

Abstract
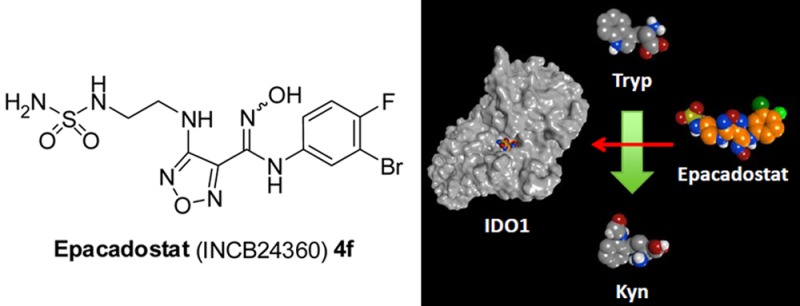
A data-centric medicinal chemistry approach led to the invention of a potent and selective IDO1 inhibitor 4f, INCB24360 (epacadostat). The molecular structure of INCB24360 contains several previously unknown or underutilized functional groups in drug substances, including a hydroxyamidine, furazan, bromide, and sulfamide. These moieties taken together in a single structure afford a compound that falls outside of “drug-like” space. Nevertheless, the in vitro ADME data is consistent with the good cell permeability and oral bioavailability observed in all species (rat, dog, monkey) tested. The extensive intramolecular hydrogen bonding observed in the small molecule crystal structure of 4f is believed to significantly contribute to the observed permeability and PK. Epacadostat in combination with anti-PD1 mAb pembrolizumab is currently being studied in a phase 3 clinical trial in patients with unresectable or metastatic melanoma.
Keywords: Epacadostat, INCB24360, IDO1, data-centric medicinal chemistry, immuno-oncology
The central tenet of cancer immunotherapy is that the immune system can be induced to recognize and eliminate malignant cells within the human body. The harnessing of our innate immune system to treat a wide variety of cancers has been proposed for decades. The recent unprecedented durable responses observed in melanoma patients when treated with anti-CTLA4 mAb (ipilimumab),1 and more recently anti-PD1 mAb (pembrolizumab and nivolumab)2 therapies have provided the first clinical validation for this novel immune mediated mechanism of action for cancer therapy (immuno-oncology or I-O). Cancer therapy is currently undergoing a paradigm shift toward identifying agents (mAb and small molecules) that afford restoration and/or activation of the immune system, often in combination strategies.3−6
Indoleamine-2,3-dioxygenase-1 (IDO1) was first shown by Munn and Mellor to play an immunomodulatory role in fetal protection from the maternal immune system.7 Multiple tumor types, including melanoma, ovarian, and colon, overexpress IDO1 and are believed to subvert this immunomodulatory mechanism and promote tolerance local to the cancer.8,9 Since Munn and Mellor’s seminal research, numerous articles have appeared further validating IDO1 as a therapeutic target for cancer immunotherapy, including studies with si-RNA,10 IDO null mice,11,12 and small molecule inhibitors 1-methyltryptophan 1 (1-MT)13 and hydroxyamidine 2 (5l)14 (Figure 1).15 Notably, IDO1 null mice16 or IDO1 inhibitors in combination with anti-PD1 mAbs have shown synergy in efficacy models.17 These studies provide strong support for the clinical testing of IDO1 inhibitors in combination with anti-PD1 mAbs and anti-PDL1 mAbs to improve the observed low response rates while maintaining their remarkable durability.
Figure 1.

IDO1 inhibitors.
IDO1, IDO2, and tryptophan 2,3-dioxygenase (TDO) are members of the heme containing myoglobin family of enzymes and oxygen transporter proteins that catalyze the initial and rate-limiting enzymatic step in the kynurenine biochemical pathway for the metabolism of the essential amino acid tryptophan to N-formylkynurenine.18 IDO1 is expressed throughout the body where it can be locally up-regulated in response to inflammation and infection by cytokines, such as interferon gamma (IFN-γ). IDO1 overexpression is associated with several diseases, but most convincingly implicated in cancer.19 IDO1 induction in dendritic cells may suppress T-cell responses and promote tolerance through either direct effects on T-cells mediated by tryptophan depletion or cytotoxic effects on T-cells from tryptophan metabolites, such as quinolinic acid and kynurenic acid. The role of IDO2 in immunomodulation is less well understood, as there are functionally inactive polymorphisms occurring in approximately 50% of the examined population with only recent correlations to cancer.20−23 Conversely, TDO is predominantly expressed in the liver where it is responsible for the homeostasis of tryptophan levels throughout the body in response to dietary intake. Though there is recent evidence that TDO is upregulated in some tumor microenvironments,24 the vast majority of research supports the predominant role of IDO1 in regulating the immune response. A selective IDO1 inhibitor was therefore desired to avoid potential untoward toxicities associated with altering global tryptophan levels through TDO inhibition.
We previously reported the discovery of a hydroxyamidine hit with micromolar potency from a high throughput screen of our internal collection (>300,000 compounds) that was rapidly improved to a proof-of-concept (PoC) lead (2 or previously reported as 5l).14 Compound 2 demonstrated nanomolar potency in our IDO1 biochemical and cellular assays and excellent selectivity over TDO, but poor oral bioavailability in rodent pharmacokinetics (PK). The PoC study with 2 demonstrated single agent efficacy in a B16-GMCSF mouse melanoma model, although it required subcutaneous dosing due to the compounds short in vivo half-life. In comparison to 1-MT 1, the first and only other confirmed competitive IDO1 inhibitor (IDO1 Ki ≈ 34 μM) reported at the time of this research,25 hydroxyamidine lead 2 provided an excellent starting point for our medicinal chemistry program. We report herein the further optimization of the PoC lead 2 to afford our clinical candidate INCB24360, epacadostat 4f, a highly potent and selective IDO1 inhibitor with good oral bioavailability across multiple species.
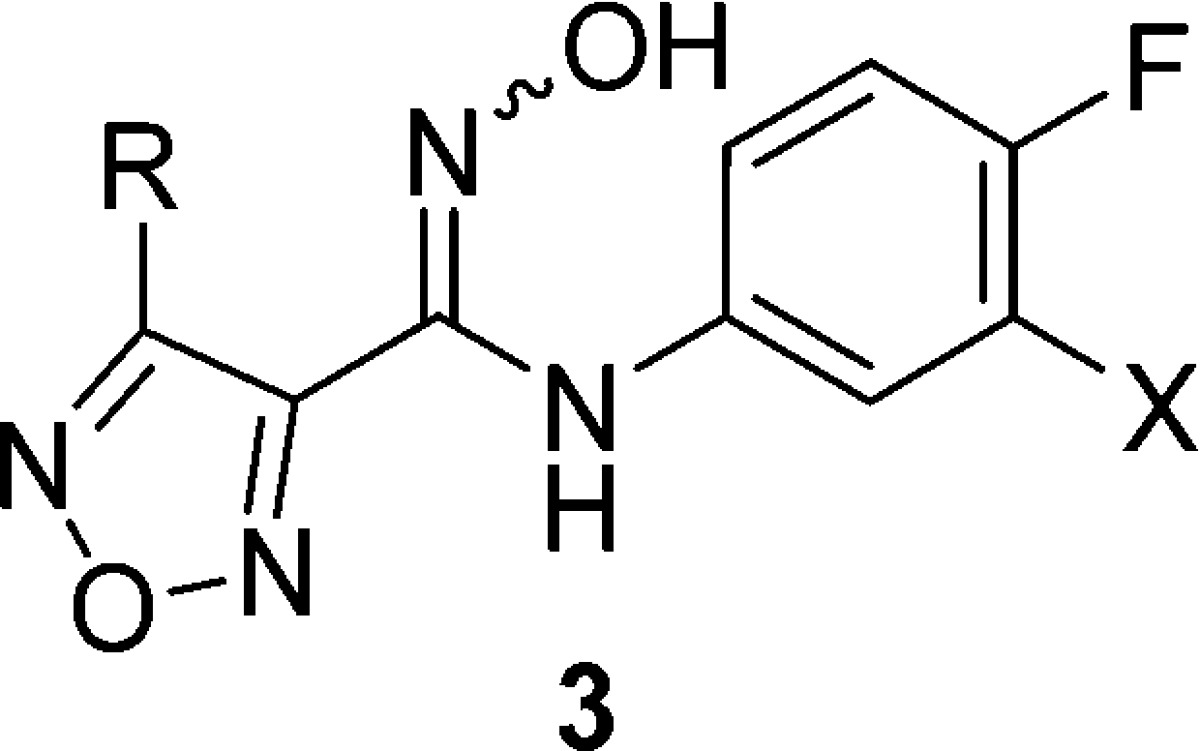
An in-depth analysis of the ADME factors that may be limiting the oral bioavailability of this novel chemotype was undertaken. The metabolism studies revealed that the oxygen of the hydroxyamidine was subject to phase two glucuronidation in vivo and was the major metabolic pathway for the hydroxyamidine class of compounds.26−28 Two in vitro clearance counter screens using liver S9 fractions were established to test our compounds’ susceptibility to glucuronidation (P2) alone and to P450 mediated metabolism plus glucuronidation (P1 + P2). Metabolic profiling of our previously reported meta-substituted phenyl derivatives of 3 did not provide significant improvements in P2 stability.14 The meta-Br-phenyl 3a was identified as a slightly more potent IDO1 inhibitor in a HeLa cellular assay compared to the meta-Cl-phenyl 2 and was therefore incorporated into most subsequent compounds within our structure–activity relationship (SAR) studies (Table 1). Replacement of the furazan by a variety of heterocycles was also investigated and revealed that the furazan was essential for potent IDO1 inhibition.29 It was hypothesized that substitution at the C3 position of the furazan might disrupt binding to the proximal glucuronidase active site through a steric and/or electronic clash and thus diminish the propensity of the hydroxyamidine to be glucuronidated. Synthesis of secondary 3-amino-furazan derivatives by direct alkylation or reductive amination of the primary 3-amino substituent were low yielding, presumably due to the electron deficiency of the furazan. A general and robust alternate route to secondary amino-furazan derivatives was developed via a Boulton–Katritzky rearrangement of the amidooxime furazan (Scheme 1).30 Syntheses and analytical data for all compounds are provided in the Supporting Information.
Table 1. Selected SAR of Furazan (3).
| cmpd | X | R | IDO IC50 (nM) | HeLa IC50 (nM) | P2 Cla | P1 + P2 Cla |
|---|---|---|---|---|---|---|
| 2 | Cl | NH2 | 75 | 19 | >2.2 | >2.2 |
| 3a | Br | NH2 | 50 | 10 | >2.2 | 1.4 |
| 3b | Br | NHMe | 100 | 14 | >2.2 | >2.2 |
| 3c | Br | NMe2 | >5000 | >5000 | >2.2 | >2.2 |
| 3d | Br | NHEt | 180 | 35 | 1.4 | >2.2 |
| 3e | Br | NHBn | 230 | 280 | 2.1 | >2.2 |
Rat IntCl (L/h/kg). Rat hepatic blood flow = 3.3 L/h/kg.
Scheme 1. Synthesis of Substituted 3-Amino Furazans.
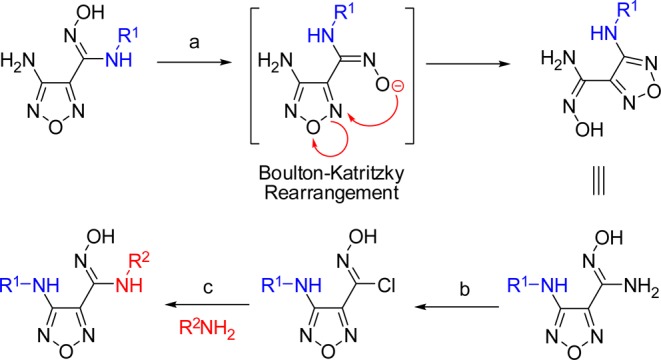
Reagents: (a) KOH, 100 °C, 15 h, 19–99%; (b) NaNO2, NaCl, HCl, H2O, 0 °C, 1.5 h, 7–99%; (c) R2NH2, Et3N, EtOH, 0 → 20 °C, 62–99%.
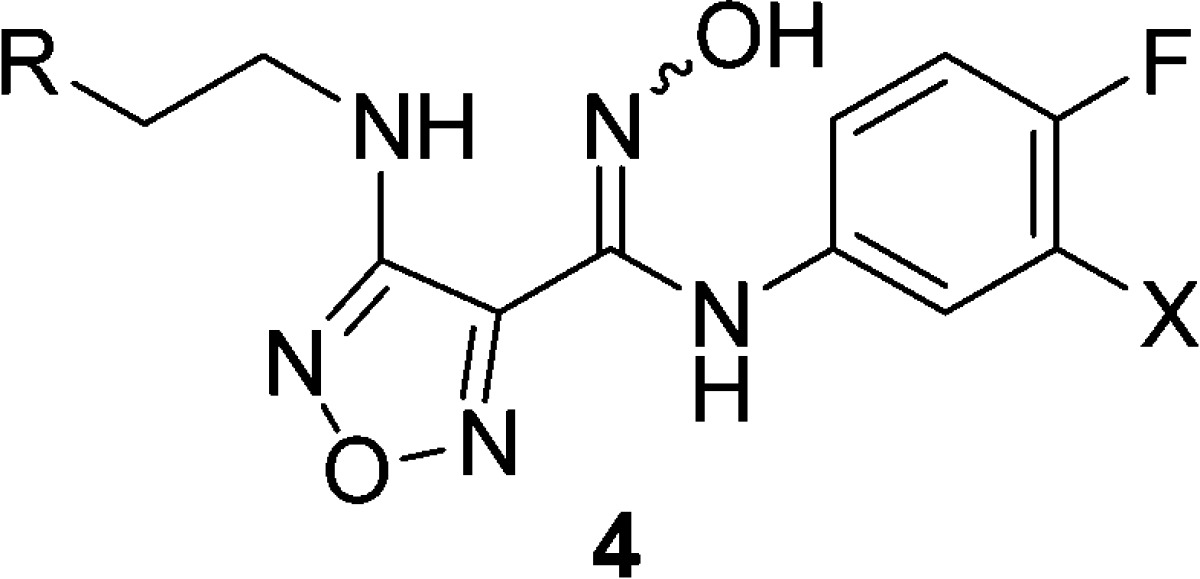
Initial SAR at the 3-amino position of the furazan demonstrated that a variety of secondary amino substituents, such as 3b and 3d, were tolerated in the biochemical and cellular assays, but did not improve the in vitro measured P2 clearance (Table 1). The tertiary amino derivatives, such as 3c, were inactive. Extension of the secondary amino side-chain to a variety of larger, more hydrophobic substituents, such as benzyl derivative 3e, maintained similar biochemical potency, and provided the initial evidence that C3 substituents on the furazan are projected into a solvent exposed region (see Figures S8 and S9) when bound to IDO1. Unfortunately, no improvements in in vitro clearance were observed and cellular activity was severely diminished, presumably due to the very high protein binding (3e: fu < 0.5%) of these lipophilic compounds. In an attempt to reduce the protein binding and restore the potent cellular activity, a series of polar side-chains at C3 of the amino-furazan were designed and synthesized. The addition of polar capping groups to an amino-ethyl C3 substituent provided a highly potent series of IDO1 inhibitors 4a–g (Table 2).
Table 2. Selected SAR of Amino-Furazan (4).
|
in vitro ADME |
calculated
propertiesb and ligand efficiency metrics |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | X | R | IDO IC50 (nM) | HeLa IC50 (nM) | P2 Cla | P1 + P2 Cla | Caco2 Pmc | FF (%)d | PSAe | cLogP | HBD | HBAf | MW | LEg | LLEg |
| 4a | Br | OMe | 200 | 100 | >2.2 | >2.2 | 44 | 0.9 | 100 | 2.7 | 3 | 8 | 374 | 0.45 | 4.3 |
| 4b | Br | OH | 170 | 22 | >2.2 | >2.2 | 31 | 2.3 | 111 | 1.8 | 4 | 8 | 360 | 0.51 | 5.8 |
| 4c | Br | NH2 | 290 | 98 | 1.1 | 0.9 | 1.8 | 6.1 | 117 | 2.0 | 5 | 8 | 359 | 0.47 | 5.0 |
| 4d | Br | NHAc | 210 | 17 | 0.9 | 2.0 | 19 | 4.4 | 120 | 1.9 | 4 | 9 | 401 | 0.45 | 5.9 |
| 4e | Br | NHSO2Me | 100 | 16 | 0.7 | 1.5 | 24 | 2.4 | 137 | 2.0 | 4 | 10 | 437 | 0.44 | 5.8 |
| 4f | Br | NHSO2NH2 | 73 | 7.4 | 0.4 | 0.7 | 4.0 | 3.1 | 163 | 1.5 | 6 | 11 | 438 | 0.46 | 6.6 |
| 4g | Cl | NHSO2NH2 | 120 | 11 | 0.5 | 0.8 | 4.3 | 3.8 | 163 | 1.4 | 6 | 11 | 394 | 0.45 | 6.6 |
Rat IntCl (L/h/kg). Rat hepatic blood flow = 3.3 L/h/kg.
ChemDraw Ultra 10.
Caco2 values Pm × (10)−6 cm/s.
Protein binding free fraction (FF).
PSA in Å2.
Total number of nitrogen (N) and oxygens (O).
Ligand efficiency (LE) and lipophilic ligand efficiency (LLE) calculated using HeLa pIC50.
The similar biochemical potencies of these polar inhibitors 4d–g compared to nonpolar analog 3e suggest that the improvements in protein binding free fractions were responsible for the dramatic improvements (up to 40-fold) observed in cellular potencies. In addition, the in vitro metabolic clearances (P1 and P2) were significantly reduced, which strongly correlated with the increasing polarity (lower cLogP, higher PSA) of compounds 4c–g.
The sulfonamide 4e and sulfamide 4f (INCB24360, epacadostat) were both highly potent IDO1 inhibitors and demonstrated moderate to low glucuronidation in vitro (0.4 and 0.2 L/h/kg, respectively). Pharmacokinetics in rats were evaluated for both compounds and demonstrated that the sulfamide 4f achieved significantly higher exposure and a longer half-life than sulfonamide 4e, in good agreement with the in vitro (P1 + P2) clearance data.
A final round of SAR at the meta-position of the phenyl ring identified the bromo 4f and chloro 4g derivatives as comparable in IDO1 enzyme and cell potency, as well as PK in both rodents and cynomologous monkeys (Table 3). A head-to-head efficacy study with oral dosing (30 mg/kg) of 4f and 4g in a CT26 tumor growth model in immunocompetent mice was designed to differentiate the two leads. The bromo 4f analog proved to be superior in efficacy (tumor growth control (TGC) = 56%) compared to the chloro 4g analog (TGC = 13%) despite the similar HeLa cellular potencies and near identical mouse PK for the two closely related analogs (Figure 2). An immune-mediated mechanism-of-action for 4f was supported by a parallel study in immune-compromised mice (nu/nu) in which no difference in tumor growth inhibition was observed between 4f treated and control animals.
Table 3. Comparisons of Bromo (4f) vs Chloro (4g).

| in vitro Potency | 4f | 4g |
|---|---|---|
| IDO1 IC50 (nM) | 73 | 120 |
| HeLa IC50 (nM) | 7.4 | 11 |
| Pharmacokinetics | ||
| rat PK AUC (μM·h) | 1.3 | 3.3 |
| t1/2 (h) | 2.2 | 2.7 |
| Cyno PK AUC (μM·h) | 9.3 | 15 |
| t1/2 (h) | 2.7 | 4.0 |
| Efficacy | ||
| TGC @ day 24 –CT26 model (%) | 56 | 13 |
| AUC (μM·h) | 15.0 | 15.1 |
Figure 2.
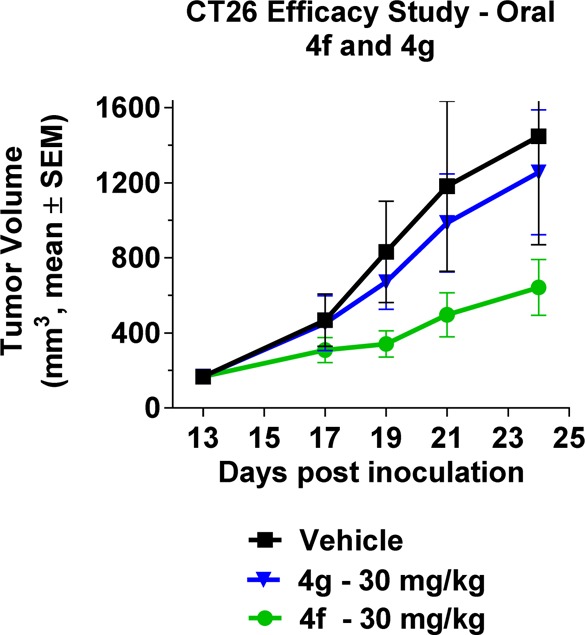
INCB24360 (4f) suppresses CT26 tumor growth in immunocompetent mice (4f) vs chloro (4g).
Based on these findings, the bromo analog 4f was fully profiled in vitro and in vivo (Table 4). In vitro characterization demonstrated 4f is a highly potent IDO1 inhibitor in cells (HeLa IC50 = 7.4 nM) and in an IFN-γ induced whole blood (WB) assay (IC50 = 125 nM).31 Absolute selectivity (>1,000-fold) was observed over the related dioxygenases, TDO and IDO2. In addition, 4f was clean in in vitro toxicology studies, including the hERG patch clamp, PXR, Cyp inhibition, and CEREP panel of over 50 receptors and enzymes (see Supporting Information).
Table 4. PK Profile of INCB24360 (4f) Across Species.
| Cl | Vss | iv | AUC | Po F | Po | |
|---|---|---|---|---|---|---|
| species | (L/h/kg) | (L/kg) | t1/2 (h) | (μM·h) | (%) | t1/2 (h) |
| rat | 1.1 | 2.0 | 1.4 | 1.3 | 11 | 2.2 |
| dog | 0.5 | 0.7 | 3.1 | 29 | 59 | 4.9 |
| cyno | 0.8 | 1.8 | 3.3 | 9.3 | 33 | 2.7 |
PK profiling of 4f in rats, dogs, and cynomologous monkeys demonstrated good exposures and oral bioavailabilities (Po F = 11%, 59%, 33%, respectively) in all species (Table 4). The PK is consistent with the moderate permeability and clearance data measured in in vitro (P1 + P2) assays (Table 4).
Pharmacokinetic/pharmacodynamic (PK/PD) studies in mice established a strong correlation between the coverage of the WB IC50 at trough and reduction in kynurenine concentrations in the plasma (Figure 3).32 Once daily oral dosing of 4f at 50 mg/kg reduced kynurenine levels in wild-type mice to basal levels present in IDO null mice (∼400 nM kynurenine). TDO metabolism is believed to be responsible for the observed baseline levels of kynurenine in these studies. Similar PK/PD correlations were observed in dogs and cynomologous monkeys. Allometric scaling using in vivo and in vitro ADME data to cover the WB IC50 at trough resulted in a predicted oral dosing of 4f in humans of 50 mg twice-a-day.
Figure 3.
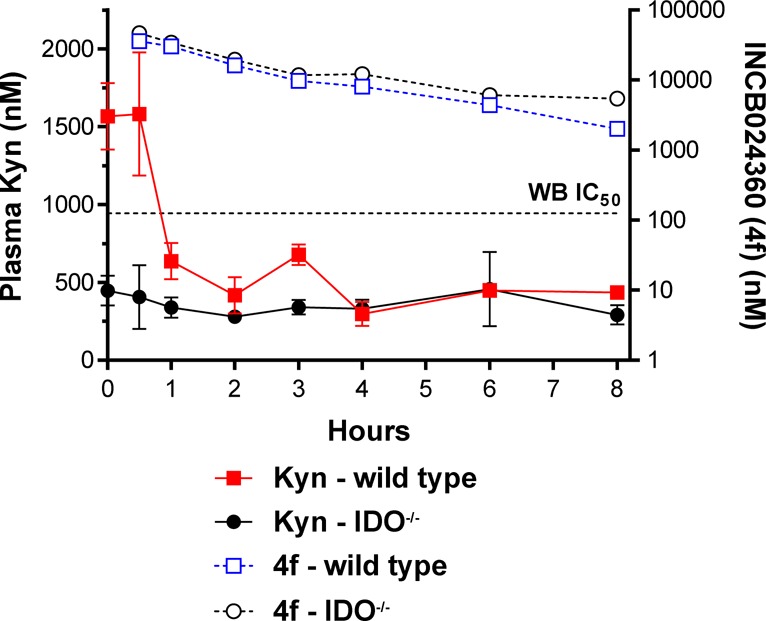
INCB24360 (4f) PK/PD in mice.
Evaluation of the physiochemical and calculated properties of 4f confirmed the molecular structure to be outside of classical “drug-like” space, including Lipinski’s rule-of-five33 (>5 HBD; 4f = 6 and >10 HBD + HBA; 4f = 11) and Veber’s permeability rules34 (PSA < 140 Å2; 4f PSA = 163 Å2) (Table 2). Contrary to the predicted low permeability of these compounds based on their high polar surface area (PSA) and large number of hydrogen bond donors (HBD), these molecules maintained moderate permeability in the Caco-2 assay. The good permeability and oral bioavailability of 4f are attributed to the two intramolecular hydrogen bonds observed in the crystal structure of 4f (Figure 4). One hydrogen bond exists between the aniline NH and the oxygen of the hydroxyamidine and the second between the imino nitrogen of the hydroxyamidine and the furazan C3 NH. This network of hydrogen bonds not only reduces the effective polarity of the compound and thus increases its permeability but also stabilizes the typically high energy cis-conformation of the amidine. This is critical since our SAR and modeling is consistent with 4f existing in the cis-conformation for binding to IDO1 (Figure 5). Further supporting evidence that 4f is significantly less polar (∼8-fold) than predicted from its calculated properties is provided by the comparison of the calculated LogP (cLogP) versus the measured LogD (@ pH 7.4) values of 2.3 and 1.5, respectively. We subscribe to the principle that ligand efficiency (LE), a measure of the free energy of binding per atom, and lipophilic ligand efficiency (LLE = pIC50 – cLogP) provide a more meaningful characterization of the quality of a clinical candidate.35 The calculated LE and LLE based on the HeLa cell assay, 0.46 and 6.6, respectively, for 4f are nearly ideal.
Figure 4.
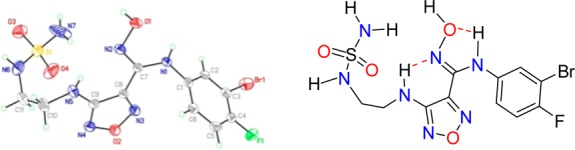
Crystal structure of INCB24360 (4f). Intramolecular hydrogen bonds stabilize the cis-conformation.
Figure 5.
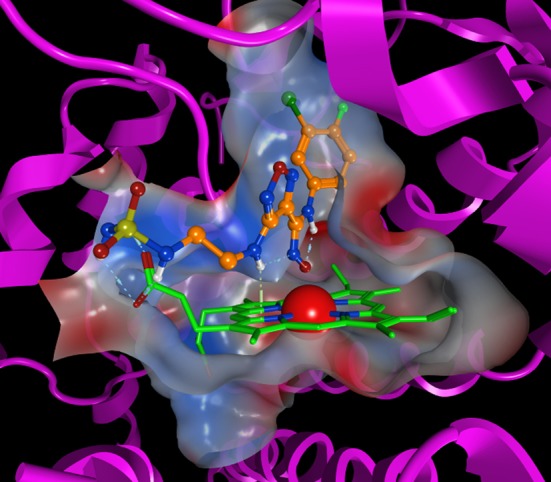
Model of INCB24360 (4f) bound to IDO1. The hydroxyl of the requisite hydroxyamidine forms a coordinate covalent bond with the ferrous iron of the heme, while the m-Br-phenyl group binds deep into the active site (pocket A) of IDO1. The aminoethyl-sulfamide substituent projects out of the active site toward solvent (pocket B). IDO1 crystal structure used in the modeling was PDB entry 4PK5.
Preclinical 28-day IND toxicology studies in mice and dogs demonstrated 4f is well-tolerated, in accord with the high selectivity observed in the in vitro profiling data. No adverse findings were identified in any parameter (clinical observations and pathology, histopathology, body weight, food consumption), and there were no signs of autoimmunity. A maximum tolerated dose (MTD) was not established at doses up to 2000 mg/kg/d in mice and 500 mg/kg/d in dogs. Plasma levels of 4f achieved multiples of the WB IC90 at trough during these safety studies. The FDA granted a clinical safe starting dose of 960 mg based on these IND safety study results.
In summary, a potent and selective IDO1 inhibitor 4f (INCB24360, epacadostat) was invented through a series of SAR studies primarily focused on improving the PK by reducing the rate of glucuronidation while maintaining cell potency. A data-centric medicinal chemistry approach was taken, and therefore calculated properties and/or perceived “drug-likeness” did not dictate the design of new molecules. Ultimately, INCB24360 4f was identified as a novel chemotype containing several previously unidentified and/or underutilized substituents (furazan, hydroxyamidine, sulfamide, bromide) in known drug substances. INCB24360 4f proved to be efficacious in rodent models of melanoma and well-tolerated in preclinical IND toxicology studies. Epacadostat (INCB24360) 4f in combination with anti-PD1 mAb, pembrolizumab, is currently being tested in a pivotal trial for the treatment of patients with unresectable or metastatic melanoma.36,37
Acknowledgments
We thank Laurine Galya, Mei Li, James Doughty, and Karl Blom for their analytical assistance. We thank Mary Becker-Pasha and Mark Rupar for production of the enzyme and development of the IDO and TDO enzyme assays, respectively. We thank Ravi Jalluri for creating the graphics of the epacadostat/IDO1 model.
Glossary
ABBREVIATIONS
- IDO1
indoleamine-2,3-dioxygenase-1
- TDO
tryptophan 2,3-dioxygenase
- I-O
immuno-oncology
- WB
whole blood
- PK/PD
pharmacokinetics/pharmacodynamics
- TGC
tumor growth control
- LE
ligand efficiency
- LLE
lipophilic ligand efficiency
- HBD
hydrogen bond donor
- HBA
hydrogen bond acceptor
- PSA
polar surface area
- MW
molecular weight
- IND
investigational new drug
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00391.
Experimental protocols and characterization data(PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hodi F. S.; O’Day S. J.; McDermott D. F.; Weber R. W.; Sosman J. A.; Haanen J. B.; Gonzalez R.; Robert C.; Schadendorf D.; Hassel J. C.; Akerley W.; van den Eertwegh A. J. M.; Lutzky J.; Lorigan P.; Vaubel J. M.; Linette G. P.; Hogg D.; Ottensmeier C. H.; Lebbe C.; Peschel C.; Quirt I.; Clark J. I.; Wolchok J. D.; Weber J. S.; Tian J.; Yellin M. J.; Nichol G. M.; Hoos A.; Urba W. J. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C.; Schachter J.; Long G. V.; Arance A.; Grob J. J.; Mortier L.; Daud A.; Carlino M. S.; McNeil C.; Lotem M.; Larkin J.; Lorigan P.; Neyns B.; Blank C. U.; Hamid O.; Mateus C.; Shapira-Frommer R.; Kosh M.; Zhou H.; Ibrahim N.; Ebbinghaus S.; Ribas A. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- Larkin J.; Chiarion-Sileni V.; Gonzalez R.; Grob J. J.; Cowey C. L.; Lao C. D.; Schadendorf D.; Dummer R.; Smylie M.; Rutkowski P.; Ferrucci P. F.; Hill A.; Wagstaff J.; Carlino M. S.; Haanen J. B.; Maio M.; Marquez-Rodas I.; McArthur G. A.; Ascierto P. A.; Long G. V.; Callahan M. K.; Postow M. A.; Grossmann K.; Sznol M.; Dreno B.; Bastholt L.; Yang A.; Rollin L. M.; Horak C.; Hodi F. S.; Wolchok J. D. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmann H. Cancer Immunotherapy: Selected Targets and Small-Molecule Modulators. ChemMedChem 2016, 11, 1576. 10.1002/cmdc.201600319. [DOI] [PubMed] [Google Scholar]; b Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Melero I.; Berman D. M.; Aznar M. A.; Korman A. J.; Gracia J. L. P.; Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. 10.1038/nrc3973. [DOI] [PubMed] [Google Scholar]
- Antonia S. J.; Larkin J.; Ascierto P. A. Immuno-oncology Combinations: A Review of Clinical Experience and Future Prospects. Clin. Cancer Res. 2014, 20, 6258–6268. 10.1158/1078-0432.CCR-14-1457. [DOI] [PubMed] [Google Scholar]
- Munn D. H.; Zhou M.; Attwood J. T.; Bondarev I.; Conway S. J.; Marshall B.; Brown C.; Mellor A. L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. IDO and tolerance to tumors. Trends Mol. Med. 2004, 10 (1), 15–18. 10.1016/j.molmed.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Uyttenhove C.; Pilotte L.; Theate I.; Stroobant V.; Colau D.; Parmentier N.; Boon T.; Van den Eynde B. J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Koropatnick J.; Li M.; Zhang X.; Ling F.; Ren X.; Hao X.; Sun H.; Vladau C.; Franek J. A.; Feng B.; Urquhart B. L.; Zhong R.; Freeman D. J.; Garcia B.; Min W.-P. Reinstalling Antitumor Immunity by Inhibiting Tumor-Derived Immunosuppressive Molecule IDO through RNA Interference. J. Immunol. 2006, 177 (8), 5639–5646. 10.4049/jimmunol.177.8.5639. [DOI] [PubMed] [Google Scholar]
- Muller A. J.; DuHadaway J. B.; Chang M. Y.; Ramalingam A.; Sutanto-Ward E.; Boulden J.; Soler A. P.; Mandik-Nayak L.; Gilmour S. K.; Prendergast G. C. Non-hematopoietic expression of IDO is integrally required for inflammatory tumor promotion. Cancer Immunol. Immunother. 2010, 59, 1655–1663. 10.1007/s00262-010-0891-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baban B.; Chandler P.; McCool D.; Marshall B.; Munn D. H.; Mellor A. L. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J. Reprod. Immunol. 2004, 61 (2), 67–77. 10.1016/j.jri.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Hou D.-Y.; Muller A. J.; Sharma M. D.; DuHadaway J.; Banerjee T.; Johnson M.; Mellor A. L.; Prendergast G. C.; Munn D. H. Inhibition of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Stereoisomers of 1-Methyl-Tryptophan Correlates with Antitumor Responses. Cancer Res. 2007, 67 (2), 792–801. 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- Yue E. W.; Douty B.; Wayland B.; Bower M.; Liu X.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Liu C.; Wei M.; Li Y.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Metcalf B.; Scherle P. A.; Combs A. P. Discovery of Potent Competitive Inhibitors of Indoleamine 2,3-Dioxygenase with in Vivo Pharmacodynamic Activity and Efficacy in a Mouse Melanoma Model. J. Med. Chem. 2009, 52, 7364–7367. 10.1021/jm900518f. [DOI] [PubMed] [Google Scholar]
- Liu X.; Newton R. C.; Friedman S. M.; Scherle P. A. Indoleamine 2,3-dioxygenase, an emerging target for anti-cancer therapy. Curr. Cancer Drug Targets 2009, 9, 938–952. 10.2174/156800909790192374. [DOI] [PubMed] [Google Scholar]
- Holmgaard R. B.; Zamarin D.; Munn D. H.; Wolchok J. D.; Allison J. P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S.; Horton B.; Koblish H. K.; Scherle P. A.; Newton R.; Gajewski T. F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest. 2007, 117 (5), 1147–1154. 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated L-tryptophan metabolism. Biochem. Biophys. Res. Commun. 2005, 338 (1), 12–19. 10.1016/j.bbrc.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Metz R.; DuHadaway J. B.; Kamasani U.; Laury-Kleintop L.; Muller A. J.; Prendergast G. C. Novel Tryptophan Catabolic Enzyme IDO2 Is the Preferred Biochemical Target of the Antitumor Indoleamine 2,3-Dioxygenase Inhibitory Compound D-1-Methyl-Tryptophan. Cancer Res. 2007, 67 (15), 7082–7087. 10.1158/0008-5472.CAN-07-1872. [DOI] [PubMed] [Google Scholar]
- Fatokun A. A.; Hunt N. H.; Ball H. J. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: characteristics and potential roles in health and disease. Amino Acids 2013, 45, 1319–1329. 10.1007/s00726-013-1602-1. [DOI] [PubMed] [Google Scholar]
- Prendergast G. C.; Metz R.; Muller A. J.; Mandik-Nayak L.; Merlo L. M. F. IDO2 in Immunomodulation and Autoimmune Disease. Front. Immunol. 2014, 5, 585. 10.3389/fimmu.2014.00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball H. J.; Yuasa H. J.; Austin C. J. D.; Weiser S.; Hunt N. H. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int. J. Biochem. Cell Biol. 2009, 41 (3), 467–471. 10.1016/j.biocel.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Pilotte L.; Larrieu P.; Stroobant V.; Colau D.; Dolusic E.; Frederick R.; De Plaen E.; Uyttenhove C.; Wouters J.; Masereel B.; Vand den Eynde B. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2497–2502. S2497/1-S2497/7 10.1073/pnas.1113873109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review of recent IDO1 inhibitors:Qian S.; Zhang M.; Chen Q.; He Y.; Wang W.; Wang Z. IDO as a drug target for cancer immunotherapy: recent developments in IDO inhibitors discovery. RSC Adv. 2016, 6, 7575–7581. 10.1039/C5RA25046C. [DOI] [Google Scholar]
- Obach R. S.; Baxter J. G.; Liston T. E.; Silber B. M.; Jones B. C.; Macintyre F.; Rance D. J.; Wastall P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. Journal of Pharmacology and Experimental Therapeutics 1997, 283, 46–58. [PubMed] [Google Scholar]
- Li P.; Wang G.-J.; Li J.; Zhang Q.; Liu X.; Khlentzos A.; Roberts M. S. The prediction of the hepatic clearance of tanshinone IIA in rat liver subcellular fractions: accuracy improvement. Curr. Drug Metab. 2008, 9, 39–45. 10.2174/138920008783331103. [DOI] [PubMed] [Google Scholar]
- Froehlich A. K.; Girreser U.; Clement B. Metabolism of benzamidoxime (N-hydroxyamidine) in human hepatocytes and role of UDP-glucuronosyltransferases. Xenobiotica 2005, 35, 17–25. 10.1080/00498250400021895. [DOI] [PubMed] [Google Scholar]
- Complete SAR for the furazan and other substituents will be presented in a subsequent full paper.
- Boulton A. J.; Katritzky A. R. N-Oxides and related compounds. XXII. The rearrangement of 4-nitrobenzofuroxan to 7-nitrobenzofuroxan. Rev. Chim., Acad. Rep. Populaire Roumaine 1962, 7, 691–7. [Google Scholar]
- Liu X.; Shin N.; Koblish H. K.; Yang G.; Wang Q.; Wang K.; Leffet L.; Hansbury M. J.; Thomas B.; Rupar M.; Waeltz P.; Bowman K. J.; Polam P.; Sparks R. B.; Yue E. W.; Li Y.; Wynn R.; Fridman J. S.; Burn T. C.; Combs A. P.; Newton R. C.; Scherle P. A. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010, 115, 3520–3530. 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- Koblish H. K.; Hansbury M. J.; Bowman K. J.; Yang G.; Neilan C. L.; Haley P. J.; Burn T. C.; Waeltz P.; Sparks R. B.; Yue E. W.; Combs A. P.; Scherle P. A.; Vaddi K.; Fridman J. S. Hydroxyamidine Inhibitors of Indoleamine-2,3-dioxygenase Potently Suppress Systemic Tryptophan Catabolism and the Growth of IDO-Expressing Tumors. Mol. Cancer Ther. 2010, 9, 489–498. 10.1158/1535-7163.MCT-09-0628. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserue G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Gangadhar T. C.; Hamid O.; Smith D. C.; Bauer T. M.; Waser J. S.; Luke J. J.; Balmanoukian A. S.; Kaufman D. R.; Zhao Y.; Maleski J.; Leopold L.; Gajewski T. F. Preliminary results from a phase I/II study of epacadostat (INCB024360) in combination with pembrolizumab in patients with selected advanced cancers. J. for ImmunoTherapy of Cancer 2015, 2 (suppl 2), 07. 10.1186/2051-1426-3-S2-O7. [DOI] [Google Scholar]
- NCT02752074: A Phase 3 Study of Pembrolizumab + Epacadostat or Placebo in Subjects With Unresectable or Metastatic Melanoma. www.ClincalTrials.gov.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.