

Abstract
Brown adipose tissue (BAT) is regulated by the sympathetic nervous system via β3-adrenergic receptors (β3-AR). Here we tested the hypothesis that pharmacological stimulation of β3-ARs leads to increased sleep in mice and if this change is BAT dependent. In wild-type (WT) animals, administration of CL-316,243, a selective β3-AR agonist, induced significant increases in non-rapid-eye movement sleep (NREMS) lasting for 4–10 h. Simultaneously, electroencephalographic slow-wave activity (SWA) was significantly decreased and body temperature was increased with a delay of 5–6 h. In uncoupling protein 1 (UCP-1) knockout mice, the middle and highest doses of the β3-AR agonist increased sleep and suppressed SWA, however, these effects were significantly attenuated and shorter-lasting as compared to WT animals. To determine if somnogenic signals arising from BAT in response to β3-AR stimulation are mediated by the sensory afferents of BAT, we tested the effects of CL-316,243 in mice with the chemical deafferentation of the intra-scapular BAT pads. Sleep responses to CL-316,243 were attenuated by ~50% in intra-BAT capsaicin-treated mice. Present findings indicate that the activation of BAT via β3-AR leads to increased sleep in mice and that this effect is dependent on the presence of UCP-1 protein and sleep responses require the intact sensory innervation of BAT.
Introduction
Adipose tissue plays a central role in the interplay between nutrition, energy balance, and health. Two distinct types of adipose tissue with fundamentally different functions exist – brown and white adipose tissue1. The tightly regulated balance between the activities of the two fat tissues is critical in maintaining metabolic homeostasis. White adipose tissue (WAT) stores excess energy in the form of fat, whereas brown adipose tissue (BAT) dissipates energy as heat. BAT was initially believed to play a role in hibernating animals, but its presence in non-hibernating animals and humans suggests a wider array of functions. The largest BAT depot in rodents is located in the interscapular region (iBAT). Other depots are present in the axillary, cervical and paravertebral regions as well as around thoracic and abdominal viscera.
BAT controls energy balance via regulated (adaptive) heat production. The thermogenic property of BAT is conferred by the tissue-specific presence of uncoupling protein 1 (UCP-1). BAT thermogenesis is under the control of the sympathetic nervous system2. Norepinephrine, released from the nerve terminals, facilitates lipolysis in BAT by acting on β3-adrenergic receptors (β3-ARs)3.
β3-ARs are expressed predominantly by white and brown adipocytes, and smooth muscle cells of various organs4–6. Selective, pharmacological activation of β3-ARs has been shown to have profound effects on adipose tissue morphology and metabolism. Administration of CL-316,243, a potent and highly selective β3-AR agonist7, leads to marked increases in thermogenesis by BAT, lipolysis in WAT, and an acute decrease in food consumption8–10. Long-term treatment of obese rodents with β3-AR agonists reduces fat stores and improves obesity-induced insulin resistance as well as facilitates the appearance of brown adipocytes in WAT tissue (“browning”)11, 12. β3-AR knockout (KO) mice are completely resistant to the effects of CL-316,243 indicating that these effects are mediated exclusively by β3-AR8.
Previous studies in our laboratory showed a role for BAT in sleep regulation13. Sleep loss stimulates BAT thermogenesis as shown by increased BAT temperature and 6-fold increases in UCP-1 mRNA. Recovery sleep after sleep deprivation is significantly attenuated in UCP-1 KO mice indicating that BAT heat production is required for rebound sleep after sleep loss.
BAT is supplied by rich afferent innervation14–16. Anterograde transneuronal viral tracing showed that sensory information reaches the brain via ascending spinal pathways in the anterolateral system. The afferents are sensitive to changes in local BAT temperature17, thus they are well positioned to convey temperature-related signals associated with BAT activation to brain sleep centers. Hypothalamic nuclei, periaqueductal gray, parabrachial nuclei, raphe nuclei, and the nucleus of the solitary tract are main elements of the BAT central sensory circuits14, 16. Sensory denervation of BAT by capsaicin suppressed rebound sleep increases after sleep deprivation by ~50% in mice13. These results indicate that sleep-promoting signals arising from BAT in response to sleep loss are mediated by small-diameter sensory afferents.
In the present study we tested the effects of acute, pharmacological stimulation of β3-ARs on sleep, body temperature and metabolic parameters in mice. We report that CL-316,243 induced immediate and long-lasting increases in non-rapid-eye movement sleep (NREMS) as well as increases in body temperature in wild-type (WT) mice. To establish the role of BAT in β3-AR stimulation-induced sleep responses, we also studied the sleep and body temperature-modulating effects of the β3-AR agonist in UCP-1 KO mice and in mice after the sensory deafferentation of the iBAT. We report that CL-316,243-induced sleep requires the intact thermogenic activity of BAT, and sleep-promoting signals that arise from activated BAT require the intact sensory innervation of the tissue to enhance sleep.
Results
The effect of CL-316,243 on sleep-wake activity in WT and UCP-1 KO mice
The behavioral effects of CL-316,243 were not quantified, but on-line video observation of the animals did not reveal any gross behavioral effects. The animals moved around in their cages after the treatment and most, especially in the WT group, assumed typical sleeping position within 10–20 min.
CL-316,243 induced immediate increases in NREMS in WT mice (Fig. 1, Table 1). NREMS was significantly elevated for 4 hours after the 0.04 and 0.2 mg/kg doses of CL-316,243 by 60 and 65%, respectively. The increases lasted for 6 hours after the 1 mg/kg dose. After 4–6 hours, NREMS amounts returned to baseline.
Figure 1.
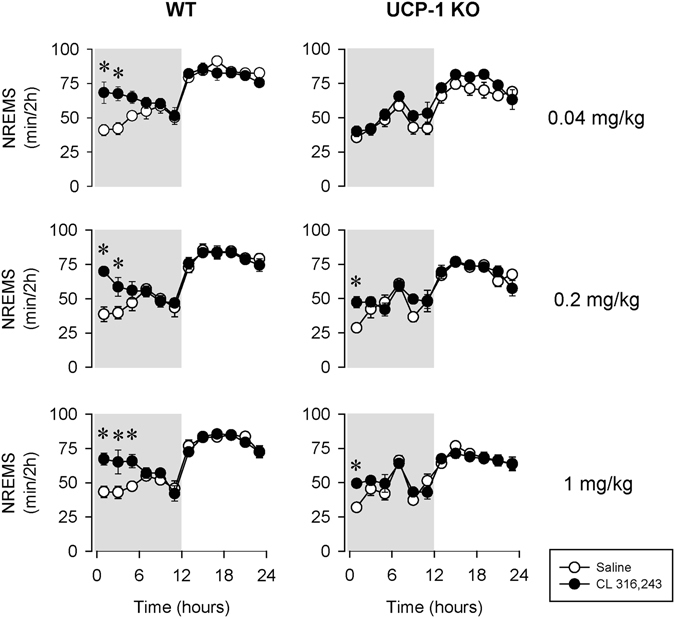
The effects of the selective β3-adrenergic receptor (β3-AR) agonist CL-316,243 (black symbols) on non-rapid-eye movement sleep (NREMS) in wild-type (WT; left panels) and uncoupling protein-1 knockout (UCP-1 KO; right panels) mice. On the baseline day saline (open symbols) was administered subcutaneously 10–15 min before the onset of the dark phase. On the experimental day, one dose of CL-316,243 (0.04, 0.2 or 1 mg/kg; top, middle and bottom panels, respectively) was injected prior dark onset. Asterisk (*) denotes significant difference from baseline, p < 0.05. Error bars: standard error. Grey areas represent the dark phase.
Table 1.
Effects of CL-316,243 on NREMS in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL-316,243- NREMS | df | F | p |
|---|---|---|---|
| Genotype | 1,11 | 20.9 | <0.05 |
| Treatment | 1,11 | 7.2 | <0.05 |
| Treatment × genotype | 1,11 | 0.5 | n.s |
| Time | 11,121 | 41.3 | <0.05 |
| Time × genotype | 11,121 | 1.3 | n.s |
| Treatment × time | 11,121 | 2.0 | <0.05 |
| Treatment × time × genotype | 11,121 | 2.9 | <0.05 |
| 0.2 mg/kg CL-316,243 - NREMS | df | F | p |
| Genotype | 1,11 | 19.8 | <0.05 |
| Treatment | 1,11 | 10.5 | <0.05 |
| Treatment × genotype | 1,11 | 0.8 | n.s |
| Time | 11,121 | 39.9 | <0.05 |
| Time × genotype | 11,121 | 1.8 | n.s |
| Treatment × time | 11,121 | 4.8 | <0.05 |
| Treatment × time × genotype | 11,121 | 1.3 | n.s |
| 1 mg/kg CL-316,243 - NREMS | df | F | p |
| Genotype | 1,10 | 33.8 | <0.05 |
| Treatment | 1,10 | 13.4 | <0.05 |
| Treatment × genotype | 1,10 | 3.3 | n.s |
| Time | 10,110 | 38.5 | <0.05 |
| Time × genotype | 10,110 | 3.5 | <0.05 |
| Treatment × time | 10,110 | 6.8 | <0.05 |
| Treatment × time × genotype | 10,110 | 1.0 | n.s |
In UCP-1 KO mice, the lowest, 0.04 mg/kg, dose of CL-316,243 failed to induce any changes in NREMS. The 0.2 and 1 mg/kg doses of CL-316,243 were followed by significant increases in NREMS, however, these changes were significantly attenuated and shorter-lasting compared to control mice. In the first 4 hours after 0.2 mg/kg CL-316,243 injection, NREMS increase in UCP-1 KO mice was significantly, by ~50% less than in controls (23.8 ± 8.5 min in UCP-1 KO mice vs. 50.2 ± 8.0 min in WT, p < 0.05). Similarly, 1 mg/kg CL-316,243 was followed by a significantly attenuated NREMS increase in UCP-1 KO mice (31.1 ± 10.5 min in UCP-1 KO mice vs. 64.2 ± 8.7 min in WT, p < 0.05). While the effects of CL-316,243 on NREMS were significantly attenuated, they still showed dose-dependency (one-way ANOVA on NREMS amounts in h 1–6 across all doses: F(3, 20) = 3.59, p < 0.05). CL-316,243 had no effect on the amounts of rapid-eye movement sleep (REMS) in WT or UCP-1 KO mice (Fig. 2, Table 2).
Figure 2.
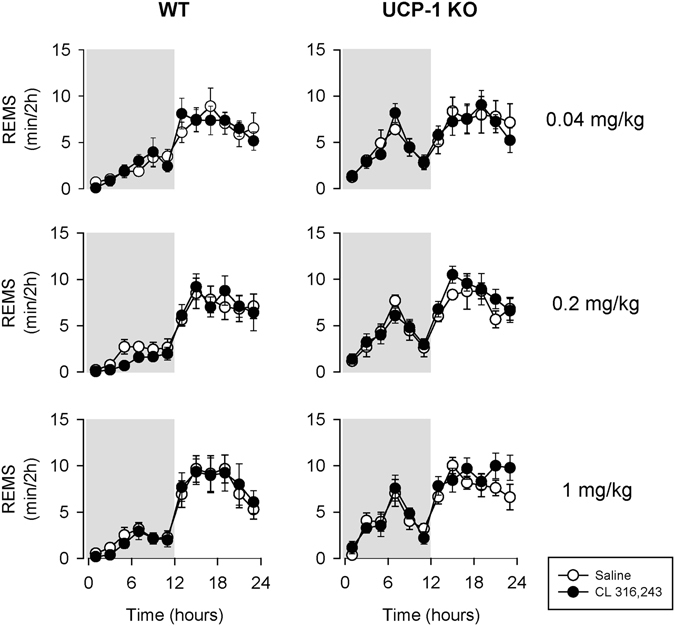
The effects of the selective β3-AR agonist CL-316,243 on rapid-eye movement sleep (REMS) in WT (left panels) and UCP-1 KO (right panels) mice. See Fig. 1 for details.
Table 2.
Effects of CL-316,243 on REMS in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL-316,243 - REMS | df | F | p |
|---|---|---|---|
| Genotype | 1,11 | 1.0 | n.s. |
| Treatment | 1,11 | 0.04 | n.s. |
| Treatment × genotype | 1,11 | 0.03 | n.s. |
| Time | 11,121 | 19.1 | <0.05 |
| Time × genotype | 11,121 | 2.2 | <0.05 |
| Treatment × time | 11,121 | 1.5 | n.s. |
| Treatment × time × genotype | 11,121 | 0.5 | n.s. |
| 0.2 mg/kg CL-316,243 - REMS | df | F | p |
| Genotype | 1,11 | 3.0 | n.s. |
| Treatment | 1,11 | 0.2 | n.s. |
| Treatment × genotype | 1,11 | 6.7 | <0.05 |
| Time | 11,121 | 32.9 | <0.05 |
| Time × genotype | 11,121 | 2.0 | <0.05 |
| Treatment × time | 11,121 | 1.3 | n.s. |
| Treatment × time × genotype | 11,121 | 0.4 | n.s. |
| 1 mg/kg CL-316,243 - REMS | df | F | p |
| Genotype | 1,10 | 1.5 | n.s. |
| Treatment | 1,10 | 1.7 | n.s. |
| Treatment × genotype | 1,10 | 3.0 | n.s. |
| Time | 10,110 | 36.4 | <0.05 |
| Time × genotype | 10,110 | 2.4 | <0.05 |
| Treatment × time | 10,110 | 1.3 | n.s. |
| Treatment × time × genotype | 10,110 | 0.3 | n.s. |
In WT animals, the increases in NREMS time were accompanied by increased number of NREMS episodes after all three doses of CL-316,243, and decreased average duration of the episodes after the middle and high doses (Fig. 3). In UCP-1 KO mice, there was a tendency towards increased NREMS episode numbers and durations, but the changes were not significant. CL-316,243 treatment did not affect REMS episode numbers or durations (data not shown).
Figure 3.
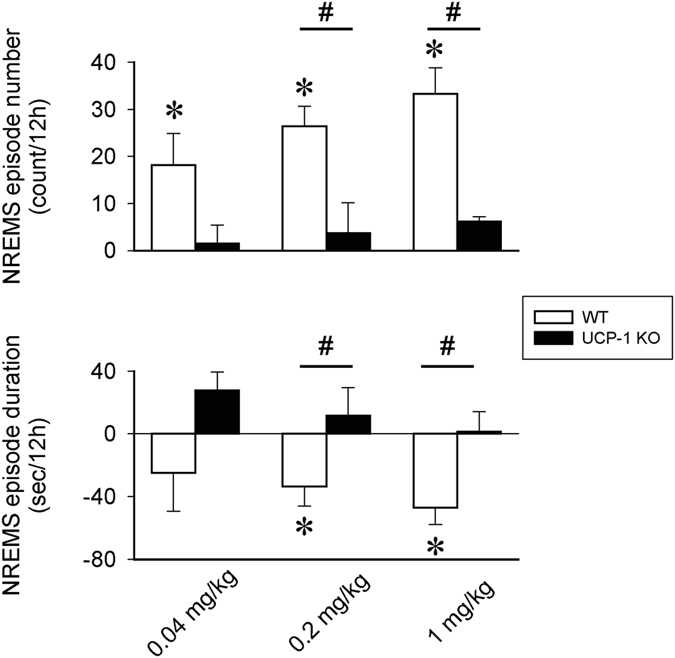
The effects the selective β3-AR agonist CL-316,243 on the number and average duration of NREMS episodes in the first 12 h after treatment in WT (white bars) and UCP-1 KO mice (black bars). Data represent differences from baseline (vehicle). Error bars: standard error. Asterisk (*) denotes significant difference from baseline, # denotes significant difference between genotypes, p < 0.05.
Electroencephalographic (EEG) slow-wave activity (SWA) was significantly reduced for 2–6 h after all three doses of CL-316,243 in WT mice (Fig. 4, Table 3). The lowest 0.04 mg/kg CL-316,243 failed to induce any changes on EEG SWA in UCP-1 KO mice. The middle and high doses of CL-316,243 were followed by slight but significant decreases in EEG SWA. Analysis of the higher EEG frequencies (4.5–25 Hz) revealed that the suppression of EEG power was not restricted to the slow-wave band (Fig. 5, Table 4).
Figure 4.
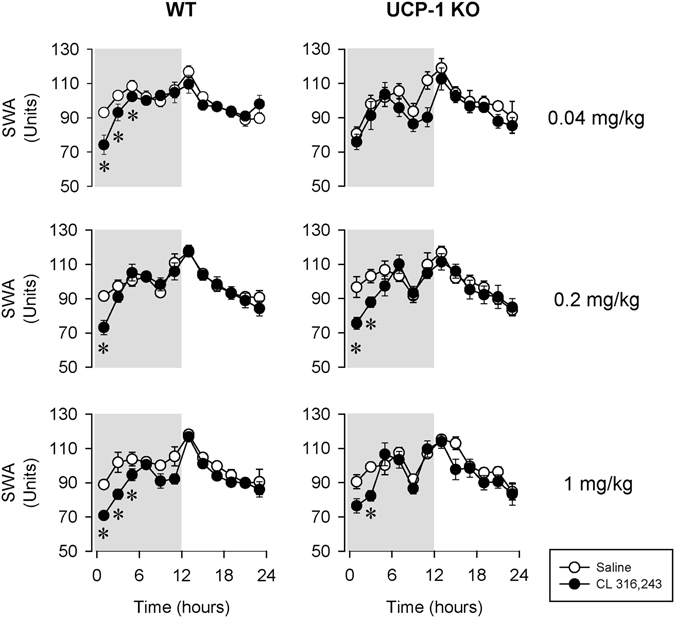
The effects of the selective β3-AR agonist, CL-316,243 on electroencephalographic slow-wave activity (EEG SWA) in WT (left panels) and UCP-1 KO (right panels) mice. See Fig. 1 for details.
Table 3.
Effects of CL-316,243 on EEG SWA in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL-316,243 – EEG SWA | df | F | p |
|---|---|---|---|
| Genotype | 1,11 | 1.4 | n.s |
| Treatment | 1,11 | 11.5 | <0.05 |
| Treatment × genotype | 1,11 | 1.4 | n.s |
| Time | 11,121 | 11.0 | <0.05 |
| Time × genotype | 11,121 | 0.8 | n.s |
| Treatment × time | 11,121 | 1.5 | n.s. |
| Treatment × time × genotype | 11,121 | 2.0 | <0.05 |
| 0.2 mg/kg CL-316,243 - EEG SWA | df | F | p |
| Genotype | 1,11 | 0.1 | n.s |
| Treatment | 1,11 | 4.4 | n.s |
| Treatment × genotype | 1,11 | 0.1 | n.s |
| Time | 11,121 | 19.0 | <0.05 |
| Time × genotype | 11,121 | 0.5 | n.s |
| Treatment × time | 11,121 | 4.0 | <0.05 |
| Treatment × time × genotype | 11,121 | 1.5 | n.s. |
| 1 mg/kg CL-316,243 - EEG SWA | df | F | p |
| Genotype | 1,10 | 0.4 | n.s. |
| Treatment | 1,10 | 12.0 | <0.05 |
| Treatment × genotype | 1,10 | 0.3 | n.s. |
| Time | 10,110 | 22.3 | <0.05 |
| Time × genotype | 10,110 | 1.2 | n.s. |
| Treatment × time | 10,110 | 3.5 | <0.05 |
| Treatment × time × genotype | 10,110 | 1.8 | n.s. |
Figure 5.
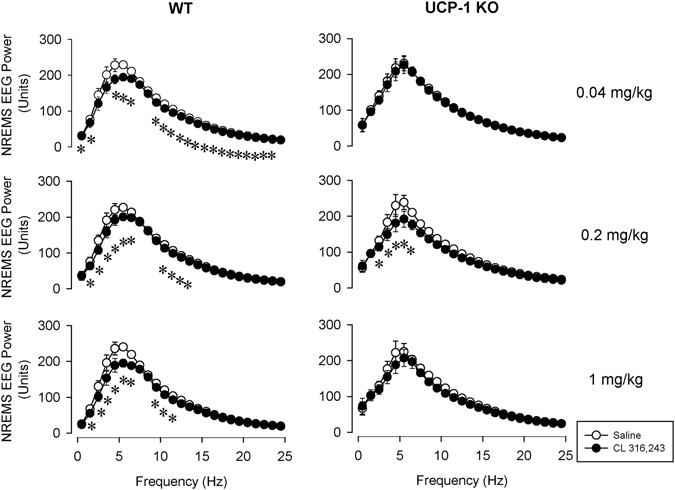
The effects of the selective β3-AR agonist, CL-316,243 on EEG power in the 0.5–25 Hz range in WT (left panels) and UCP-1 KO (right panels) mice. Power values represent the average in the first 6 h after the treatments. See Fig. 1 for details.
Table 4.
Effects of CL-316,243 on the NREMS EEG power spectrum averaged for the first 6 h in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL – EEG power 0.5–25 Hz | df | F | p |
|---|---|---|---|
| Genotype | 1,11 | 2.8 | n.s |
| Treatment | 1,11 | 12.1 | <0.05 |
| Treatment × genotype | 1,11 | 0.3 | n.s |
| Frequency | 24,264 | 87.5 | <0.05 |
| Frequency × genotype | 24,264 | 0.4 | n.s |
| Treatment × Frequency | 24,264 | 9.5 | <0.05 |
| Treatment × Frequency × genotype | 24,264 | 1.5 | <0.05 |
| 0.2 mg/kg CL - EEG power 0.5–25 Hz | df | F | p |
| Genotype | 1,11 | 0.1 | n.s |
| Treatment | 1,11 | 20.3 | <0.05 |
| Treatment × genotype | 1,11 | 1.3 | n.s |
| Frequency | 24,264 | 98.7 | <0.05 |
| Frequency × genotype | 24,264 | 0.6 | n.s |
| Treatment × Frequency | 24,264 | 12.6 | <0.05 |
| Treatment × Frequency × genotype | 24,264 | 2.2 | <0.05 |
| 1 mg/kg CL - EEG power 0.5–25 Hz | df | F | p |
| Genotype | 1,10 | 2.8 | n.s |
| Treatment | 1,10 | 11.3 | <0.05 |
| Treatment × genotype | 1,10 | 0.4 | n.s |
| Frequency | 24,240 | 91.7 | <0.05 |
| Frequency × genotype | 24,240 | 0.9 | n.s |
| Treatment × Frequency | 24,240 | 10.3 | <0.05 |
| Treatment × Frequency × genotype | 24,240 | 2.2 | <0.05 |
Activation of β3-ARs stimulates thermogenesis (Fig. 6, Table 5). Core body temperature rose with a delay of 5 h after the 0.04 and 0.2 mg/kg doses of CL-316,243 in WT mice. The highest dose of CL-316,243 increased body temperature immediately after injection. Compared to controls, increases in body temperature were more delayed and significantly attenuated in UCP-1 KO mice after the 0.04 and 1 mg/kg doses (Student’s t-test on 12-h temperature averages; difference from baseline averaged for the first 12 h in WT: 0.36 ± 0.07 °C, 0.28 ± 0.05 °C, 0.37 ± 0.11 °C and in UCP-1 KO: 0.18 ± 0.04 °C, 0.31 ± 0.12 °C, 0.15 ± 0.04 °C after the 0.04, 0.2 and 1 mg/kg doses, respectively).
Figure 6.
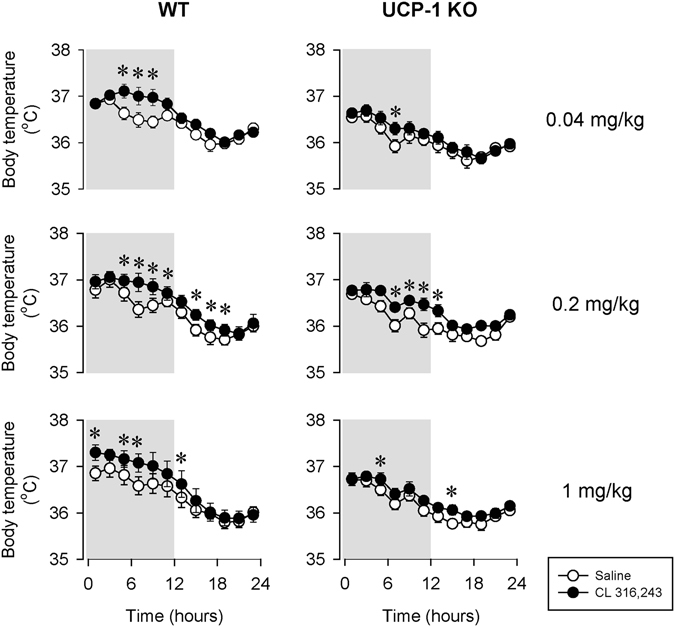
The effects of the selective β3-AR agonist CL-316,243 on body temperature in WT (left panels) and UCP-1 KO (right panels) mice. See Fig. 1 for details.
Table 5.
Effects of CL-316,243 on body temperature in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL-316,243 – Body Temperature | df | F | p |
|---|---|---|---|
| Genotype | 1,11 | 8.1 | <0.05 |
| Treatment | 1,11 | 24.1 | <0.05 |
| Treatment × genotype | 1,11 | 1.5 | n.s. |
| Time | 11,121 | 55.9 | <0.05 |
| Time × genotype | 11,121 | 1.6 | n.s. |
| Treatment × time | 11,121 | 3.5 | <0.05 |
| Treatment × time × genotype | 11,121 | 1.1 | n.s. |
| 0.2 mg/kg CL-316,243 – Body Temperature | df | F | p |
| Genotype | 1,11 | 0.8 | n.s. |
| Treatment | 1,11 | 17.9 | <0.05 |
| Treatment × genotype | 1,11 | 0.0 | n.s. |
| Time | 11,121 | 79.8 | <0.05 |
| Time × genotype | 11,121 | 6.5 | <0.05 |
| Treatment × time | 11,121 | 3.5 | <0.05 |
| Treatment × time × genotype | 11,121 | 1.9 | <0.05 |
| 1 mg/kg CL-316,243 – Body Temperature | df | F | p |
| Genotype | 1,10 | 2.3 | n.s. |
| Treatment | 1,10 | 16.1 | <0.05 |
| Treatment × genotype | 1,10 | 0.3 | n.s. |
| Time | 10,110 | 61.9 | <0.05 |
| Time × genotype | 10,110 | 5.9 | <0.05 |
| Treatment × time | 10,110 | 2.4 | <0.05 |
| Treatment × time × genotype | 10,110 | 2.6 | <0.05 |
All three doses of CL-316,243 induced increases in heat production, oxygen uptake (VO2) and suppression of respiratory exchange ratio (RER) and feeding in both genotypes (Fig. 7, Tables 6, 7, 8 and 9). Changes in heat production and VO2 in UCP-1 KO mice, however, were significantly attenuated compared to WTs. There was a tendency towards attenuated RER response to CL-316,243 in the KO animals, but the differences between the two genotypes did not reach the level of significance. Decreases in 24-h food intake was also significantly different between WT and UCP-1 KO mice with KO mice showing smaller decreases after the 0.04 and 1 mg/kg doses of CL-316,243.
Figure 7.
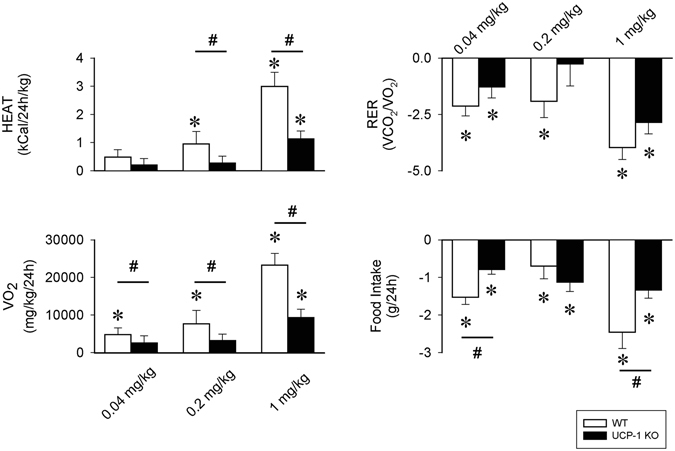
The effects of the selective β3-AR agonist CL-316,243 on heat production (HEAT), O2 consumption (VO2), respiratory exchange ratio (RER) and 24-h food intake in WT (white bars) and UCP-1 KO mice (black bars). Data represent differences from baseline (vehicle). Error bars: standard error. Asterisk (*) denotes significant difference from baseline, # denotes significant difference between genotypes, p < 0.05.
Table 6.
Effects of CL-316,243 on heat production in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL-316,243 - HEAT | df | F | p |
|---|---|---|---|
| Genotype | 1,14 | 12.7 | <0.05 |
| Treatment | 1,14 | 3.6 | n.s. |
| Treatment × genotype | 1,14 | 0.5 | n.s. |
| 0.2 mg/kg CL-316,243 - HEAT | df | F | p |
| Genotype | 1,14 | 6.9 | <0.05 |
| Treatment | 1,14 | 23.9 | <0.05 |
| Treatment × genotype | 1,14 | 13.5 | <0.05 |
| 1 mg/kg CL-316,243 - HEAT | df | F | p |
| Genotype | 1,14 | 21.6 | <0.05 |
| Treatment | 1,14 | 51.3 | <0.05 |
| Treatment × genotype | 1,14 | 10.5 | <0.05 |
Table 7.
Effects of CL-316,243 on respiratory exchange ratio in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL-316,243 - RER | df | F | p |
|---|---|---|---|
| Genotype | 1,14 | 0.3 | n.s. |
| Treatment | 1,14 | 33.2 | <0.05 |
| Treatment × genotype | 1,14 | 1.8 | n.s. |
| 0.2 mg/kg CL-316,243 - RER | df | F | p |
| Genotype | 1,14 | 0.9 | n.s. |
| Treatment | 1,14 | 3.4 | n.s. |
| Treatment × genotype | 1,14 | 2.5 | n.s. |
| 1 mg/kg CL-316,243 - RER | df | F | p |
| Genotype | 1,14 | 0.1 | n.s |
| Treatment | 1,14 | 88.6 | <0.05 |
| Treatment × genotype | 1,14 | 2.3 | n.s |
Table 8.
Effects of CL-316,243 3 on VO2 in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL-316,243 – VO2 | df | F | p |
|---|---|---|---|
| Genotype | 1,14 | 7.3 | <0.05 |
| Treatment | 1,14 | 8.1 | <0.05 |
| Treatment × genotype | 1,14 | 0.7 | n.s. |
| 0.2 mg/kg CL-316,243 - VO 2 | df | F | p |
| Genotype | 1,14 | 4.2 | n.s. |
| Treatment | 1,14 | 30.7 | <0.05 |
| Treatment × genotype | 1,14 | 12.3 | <0.05 |
| 1 mg/kg CL-316,243 - VO 2 | df | F | p |
| Genotype | 1,14 | 9.2 | <0.05 |
| Treatment | 1,14 | 70.7 | <0.05 |
| Treatment × genotype | 1,14 | 13.0 | <0.05 |
Table 9.
Effects of CL-316,243 on food intake in WT and UCP-1 KO mice – statistical results.
| 0.04 mg/kg CL-316,243 - Food Intake | df | F | p |
|---|---|---|---|
| Genotype | 1,14 | 8.8 | <0.05 |
| Treatment | 1,14 | 98.9 | <0.05 |
| Treatment × genotype | 1,14 | 10.6 | <0.05 |
| 0.2 mg/kg CL-316,243 - Food Intake | df | F | p |
| Genotype | 1,14 | 2.0 | n.s. |
| Treatment | 1,14 | 56.4 | <0.05 |
| Treatment × genotype | 1,14 | 1.4 | n.s |
| 1 mg/kg CL-316,243 – Food Intake | df | F | p |
| Genotype | 1,14 | 1.8 | n.s |
| Treatment | 1,14 | 60.3 | <0.05 |
| Treatment × genotype | 1,14 | 5.3 | <0.05 |
The effects of CL-316,243 on sleep-wake activity and body temperature in control and mice with iBAT sensory afferents denervation
BAT-CAPS mice had significantly lower iBAT CGRP levels compared to controls (BAT-CAPS: 4.05 ± 0.21 pg/mg vs. BAT-VEH 12.30 ± 1.23 pg/mg; p < 0.001, Student’s t-test), indicating a ~70% decrease and confirming the efficiency of intra-BAT capsaicin treatment to reduce the sensory innervation of the organ.
To test the involvement of BAT sensory innervation in β3-AR-induced sleep, 0.2 mg/kg CL-316,243 was injected to intra-BAT CAPS-treated mice (Fig. 8). Sleep responses to CL-316,243 were attenuated by ~50% in intra-BAT CAPS-treated mice as compared to vehicle-injected controls. NREMS increase in the first 12-h after CL injection in the control mice was 92.4 ± 11 min while it was 57.3 ± 11.6 min in BAT-CAPS animals (p < 0.05, Student’s t-test). The effects on body temperature tended to be shorter lasting in BAT-CAPS animals, but did not differ significantly from those in BAT-VEH mice.
Figure 8.
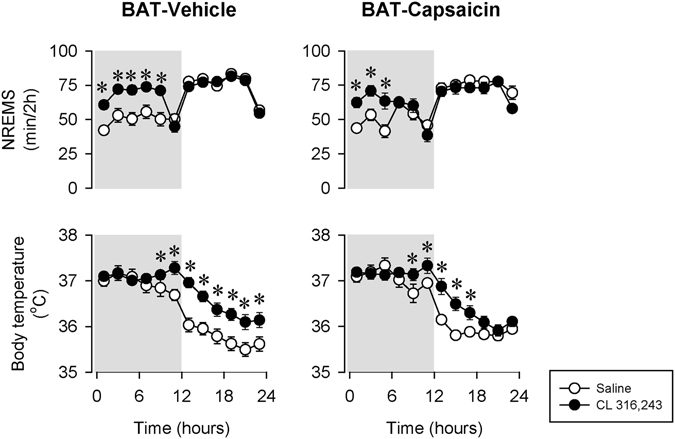
The effects of the selective β3-AR agonist CL-316,243 on NREMS and body temperature in mice with the chemical deafferentation of the intra-scapular BAT pads (BAT-Capsaicin; black symbols) and control mice (BAT-Vehicle; open symbols). See Fig. 1 for details.
Discussion
Activation of β3-ARs brings about an array of metabolic changes characterized by suppression of feeding, elevated energy expenditure, increased lipolysis, remodeling of WAT, thermogenic activation of BAT and the proliferation of brite, UCP-1 positive, adipocytes in the WAT. We report here that these profound metabolic effects are also accompanied by robust increases in NREMS in mice. Systemic injection of CL-316,243, a selective β3-AR agonist, produced rapid and marked increases in NREMS, increased body temperature and energy expenditure, decreased RER and suppressed feeding. The effects were abolished or attenuated in UCP-1 KO mice indicating that intact thermogenesis by BAT is required for the somnogenic and metabolic effects of β3-AR activation. Further, sensory denervation of the iBAT suppressed CL-316,243-induced sleep indicating the involvement of BAT afferents in the somnogenic response.
The effects of β3-AR activation on sleep have been addressed in two previous studies, but significant limitations of those experiments prevent the valid interpretation of the findings. In one study, light onset injection of 3 and 10 mg/kg CL-316,243 suppressed REMS in rats, while 0.1 mg/kg was without significant effect18. Unfortunately, the effects on NREMS were not reported in that paper. Also, the CL-316,243 doses were 10–100-fold higher that in the present experiments thus non-specific effects of the drug were more likely to manifest. In another study, high doses of SR58611A, a brain-penetrant β3-AR agonist, suppressed REMS when injected during daytime in rats19. In that study, sleep was recorded only for 6 h after treatments, and vigilance states were determined by simple spectral analysis of the EEG. While spectral components of the EEG show correlation with vigilance states, spectral analysis by itself is insufficient to reliably determine sleep-wake states. In our experiments, we did not observe any effect on REMS. The differences between our findings and previous reports may be due to the fact that we injected CL-316,243 at dark onset, when spontaneous REMS is minimal in mice thus further suppression is difficult to achieve. Alternatively, there could be a species specific difference in the REMS responses between rats and mice.
The best characterized and most abundant expression sites for β3-AR are the brown and white adipose tissues4, 5. Both adipose tissues are sources of peripheral, sleep-inducing signals. For example, WAT is a major source of leptin, interleukin 6 (IL-6), and tumor necrosis factor-α (TNFα)20–22, hormones with potent sleep-promoting activities23–25. Also, we have previously reported that sleep loss activates the thermogenic machinery in BAT and BAT thermogenesis is required for the full expression of homeostatic sleep rebound after sleep loss13. Since BAT abundantly expresses β3-ARs and β3-AR stimulation activates BAT thermogenesis8–10, we tested if BAT thermogenesis is a contributing factor in CL-316,243-induced sleep. The thermogenic capacity of BAT is conferred by the presence of UCP-1, a mitochondrial proton carrier26. Systemic injection of CL-316,243 stimulates BAT thermogenesis and increases energy expenditure. These effects are abolished in UCP-1 KO mice indicating that BAT is a main effector of the metabolic actions of CL-316,24327–29. The NREMS-promoting effects of 0.04 mg/kg CL-316,243 were completely abolished in UCP-1 KO mice, while the somnogenic effects of the two higher doses were attenuated by ~50–70%. These findings indicate that intact BAT thermogenesis a key factor in CL-316,243-induced sleep.
There are several possible mechanisms that may mediate the effects of activated BAT on sleep. One, BAT is a main effector organ for non-shivering thermogenesis and BAT activation may lead to increased body temperature3. Elevated body temperature is often associated with increased NREMS, e.g., during the exposure to warm ambient temperature30 or after the injection of pyrogenic cytokines, such as IL-1β and TNFα25, 31, 32. It is unlikely, however, that CL-316,243-induced sleep is due to changes in core body temperature as indicated by the clear dissociation between the time courses of the sleep and temperature effects. The sleep-promoting effects of CL-316,243 were immediate; in the first hour after the β3-AR agonist treatment, NREMS was already elevated by ~50%. Body temperature, however, became elevated only in hours 5–6 after the treatment. Two, BAT is innervated by thermo-sensitive afferents17. Signaling via these afferents is key to sleep loss-induced rebound sleep13. In the present study, lesion of the capsaicin-sensitive BAT afferents attenuated CL-316,243-induced sleep about the same degree as the lack of thermogenic protein, UCP-1. This strongly suggests that neuronal signaling from the activated BAT is a major component of BAT-induced sleep.
While the somnogenic effects of the lowest dose of CL-316,243 were completely abolished in UCP-1 KO mice, the effects of higher doses were not completely suppressed in UCP-1 KO or in BAT deafferented animals. This suggests that in high doses, CL-316,243 also activates somnogenic mechanisms that are not related to BAT thermogenesis. It is unlikely that the BAT-independent component of CL-316,243-induced sleep is due to the activation of central β3-ARs. β3-AR expression in the brain is low33 and central receptors are not reached by systemically administered CL-316,243 due to its inability to cross the blood-brain barrier34. Similarly to the sleep-inducing effects, the stimulatory effects of 1 mg/kg CL-316,243 on VO2 were not completely abolished in UCP-1 KO animals. This strongly suggests that relatively high doses of the β3-AR agonist activate targets, e.g., the white adipose tissue, where heat production is independent of UCP-1 expression.
CL-316,243 induces the production and release of several humoral factors from peripheral tissues that are known to induce NREMS. CL-316,243 induces 2–4-fold increases in circulating fatty acid levels with a latency of only a few minutes8, 9, 35. Various forms of fatty acids are known to induce36–38, while inhibition of fatty acid synthase to suppresses sleep39. Also, fatty acids are activators of UCP-1 in brown adipocytes and BAT thermogenesis40, 41 thus fatty acids may play a role in both the BAT-dependent as well as the BAT- independent components of the sleep-inducing effects of CL-316,243. A role for fatty acids in mediating the effects of CL-316,243 on metabolism in mice was previously proposed42. Further, β3-AR activation stimulates IL-642–45 and insulin secretion8, 9, 46 resulting in a 6 and 50–140 fold increase in their respective plasma levels. Both insulin and IL-6 induce sleep when administered in bolus24, 47–49. Finally, bolus injection of CL-316,243 causes a 2–15 fold increase in TNFα mRNA expression in the WAT45, 50, 51. TNF is one of the best characterized NREMS-inducing hormone/adipokine52. It is possible that the combination of these, and potentially other, sleep-promoting molecules is responsible for the UCP-1-independent component of CL-316,243-induced NREMS.
The two-process model is a widely accepted model of sleep regulation. It describes sleep as the function of a circadian and a homeostatic factor53. The circadian component is driven by the biological clock in the suprachiasmatic nucleus. The homeostatic component is determined by the duration of prior wakefulness; prolonged wakefulness, such as occurring during sleep deprivation, is followed by longer and more intense/deeper NREMS periods. SWA of the EEG is commonly regarded as the indicator of NREMS intensity and the reflection of the activity of the homeostatic process53. EEG SWA is elevated after sleep loss indicating an increased homeostatic sleep pressure due to unfulfilled sleep need. In our studies, increased NREMS after β3-AR agonist treatment was accompanied by significant decreases in SWA. This decrease, however, was not specific to the SWA (0.5–4.5 Hz) range, but it was also present in the higher frequencies. Therefore, it is likely that suppressed SWA in response to CL-316,243 is not a reflection of decreased NREMS intensity, but it is part of a broader suppressive effect on sleep EEG.
Our findings confirm previous reports that systemic bolus injection of CL-316,243 leads to elevated body temperature, energy expenditure and suppressed feeding8–10. These effects were significantly attenuated in UCP-1 KO mice, but, similarly to the sleep-promoting actions, were not completely abolished. This suggests that thermogenic activation of BAT is a major component in these effects, but other, UCP-1-independent mechanisms also play a role. These mechanisms could be shared with the UCP-1-independent sleep-inducing machinery, as several of the bioactive molecules, such as IL-6 and TNFα, which are released in response to β3-AR, not only produce sleep but also elicit fever and anorexia54. Our findings give support to the idea that WAT directly or indirectly participates in the overall thermogenic response to acute β3-AR stimulation and that a component of thermogenesis is UCP-1 independent27.
In summary, we demonstrated that activation of β3-ARs by systemic injection of a selective ligand elicits robust sleep increases, as well as coordinated metabolic responses. Major component of these actions requires the intact thermogenic capacity of the brown adipose tissue. These findings confirm the importance of systemic signals arising from metabolic organs in sleep regulation and gives support to the notion that regulation of sleep and metabolism is highly integrated and are consistent with our multi-process model that in addition to the homeostatic and circadian processes, other, mainly metabolically driven process(es) are integral part of the complex sleep-regulatory mechanism.
Methods
Animals and surgery
Male, 5–6 months old WT C57BL/6 and congenic UCP-1 KO mice were used in the experiments. Breeding pairs of the UCP-1 KO animals were obtained from Dr. Leslie P. Kozak of Pennington Research Institute (Baton Rouge, LA) and were further bred at Washington State University. Each mouse used in the experiments was genotyped (Transnetyx, Cordova, TN). All animal work was conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Washington State University.
The body weight of the WT and UCP-1 KO mice at the time of surgery was 31.3 ± 0.9 g and 31.2 ± 0.4 g, respectively. All surgical procedures were performed with the mice under ketamine-xylazine anesthesia (87 and 13 mg/kg, respectively). For polysomnography recordings, the animals were implanted with cortical EEG electrodes, placed over the frontal and parietal cortices, and nuchal electromyographic (EMG) electrodes. The EEG and EMG electrodes were anchored to the skull with dental cement. Telemetry transmitters were implanted intraperitoneally for body temperature and motor activity recordings. To lesion the small-diameter sensory afferents arising from the iBAT selectively, anesthetized mice were injected with 20 microinjections (1 µl/injection) of 20 µg/µl of capsaicin into the iBAT (BAT-CAPS, n = 7, BW = 37.9 ± 1.1 g) to cover the full extent of both iBAT pads16. Mice in the control group were injected with vehicle (10% ethanol, 10% Tween 80, in isotonic NaCl, BAT-VEH, n = 8, BW = 34.7 ± 1.7 g). These animals were instrumented for sleep recordings 3 weeks after BAT treatments. All mice were allowed to recover from surgery for at least 10 days before any experimental manipulation started. During the recovery and experimental periods, mice were housed individually in a sound-attenuated environmental chamber at controlled light–dark cycles (12–12 h, lights on: 4 am) at 30 ± 1 °C ambient temperature. Food (regular lab chow; Harlan Teklad, Product No. 8460) and water were available ad libitum throughout the experiments.
Data collection and analyses
The animals were tethered to commutators, which were further routed to Grass Model 15 Neurodata amplifier system (Grass Instrument Division of Astro-Med, Inc., West Warwick, RI). The amplified EEG and EMG signals were digitized at 256 Hz and recorded by computer. The high-pass and low-pass filters for EEG signals were 0.5 and 30.0 Hz, respectively. The EMG signals were filtered with low and high cut-off frequencies at 100 and 10,000 Hz, respectively. The outputs from the 12A5 amplifiers were fed into an analog-to-digital converter and collected by computer using SleepWave software (Biosoft Studio, Hersey, PA). Sleep-wake states were scored visually off-line in 10-s segments. The vigilance states were defined as NREMS, REMS and wakefulness (W) according to standard criteria as described previously39. Time spent in W, NREMS and REMS was calculated in 2-, 4-, 6-, 12- and 24-h blocks. EEG power data from each artifact free 10-s segment were subjected to off-line spectral analysis by fast Fourier transformation. EEG power data in the range of 0.5 to 4.0 Hz during NREMS were used to compute EEG SWA. EEG SWA data were normalized for each animal by using the average EEG SWA across 24 h on the baseline day as 100. The results were further averaged in 2-h bins.
Telemetry recordings
For the sleep experiments, core body temperature was also recorded by MiniMitter telemetry system (Philips Respironics, Bend, OR) using VitalView software. Temperature values were collected every 1 min throughout the experiment and were averaged over 2-h time blocks.
Experimental procedures
Experiment 1: The effects of CL-316,243 on sleep-wake activity, body temperature and metabolic parameters in WT and UCP-1 KO mice
Mice (n = 7 for WT and n = 6 for UCP-1 KO mice) received subcutaneous injections of CL-316,243 (Sigma-Aldrich) or saline 10–15 min before the onset of the dark period. The three doses of CL-316,243 were of 0.04, 0.2, and 1 mg/kg. All the mice were injected with all three doses of CL-316,243 with at least 7 days between the CL-316,243 injection days.
In a separate group of mice (n = 8 for both WT and UCP-1 KO mice) oxygen consumption (VO2, ml/kg/h) and carbon dioxide production (VCO2, ml/kg/h) were measured with indirect calorimetry (Oxymax System, Columbus Instruments, Columbus, OH). Twenty-four hour food intake (g/kg/h) was also recorded. All three measurements were taken every 10 min and the data were collapsed into 24-h bins. Respiratory exchange ratio (RER; VCO2/VO2) was calculated. The animals were habituated to the calorimetry cages for three days before the experiments. On the fourth day mice were injected with saline, on the fifth day they received one of the three doses of CL-316,243.
Experiment 2: The effects of CL-316,243 on sleep-wake activity and body temperature in control and mice with sensory afferents denervation
To determine if somnogenic signals arising from BAT in response to β3-AR stimulation are mediated by the sensory afferents of BAT, we tested the effects of CL-316,243 in C57/BL6 mice with the chemical deafferentation of the iBAT. Both BAT-VEH (n = 8) and BAT-CAPS (n = 7) mice were injected with 0.2 mg/kg CL-316,243 10–15 min before the onset of the dark phase. Each mouse was recorded for 24 hours starting after injections. To verify that intra-BAT injection of capsaicin did not cause the degeneration of small-diameter sensory nerves systemically, eye-wiping test was performed using 0.01% capsaicin solution55. All BAT-CAPS mice showed positive eye-wiping test indicating intact nociceptive innervation of the cornea, thus capsaicin injections remained localized in the BAT. At the end of the experiment, iBATs were removed, weighed, and stored at −80 °C.
Verification of iBAT sensory denervation. iBAT samples were homogenized in 2 N acetic acid. Calcitonin gene-related peptide (CGRP) levels in iBAT were measured using a commercially available EIA kit (Bertin Pharma/SPI-Bio, Montigny-le-Bretonneux, France) according to the manufacturer’s instructions. For the CGRP assays, the correlation coefficient was 99.8%.
Statistics
The amounts of NREMS, REMS, wakefulness, as well as EEG SWA, body temperature, VO2, and RER data in Experiment 1 were calculated in 2-h blocks. Three-way mixed ANOVA was performed separately for each dose of CL-316,243 across 24 h (independent measure: genotype, repeated measures: time and treatment). Daily food intake, VCO2, VO2 and RER were analyzed by using two-way mixed ANOVA (independent measure: genotype, repeated measure: treatment). When ANOVA indicated significant effects, post hoc paired t-tests (for repeated measures) or Student’s t-tests (for independent measures) were performed for all experiments.
Acknowledgements
We thank the excellent technical help of Conner Ames. This work was supported by the National Institute of Health, National Heart, Lung, and Blood Institute, grant number R01HL122390, to ES.
Author Contributions
É.S. and L.K. conceived, designed and performed the experiments, analyzed the data and wrote the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cinti S. The adipose organ: morphological perspectives of adipose tissues. Proc Nutr Soc. 2001;60(3):319–28. doi: 10.1079/PNS200192. [DOI] [PubMed] [Google Scholar]
- 2.Richard, D., Monge-Roffarello, B., Chechi, K., Labbe, S. M. & Turcotte, E. E. Control and physiological determinants of sympathetically mediated brown adipose tissue thermogenesis. Front Endocrinol (Lausanne) 3, Article 36 (2012). [DOI] [PMC free article] [PubMed]
- 3.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84(1):277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 4.Nahmias C, Blin N, Elalouf JM, Mattei MG, Strosberg AD, Emorine LJ. Molecular characterization of the mouse β3-adrenergic receptor: relationship with the atypical receptor of adipocytes. EMBO J. 1991;10(12):3721–7. doi: 10.1002/j.1460-2075.1991.tb04940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langin D, Portillo MP, Saulnier-Blache JS, Lafontan M. Coexistence of three β-adrenoceptor subtypes in white fat cells of various mammalian species. Eur J Pharmacol. 1991;199(3):291–301. doi: 10.1016/0014-2999(91)90492-9. [DOI] [PubMed] [Google Scholar]
- 6.Evans BA, Papaioannou M, Bonazzi VR, Summers RJ. Expression of β3-adrenoceptor mRNA in rat tissues. Br J Pharmacol. 1996;117(1):210–6. doi: 10.1111/j.1476-5381.1996.tb15176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bloom JD, et al. Disodium (R,R)-5-[2-[[2-(3-chlorophenyl)-2-hydroxyethyl]-amino] propyl]-1,3-benzodioxole-2,2-dicarboxylate (CL 316,243). A potent β-adrenergic agonist virtually specific for β3 receptors. A promising antidiabetic and antiobesity agent. J Med Chem. 1992;35(16):3081–4. doi: 10.1021/jm00094a025. [DOI] [PubMed] [Google Scholar]
- 8.Susulic VS, et al. Targeted disruption of the β3-adrenergic receptor gene. J Biol Chem. 1995;270(49):29483–92. doi: 10.1074/jbc.270.49.29483. [DOI] [PubMed] [Google Scholar]
- 9.Grujic D, et al. β3-adrenergic receptors on white and brown adipocytes mediate β3-selective agonist-induced effects on energy expenditure, insulin secretion, and food intake. A study using transgenic and gene knockout mice. J Biol Chem. 1997;272(28):17686–93. doi: 10.1074/jbc.272.28.17686. [DOI] [PubMed] [Google Scholar]
- 10.Atgié C, D’Allaire F, Bukowiecki LJ. Role of β1- and β3-adrenoceptors in the regulation of lipolysis and thermogenesis in rat brown adipocytes. Am J Physiol. 1997;273(4 Pt 1):C1136–42. doi: 10.1152/ajpcell.1997.273.4.C1136. [DOI] [PubMed] [Google Scholar]
- 11.Himms-Hagen J, et al. Effect of CL-316,243, a thermogenic β3-agonist, on energy balance and brown and white adipose tissues in rats. Am J Physiol. 1994;266(4 Pt 2):R1371–82. doi: 10.1152/ajpregu.1994.266.4.R1371. [DOI] [PubMed] [Google Scholar]
- 12.Ghorbani M, Claus TH, Himms-Hagen J. Hypertrophy of brown adipocytes in brown and white adipose tissues and reversal of diet-induced obesity in rats treated with a β3-adrenoceptor agonist. Biochem Pharmacol. 1997;54(1):121–31. doi: 10.1016/S0006-2952(97)00162-7. [DOI] [PubMed] [Google Scholar]
- 13.Szentirmai É, Kapás L. Intact brown adipose tissue thermogenesis is required for restorative sleep responses after sleep loss. Eur J Neurosci. 2014;39(6):984–98. doi: 10.1111/ejn.12463. [DOI] [PubMed] [Google Scholar]
- 14.Bartness TJ, Vaughan CH, Song CK. Sympathetic and sensory innervation of brown adipose tissue. Int J Obes (Lond) 2010;34(Suppl 1):S36–S42. doi: 10.1038/ijo.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartness TJ, Shrestham YB, Vaughan CH, Schwartz GJ, Song CK. Sensory and sympathetic nervous system control of white adipose tissue lipolysis. Mol Cell Endocrinol. 2010;318:34–43. doi: 10.1016/j.mce.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaughan CH, Bartness TJ. Anterograde transneuronal viral tract tracing reveals central sensory circuits from brown fat and sensory denervation alters its thermogenic responses. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1049–R1058. doi: 10.1152/ajpregu.00640.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osaka T, et al. Temperature- and capsaicin-sensitive nerve fibers in brown adipose tissue attenuate thermogenesis in the rat. Pflugers Arch. 1998;437:36–42. doi: 10.1007/s004240050743. [DOI] [PubMed] [Google Scholar]
- 18.Conley RK, Li J, Ivarsson M, Hutson PH. Effect of the β3 adrenoceptor agonist CL 316243 on hypothalamic 5-HT synthesis and suppression of REM sleep in the rat. J Psychopharmacol. 2007;21(6):597–602. doi: 10.1177/0269881106070424. [DOI] [PubMed] [Google Scholar]
- 19.Stemmelin, J. et al. Stimulation of the β3-Adrenoceptor as a novel treatment strategy for anxiety and depressive disorders. Neuropsychopharmacology. 2008, 33(3), 574–87 (2008). [DOI] [PubMed]
- 20.Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab. 1998;83(3):847–50. doi: 10.1210/jcem.83.3.4660. [DOI] [PubMed] [Google Scholar]
- 21.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L. & Friedman, J. M. Positional cloning of the mouse obese gene and its human homologue. Nature1, 372(6505), 425–32 (1994). [DOI] [PubMed]
- 23.Sinton CM, Fitch TE, Gershenfeld HK. The effects of leptin on REM sleep and slow wave delta in rats are reversed by food deprivation. J Sleep Res. 1999;8(3):197–203. doi: 10.1046/j.1365-2869.1999.00158.x. [DOI] [PubMed] [Google Scholar]
- 24.Hogan D, Morrow JD, Smith EM, Opp MR. Interleukin-6 alters sleep of rats. J Neuroimmunol. 2003;137(1–2):59–66. doi: 10.1016/S0165-5728(03)00038-9. [DOI] [PubMed] [Google Scholar]
- 25.Jewett KA, Krueger JM. Humoral sleep regulation; interleukin-1 and tumor necrosis factor. Vitam Horm. 2012;89:241–57. doi: 10.1016/B978-0-12-394623-2.00013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapás L, et al. Somnogenic, pyrogenic, and anorectic activities of tumor necrosis factor-α and TNF-α fragments. Am J Physiol. 1992;263(3 Pt 2):R708–15. doi: 10.1152/ajpregu.1992.263.3.R708. [DOI] [PubMed] [Google Scholar]
- 27.Crichton PG, Lee Y, Kunji ERS. The molecular features of uncoupling protein 1 support a conventional mitochondrial carrier-like mechanism. Biochimie. 2017;134:35–50. doi: 10.1016/j.biochi.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Granneman JG, Burnazi M, Zhu Z, Schwamb LA. White adipose tissue contributes to UCP1-independent thermogenesis. Am J Physiol Endocrinol Metab. 2003;285(6):E1230–6. doi: 10.1152/ajpendo.00197.2003. [DOI] [PubMed] [Google Scholar]
- 29.Inokuma K, et al. Indispensable role of mitochondrial UCP1 for antiobesity effect of β3-adrenergic stimulation. Am J Physiol Endocrinol Metab. 2006;290(5):E1014–21. doi: 10.1152/ajpendo.00105.2005. [DOI] [PubMed] [Google Scholar]
- 30.Hankir, M. K. et al. Dissociation between brown adipose tissue 18F-FDG uptake and thermogenesis in uncoupling protein 1 deficient mice. J Nucl Med pii: jnumed.116.186460. doi:10.2967/jnumed.116.186460 (2017). [DOI] [PubMed]
- 31.Obál F, Jr., Tobler I, Borbély AA. Effect of ambient temperature on the 24-hour sleep-wake cycle in normal and capsaicin-treated rats. Physiol Behav. 1983;30(3):425–30. doi: 10.1016/0031-9384(83)90148-8. [DOI] [PubMed] [Google Scholar]
- 32.Krueger JM, Walter J, Dinarello CA, Wolff SM, Chedid L. Sleep-promoting effects of endogenous pyrogen (interleukin-1) Am J Physiol. 1984;246(6 Pt 2):R994–9. doi: 10.1152/ajpregu.1984.246.6.R994. [DOI] [PubMed] [Google Scholar]
- 33.Opp MR, Obál F, Jr., Krueger JM. Interleukin 1 alters rat sleep: temporal and dose-related effects. Am J Physiol. 1991;260(1 Pt 2):R52–8. doi: 10.1152/ajpregu.1991.260.1.R52. [DOI] [PubMed] [Google Scholar]
- 34.Summers RJ, Papaioannou M, Harris S, Evans BA. Expression of β3-adrenoceptor mRNA in rat brain. Br J Pharmacol. 1995;116(6):2547–8. doi: 10.1111/j.1476-5381.1995.tb17205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mirbolooki MR, Constantinescu CC, Pan ML, Mukherjee J. Quantitative assessment of brown adipose tissue metabolic activity and volume using 18F-FDG PET/CT and β3-adrenergic receptor activation. EJNMMI Res. 2011;1(1):30. doi: 10.1186/2191-219X-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gavrilova O, Marcus-Samuels B, Reitman ML. Lack of responses to a β3-adrenergic agonist in lipoatrophic A-ZIP/F-1 mice. Diabetes. 2000;49(11):1910–6. doi: 10.2337/diabetes.49.11.1910. [DOI] [PubMed] [Google Scholar]
- 37.Samson FE, Jr., White RP. Effects of fatty acid anions on the electroencephalogram of unanesthetized rabbits. Am J Physiol. 1956;186(2):271–4. doi: 10.1152/ajplegacy.1956.186.2.271. [DOI] [PubMed] [Google Scholar]
- 38.Matsuzaki M, Takagi H, Tokizane T. Paradoxical phase of sleep: its artificial induction in the cat by sodium butyrate. Science. 1964;146(3649):1328–9. doi: 10.1126/science.146.3649.1328. [DOI] [PubMed] [Google Scholar]
- 39.Mendelson WB, Basile AS. The hypnotic actions of the fatty acid amide, oleamide. Neuropsychopharmacology. 2001;25(5 Suppl):S36–9. doi: 10.1016/S0893-133X(01)00341-4. [DOI] [PubMed] [Google Scholar]
- 40.Pellinen J, Szentirmai É. The effects of C75, an inhibitor of fatty acid synthase, on sleep and metabolism in mice. PLoS One. 2012;7(2):e30651. doi: 10.1371/journal.pone.0030651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Divakaruni AS, Humphrey DM, Brand MD. Fatty acids change the conformation of uncoupling protein 1 (UCP1) J Biol Chem. 2012;287(44):36845–53. doi: 10.1074/jbc.M112.381780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell. 2012;151(2):400–13. doi: 10.1016/j.cell.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacPherson RE, Castellani L, Beaudoin MS, Wright DC. Evidence for fatty acids mediating CL 316,243-induced reductions in blood glucose in mice. Am J Physiol Endocrinol Metab. 2014;307(7):E563–70. doi: 10.1152/ajpendo.00287.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burýsek L, Houstek J. β-adrenergic stimulation of interleukin-1α and interleukin-6 expression in mouse brown adipocytes. FEBS Lett. 1997;411(1):83–6. doi: 10.1016/S0014-5793(97)00671-6. [DOI] [PubMed] [Google Scholar]
- 45.Mohamed-Ali V, et al. β-adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies. J Clin Endocrinol Metab. 2001;86(12):5864–9. doi: 10.1210/jcem.86.12.8104. [DOI] [PubMed] [Google Scholar]
- 46.Castellani L, Root-Mccaig J, Frendo-Cumbo S, Beaudoin MS, Wright DC. Exercise training protects against an acute inflammatory insult in mouse epididymal adipose tissue. J Appl Physiol. 2014;116(10):1272–80. doi: 10.1152/japplphysiol.00074.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida T. The antidiabetic β3-adrenoceptor agonist BRL 26830A works by release of endogenous insulin. Am J Clin Nutr. 1992;55(1 Suppl):237S–241S. doi: 10.1093/ajcn/55.1.237s. [DOI] [PubMed] [Google Scholar]
- 48.Späth-Schwalbe E, et al. Acute effects of recombinant human interleukin-6 on endocrine and central nervous sleep functions in healthy men. J Clin Endocrinol Metab. 1998;83(5):1573–9. doi: 10.1210/jcem.83.5.4795. [DOI] [PubMed] [Google Scholar]
- 49.Danguir J, Nicolaidis S. Chronic intracerebroventricular infusion of insulin causes selective increase of slow wave sleep in rats. Brain Res. 1984;306(1–2):97–103. doi: 10.1016/0006-8993(84)90359-7. [DOI] [PubMed] [Google Scholar]
- 50.Sangiah S, Caldwell DF, Villeneuve MJ, Clancy JJ. Sleep: sequential reduction of paradoxical (REM) and elevation of slow-wave (NREM) sleep by a non-convulsive dose of insulin in rats. Life Sci. 1982;31(8):763–9. doi: 10.1016/0024-3205(82)90702-0. [DOI] [PubMed] [Google Scholar]
- 51.Fu L, Isobe K, Zeng Q, Suzukawa K, Takekoshi K, Kawakami Y. β-adrenoceptor agonists downregulate adiponectin, but upregulate adiponectin receptor 2 and tumor necrosis factor-α expression in adipocytes. Eur J Pharmacol. 2007;569(1–2):155–62. doi: 10.1016/j.ejphar.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 52.Roth Flach RJ, Matevossian A, Akie TE, Negrin KA, Paul MT, Czech MP. β3-Adrenergic receptor stimulation induces E-selectin-mediated adipose tissue inflammation. J Biol Chem. 2013;288(4):2882–92. doi: 10.1074/jbc.M112.412346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Borbély AA. From slow waves to sleep homeostasis: new perspectives. Arch Ital Biol. 2001;139(1–2):53–61. [PubMed] [Google Scholar]
- 54.Grossberg AJ, Scarlett JM, Marks DL. Hypothalamic mechanisms in cachexia. Physiol Behav. 2010;100(5):478–89. doi: 10.1016/j.physbeh.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakao A, Takahashi Y, Nagase M, Ikeda R, Kato F. Role of capsaicin-sensitive C-fiber afferents in neuropathic pain-induced synaptic potentiation in the nociceptive amygdala. Mol Pain. 2012;8:51. doi: 10.1186/1744-8069-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]