety of sulfonylureas: the role of KATP channels in neuroprotection and the risk of stroke in patients with type 2 diabetes. Diabetes 65, 2795–2809. [DOI] [PubMed] [Google Scholar]
- Liu XH, Yan H, Xu M, Zhao YL, Li LM, Zhou XH, Wang MX & Ma L (2013). Hyperbaric oxygenation reduces long‐term brain injury and ameliorates behavioral function by suppression of apoptosis in a rat model of neonatal hypoxia–ischemia. Neurochem Int 62, 922–930. [DOI] [PubMed] [Google Scholar]
- Minke B, Wu C & Pak WL (1975). Induction of photoreceptor voltage noise in the dark in Drosophila mutant. Nature 258, 84–87. [DOI] [PubMed] [Google Scholar]
- Monteilh‐Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R & Fleig A (2003). TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol 121, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN & O'Keefe J (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297, 681–683. [DOI] [PubMed] [Google Scholar]
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de FS, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW & Turner MB (2016). Heart disease and stroke statistics – 2016 update: A report from the American Heart Association. Circulation 133, e38–e60. [DOI] [PubMed] [Google Scholar]
- Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM & Fleig A (2001). LTRPC7 is a Mg.ATP‐regulated divalent cation channel required for cell viability. Nature 411, 590–595. [DOI] [PubMed] [Google Scholar]
- Nelson KB & Lynch JK (2004). Stroke in newborn infants. Lancet Neurol 3, 150–158. [DOI] [PubMed] [Google Scholar]
- Oancea E, Wolfe JT & Clapham DE (2006). Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res 98, 245–253. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Owsianik G & Nilius B (2005). TRP channels: an overview. Cell Calcium 38, 233–252. [DOI] [PubMed] [Google Scholar]
- Penner R & Fleig A (2007). The Mg2+ and Mg2+‐nucleotide‐regulated channel‐kinase TRPM7. Handb Exp Pharmacol 313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignataro G, Tortiglione A, Scorziello A, Giaccio L, Secondo A, Severino B, Santagada V, Caliendo G, Amoroso S, di RG & Annunziato L (2004). Evidence for a protective role played by the Na+/Ca2+ exchanger in cerebral ischemia induced by middle cerebral artery occlusion in male rats. Neuropharmacology 46, 439–448. [DOI] [PubMed] [Google Scholar]
- Rehncrona S (1985). Brain acidosis. Ann Emerg Med 14, 770–776. [DOI] [PubMed] [Google Scholar]
- Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA & Gerzanich V (2006). Newly expressed SUR1‐regulated NC(Ca‐ATP) channel mediates cerebral edema after ischemic stroke. Nat Med 12, 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroke Therapy Academic Industry Roundtable (STAIR) (1999). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 30, 2752–2758. [DOI] [PubMed] [Google Scholar]
- Su LT, Agapito MA, Li M, Simonson WT, Huttenlocher A, Habas R, Yue L & Runnels LW (2006). TRPM7 regulates cell adhesion by controlling the calcium‐dependent protease calpain. J Biol Chem 281, 11260–11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, Barber PA, Buchan AM & French RJ (2007). Kir6.2‐containing ATP‐sensitive potassium channels protect cortical neurons from ischemic/anoxic injury in vitro and in vivo . Neuroscience 144, 1509–1515. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, Miki T, Seino S & French RJ (2006). Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2‐containing ATP‐sensitive K+ channels. J Neurophysiol 95, 2590–2601. [DOI] [PubMed] [Google Scholar]
- Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, Kiyonaka S, Mori Y, Jones M, Forder JP, Golde TE, Orser BA, Macdonald JF & Tymianski M (2009). Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci 12, 1300–1307. [DOI] [PubMed] [Google Scholar]
- Sun HS, Xu B, Chen W, Xiao A, Turlova E, Alibraham A, Barszczyk A, Bae CY, Quan Y, Liu B, Pei L, Sun CL, Deurloo M & Feng ZP (2015). Neuronal KATP channels mediate hypoxic preconditioning and reduce subsequent neonatal hypoxic–ischemic brain injury. Exp Neurol 263, 161–171. [DOI] [PubMed] [Google Scholar]
- Takezawa R, Schmitz C, Demeuse P, Scharenberg AM, Penner R & Fleig A (2004). Receptor‐mediated regulation of the TRPM7 channel through its endogenous protein kinase domain. Proc Natl Acad Sci USA 101, 6009–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten VS, Bradley‐Moore M, Gingrich JA, Stark RI & Pinsky DJ (2003). Brain injury and neurofunctional deficit in neonatal mice with hypoxic–ischemic encephalopathy. Behav Brain Res 145, 209–219. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Zhou N & MacVicar BA (2006). Ischemia opens neuronal gap junction hemichannels. Science 312, 924–927. [DOI] [PubMed] [Google Scholar]
- Turlova E, Bae CY, Deurloo M, Chen W, Barszczyk A, Horgen FD, Fleig A, Feng ZP & Sun HS (2016). TRPM7 regulates axonal outgrowth and maturation of primary hippocampal neurons. Mol Neurobiol 53, 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M (2011). Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci 14, 1369–1373. [DOI] [PubMed] [Google Scholar]
- Vannucci RC (2000). Hypoxic–ischemic encephalopathy. Am J Perinatol 17, 113–120. [DOI] [PubMed] [Google Scholar]
- WHO . WHO (2014). The top 10 causes of death. Retrieved from http://www.who.int/mediacentre/factsheets/fs310/en/.
- Wu LJ, Sweet TB & Clapham DE (2010). International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev 62, 381–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, Macdonald JF, Wemmie JA, Price MP, Welsh MJ & Simon RP (2004). Neuroprotection in ischemia: blocking calcium‐permeable acid‐sensing ion channels. Cell 118, 687–698. [DOI] [PubMed] [Google Scholar]
- Zierler S, Yao G, Zhang Z, Kuo WC, Porzgen P, Penner R, Horgen FD & Fleig A (2011). Waixenicin A inhibits cell proliferation through magnesium‐dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J Biol Chem 286, 39328–39335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivin JA (2009). Acute stroke therapy with tissue plasminogen activator (tPA) since it was approved by the U.S. Food and Drug Administration (FDA). Ann Neurol 66, 6–10. [DOI] [PubMed] [Google Scholar]