

Abstract
Biochemistry textbooks and cell culture experiments seem to be telling us two different things about the significance of external glutamine supply for mammalian cell growth and proliferation. Despite the fact that glutamine is a nonessential amino acid that can be synthesized by cells from glucose‐derived carbons and amino acid‐derived ammonia, most mammalian cells in tissue culture cannot proliferate or even survive in an environment that does not contain millimolar levels of glutamine. Not only are the levels of glutamine in standard tissue culture media at least ten‐fold higher than other amino acids, but glutamine is also the most abundant amino acid in the human bloodstream, where it is assiduously maintained at approximately 0.5 mM through a combination of dietary uptake, de novo synthesis, and muscle protein catabolism. The complex metabolic logic of the proliferating cancer cells' appetite for glutamine—which goes far beyond satisfying their protein synthesis requirements—has only recently come into focus. In this review, we examine the diversity of biosynthetic and regulatory uses of glutamine and their role in proliferation, stress resistance, and cellular identity, as well as discuss the mechanisms that cells utilize in order to adapt to glutamine limitation.
Keywords: cancer, glutamine metabolism, proliferation, stress response
Subject Categories: Cancer, Metabolism
Introduction
With the exception of a few instances, such as cleavage divisions of a fertilized zygote in early embryogenesis, cell proliferation is linked to the biomass accumulation. In the past decade, studies in cancer cell metabolism revealed the central role of numerous metabolic pathways and metabolites in facilitating biosynthesis and bioenergetics required for cell growth and proliferation. Thus, to ensure accumulation of biomass necessary for proliferation, deregulated pro‐proliferative and pro‐survival signals of cancer cells rewire metabolism to support biosynthesis of proteins, nucleotides, glycans, and lipids, as well as production of energy and NADPH.
In this review, we will first describe the many uses of glutamine and its products in proliferating cells, including its role in supplying carbon and nitrogen atoms for construction of a variety of macromolecular precursors, as well as its significance as a regulator of biosynthesis and bioenergetics, anti‐oxidative defense, and gene expression. The consequence of the high demand of proliferating cells for glutamine is the disproportionate depletion of the latter from the surrounding environment. To this end, we will discuss the adaptations that cells use to deal with glutamine limitation, including de novo biosynthesis and proteolytic scavenging.
Glutamine utilization beyond protein synthesis
Along with the rest of the proteinogenic amino acids, glutamine is incorporated into proteins. It is estimated that glutamine accounts for approximately 4.7% of all amino acid residues in human proteome, while in select proteins, for instance, in the structural component of skin epidermal barrier, involucrin, the representation of glutamine residues can reach 25%. However, consumption of glutamine in proliferating cells far exceeds the demands imposed by protein synthesis.
Amino acids contribute to the majority of biomass accumulation in proliferating mammalian cells (Hosios et al, 2016). In contrast to unicellular organisms, mammals cannot synthesize all the necessary amino acids for protein synthesis, and must acquire nine out of 21 amino acids from the diet. Notably, the biosynthesis of the rest of the amino acids, which are regarded to as nonessential, is heavily dependent on glutamine. Thus, glutamine deamidation, performed by numerous enzymes in the cells, yields glutamate, which can further be transformed into proline through a series of reductive steps, as well as into aspartate and asparagine, via the utilization of oxidative reactions of the tricarboxylic acid (TCA) cycle. Glutamine‐derived glutamate also donates its amine nitrogen toward the biosynthesis of alanine and serine and, by extension, glycine.
Glutamine as a nitrogen donor and amino acid precursor
Another dominant class of nitrogenous compounds that are required for cell proliferation is nucleotides. Notably, glutamine is an indispensable donor of reduced nitrogen for building both purine and pyrimidine bases (Wise & Thompson, 2010). In purine biosynthesis, two glutamine nitrogens are consumed in the biosynthesis of inosine monophosphate (IMP), which gives rise to both AMP and GMP. A third nitrogen from glutamine is required to produce guanosine monophosphate (GMP) from inosine monophosphate (IMP). Likewise, the initiating step of pyrimidine biosynthesis involves condensation of glutamine‐derived nitrogen with bicarbonate and ATP to generate carbamoyl phosphate. Finally, one more glutamine is consumed in the synthesis of cytidine triphosphate (CTP) from uridine triphosphate (UTP).
Notably, the increased utilization of glutamine nitrogen in nucleotide production is facilitated by the growth‐promoting signals. For instance, elevated levels of c‐Myc induce the expression of a number of enzymes in the nucleotide biosynthetic pathways, including phosphoribosyl pyrophosphate synthetase 2 (PRPS2), carbamoyl phosphate synthetase II (CAD), thymidylate synthase (TS), inosine monophosphate dehydrogenase 1/2 (IMPDH1/2), and others (Eberhardy & Farnham, 2001; Liu et al, 2008; Mannava et al, 2008; Cunningham et al, 2014). Similarly, loss of Rb and E2F upregulation induces nucleotide biosynthesis enzymes as well (Nicolay & Dyson, 2013). In addition, cancer cells display increased expression of phosphoribosyl amidotransferase (PPAT), the enzyme that transfers amide nitrogen from glutamine to 5‐phosphoribosyl pyrophosphate (PRPP), a key reaction in purine biosynthesis (Goswami et al, 2015). In addition to being a c‐Myc target gene, the CAD enzyme, which generates carbamoyl phosphate in the initiating reaction in the pyrimidine biosynthesis cascade, is positively regulated via phosphorylation by MAP kinase or by S6 kinase downstream of mTORC1 (Graves et al, 2000; Ben‐Sahra et al, 2013; Robitaille et al, 2013). The transcriptional induction of genes induced in nucleotide biosynthesis is also observed in cancer cells harboring mutant p53 (Kollareddy et al, 2015).
The above results suggest a potential mechanism that proliferating cells use to coordinate growth‐promoting signals with glutamine utilization to drive nucleotide biosynthesis. Interestingly, the five reactions in nucleotide biosynthesis that directly utilize glutamine as a substrate exclusively use the γ‐nitrogen (amide group) of glutamine (Fig 1A). In addition to nucleotide biosynthesis, the γ‐nitrogen of glutamine in mammalian cells is also required to synthesize NAD, glucosamine‐6‐phosphate, a precursor for protein glycosylation, and asparagine, another nonessential amino acid (Richards & Schuster, 1998; Wellen et al, 2010; Fig 1B).
Figure 1. Glutamine supplies nitrogen and carbon for biosynthetic reactions.
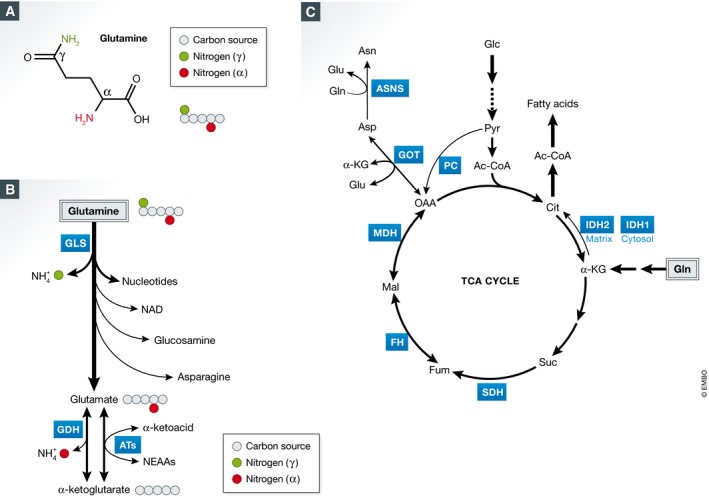
(A) Chemical structure of glutamine. (B) Usage of γ‐ and α‐nitrogen of glutamine in mammalian cells. GLS: glutaminase; GDH: glutamate dehydrogenase; ATs: aminotransferases. (C) Glutamine‐derived carbon enters the TCA cycle through α‐KG to supply anaplerotic substrates. Glucose‐derived pyruvate can enter the TCA cycle through OAA. This reaction is mediated by PC, which is suppressed when glutamine‐derived carbon enters the TCA cycle. Gln: glutamine; α‐KG: α‐ketoglutarate; Suc: succinate; Fum: fumarate; Mal: malate; OAA: oxaloacetate; Cit: citrate; Glu: glutamate; Asp: aspartate; Asn: asparagine; Glc: glucose; Pyr: pyruvate; Ac‐CoA: acetyl‐CoA; SDH: succinate dehydrogenase; FH: fumarase; MDH: malate dehydrogenase; GOT: aspartate aminotransferase; ASNS: asparagine synthetase; PC: pyruvate carboxylase; IDH1/2: isocitrate dehydrogenase 1/2. IDH1 is localized in cytosol.
In addition to being incorporated into nucleotides, amino acids, and glucosamine‐6‐phosphate, the γ‐nitrogen of glutamine is subject to a direct cleavage by glutaminase (GLS) enzymes, producing glutamate and free ammonia. GLS is frequently deregulated in cancer (Gao et al, 2009; Hu et al, 2010; Suzuki et al, 2010; Lukey et al, 2016), and loss of a single copy of Gls1 delays tumorigenesis in a mouse model of hepatocellular carcinoma (Xiang et al, 2015). It was proposed that the deamidation of γ‐nitrogen by GLS contributes to the major intracellular pool of glutamate, another NEAA that can continue to supply both nitrogen and carbon for other biosynthetic reactions. The existence of both mitochondrial and cytosolic isoforms of GLS suggests the importance of compartmentalized production of glutamate, such that the two segregated pools can be used for distinct biosynthetic processes (Cassago et al, 2012). In this regard, a number of glutaminase inhibitors have been developed and show tumor‐suppressive activities in preclinical models (Wang et al, 2010; Gross et al, 2014; Shroff et al, 2015; Xiang et al, 2015). However, in certain cases GLS inhibitors do not have a therapeutic effect (Davidson et al, 2016). One possible interpretation is that beside GLS‐catalyzed reaction, other biochemical reactions that use γ‐nitrogen of glutamine, described above, release glutamate as a product as well. Thus, as GLS activity is inhibited, more glutamine may become funneled into other glutamine‐utilizing pathways. Another explanation is that in certain cases, cancer cells may use glucose‐derived carbon to maintain TCA cycle intermediates and produce glutamate, therefore diminishing the contribution of the GLS‐catalyzed reaction for these processes.
One advantage that proliferating cells that rely on GLS to produce glutamate from glutamine may have is the ability to maintain a high ratio of glutamate to α‐ketoglutarate, which is necessary for driving the biosynthesis of other NEAAs. To this end, the remaining nitrogen of glutamate, which resides at the α‐position of glutamine carbon chain (the amine group, Fig 1A), can be transferred to different α‐ketoacids by a family of aminotransferases to produce other NEAAs, among which are alanine, aspartate, serine, and ornithine. Recently, alanine aminotransferase 2 (GPT2) has been found to be upregulated by PIK3CA mutation in colorectal cancer, as well as by liver receptor homolog 1 (LRH‐1) in liver cancer (Hao et al, 2016a; Xu et al, 2016). Hao et al (2016a) show that PIK3CA mutation causes elevated expression of GPT2, an aminotransferase that transfers amino group from glutamate to pyruvate to generate alanine. Similarly, in liver cancer, both GPT2 and aspartate aminotransferase 1 (GOT1), which transfers an amino group from glutamate to oxaloacetate to generate aspartate, are transcriptionally induced by LRH‐1 (Xu et al, 2016). Furthermore, phosphoserine aminotransferase (PSAT1) transfers amino group from glutamate to 3‐phosphohydroxypyruvate to generate 3‐phosphoserine, the precursor of serine. Overexpression of PSAT1 has been found to confer growth advantage and resistance to chemotherapy in colorectal cancer (Vie et al, 2008). In addition, elevated expression of PSAT1 correlates with poor prognosis in patients with esophageal squamous cell carcinoma (ESCC) (Liu et al, 2016). Indeed, multiple studies show that aminooxyacetate (AOA), a general inhibitor of cellular aminotransferases, profoundly inhibits tumor growth in vitro and in vivo (Thornburg et al, 2008; Korangath et al, 2015; Hao et al, 2016a). Finally, glutamate supplies both the carbon backbone and the nitrogen for proline biosynthesis. Notably, oncogenic c‐Myc induces the expression of proline biosynthesis enzymes while suppressing proline dehydrogenase (POX/PRODH), the first enzyme for proline catabolism (Liu et al, 2012). These studies implicate glutamate as a critical product of glutamine catabolism, which is used to synthesize several other NEAAs.
Glutamine as a carbon donor
Beyond its role as a nitrogen donor, glutamine serves as an important source of carbon for cellular bioenergetic and biosynthetic needs. Indeed, cell proliferation is associated with the high influx of glutamine‐derived carbon into the TCA cycle (DeBerardinis et al, 2007). Why do proliferating cells require the continuous replenishing of the TCA cycle? Indeed, as demonstrated by DeBerardinis et al, proliferating cells utilize the TCA cycle as the source of not only bioenergetic NADH and FADH2 equivalents, but also biosynthetic precursors as well. Thus, most of the citrate generated in the TCA cycle in proliferating cells becomes exported into cytosol, where it is converted into acetyl‐CoA, a precursor for the biosynthesis of fatty acids and cholesterol. Furthermore, TCA cycle‐derived oxaloacetate is used to synthesize aspartate and asparagine.
In many cell types, pyruvate carboxylation to oxaloacetate is suppressed, thereby rendering them reliant on glutamine catabolism to replenish the oxaloacetate that can condense with acetyl‐CoA to produce citrate and drive the TCA cycle (DeBerardinis et al, 2007). The efflux of carbon precursors away from the TCA cycle must be balanced by the influx of carbons elsewhere. A route of entry for the glutamine‐derived carbon into the TCA cycle is via the conversion of glutamate into its α‐ketoacid form, α‐ketoglutarate (α‐KG; Fig 1C). In agreement with this notion, GLS1 expression itself is under positive control by c‐Myc (Gao et al, 2009). In addition, a cell‐permeable form of α‐KG (dimethyl‐α‐KG) can completely suppress glutamine‐depletion‐induced apoptosis in MYC‐transformed cells (Yuneva et al, 2007; Wise et al, 2008). As far as the conversion of glutamate to α‐ketoglutarate goes, this reaction can be catalyzed either by glutamate dehydrogenase (GDH), which releases free ammonia, or by a family of aminotransferases, which transfer the α‐amine to α‐ketoacids, expanding the nonessential amino acid pool. These aminotransferases may play a dominant role to replenish the cellular pool of α‐KG, as their inhibition induces cell death, which can be rescued by dimethyl‐α‐KG (Wise et al, 2008). However, when glycolysis is perturbed, GDH is required for cell survival, suggesting a potential compensation due to the lack of glucose‐derived carbon influx into the TCA cycle (Yang et al, 2009).
In addition to its role in providing carbon and nitrogen toward the biosynthesis of diverse biosynthetic precursors, glutamine carbons also contribute to ATP production through their oxidation in the TCA cycle. Indeed, in proliferating cells, glutamine depletion markedly reduces NADH/NAD+ ratio and inhibits oxygen consumption (Fan et al, 2013). This study concludes that glycolysis alone is not sufficient to sustain ATP production, even though the inhibition of complex I of the respiratory chain is not able to alter the cellular ATP level when glucose is unlimited (Javeshghani et al, 2012). However, whether ATP is a limiting factor for cell proliferation has been controversial until recently. Two recent studies shed light on this question. Birsoy et al (2015) and Sullivan et al (2015) have demonstrated that the inhibition of cell proliferation by oligomycin, an ATP synthase inhibitor, can be rescued by an uncoupling agent FCCP, which dissipates the electrochemical gradient across the mitochondrial membrane, resetting the NADH/NAD+ ratio to normal. In this study, the authors show that it is not the mitochondrial ATP production, but the uninterrupted flux through the TCA cycle, enabled by the low NADH/NAD+ ratio, is the major limiting factor for cell proliferation. Thus, continued transfer of electrons from NADH to molecular oxygen is necessary for maintaining the low NADH/NAD+ ratio, which, in turn, allows the TCA cycle flux toward the biosynthesis of oxaloacetate. Oxaloacetate is an immediate precursor of aspartate, a proteinogenic amino acid, as well as a precursor for the biosynthesis of nucleotides and asparagine. Together, these results suggest that despite the fact that glutamine carbons contribute to a significant fraction of cellular energy production, the remaining ATP production via glycolysis is sufficient to maintain the energy needed for a cell to proliferate even when the electron transport is compromised.
Therapeutic application of glutamine catabolism in cancer
Given a versatile usage of glutamine in proliferating cells, a number of glutamine‐mimetic compounds, including 6‐diazo‐5‐oxo‐L‐norleucine (DON), acivicin, and azaserine, have been evaluated in preclinical and clinical settings for their anti‐tumor activities. Despite their promising tumor‐suppressive activities in vitro, all of these compounds displayed significant toxicity toward the gastrointestinal tract, immune cells, and central nerve system due to their nonselective inhibition of glutamine metabolism (Ahluwalia et al, 1990). In search for more selective inhibitors of glutamine catabolism, extensive efforts have been focused on glutaminase (GLS), the activity of which is dysregulated in variety of cancers. In this regard, several glutaminase inhibitors have been developed, including 968, BPTES, and CB‐389 (Le et al, 2012; Gross et al, 2014; Stalnecker et al, 2015). Among these, CB‐389 is currently being tested in a number of phase I clinical trials, where its efficacy is being evaluated in patients with both solid tumors and hematological malignancies.
In addition to the GLS‐targeted compounds, inhibitors that target the conversion of glutamate to α‐KG have also been tested in preclinical models of breast cancer and neuroblastoma (Qing et al, 2012; Korangath et al, 2015). In this regard, green tea catechin EGCG, which is a GDH inhibitor, and aminooxyacetate (AOA), a nonselective aminotransferase inhibitor, have been evaluated. The anti‐tumor efficacy of these inhibitors may depend upon the relative dependency of a tumor on the activity of GDH or on aminotransferases as means of replenishing the intracellular pool of α‐KG from glutamate. Indeed, AOA has been approved for the treatment of tinnitus (Guth et al, 1990). Its application in cancer deserves further exploration in light of the critical role of aminotransferases in mediating glutamine‐dependent biosynthesis.
Role of glutamine in redox control
Tumor cells encounter oxidative stress during their initiation, progression, metastatic colonization, and following the exposure to anti‐tumor therapeutics, which increases their dependence on anti‐oxidative responses (Gorrini et al, 2013). Products of glutamine metabolism in particular play an essential role in facilitating cellular anti‐oxidative defenses. First, glutamine‐derived glutamate is utilized in the de novo biosynthesis of glutathione, a primary cellular antioxidant. Glutathione is a tripeptide comprised of three NEAAs: glutamate, cysteine, and glycine (Lu, 2009). Glutamate–cysteine ligase (GCL) condenses glutamate with cysteine in an ATP‐dependent manner to generate γ‐glutamylcysteine, which is further condensed with glycine through glutathione synthetase (GS), generating glutathione (Fig 2). In agreement with glutamine being the primary source of glutamate in the cell, exposure of cells to the uniformly labeled 13C‐glutamine shows a pattern of enrichment of five 13C carbons in glutathione. Glutamine starvation of transformed cells reduces their glutathione pool (Yuneva et al, 2007). Human primary acute myeloid leukemia (AML) and metastatic liver cancer are characterized by significant elevation of the enzymes for glutathione biosynthesis, including GCL and GS (Pei et al, 2013; Nguyen et al, 2016a), suggesting that these tumors might be sensitive to inhibitors of glutathione biosynthesis.
Figure 2. The key role of glutamine‐derived glutamate in glutathione biosynthesis.
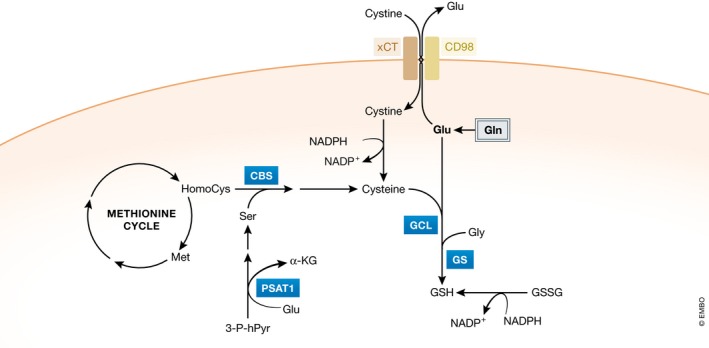
Glutamine‐derived glutamate is a necessary substrate to synthesize glutathione. In addition, glutamate functions as an exchanging counter ion to import extracellular cystine through the xCT transporter. In the cell, cystine is converted to cysteine that is used as a second substrate for glutathione biosynthesis. Gln: glutamine; Glu: glutamate; Gly: glycine; GSH: glutathione; GSSG: glutathione disulfide; α‐KG: α‐ketoglutarate: Ser: serine; Met: methionine; HomoCys: homocysteine; GCL: glutamate–cysteine ligase; GS: glutathione synthetase; CBS: cystathionine beta‐synthase; PSAT1: phosphoserine aminotransferase 1.
A second way in which glutamine‐derived glutamate contributes to glutathione biosynthesis is through facilitating the uptake of cystine via the xCT transporter, which is coupled to the efflux of glutamate (Fig 2). Once inside the cell, cystine is converted to cysteine, which can then be incorporated into glutathione. Glutamine‐starved breast cancer cells display a defect in cystine uptake through the xCT antiporter (Timmerman et al, 2013). In addition, pharmacological inhibition of xCT elevates cellular reactive oxygen species (ROS) levels and suppresses tumor growth, making it a potential therapeutic target (Timmerman et al, 2013; Lanzardo et al, 2016; Tsuchihashi et al, 2016). Interestingly, cysteine itself can be synthesized in the cell from homocysteine, an intermediate of methionine catabolism (Lu, 2009). It will be interesting to know whether the resistance of some tumor cells to xCT inhibitors is at least partially due to their capacity to synthesize cysteine de novo.
The third way in which glutamine contributes to the cellular redox balance is via support of NADPH production. In a proliferating cell, reducing equivalent donor NADPH is utilized not only in the biosynthesis of fatty acids and cholesterol, but also to revert oxidized glutathione (GSSG), as well as thioredoxins, a class of cysteine‐containing antioxidant proteins, back to their reduced states. Son et al showed that pancreatic ductal adenocarcinoma (PDAC) cells rely on glutamine to maintain the cytosolic NADPH pool (Son et al, 2013). In this paper, the authors show that glutamine‐derived aspartate is converted to oxaloacetate (OAA) in the cytosol through GOT1, which is transcriptionally induced by mutant KRas, a common oncogenic lesion in PDAC. Subsequently, OAA is converted to malate by malate dehydrogenase 1 (MDH1), and malate is converted to pyruvate to generate NADPH through malic enzyme 1 (ME1).
Glutamine metabolism contributes to chromatin organization
Multiple lines of evidence indicate that select cellular metabolites are not only used to generate macromolecular building blocks or extract energy, but also serve as co‐factors or substrates in a variety of cellular regulatory cascades, including those that directly modify histones and DNA. Thus, levels of certain metabolites are continuously monitored by a cell, directly informing cellular decisions on gene expression, affecting cellular differentiation as a result (Pavlova & Thompson, 2016). The glutamine‐derived metabolite α‐KG has been implicated in modulating cellular histone and DNA methylation levels. α‐KG serves as a co‐substrate for a class of dioxygenase enzymes, among which are Jumonji C domain‐containing histone demethylases and TET family DNA demethylases, which catalyze the oxidation of methyllysine residues of histones to hydroxymethyllysine and oxidation of 5‐methylcytosine to 5‐hydroxymethylcytosine, respectively (Fig 3). In these reactions, α‐KG itself is oxidized to succinate, and the rising levels of the latter can inhibit the progression of α‐KG‐dependent histone or DNA demethylase reactions. Indeed, in murine embryonic stem cells (mESC), elevated α‐KG/succinate ratios are associated with the naïve pluripotent state, and the direct manipulation of intracellular α‐KG by either addition of cell‐permeable form of α‐KG or via glutamine withdrawal is sufficient to modulate H3K27me3 and TET‐dependent DNA methylation and affect differentiation (Carey et al, 2015). Glutamine depletion can also promote differentiation of naïve CD4+ T cells into immunosuppressive Foxp3+ regulatory T (Treg) cells even in the presence of cytokines that typically promote the induction of T helper 1 (TH1) cells (Klysz et al, 2015). Accordingly, addition of dimethyl‐α‐KG restores the intracellular α‐KG levels and enables TH1 cell differentiation under glutamine deprivation. In cancer cells, loss‐of‐function mutations of succinate dehydrogenase (SDH) subunits have been found in familial paragangliomas and pheochromocytomas, as well as in a subset of sporadic gastrointestinal stromal tumors (Astuti et al, 2001; Janeway et al, 2011). Accumulation of intracellular succinate in these tumors as a consequence of SDH loss is associated with a global inhibition of DNA demethylation, which contributes to their tumorigenic state (Xiao et al, 2012; Killian et al, 2013; Letouze et al, 2013). Recently, Pan et al (2016) showed that glutamine deficiency in the core region of solid tumors correlates with histone hypermethylation, dedifferentiation, and therapeutic resistance in a melanoma model.
Figure 3. Glutamine‐derived α‐KG is a substrate for α‐KG‐dependent dioxygenases.
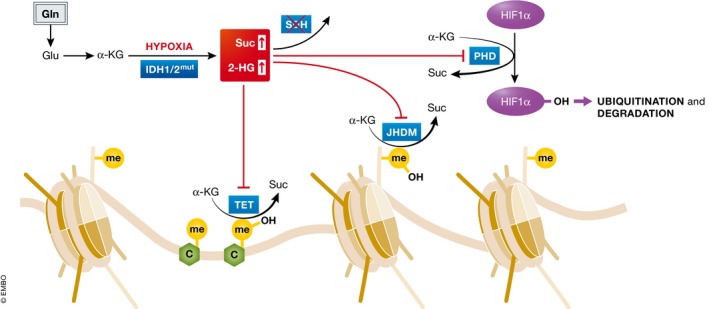
α‐KG is the substrate of Jumonji C histone demethylases (JHDM) and TET DNA demethylases and therefore mediates histone and DNA demethylation. In addition, α‐KG is the substrate of prolyl hydroxylase (PHD) that mediates HIF1α ubiquitination and degradation. These α‐KG‐dependent dioxygenases convert α‐KG to succinate that can feedback inhibit their dioxygenase activity. Either cancer‐associated IDH1/2 mutations or oxygen limitation can cause accumulation of 2‐HG, which can competitively inhibit α‐KG‐dependent dioxygenases.
The central role of glutamine‐derived α‐KG in modulating histone and DNA methylation is also exemplified by the gain‐of‐function mutations in IDH1 and IDH2, which have been identified in glioma, chondrosarcoma, cholangiocarcinoma, acute myeloid leukemia (AML), and a small portion of adult T‐cell acute lymphoblastic leukemia (T‐ALL) (Balss et al, 2008; Parsons et al, 2008; Mardis et al, 2009; Paschka et al, 2010; Borger et al, 2012; Cohen et al, 2013; Van Vlierberghe et al, 2013). The mutations of IDH1 or IDH2 exhibit a neomorphic activity by converting glutamine‐derived α‐KG to 2‐hydroxyglutarate (2‐HG), which competitively inhibits α‐KG‐dependent histone and DNA demethylases (Dang et al, 2010; Figueroa et al, 2010; Ward et al, 2010; Lu et al, 2012; Turcan et al, 2012; Fig 3). Consistently, glioma cells with mutant IDH1 display an elevated dependency on glutamine, rerouting the entry of pyruvate into the TCA cycle through PC (Seltzer et al, 2010; Izquierdo‐Garcia et al, 2014). In a mouse model of T‐ALL, mutation of IDH1 increases the sensitivity of leukemic cells to glutamine depletion (Hao et al, 2016b), indicating a potential therapeutic vulnerability in tumors harboring IDH mutations.
In addition to dioxygenase enzymes that drive histone and DNA demethylation, α‐KG also serves as a substrate for a class of prolyl 4‐hydroxylase enzymes, which mediate the ubiquitination and degradation of hypoxia‐inducible factor 1 α (HIF1α), a key transcription factor that facilitates the cellular adaptation to low oxygen levels (Fig 3). Despite the fact that IDH1 mutations were originally found to result in an increased level of HIF1α (Zhao et al, 2009), Kuivonen et al showed that prolyl 4‐hydroxylases are only sensitive to the L‐enantiomer (Koivunen et al, 2012). Further exploration is necessary to determine the contribution of glutamine to the gene expression as a consequence of the alteration of intracellular α‐KG, 2‐HG, and succinate, which can affect global chromatin modification patterns or levels of specific transcriptional regulators, such as HIF, depending on the cellular context.
Cellular adaptations to glutamine limitation
Increased consumption of glutamine by proliferating tumor cells, coupled with inadequacies of tumor vascular supply, results in selective depletion of glutamine from the microenvironment (Vaupel et al, 1989). In tumors, glutamine levels can be profoundly reduced when compared to the surrounding normal tissues and plasma (Roberts et al, 1956; Rivera et al, 1988; Marquez et al, 1989). In addition, a spatial examination of amino acid levels in cancer cell xenografts has found glutamine to be among the most depleted in the xenograft core, when compared to the periphery of the tumor (Pan et al, 2016). Furthermore, metabolomic analysis from primary human pancreatic ductal adenocarcinoma (PDAC) tissue revealed a significant reduction of glutamine and several other NEAAs in the tumor tissues relative to the adjacent normal tissues as well (Kamphorst et al, 2015). These findings warrant further investigation of the adaptations that various tumors may employ to augment limited glutamine levels. Understanding these adaptations may not only uncover tumor metabolic vulnerabilities that can be exploited in anti‐tumor therapy, but also provide guidance for more effective immunotherapy as well, as the tumor‐associated T lymphocytes may compete with tumor cells for glutamine.
Glutamine uptake
One way to increase glutamine acquisition within the tumor environment is through the induction of glutamine uptake. In most cells, ASCT2 is the major transporter for glutamine uptake. Its expression is upregulated by oncogenic MYC or E2F3, consistent with the role of these signaling molecules in directly increasing glutamine uptake (Wise et al, 2008; Reynolds et al, 2014). Elevated expression of ASCT2 was found in triple‐negative breast cancer patients, correlating with poor survival in xenograft mouse models (van Geldermalsen et al, 2016). Gamma‐L‐glutamyl‐p‐nitroanilide (GPNA), an ASCT2 inhibitor, has been shown to suppress glutamine uptake and cell growth in lung cancer cells (Hassanein et al, 2013). In mice, ASCT2 deficiency impairs the induction of TH1 and TH17 cells, due to a defect of glutamine uptake and mTORC1 activation (Nakaya et al, 2014). However, ASCT2 is not the only transporter for glutamine uptake. It was recently reported that depletion of ASCT2 leads to the induction of SNAT1 and SNAT2, two other sodium‐neutral amino acid transporters, which is sufficient to compensate for glutamine uptake (Broer et al, 2016; Fig 4A). Glutamine transporters are potential cancer therapeutic targets, but the therapeutic effects associated with their inhibition may be affected by the effects of this inhibition on the transport of other amino acids as well.
Figure 4. Glutamine uptake and de novo biosynthesis.
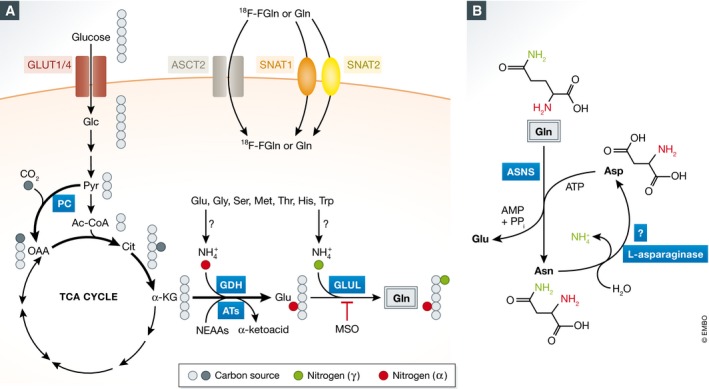
(A) Glutamine uptake can be mediated by ASCT2 and SNAT1/2 transporters, allowing the application of 18F‐FGln‐based PET imaging as a diagnostic tool for brain tumors. Furthermore, glucose‐derived carbon can be used as precursors to synthesize glutamine de novo. (B) A potential mechanism for asparagine as the nitrogen source for glutamine biosynthesis. However, this activity has only been reliably observed in unicellular organisms thus far. GLUL: glutamate‐ammonia ligase (glutamine synthetase); MSO: methionine sulfoximine.
In addition to its therapeutic implications, the increased uptake of glutamine by some tumors provides a unique potential for tumor imaging. Indeed, glucose‐based [18F] fluorodeoxyglucose positron emission tomography (FDG‐PET) has been used for thirty years to detect and monitor tumors in clinic, exploiting the phenomenon of the markedly elevated uptake of glucose by a wide variety of tumors (Kelloff et al, 2005). However, FDG‐PET imaging is not effective in brain tumors, as normal brain tissue takes up large quantities of glucose as well. In contrast to FDG‐based imaging, [18F] fluorinated glutamine (18F‐FGln) displays low background uptake in brain tissues, and has been successfully used for tumor detection in mouse models of glioma and in glioma patients (Lieberman et al, 2011; Ploessl et al, 2012), validating it as a diagnostic tool (Venneti et al, 2015).
Glutamine biosynthesis de novo
The fact that most cancer cells cannot proliferate or even survive in the absence of exogenous glutamine is surprising, because mammalian cells possess all the necessary enzymatic machinery to synthesize glutamine de novo. In particular, glutamine synthetase (GLUL) enzyme catalyzes the condensation reaction between glutamate and ammonia in an ATP‐dependent manner, which generates glutamine. In mammalian tissues, GLUL is ubiquitously expressed but is particularly enriched in the liver, brain, and muscle (Haberle et al, 2006). Multiple growth factors and oncogenic signals positively regulate GLUL transcription (van der Vos et al, 2012; Bott et al, 2015; Cox et al, 2016), and exogenous glutamine was found to directly destabilize GLUL protein through facilitating its ubiquitination and degradation (Nguyen et al, 2016b). Elevated GLUL expression was found to be an early marker of hepatocellular carcinoma (Long et al, 2010), and is a predictor of poor survival in patients with glioma and liver cancer (Osada et al, 2000; Rosati et al, 2013). However, despite expressing GLUL, most cancer cells in culture require exogenous glutamine for growth and survival. One simple interpretation is that at least in tissue culture, de novo biosynthesis of glutamine is not sufficient to accommodate the demands of the great variety of glutamine‐utilizing enzymes. However, the limiting factors for cellular adaptation to the deficit of exogenous glutamine have yet to be elucidated. One potential lead is a recent report demonstrating that mESC can proliferate without exogenous glutamine when treated with both a MEK and a GSK‐3β inhibitor (Carey et al, 2015). The fact that the MEK pathway and/or GSK‐3β are activated in most malignant cells suggests that their activation may be responsible for the inhibition of cancer cell to upregulate glutamine synthesis to levels that support cell growth.
Second, tumors have been reported to synthesize some glutamine. Kung et al (2011) showed that luminal breast cancer cells are resistant to glutamine‐depletion‐induced growth inhibition and apoptosis, when compared to the basal breast cancer cells. Consistently, luminal breast cancer cells display high levels of GLUL expression. In a mutant KRas‐driven mouse pancreatic tumor model, oncogenic MYC enhances GLUL expression (Bott et al, 2015). In this study, the authors show that inhibition of GLUL suppresses tumor growth in vivo. Furthermore, glioma cells were shown to utilize glucose carbons to maintain TCA cycle anaplerosis, and consequently, glutamate and glutamine biosynthesis under glutamine limitation (Tardito et al, 2015; Fig 4A). However, the source of the free ammonia that is necessary for glutamine biosynthesis through the GLUL remains more of a mystery. Together, these studies suggest that the dependency of tumor cells on glutamine can be dictated both by the oncogenic signals and by the tissue of origin.
In some cell types, asparagine is sufficient to suppress glutamine‐depletion‐induced apoptosis (Zhang et al, 2014). However, despite the fact that most cells synthesize asparagine from glutamine (Balasubramanian et al, 2013), its catabolism has only been reliably reported in unicellular organisms (Peterson & Ciegler, 1969; Jones & Mortimer, 1970; Dunlop & Roon, 1975; Fig 4B). Recently, it was reported that asparagine may rescue cells by replacing a required role for glutamine as a counter ion in the import of extracellular amino acids, which is essential to maintain mTOR activation and protein translation (Nicklin et al, 2009; Krall et al, 2016). Whether asparagine also supplies reduced nitrogen for glutamine biosynthesis, or plays other regulatory roles to mediate cellular adaptation to glutamine limitation in mammalian cells remains to be determined.
Glutamine catabolism in vivo
As mentioned above, the ability of some tumors to synthesize glutamine de novo to meet biosynthetic needs suggests that exogenous glutamine may not be required by all proliferating cells in vivo. In an orthotopic mouse model of primary human glioma and in a mouse model of lung cancer driven by mutant KRas, it has been shown that the tumor cells can utilize glucose as a preferred anaplerotic entry port into the TCA cycle via the action of pyruvate carboxylase (PC; Marin‐Valencia et al, 2012; Davidson et al, 2016). However, PC was found to be dispensable for the cancer cells in vitro, when glutamine is supplied in the culture medium. These two studies challenge the importance of glutamine catabolism in replenishing the TCA cycle intermediates, a phenomenon observed in most of the cultured cells. These results also put into question the broader applicability of the therapeutics that target glutamine entry into the TCA cycle or its catabolism via glutaminase. Further exploration of other in vivo tumor models as well as clinical studies is necessary to determine whether the limited dependence of some tumor cells on glutamine anaplerosis is associated with select oncogenic contexts or tissues of origin, or whether it reflects glutamine availability in the specific tissue environments.
As mentioned above, availability of glutamine in the milieu of the tumor is often limited (Roberts et al, 1956; Rivera et al, 1988). The fact that cancer cells can use glutamine‐derived carbons to fuel the TCA cycle, or use glucose‐derived carbons to both replenish the TCA cycle and synthesize glutamine may reflect a certain degree of metabolic plasticity of tumor cells, aimed at optimizing their growth in shifting nutrient conditions. Furthermore, investigating the relative degree of tumor dependency on exogenous glutamine at distinct stages of tumorigenesis—that is, in the primary tumor initiation and expansion, local tissue invasion, survival in the circulation and seeding, and colonization of distant organs—may provide further insights into the therapeutic potential of targeting glutamine metabolism in cancer therapy.
To evaluate how tumor glutamine dependency is influenced by the concentration of amino acids and other nutrients that are typically encountered by cells in vivo, Tardito et al have designed a serum‐like modified Eagle's medium (SMEM), which contains nutrient concentrations at levels close to those found in human plasma, and assessed the response to glutamine limitation of glioma cells under these conditions. Indeed, they found that the cell proliferation in SMEM medium was largely unaffected by the presence or absence of exogenous glutamine; furthermore, GLUL was found to be both necessary and sufficient for the proliferation in the absence of exogenous glutamine (Tardito et al, 2015). In turn, newly synthesized glutamine was then utilized for nucleotide biosynthesis to support the growth of glioma cells. This study is consistent with the fact that glucose‐derived carbon is the main supply of the TCA cycle intermediates in vivo, which can be used as precursors to synthesize glutamine and other NEAAs. However, the source of reduced nitrogen for the de novo glutamine biosynthesis remains more of a mystery. As glutamine biosynthesis through GLUL enzyme requires free ammonia, which is maintained at rather low levels in plasma (less than 35 μM), and is absent in standard tissue culture media formulations, ammonia can be generated through the catabolism of glutamate, glycine, serine, methionine, threonine, histidine, and tryptophan (Fig 4A); however, the relative contribution of these sources to the ammonia supply for glutamine biosynthesis is in need of further investigation. Insights into the sources of ammonia for the de novo glutamine biosynthesis may unveil novel therapeutic targets aimed at the inhibition of glutamine biosynthesis.
Protein and cell corpses can supply glutamine
In addition to the upregulation of uptake from the extracellular fluid and de novo biosynthesis in cell, some tumor cells can use alternative ways to obtain glutamine and other amino acids via the breakdown of engulfed extracellular proteins, apoptotic bodies, or even living cells. Such unconventional nutrient acquisition strategies can play a critical adaptive role in conditions when glutamine is limited. For example, extracellular proteins are abundant in plasma and tumor environment, but are not typically considered as a nutrient source. However, it was demonstrated that extracellular proteins can be taken up by cells through macropinocytosis, a process that involves a nonselective engulfment of the extracellular fluid phase, which gives rise to giant vesicles termed macropinosomes (Kerr & Teasdale, 2009). Engulfed proteins, as well as larger macromolecular structures, can then be digested through the action of lysosomal proteases as means of recovering free amino acids (Fig 5).
Figure 5. Glutamine acquisition through proteolytic scavenging.
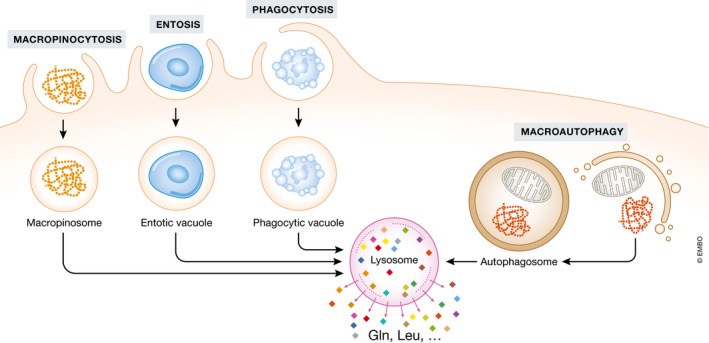
Extracellular proteins (macropinocytosis) and live/dead cells (entosis and phagocytosis) can be engulfed and digested in lysosomes to release free amino acids, including glutamine. In addition, intracellular proteins and organelles can also be digested in lysosomes to release free amino acids via macroautophagy.
Macropinocytosis has been described in normal, growth factor‐stimulated cells, but it becomes markedly enhanced by oncogenic Ras signaling (Bar‐Sagi & Feramisco, 1986). Indeed, incubation of KRas‐transformed cells with 13C‐labeled soluble proteins under low glutamine conditions restored labeled free amino acids and TCA cycle intermediates, indicating a recovery of sufficient quantities of glutamine to replenish the TCA cycle (Commisso et al, 2013). In this study, the authors show that soluble albumin can rescue proliferation of KRas‐transformed cells in glutamine‐limited conditions. Furthermore, 5‐(N‐ethyl‐N‐isopropyl) amiloride (EIPA), an inhibitor of macropinocytosis, suppresses xenograft tumor growth, indicating a critical role of macropinocytosis to supply amino acid in vivo (Commisso et al, 2013). While not specific for glutamine, the catabolism of proteins increases not only glutamine but other amino acids essential and nonessential as well (Kamphorst et al, 2015; Palm et al, 2015).
In addition to the extracellular proteins, the engulfment and digestion of entire living cells or apoptotic cells can be also employed as a way of recovering free amino acids (Fig 5). Thus, the growth of MCF10A mammary epithelial cells under amino acid‐free conditions can sustain through the engulfment of living cells via the process of entosis, and this effect can be reversed by blocking of lysosomal‐mediated digestion (Krajcovic et al, 2013). In this study, similar effect was observed in macrophages engulfing apoptotic bodies (Krajcovic et al, 2013). Notably, entosis represents an interesting example of oncogene‐driven cell–cell competition, as KRas‐transformed cells were found to be more likely to consume their nontransformed neighbors than be engulfed themselves (Sun et al, 2014). Amino acids can also be recovered from the intracellular sources, such as proteins and organelles, via a process of autophagy (Jiang et al, 2015). During autophagy, intracellular proteins and organelles are enwrapped by a double‐membrane structure that is eventually fused with the lysosome for digestion (Fig 5). Since autophagy recycles intracellular materials, it cannot give rise to the biomass accumulation that is necessary for proliferation; however, it can sustain energy production to support long‐term cell survival (Lum et al, 2005). Similar to the other amino acid scavenging pathways, autophagy recycles intracellular amino acids nonselectively.
Summary
Glutamine has been the most widely studied nutrient other than glucose in the field of cancer cell metabolism during the past decade. The central function of glutamine in cell proliferation can be attributed to its role in numerous biological processes, including its role in biosynthesis and bioenergetics, anti‐oxidative defense, chromatin modification/gene transcription, facilitation of transport of other amino acids across the plasma membrane, and regulation of cell signaling (Gonzalez & Hall, 2017). The relative effects of glutamine as well as glutamine‐derived metabolites can be dictated by both the tissue and the oncogenic context. A number of pharmacological inhibitors of glutamine uptake and catabolism have been developed. Some of them have entered clinical trials or have been FDA‐approved to be used in patients with cancer or other diseases (Altman et al, 2016). One of the first successful metabolic therapies, L‐asparaginase has been used in the clinic to treat acute lymphoblastic leukemia (ALL) for close to three decades. L‐asparaginase functions by depleting plasma asparagine and glutamine, such that the ALL cells, which are auxotrophic for asparagine and require large amounts of glutamine, are selectively affected by this treatment (van den Berg, 2011). However, L‐asparaginase has only been proven to be effective in ALL and some NK/T‐cell lymphomas. Its application in AML, non‐Hodgkin's lymphoma (NHL), and solid tumor has not been found to be successful (Clarkson et al, 1970; Jaffe et al, 1971). Its use in other cancers has been limited by its immunogenicity (van den Berg, 2011).
On the other hand, growing evidence suggests that various tumor types may reside in an environment where glutamine is profoundly limited. Therefore, tumor cells have to develop adaptive strategies that would allow them to sustain their growth and survival. In this regard, induction of de novo biosynthesis of glutamine or acquisition of glutamine through catabolism of extracellular and intracellular proteins has been shown to provide a source of missing glutamine for cells. For example, in a KRasG12D‐driven mouse pancreatic cancer model, inhibition of mTORC1 remarkably enhances the capacity of tumor cells to use extracellular protein to restore their amino acid pools, leading to increase in the tumor burden (Palm et al, 2015). Even though the phenotype is unlikely to be solely due to the augmented glutamine acquisition, this study revealed a fundamentally opposing strategy of nutrient acquisition in amino acid‐replete and amino acid‐starved settings, which warrants reconsideration of the current therapeutic approaches that are based on mTORC1 inhibition in at least some oncogenic contexts.
In addition, tumor cells can adapt to the limitation of glutamine through the inhibition of global protein translation. In yeast and in mammalian cells alike, glutamine was shown to activate mTORC1 in a RagA/B‐independent manner (Stracka et al, 2014; Jewell et al, 2015). Accordingly, depletion of glutamine may result in suppressed mTORC1 signaling, especially when other amino acids are limiting, too. In addition, depletion of glutamine, along with other amino acids, triggers the inhibitory eIF2α phosphorylation through GCN2 kinase (Harding et al, 2000). Both of these signaling modulations result in the inhibition of translation initiation, acting together to preserve the amino acid pools while allowing selective translation of proteins necessary for the adaptive responses. For instance, the eIF2α phosphorylation promotes the translation of the transcription factor ATF4, which functions as a master regulator of a set of genes involved in adaption to starvation. Accordingly, the inhibition of GCN2 or of ATF4 prevents xenograft growth in vivo (Ye et al, 2010; Horiguchi et al, 2012), suggesting a potential to target this pathway as a cancer therapeutic.
In conclusion, glutamine metabolism is key to the survival, proliferation, differentiation state, and stress resilience in normal proliferating cells as well as in the context of tumorigenesis. Further exploration of the ways in which cellular glutamine status affects these diverse processes, as well as the investigation of strategies that cells may adopt to withstand glutamine limitation, may uncover new intersections between metabolism and disease, revealing novel opportunities for therapeutic intervention.
Conflict of interest
C.B.T. is a co‐founder of Agios Pharmaceuticals and a member of the board of directors of Merck and Charles River Laboratories.
Acknowledgements
N.P. is supported by the postdoctoral fellowship from the Terri Brodeur Breast Cancer Foundation. This work was funded by grants from NCI (C.B.T.), including the MSKCC cancer center support grant P30 CA008748 and the P01 CA104838 (University of Pennsylvania).
The EMBO Journal (2017) 36: 1302–1315
References
- Ahluwalia GS, Grem JL, Hao Z, Cooney DA (1990) Metabolism and action of amino acid analog anti‐cancer agents. Pharmacol Ther 46: 243–271 [DOI] [PubMed] [Google Scholar]
- Altman BJ, Stine ZE, Dang CV (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16: 619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER (2001) Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69: 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian MN, Butterworth EA, Kilberg MS (2013) Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am J Physiol Endocrinol Metab 304: E789–E799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116: 597–602 [DOI] [PubMed] [Google Scholar]
- Bar‐Sagi D, Feramisco JR (1986) Induction of membrane ruffling and fluid‐phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233: 1061–1068 [DOI] [PubMed] [Google Scholar]
- Ben‐Sahra I, Howell JJ, Asara JM, Manning BD (2013) Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339: 1323–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg H (2011) Asparaginase revisited. Leuk Lymphoma 52: 168–178 [DOI] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu‐Remaileh M, Sabatini DM (2015) An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162: 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias‐Santagata D, Ellisen LW, Zhu AX, Iafrate AJ (2012) Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad‐based tumor genotyping. Oncologist 17: 72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D, Lu W, Pan JA, Powers S, Rabinowitz J, Hatzoglou M, Murphy DJ, Jones R, Wu S, Girnun G, Zong WX (2015) Oncogenic myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab 22: 1068–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broer A, Rahimi F, Broer S (2016) Deletion of amino acid transporter ASCT2 (SLC1A5) reveals an essential role for transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to sustain glutaminolysis in cancer cells. J Biol Chem 291: 13194–13205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB (2015) Intracellular alpha‐ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518: 413–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS, Pereira HM, Garratt RC, Dias SM, Ambrosio AL (2012) Mitochondrial localization and structure‐based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci USA 109: 1092–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson B, Krakoff I, Burchenal J, Karnofsky D, Golbey R, Dowling M, Oettgen H, Lipton A (1970) Clinical results of treatment with E. coli L‐asparaginase in adults with leukemia, lymphoma, and solid tumors. Cancer 25: 279–305 [DOI] [PubMed] [Google Scholar]
- Cohen AL, Holmen SL, Colman H (2013) IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 13: 345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner‐Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, Bar‐Sagi D (2013) Macropinocytosis of protein is an amino acid supply route in Ras‐transformed cells. Nature 497: 633–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AG, Hwang KL, Brown KK, Evason KJ, Beltz S, Tsomides A, O'Connor K, Galli GG, Yimlamai D, Chhangawala S, Yuan M, Lien EC, Wucherpfennig J, Nissim S, Minami A, Cohen DE, Camargo FD, Asara JM, Houvras Y, Stainier DY et al (2016) Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat Cell Biol 18: 886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Moreno MV, Lodi A, Ronen SM, Ruggero D (2014) Protein and nucleotide biosynthesis are coupled by a single rate‐limiting enzyme, PRPS2, to drive cancer. Cell 157: 1088–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM (2010) Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature 465: 966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O'Brien JP, Pierce KA, Gui DY, Sullivan LB, Wasylenko TM, Subbaraj L, Chin CR, Stephanopolous G, Mott BT, Jacks T, Clish CB, Vander Heiden MG (2016) Environment impacts the metabolic dependencies of ras‐driven non‐small cell lung cancer. Cell Metab 23: 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104: 19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop PC, Roon RJ (1975) L‐asparaginase of Saccharomyces cerevisiae: an extracellular enzyme. J Bacteriol 122: 1017–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ (2001) c‐Myc mediates activation of the cad promoter via a post‐RNA polymerase II recruitment mechanism. J Biol Chem 276: 48562–48571 [DOI] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD (2013) Glutamine‐driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol 9: 712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel‐Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18: 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV (2009) c‐Myc suppression of miR‐23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, Ritchie W, Feng Y, Bailey CG, Deng N, Harvey K, Beith JM, Selinger CI, O'Toole SA, Rasko JE, Holst J (2016) ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple‐negative basal‐like breast cancer. Oncogene 35: 3201–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Hall MN (2017) Nutrient sensing and TOR signaling in yeast and mammals. EMBO J 36: 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947 [DOI] [PubMed] [Google Scholar]
- Goswami MT, Chen G, Chakravarthi BV, Pathi SS, Anand SK, Carskadon SL, Giordano TJ, Chinnaiyan AM, Thomas DG, Palanisamy N, Beer DG, Varambally S (2015) Role and regulation of coordinately expressed de novo purine biosynthetic enzymes PPAT and PAICS in lung cancer. Oncotarget 6: 23445–23461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, Collins MA, Dahlstrand EN, Earp HS III, Evans DR (2000) Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 403: 328–332 [DOI] [PubMed] [Google Scholar]
- Gross MI, Demo SD, Dennison JB, Chen L, Chernov‐Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, Mackinnon AL, Parlati F, Rodriguez ML, Shwonek PJ, Sjogren EB, Stanton TF, Wang T, Yang J, Zhao F, Bennett MK (2014) Antitumor activity of the glutaminase inhibitor CB‐839 in triple‐negative breast cancer. Mol Cancer Ther 13: 890–901 [DOI] [PubMed] [Google Scholar]
- Guth PS, Risey J, Briner W, Blair P, Reed HT, Bryant G, Norris C, Housley G, Miller R (1990) Evaluation of amino‐oxyacetic acid as a palliative in tinnitus. Ann Otol Rhinol Laryngol 99: 74–79 [DOI] [PubMed] [Google Scholar]
- Haberle J, Gorg B, Toutain A, Rutsch F, Benoist JF, Gelot A, Suc AL, Koch HG, Schliess F, Haussinger D (2006) Inborn error of amino acid synthesis: human glutamine synthetase deficiency. J Inherit Metab Dis 29: 352–358 [DOI] [PubMed] [Google Scholar]
- Hao Y, Samuels Y, Li Q, Krokowski D, Guan BJ, Wang C, Jin Z, Dong B, Cao B, Feng X, Xiang M, Xu C, Fink S, Meropol NJ, Xu Y, Conlon RA, Markowitz S, Kinzler KW, Velculescu VE, Brunengraber H et al (2016a) Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat Commun 7: 11971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z, Cairns RA, Inoue S, Li WY, Sheng Y, Lemonnier F, Wakeham A, Snow BE, Dominguez‐Brauer C, Ye J, Larsen DM, Straley KS, Tobin ER, Narayanaswamy R, Gaulard P, Mak TW (2016b) Idh1 mutations contribute to the development of T‐cell malignancies in genetically engineered mice. Proc Natl Acad Sci USA 113: 1387–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, Clark JE, Alborn WE, Eisenberg R, Massion PP (2013) SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res 19: 560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi M, Koyanagi S, Okamoto A, Suzuki SO, Matsunaga N, Ohdo S (2012) Stress‐regulated transcription factor ATF4 promotes neoplastic transformation by suppressing expression of the INK4a/ARF cell senescence factors. Cancer Res 72: 395–401 [DOI] [PubMed] [Google Scholar]
- Hosios AM, Hecht VC, Danai LV, Johnson MO, Rathmell JC, Steinhauser ML, Manalis SR, Vander Heiden MG (2016) Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell 36: 540–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z (2010) Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA 107: 7455–7460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo‐Garcia JL, Cai LM, Chaumeil MM, Eriksson P, Robinson AE, Pieper RO, Phillips JJ, Ronen SM (2014) Glioma cells with the IDH1 mutation modulate metabolic fractional flux through pyruvate carboxylase. PLoS One 9: e108289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe N, Traggis D, Das L, Moloney WC, Hann HW, Kim BS, Nair R (1971) L‐asparaginase in the treatment of neoplastic diseases in children. Cancer Res 31: 942–949 [PubMed] [Google Scholar]
- Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, Lai AH, Kelly L, Hornick JL, O'Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L et al (2011) Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA 108: 314–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javeshghani S, Zakikhani M, Austin S, Bazile M, Blouin MJ, Topisirovic I, St‐Pierre J, Pollak MN (2012) Carbon source and myc expression influence the antiproliferative actions of metformin. Cancer Res 72: 6257–6267 [DOI] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL (2015) Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347: 194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Overholtzer M, Thompson CB (2015) Autophagy in cellular metabolism and cancer. J Clin Invest 125: 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GE, Mortimer RK (1970) L‐asparaginase‐deficient mutants of yeast. Science 167: 181–182 [DOI] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar‐Sagi D, Thompson CB, Rabinowitz JD (2015) Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75: 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, Cheson BD, O'Shaughnessy J, Guyton KZ, Mankoff DA, Shankar L, Larson SM, Sigman CC, Schilsky RL, Sullivan DC (2005) Progress and promise of FDG‐PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res 11: 2785–2808 [DOI] [PubMed] [Google Scholar]
- Kerr MC, Teasdale RD (2009) Defining macropinocytosis. Traffic 10: 364–371 [DOI] [PubMed] [Google Scholar]
- Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI Jr, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ et al (2013) Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov 3: 648–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, Yong C, Surh N, Marie JC, Huehn J, Zimmermann V, Kinet S, Dardalhon V, Taylor N (2015) Glutamine‐dependent alpha‐ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal 8: ra97. [DOI] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG Jr (2012) Transformation by the (R)‐enantiomer of 2‐hydroxyglutarate linked to EGLN activation. Nature 483: 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, Carrero ZI, Ramakrishnan G, Watabe K, Haupt Y, Haupt S, Pochampally R, Boss GR, Romero DG, Radu CG, Martinez LA (2015) Regulation of nucleotide metabolism by mutant p53 contributes to its gain‐of‐function activities. Nat Commun 6: 7389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korangath P, Teo WW, Sadik H, Han L, Mori N, Huijts CM, Wildes F, Bharti S, Zhang Z, Santa‐Maria CA, Tsai H, Dang CV, Stearns V, Bhujwalla ZM, Sukumar S (2015) Targeting glutamine metabolism in breast cancer with Aminooxyacetate. Clin Cancer Res 21: 3263–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajcovic M, Krishna S, Akkari L, Joyce JA, Overholtzer M (2013) mTOR regulates phagosome and entotic vacuole fission. Mol Biol Cell 24: 3736–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Xu S, Graeber TG, Braas D, Christofk HR (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7: 11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung HN, Marks JR, Chi JT (2011) Glutamine synthetase is a genetic determinant of cell type‐specific glutamine independence in breast epithelia. PLoS Genet 7: e1002229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzardo S, Conti L, Rooke R, Ruiu R, Accart N, Bolli E, Arigoni M, Macagno M, Barrera G, Pizzimenti S, Aurisicchio L, Calogero RA, Cavallo F (2016) Immunotargeting of antigen xCT attenuates stem‐like cell behavior and metastatic progression in breast cancer. Cancer Res 76: 62–72 [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJ, Lorkiewicz PK, Higashi RM, Fan TW, Dang CV (2012) Glucose‐independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reynies A, Gimenez‐Roqueplo AP, Favier J (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23: 739–752 [DOI] [PubMed] [Google Scholar]
- Lieberman BP, Ploessl K, Wang L, Qu W, Zha Z, Wise DR, Chodosh LA, Belka G, Thompson CB, Kung HF (2011) PET imaging of glutaminolysis in tumors by 18F‐(2S,4R)4‐fluoroglutamine. J Nucl Med 52: 1947–1955 [DOI] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, Dang CV (2008) Global regulation of nucleotide biosynthetic genes by c‐Myc. PLoS One 3: e2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM (2012) Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c‐MYC. Proc Natl Acad Sci USA 109: 8983–8988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Jia Y, Cao Y, Wu S, Jiang H, Sun X, Ma J, Yin X, Mao A, Shang M (2016) Overexpression of phosphoserine aminotransferase 1 (PSAT1) predicts poor prognosis and associates with tumor progression in human esophageal squamous cell carcinoma. Cell Physiol Biochem 39: 395–406 [DOI] [PubMed] [Google Scholar]
- Long J, Lang ZW, Wang HG, Wang TL, Wang BE, Liu SQ (2010) Glutamine synthetase as an early marker for hepatocellular carcinoma based on proteomic analysis of resected small hepatocellular carcinomas. Hepatobiliary Pancreat Dis Int 9: 296–305 [PubMed] [Google Scholar]
- Lu SC (2009) Regulation of glutathione synthesis. Mol Aspects Med 30: 42–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel‐Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O'Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483: 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukey MJ, Greene KS, Erickson JW, Wilson KF, Cerione RA (2016) The oncogenic transcription factor c‐Jun regulates glutaminase expression and sensitizes cells to glutaminase‐targeted therapy. Nat Commun 7: 11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120: 237–248 [DOI] [PubMed] [Google Scholar]
- Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, Mathews CK, Shewach DS, Nikiforov MA (2008) Direct role of nucleotide metabolism in C‐MYC‐dependent proliferation of melanoma cells. Cell Cycle 7: 2392–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361: 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin‐Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, Cai L, Good L, Tu BP, Hatanpaa KJ, Mickey BE, Mates JM, Pascual JM, Maher EA, Malloy CR, Deberardinis RJ et al (2012) Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo . Cell Metab 15: 827–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez J, Sanchez‐Jimenez F, Medina MA, Quesada AR, Nunez de Castro I (1989) Nitrogen metabolism in tumor bearing mice. Arch Biochem Biophys 268: 667–675 [DOI] [PubMed] [Google Scholar]
- Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X, Sun SC (2014) Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40: 692–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A, Loo JM, Mital R, Weinberg EM, Man FY, Zeng Z, Paty PB, Saltz L, Janjigian YY, de Stanchina E, Tavazoie SF (2016a) PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J Clin Invest 126: 681–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Lee JE, Sweredoski MJ, Yang SJ, Jeon SJ, Harrison JS, Yim JH, Lee SG, Handa H, Kuhlman B, Jeong JS, Reitsma JM, Park CS, Hess S, Deshaies RJ (2016b) Glutamine triggers acetylation‐dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol Cell 61: 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136: 521–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolay BN, Dyson NJ (2013) The multiple connections between pRB and cell metabolism. Curr Opin Cell Biol 25: 735–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada T, Nagashima I, Tsuno NH, Kitayama J, Nagawa H (2000) Prognostic significance of glutamine synthetase expression in unifocal advanced hepatocellular carcinoma. J Hepatol 33: 247–253 [DOI] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB (2015) The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162: 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez‐Davies JE, Rosales KK, Li H, Hugo W, Song C, Xu X, Schones DE, Ann DK, Gradinaru V, Lo RS, Locasale JW, Kong M (2016) Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18: 1090–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L, Spath D, Kayser S, Zucknick M, Gotze K, Horst HA, Germing U, Dohner H, Dohner K (2010) IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28: 3636–3643 [DOI] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23: 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, O'Dwyer KM, Li Z, Shi L, Greninger P, Settleman J, Benes C, Hagen FK, Munger J, Crooks PA, Becker MW et al (2013) Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem 288: 33542–33558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RE, Ciegler A (1969) L‐asparaginase production by various bacteria. Appl Microbiol 17: 929–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploessl K, Wang L, Lieberman BP, Qu W, Kung HF (2012) Comparative evaluation of 18F‐labeled glutamic acid and glutamine as tumor metabolic imaging agents. J Nucl Med 53: 1616–1624 [DOI] [PubMed] [Google Scholar]
- Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, Mayes PA, Wise DR, Thompson CB, Maris JM, Hogarty MD, Simon MC (2012) ATF4 regulates MYC‐mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 22: 631–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, Dean DC, Clem BF (2014) Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 33: 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards NG, Schuster SM (1998) Mechanistic issues in asparagine synthetase catalysis. Adv Enzymol Relat Areas Mol Biol 72: 145–198 [DOI] [PubMed] [Google Scholar]
- Rivera S, Azcon‐Bieto J, Lopez‐Soriano FJ, Miralpeix M, Argiles JM (1988) Amino acid metabolism in tumour‐bearing mice. Biochem J 249: 443–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E, Simonsen DG, Tanaka KK, Tanaka T (1956) Free amino acids in growing and regressing ascites cell tumors: host resistance and chemical agents. Cancer Res 16: 970–978 [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto‐Baschong C, Sauer U, Jenoe P, Hall MN (2013) Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339: 1320–1323 [DOI] [PubMed] [Google Scholar]
- Rosati A, Poliani PL, Todeschini A, Cominelli M, Medicina D, Cenzato M, Simoncini EL, Magrini SM, Buglione M, Grisanti S, Padovani A (2013) Glutamine synthetase expression as a valuable marker of epilepsy and longer survival in newly diagnosed glioblastoma multiforme. Neuro Oncol 15: 618–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, Dang CV, Riggins GJ (2010) Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 70: 8981–8987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ, Bellovin DI, Tran PT, Philbrick WM, Garcia‐Ocana A, Casey SC, Li Y, Dang CV, Zare RN, Felsher DW (2015) MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci USA 112: 6539–6544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh‐Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC (2013) Glutamine supports pancreatic cancer growth through a KRAS‐regulated metabolic pathway. Nature 496: 101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalnecker CA, Ulrich SM, Li Y, Ramachandran S, McBrayer MK, DeBerardinis RJ, Cerione RA, Erickson JW (2015) Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc Natl Acad Sci USA 112: 394–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracka D, Jozefczuk S, Rudroff F, Sauer U, Hall MN (2014) Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J Biol Chem 289: 25010–25020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG (2015) Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162: 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Luo T, Ren Y, Florey O, Shirasawa S, Sasazuki T, Robinson DN, Overholtzer M (2014) Competition between human cells by entosis. Cell Res 24: 1299–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, Sugano S, Sato E, Nagao T, Yokote K, Tatsuno I, Prives C (2010) Phosphate‐activated glutaminase (GLS2), a p53‐inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA 107: 7461–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PO, Weinstock A, Wagner A, Lindsay SL, Hock AK, Barnett SC, Ruppin E, Morkve SH, Lund‐Johansen M, Chalmers AJ, Bjerkvig R, Niclou SP, Gottlieb E (2015) Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine‐restricted glioblastoma. Nat Cell Biol 17: 1556–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J (2008) Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res 10: R84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, Hu M, Chan DA, Ethier SP, van‘t Veer LJ, Polyak K, McCormick F, Gray JW (2013) Glutamine sensitivity analysis identifies the xCT antiporter as a common triple‐negative breast tumor therapeutic target. Cancer Cell 24: 450–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchihashi K, Okazaki S, Ohmura M, Ishikawa M, Sampetrean O, Onishi N, Wakimoto H, Yoshikawa M, Seishima R, Iwasaki Y, Morikawa T, Abe S, Takao A, Shimizu M, Masuko T, Nagane M, Furnari FB, Akiyama T, Suematsu M, Baba E et al (2016) The EGF receptor promotes the malignant potential of glioma by regulating amino acid transport system xc(‐). Cancer Res 76: 2954–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483: 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P, Ambesi‐Impiombato A, De Keersmaecker K, Hadler M, Paietta E, Tallman MS, Rowe JM, Forne C, Rue M, Ferrando AA (2013) Prognostic relevance of integrated genetic profiling in adult T‐cell acute lymphoblastic leukemia. Blood 122: 74–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P, Kallinowski F, Okunieff P (1989) Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 49: 6449–6465 [PubMed] [Google Scholar]
- Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C, Carlin SD, La Rocca G, Lyashchenko S, Ploessl K, Rohle D, Omuro AM, Cross JR, Brennan CW, Weber WA, Holland EC, Mellinghoff IK, Kung HF, Lewis JS, Thompson CB (2015) Glutamine‐based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo . Sci Transl Med 7: 274ra217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vie N, Copois V, Bascoul‐Mollevi C, Denis V, Bec N, Robert B, Fraslon C, Conseiller E, Molina F, Larroque C, Martineau P, Del Rio M, Gongora C (2008) Overexpression of phosphoserine aminotransferase PSAT1 stimulates cell growth and increases chemoresistance of colon cancer cells. Mol Cancer 7: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vos KE, Eliasson P, Proikas‐Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, Verhagen LP, Groot Koerkamp MJ, Braat AK, Dansen TB, Holstege FC, Gebhardt R, Burgering BM, Coffer PJ (2012) Modulation of glutamine metabolism by the PI(3)K‐PKB‐FOXO network regulates autophagy. Nat Cell Biol 14: 829–837 [DOI] [PubMed] [Google Scholar]
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, Cerione RA (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18: 207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel‐Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB (2010) The common feature of leukemia‐associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha‐ketoglutarate to 2‐hydroxyglutarate. Cancer Cell 17: 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB (2010) The hexosamine biosynthetic pathway couples growth factor‐induced glutamine uptake to glucose metabolism. Genes Dev 24: 2784–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105: 18782–18787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35: 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Stine ZE, Xia J, Lu Y, O'Connor RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG, Gao P, Sun L, Song L, Yan B, Slusher BS, Zhuo J, Ooi LL, Lee CG, Mancuso A, McCallion AS, Le A et al (2015) Targeted inhibition of tumor‐specific glutaminase diminishes cell‐autonomous tumorigenesis. J Clin Invest 125: 2293–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL (2012) Inhibition of alpha‐KG‐dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26: 1326–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Oosterveer MH, Stein S, Demagny H, Ryu D, Moullan N, Wang X, Can E, Zamboni N, Comment A, Auwerx J, Schoonjans K (2016) LRH‐1‐dependent programming of mitochondrial glutamine processing drives liver cancer. Genes Dev 30: 1255–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ (2009) Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 69: 7986–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova‐Marjon E, Diehl JA, Ron D, Koumenis C (2010) The GCN2‐ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29: 2082–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y (2007) Deficiency in glutamine but not glucose induces MYC‐dependent apoptosis in human cells. J Cell Biol 178: 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B, Djaballah H, Kanai M, Cheng EH, Judkins AR, Pawel B, Baggs J, Cherry S, Rabinowitz JD, Thompson CB (2014) Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell 56: 205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y (2009) Glioma‐derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF‐1alpha. Science 324: 261–265 [DOI] [PMC free article] [PubMed] [Google Scholar]