

Abstract
Background
Melatonin can be neuroprotective in models of neurological injury, but its effects on blood vessel loss and neurological impairment following spinal cord injury (SCI) are unclear. Our goal herein was to evaluate the possible protective action of melatonin on the above SCI-induced damage in rats.
Materials and methods
Sixty-three female Sprague-Dawley rats were randomly divided into three equal groups: sham, SCI and melatonin groups. Melatonin (10 mg/kg) was injected intraperitoneally and further administered twice a day at indicated time after a moderate injury at T10 in melatonin group. Blood vessel was assessed by CD31staining and FITC-LEA, the permeability of blood–spinal cord barrier (BSCB) was detected by Evan's Blue. Neuron was assessed by NeuN staining and the expression of Nissl bodies in the neurons was assessed by Nissl staining. The expressions of brain-derived neurotrophic factor (BDNF), synapsin I, or growth associated protein-43 (GAP-43) in the spinal cord and hippocampus were evaluated by Western blotting.
Results
At 7 days post-injury, melatonin treatment rescued blood vessels, increased CD31 levels, ameliorated BSCB permeability. Additionally, melatonin significantly increased the number of neurons and the expression of Nissl bodies in neurons at the injury epicenter. Furthermore, our data showed that SCI reduced levels of the molecular substrates of neurological plasticity, including BDNF, synapsin I, or GAP-43 in the spinal cord and hippocampus. Melatonin treatment partially prevented these reductions.
Conclusion
The neuroprotective effect of melatonin was associated with melioration of the microcirculation in the spinal cord and reduction of neurological impairment in the spinal cord and brain.
Keywords: Spinal cord injury, Blood vessel loss, Neurological impairment, melatonin
Introduction
Spinal cord injury (SCI) is a serious problem afflicting persons worldwide, for which no suitable cure or therapy has thus far been developed.1 However, China is believed to have more patients with SCI than any other country in the world.2 SCI is initiated by a primary injury such as mechanical contusion of the spinal cord, and results in progressive tissue loss, in part, secondary to blood vessel dysfunction and inflammation at the epicenter.3,4 In addition, the milieu of cellular debris and blood at the epicenter is toxic, causing further cell death.5 Neurons are thought to be highly sensitive to such noxious stimuli, and are extremely vulnerable to reductions in perfusion and resultant periods of ischemia.6 Blood vessel loss and disruption of the blood-spinal cord barrier (BSCB) following SCI can also compromise neuronal functions.
Melatonin is an indoleamine naturally produced in the pineal gland and other tissues, and easily passes the blood-brain barrier.7 In an animal model of SCI, melatonin was able to reduce SCI-induced tissue injury by effectively reducing lipid peroxidation.8–10 Further work demonstrated that melatonin reduced calpain expression and attenuated cytoskeletal protein degradation, preserved the structure of axons and myelin and limited neuronal death in SCI.11 In addition, an earlier study demonstrated that melatonin protected against subarachnoid hemorrhage-induced upregulation of BSCB permeability and the associated increase in brain edema.12 Our previous studies showed that melatonin decreased inflammation-induced hyperpermeability of endothelial cells, reflecting a barrier-protective effect in vitro.13,14 Melatonin treatment protected against SCI-induced disruption of the BSCB in vivo.15,16 Furthermore, melatonin attenuated impairment of hippocampal structural neuroplasticity in OXYS rats during the active progression of an Alzheimer's disease-like pathology.17 Based on these findings, we hypothesized that melatonin would increase vascular and neurological protection. The purpose of this study was to improve our understanding of melatonin's role in affecting vascular response and neurological alteration to SCI.
Materials and methods
Animal care and injury induction
Female Sprague-Dawley rats (200 ± 20 g) were purchased from the Center of Experimental Animals, Capital Medical University (Beijing, China). Rats were maintained in an air-conditioned room with 12:12 light/dark cycles at 22 ± 2°C and 55 ± 10% relative humidity. Food and water were available ad libitum. Animal protocols followed guidelines established by the NIH, and were approved by the Animal Care and Use Committee of Capital Medical University. Rats were anesthetized with an intraperitoneal injection of phenobarbital sodium (35 mg/kg). The modified NYU weight-drop model was used to induce SCI. Briefly, a laminectomy was performed at the T10 level, and the spinous processes of T8 and T11 were clamped to stabilize the spine. Then a 10-g weight (3-mm diameter tip) was dropped from a height of 25 mm onto the dorsal surface of the dura mater. During the surgical procedure and recovery from anesthesia, rats were placed in a warming chamber and their body temperatures maintained at approximately 38°C until they were completely awake. Surgical interventions and postoperative animal care were performed in accordance with the guidelines and policies for rodent survival surgery provided by the Experimental Animal Committee of Capital Medical University.
Experiment groups
Rats were randomized into the following three groups: (a) Sham group, which underwent a T10 laminectomy without injury (n = 21); (b) Vehicle group, which was subjected to SCI and given 1% ethanol (n = 21); (c) 10 mg/kg melatonin group, which was subjected to SCI and administered 10 mg/kg melatonin (Sigma, St Louis, MO, USA, dissolved by 1% ethanol) (n = 21). Melatonin was administered by intraperitoneal injection at 30 minutes after SCI for one week, twice daily (07:00, 19:00).
Tissue processing
At 7 days post-injury, rats were perfused transcardially with 150–200 ml saline, followed by 400 ml 4% paraformaldehyde (PFA). Spinal cords were dissected out, and 1-cm segments containing the injury site were post-fixed in 4% PFA for 4 hours. Tissue was then soaked overnight in 20% sucrose followed by 30% sucrose, and transverse sections with a thickness of 20 μm were obtained using a cryostat. The sections were stored in sequence at –80°C until use.
Measurement of BSCB permeability
At 7 days after SCI, 2 ml of 2% Evan's Blue dye (Sigma, St Louis, MO, USA) solution was administered intraperitoneally. Rats were anesthetized and perfused transcardially with saline after 3 hours. The T10 spinal cord segment was removed, dried, and weighed for quantitative measurement of Evan's Blue extravasation. After being homogenized in a 50% trichloroacetic acid solution for 3 days at room temperature, samples were centrifuged at 10,000 g for 10 minutes. The supernatants were collected and fluorescence was quantified at an excitation wavelength of 620 nm and an emission wavelength of 680 nm. For qualitative examination of Evan's Blue extravasation, rats were perfused and fixed as mentioned above. The spinal cords were then sectioned at 20 μm. One in every five of the transverse sections at each rostro-caudal 1 mm level was observed and the relative fluorescence intensity of Evan's Blue was calculated with Image Pro Plus7.0. (Media Cybernetics, Silver Spring, MD, USA) (IPP 7.0).
Systemic intravascular lectin injection
To pre-label perfused blood vessel, rats were anesthetized 7 days after SCI, 200 μl of FITC-conjugated lycopersicon esculentum agglutinin lectin (LEA, Sigma, St Louis, MO, USA) was delivered systemically by intravenous injection into the jugular vein and allowed to circulate for 20 minutes prior to perfusion.18
Immunohistochemistry
Immunofluorescence
Sections (20 μm) were warmed at room temperature for 1 hour, then equilibrated in 0.1 M Tris-buffered saline for 10 minutes, and finally permeabilized with 0.3% Triton X-100 for 30 minutes. Nonspecific antibody binding was then blocked with 10% normal goat serum in PBS for 1 hour. Slides were incubated for 1 hour with appropriate dilutions of primary IgG antibodies, including rabbit polyclonal anti-CD31 (1:100, Abcam, Cambridge, MA, USA), or mouse monoclonal NeuN (1:100, Abcam, Cambridge, MA, USA). Slides were rinsed after primary incubation, then incubated with secondary antibody (fluorophore-labeled donkey anti-mouse IgG (H + L) antibodies (Alexa Fluor® 488, 1:1000 dilution; Dylight 549 rabbit anti-rabbit, 1:1000 dilution, Thermo Fisher Scientific Inc., Waltham, MA, USA).
Nissl staining
Serial sections (20 μm) were collected on gelatin-coated slides and stained with cresyl violet to observe the Nissl bodies.
Western blot analysis
A 1-cm length of spinal cord (epicenter ± 5 mm) and the hippocampus were collected. Total protein was prepared in a lysis buffer (Beyotime, China) by lysing tissue homogenates for 1 hour, and then centrifuging at 14,000 g for 8 minutes at 4°C. The protein content of the supernatant was determined using a protein assay kit (BCA, Pierce, Rockford, IN, USA). Equal amounts of total protein (50 μg) were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% non-fat skim milk in Tris-buffered saline solution with 0.05% Tween-20 (TBST) for 1 hour, and then incubated with antibodies against CD31 (1:100, Abcam), brain-derived neurotrophic factor (BDNF; 1:100, Abcam), GAP-43 (1:100, Abcam), or synapsin I (1:100, Abcam) at 4°C overnight. After washing 3 times with TBST, the primary antibodies were detected with the appropriate horseradish peroxidase-conjugated secondary antibodies, and β-actin (1:1000, Cell Signaling Technology) used as an internal control. The bands were visualized using enhanced chemiluminescence, and images were acquired with ChemiDoc MP System (Bio-Rad, Hercules, CA, USA). The relative band intensities were quantified using Quantity One (Bio-Rad, Hercules, CA, USA).
Image quantitative measurements and statistical analyses
Quantitative assessment of the number of neurons was accomplished by using methods similar to those previously described with IPP 7.0.19 Every fifth transverse sections at each rostrol-caudal 1 mm level were chosen, neurons of three microscopic fields at a magnification of 200 x in each section were counted. During quantification, overlapping cells were defined only if a clear outline of each cell was discernible. The total area of CD31-labeled blood vessels/LEA-labeled blood vessels in each image was automatically calculated using IPP 7.0, and each individual CD31-labeled blood vessels/LEA-labeled blood vessels was then measured by dividing its area by the total area. Images were acquired using identical exposure settings for each analytical group, every fifth transverse section from each cord was photographed with a 20x objective, images with identical position were taken at 1-mm rostral and caudal to the injury epicenter. The data were analyzed using the SPSS Version 17.0 statistic software package (SPSS Inc., Chicago, IL, USA). Student's t-tests were used to determine significance between vehicle and melatonin-treated groups, and one-way analysis of variance (ANOVA) followed by post-hoc Tukey's analysis was performed to compare three or more groups. Data are presented as mean ± SD, and values of P < 0.05 are considered significant.
Results
Melatonin rescued blood vessels after SCI
Spinal cord sections were stained with antibodies directed against the endothelial marker protein (CD31), and the immunolabeled area at the ventral horn of the spinal cord was analyzed (Fig. 1A-B). In the sham group (Sham), a large area of blood vessels was labeled by CD31, while in the vehicle (Veh) group, much lesser CD31-positive area was labeled (Sham, 0.17 ± 0.02 versus Veh, 0.07 ± 0.02, P < 0.01). Melatonin treatment increased the proportion of CD31-labeled blood vessels (Mel, 0.14 ± 0.02 versus Veh, 0.07 ± 0.02, P < 0.05). Moreover, the expression of CD31was significantly higher in the melatonin (Mel (10 mg/kg)) group versus the Veh group (Mel, 0.38 ± 0.02 versus Veh, 0.17 ± 0.01, P < 0.01) (Fig. 1C-D). To evaluate the extent of rescue of perfused blood vessels, cell-binding FITC-conjugated LEA lectin was injected intravenously. It was observed that LEA-labeled blood vessels in the ventral horn were less during the procedure of injury compared with the Sham group (Sham, 0.16 ± 0.02 versus Veh, 0.06 ± 0.01, P < 0.01), and melatonin treatment rescued a proportion of injured LEA-labeled blood vessels (Mel, 0.12 ± 0.02 versus Veh, 0.06 ± 0.01, P < 0.05) (Fig. 1E-F).
Figure 1.
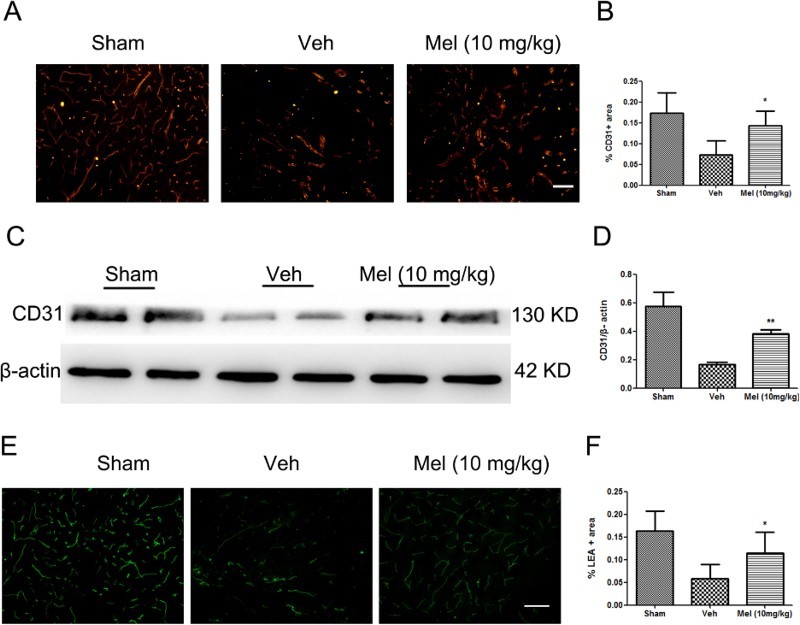
The effect of melatonin on blood vessels following spinal cord injury (SCI). (A) Blood vessels were visualized by anti-CD31 immunofluorescence in the ventral horn of the spinal cord and were presented for each of the different treatment groups (n = 4). (B) CD31-stained area was quantified in all groups. (C-D) The expression of CD31 was detected by western blot (n = 4), and the relative intensity of CD31 was analyzed in the different treatment groups. (E) Perfused blood vessels were labeled by LEA in the ventral horn of the spinal cord and were presented for each of the different treatment groups (n = 3). (F) LEA-labeled area was quantified in all groups. *P < 0.05 in Veh versus Mel (10 mg/kg), **P < 0.01 in Veh versus Mel (10 mg/kg). Scale bar = 50 μm (Colour online)
(sham group: Sham; vehicle group: Veh; melatonin group: Mel (10 mg/kg))
Melatonin treatment decreased BSCB permeability
The effect of melatonin on BSCB permeability at 7 days after SCI was examined by Evan's Blue assay. As shown in Fig. 2A-B, SCI increased the fluorescence intensity of Evan's Blue dye versus the Sham group, a result of extravasation of dye through damaged blood vessels. Melatonin significantly reduced the fluorescence intensity of the leaked Evan's Blue dye and improved the function of impaired blood vessels. Quantitative analysis showed that the fluorescence intensity/the amount of extravasated Evan's Blue dye in the Veh group was higher than that in the Sham group, which was reduced by melatonin treatment (the fluorescence intensity of extravasated Evan's Blue dye: Mel, 35.73 ± 3.42 versus Veh, 67.76 ± 2.20, P < 0.01; the amount of extravasated Evan's Blue dye: Mel, 0.12 ± 0.01 versus Veh, 0.18 ± 0.02, P < 0.05) (Fig. 2C-D).
Figure 2.
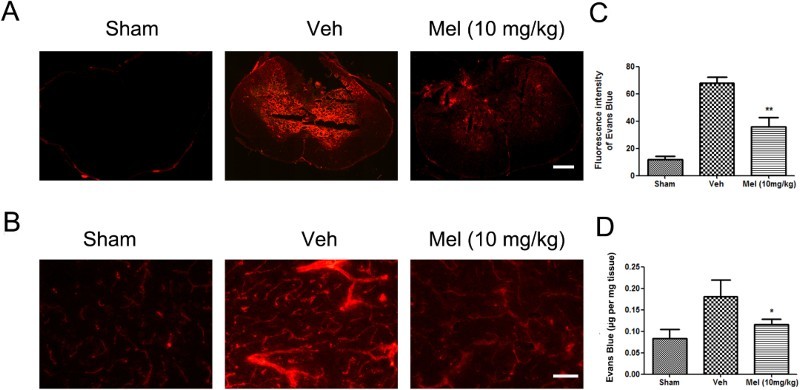
The effect of melatonin on the permeability of the blood–spinal cord barrier (BSCB) following SCI. (A-B) Representative fluorescent image of an Evan's Blue dye extravasation at the spinal parenchyma at 7 days after SCI, in an entire spinal cord transverse section, and in the ventral horn of the spinal cord (n = 6). Scale bar = 50μm. (C) Quantification of the fluorescence intensity of Evan's Blue. (D) Quantification of the amount of Evan's Blue. *P < 0.05 in Veh versus Mel (10 mg/kg), **P < 0.01 in Veh versus Mel (10 mg/kg) (Colour online)
(sham group: Sham; vehicle group: Veh; melatonin group: Mel (10 mg/kg))
Melatonin-protected neurons after SCI
To verify the neuroprotective effect of melatonin on treated animals, we analyzed spinal cord tissue for neuronal density using antibodies specific to the neuronal marker NeuN (Fig. 3). In the Sham group, neurons in the ventral horn appeared normal, with larger cell bodies and intact axons. SCI induced a significant loss of NeuN positive neurons. Whereas, the number of NeuN-positive cells was significantly higher in the Mel (10 mg/kg) group compared to the Veh group (Fig. 3A). The quantification of neuronal cell bodies across groups is provided in Fig. 3B. (Sham: 205.30 ± 25.62, Vel: 50.50 ± 5.98, and Mel: 101.00 ± 9.43, P < 0.01). The expression of Nissl bodies in the Veh group was decreased (Sham, 0.45 ± 0.02 versus Veh, 0.20 ± 0.03, P < 0.01), while melatonin treatment partially reversed this reduction (Mel, 0.35 ± 0.03 versus Veh, 0.20 ± 0.03, P < 0.01) (Fig. 3C-D). All of these data indicated the protective effect of melatonin on neurons against SCI.
Melatonin mediated the expressions of BDNF, synapsin I and GAP-43in the spinal cord and hippocampus after SCI
BDNF is a promoter of neuronal plasticity.20,21 synapsin I and GAP-43 proteins are also involved in the mechanism of BDNF-related neuronal protection.22 As shown in Fig. 4, the expression of BDNF and synapsin I were prominently decreased at 7 days after injury, to levels below those in the Sham group in the spinal cord and hippocampus (spinal cord: BDNF: Sham, 0.67 ± 0.05 versus Veh, 0.29 ± 0.04, P < 0.01; synapsin I: Sham, 0.41 ± 0.05 versus Veh, 0.12 ± 0.03, P < 0.01; hippocampus: BDNF: Sham, 0.35 ± 0.06 versus Veh, 0.12 ± 0.03, P < 0.01; synapsin I: Sham, 0.40 ± 0.05 versus Veh, 0.19 ± 0.02, P < 0.01). Melatonin administration significantly upregulated the levels of BDNF and synapsin I in the spinal cord and hippocampus at 7 days following SCI (spinal cord: BDNF: Mel, 0.64 ± 0.05 versus Veh, 0.29 ± 0.04, P < 0.01; synapsin I: Mel, 0.38 ± 0.04 versus Veh, 0.12 ± 0.03, P < 0.01; hippocampus: BDNF: Mel, 0.27 ± 0.04 versus Veh, 0.12 ± 0.03, P < 0.05; synapsin I: Mel, 0.40 ± 0.03 versus Veh, 0.19 ± 0.02, P < 0.01). Melatonin partially reversed the decrease of GAP-43 levels induced by SCI in the spinal cord (Sham: 0.80 ± 0.06, Veh: 0.34 ± 0.03, and Mel: 0.52 ± 0.05, P < 0.01). SCI did not alter GAP-43 levels in the hippocampus (Sham, 0.50 ± 0.05 versus Veh, 0.49 ± 0.05, P > 0.05), and no significant difference was shown between the Veh and Mel groups (Mel, 0.53 ± 0.06 versus Veh, 0.49 ± 0.05, P > 0.05). The data suggested that melatonin could exert protective effect on neurological plasticity after SCI.
Figure 4.
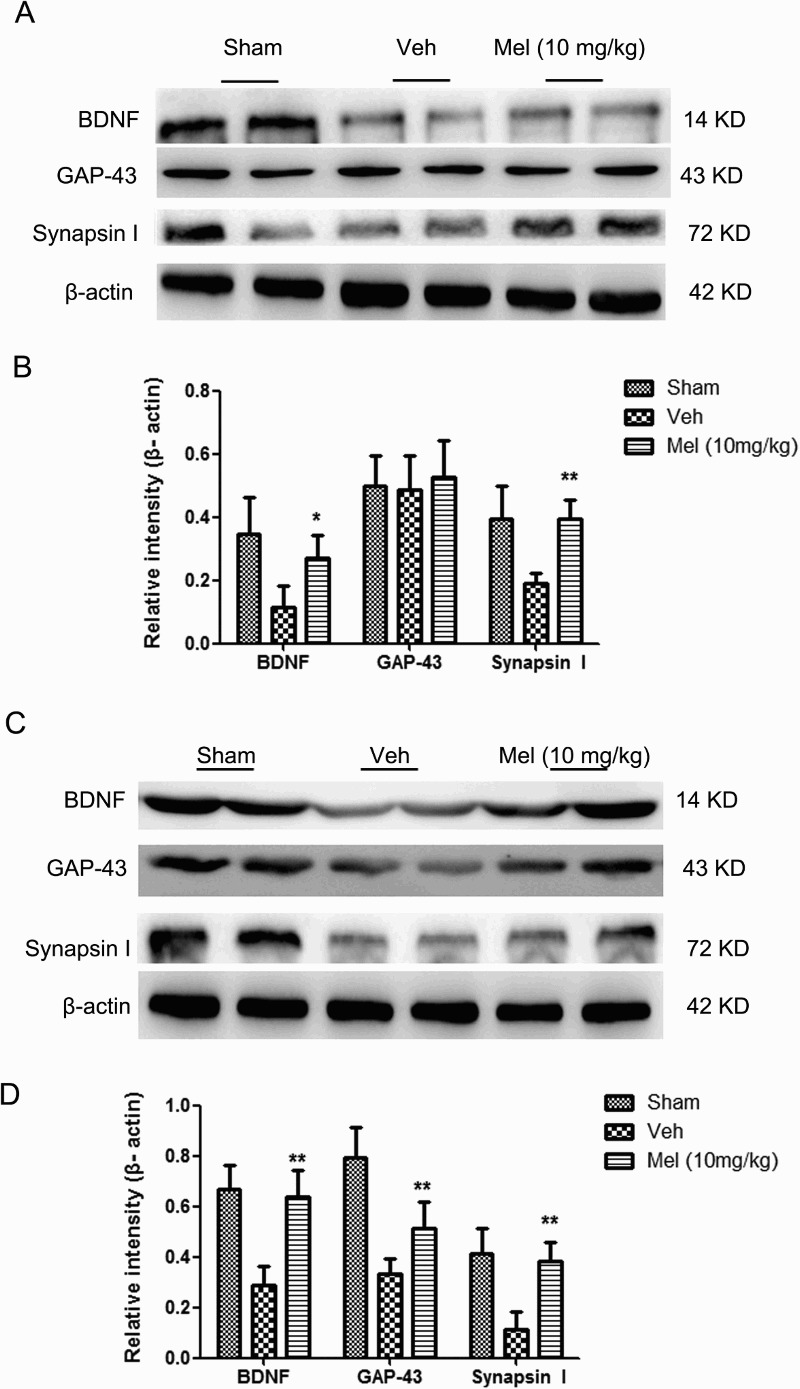
The expressions of brain-derived neurotrophic factor (BDNF), GAP-43, and synapsin I in the different treatment groups were determined in brain and spinal cord following SCI. (A) BDNF, GAP-43, and synapsin I expressions in the brain (n = 4). (B) The relative intensity of BDNF, GAP-43, and synapsin I expression across treatment groups. (C) The BDNF, GAP-43, and synapsin I expressions in the spinal cord (n = 4). (D) The relative intensity of BDNF, GAP-43, and synapsin I expressions across treatment groups. *P < 0.05 in Veh versus Mel (10 mg/kg), **P < 0.01 in Veh versus Mel (10 mg/kg)
(sham group: Sham; vehicle group: Veh; melatonin group: Mel (10 mg/kg))
Figure 3.
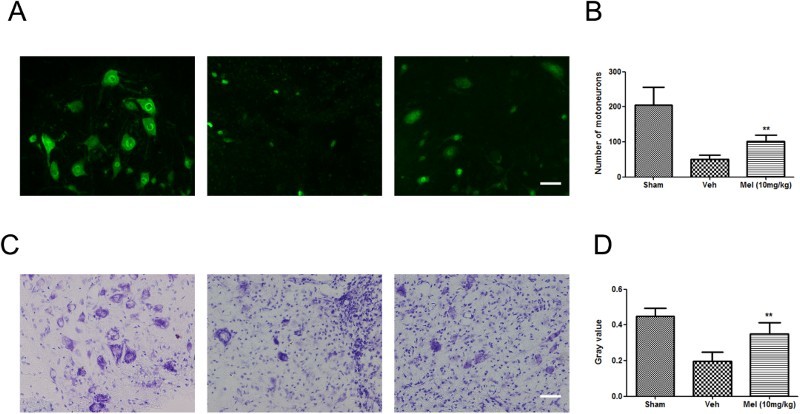
The effect of melatonin on neurons following SCI. (A) NeuN positive neurons were detected in the different treatment groups by immunofluorescence (n = 4). (B) Quantification of NeuN positive neuron cell bodies in the T10 region across groups. (C) Spinal cord transection showed Nissl substance in the control and treated groups by cresyl violet staining (n = 4). (D) Gray value of Nissl body was analyzed across groups. **P < 0.01 in Veh versus Mel (10 mg/kg). Scale bar = 50 μm. (Colour online)
(sham group: Sham; vehicle group: Veh; melatonin group: Mel (10 mg/kg))
Discussion
Spinal cord blood vessels are important for maintaining the normal function of the spinal cord, and are critical to the progressive degeneration and subsequent functional deficits post-SCI.23–25 Herein, our results showed that rats with SCI suffered from disruption of microvessels and BSCB at 7 days post-injury. The amelioration of damage to the microcirculation after melatonin treatment was observed in parallel to some restoration of neurological impairment. Recent studies have shown that the increase in blood vessels following SCI is well-correlated with the improvement of sensorimotor function.26,27 Thus, these results indicated that the rescue of blood vessel could be an essential mechanism of the neuroprotective effect for melatonin.”
In the present study, we investigated the vascular structure and function using CD31 staining and FITC-LEA. CD31 staining quantified all vascular structure without distinguishing whether the vessel had an active perfusion state. LEA has been used as a marker of microvascular perfusion in the murine spinal cord.28 Our result showed that a significant increase of blood vessel area was noted in the melatonin-treated SCI rats, based on the expression of CD31 and luminal surface of endothelial cells marked by LEA, suggesting that melatonin ameliorated damage of vascular structure and upregulated the number of functional vessel. However, we did not investigate the effect of melatonin on angiogenesis and arteriogensis which represent important potential for treatment of vascular network. Previous studies had shown that melatonin reduced the expression of vascular endothelial growth factor (VEGF)/VEGF2, protecting the integrity of the BSCB.16 Haddadi et al.29 reported that melatonin pretreatment downregulated VEGF expression in rats after radiation-induced injury to the cervical spinal cord, which increased the survival rate of the irradiated animals. Then, VEGF signaling is not only critical for inducing angiogenesis,30,31 but also critical for promoting collateral growth and de novo arteriogenesis.32 Taken together, melatonin induced VEGF reduction impacted both angiogenesis and arteriogensis processes. It is likely that melatonin rescued the injured endothelial cells, thus exerting a protective effect on the BSCB and microcirculation.
Our results showed that higher numbers of neuronal cell bodies were consistent with more blood vessels, indicating that the increase of blood vessels directly or indirectly exerted a beneficial effect on neurons. This may be due to the high metabolic requirements of these neurons, which makes them extremely vulnerable to reductions in perfusion and periods of ischemia. On the other hand, ultrastructural analysis of Nissl bodies has shown that they are related to the synthesis of proteins for intercellular use. They are also very sensitive to pathological stimuli and are therefore usually considered a good index of neurocyte injury. Melatonin treatment significantly increased the number of neurons, as well as the expression of Nissl bodies. This effect is most likely attributable to the melatonin-mediated recovery of the microfluid environment of the spinal cord, which needs to be explored in further study.
We suggested that melatonin treatment reduced neuronal impairment. In line with this assumption, our data showed that melatonin treatment in part reversed the decline of neuroplasticity-associated proteins including BDNF, synapsin I, and GAP-43 in the spinal cord following injury. BDNF appears to be an important player following SCI, as its levels are markedly decreased after injury33 and are found to be increased after rehabilitative training.34 GAP-43, an established marker for axonal regeneration, is used to visualize the regenerative process over time in the central nervous system.35 Additionally, effective intervention promotes the growth of GAP-43-labeled axonal fibers.36 A previous regression analysis showed that the expression of synapsin I was strongly correlated with the spontaneous recovery of hindlimb locomotion after spinal cord hemisection.37 Above all, melatonin treatment ameliorated impairment of neurological plasticity. Interestingly, recent studies suggest that spinal cord lesions can affect molecular systems for neurological plasticity in the brain. In present study, we noted a reduction of BDNF and synapsin I levels, but not GAP-43, induced by SCI in the hippocampus. Fumagalli et al.38 reported that the reduction of hippocampal BDNF mRNA and protein levels were persistent over at least 7 days after spinal cord damage, and that GAP-43 mRNA did not decline in the hippocampus after experimental SCI, consistent with the results herein. Furthermore, exercise provided before the injury onset increased BDNF levels in the spinal cord and hippocampus, while its production was also shown to have a protective action in the brain and spinal cord.22 Our results demonstrated that melatonin treatment upregulated the expressions of BDNF and synapsin I in the hippocampus, which might be beneficial for recovery following the injury.
Conclusion
Our results suggested that melatonin treatment ameliorated damage to microcirculation, decreased the loss of neurons following SCI. In particular, melatonin was able to counteract the injury-related reductions in BDNF, synapsin I or GAP-43 in the hippocampus and spinal cord, emphasizing the effect of melatonin on molecular systems that support neurological plasticity in the brain and spinal cord. All of above results indicated that the neuroprotective effects of melatonin may be related to its ability of preventing blood vessel loss and reducing neurological impairment. However, the molecular mechanism underlining those effects and the relationship between vascular preservation and neurological protection should be explored in further studies.
Declaration of interest
The authors report no declarations of interest.
Disclaimer statements
Contributors None.
Funding This work was funded by the Promote Scientific Research Project of Beijing Institute for Brain Disorder (PXM2014_014226_000016) and the Beijing Municipal Science & Technology Commission (BSTC, No.Z151100001615055).
Conflicts of interest None.
Ethics approval None.
References
- 1.Sharma HS. New perspectives for the treatment options in spinal cord injury. Expert Opin Pharmacother 2008;9(16):2773–800. doi: 10.1517/14656566.9.16.2773 [DOI] [PubMed] [Google Scholar]
- 2.Qiu J. China Spinal Cord Injury Network: changes from within. Lancet Neurol 2009;8(7):606–7. doi: 10.1016/S1474-4422(09)70162-0 [DOI] [PubMed] [Google Scholar]
- 3.Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res 2007;161:125–41. doi: 10.1016/S0079-6123(06)61009-1 [DOI] [PubMed] [Google Scholar]
- 4.Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, et al. . The cellular inflammatory response in human spinal cords after injury. Brain 2006;129(Pt 12):3249–69. doi: 10.1093/brain/awl296 [DOI] [PubMed] [Google Scholar]
- 5.Oudega M. Molecular and cellular mechanisms underlying the role of blood vessels in spinal cord injury and repair. Cell Tissue Res 2012;349(1):269–88. doi: 10.1007/s00441-012-1440-6 [DOI] [PubMed] [Google Scholar]
- 6.Ng MT, Stammers AT, Kwon BK. Vascular disruption and the role of angiogenic proteins after spinal cord injury. Transl Stroke Res 2011;2(4):474–91. doi: 10.1007/s12975-011-0109-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reiter RJ, Tan DX, Manchester LC, Tamura H. Melatonin defeats neurally-derived free radicals and reduces the associated neuromorphological and neurobehavioral damage. J Physiol Pharmacol 2007;58 Suppl 6:5–22. [PubMed] [Google Scholar]
- 8.Erten SF, Kocak A, Ozdemir I, Aydemir S, Colak A, Reeder BS. Protective effect of melatonin on experimental spinal cord ischemia. Spinal Cord 2003;41(10):533–8. doi: 10.1038/sj.sc.3101508 [DOI] [PubMed] [Google Scholar]
- 9.Hong Y, Palaksha KJ, Park K, Park S, Kim HD, Reiter RJ, et al. . Melatonin plus exercise-based neurorehabilitative therapy for spinal cord injury. J Pineal Res 2010;49(3):201–9. doi: 10.1111/j.1600-079X.2010.00786.x [DOI] [PubMed] [Google Scholar]
- 10.Ersahin M, Ozdemir Z, Ozsavci D, Akakin D, Yegen BC, Reiter RJ, et al. . Melatonin treatment protects against spinal cord injury induced functional and biochemical changes in rat urinary bladder. J Pineal Res 2012;52(3):340–8. doi: 10.1111/j.1600-079X.2011.00948.x [DOI] [PubMed] [Google Scholar]
- 11.Samantaray S, Sribnick EA, Das A, Knaryan VH, Matzelle DD, Yallapragada AV, et al. . Melatonin attenuates calpain upregulation, axonal damage and neuronal death in spinal cord injury in rats. J Pineal Res 2008;44(4):348–57. doi: 10.1111/j.1600-079X.2007.00534.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayer RE, Sugawara T, Chen W, Tong W, Zhang JH. Melatonin decreases mortality following severe subarachnoid hemorrhage. J Pineal Res 2008;44(2):197–204. doi: 10.1111/j.1600-079X.2007.00508.x [DOI] [PubMed] [Google Scholar]
- 13.Qin W, Lu W, Li H, Yuan X, Li B, Zhang Q, et al. . Melatonin inhibits IL1beta-induced MMP9 expression and activity in human umbilical vein endothelial cells by suppressing NF-kappaB activation. J Endocrinol 2012;214(2):145–53. doi: 10.1530/JOE-12-0147 [DOI] [PubMed] [Google Scholar]
- 14.Yuan X, Li B, Li H, Xiu R. Melatonin inhibits IL-1beta-induced monolayer permeability of human umbilical vein endothelial cells via Rac activation. J Pineal Res 2011;51(2):220–5. doi: 10.1111/j.1600-079X.2011.00882.x [DOI] [PubMed] [Google Scholar]
- 15.Jing Y, Wu Q, Yuan X, Li B, Liu M, Zhang X, et al. . Microvascular protective role of pericytes in melatonin-treated spinal cord injury in the C57BL/6 mice. Chin Med J (Engl) 2014;127(15):2808–13. [PubMed] [Google Scholar]
- 16.Wu Q, Jing Y, Yuan X, Zhang X, Li B, Liu M, et al. . Melatonin treatment protects against acute spinal cord injury-induced disruption of blood spinal cord barrier in mice. J Mol Neurosci 2014;54(4):714–22. doi: 10.1007/s12031-014-0430-4 [DOI] [PubMed] [Google Scholar]
- 17.Stefanova NA, Maksimova KY, Kiseleva E, Rudnitskaya EA, Muraleva NA, Kolosova NG. Melatonin attenuates impairments of structural hippocampal neuroplasticity in OXYS rats during active progression of Alzheimer's disease-like pathology. J Pineal Res 2015;59(2):163–77. doi: 10.1111/jpi.12248 [DOI] [PubMed] [Google Scholar]
- 18.Muradov JM, Ewan EE, Hagg T. Dorsal column sensory axons degenerate due to impaired microvascular perfusion after spinal cord injury in rats. Exp Neurol 2013;249:59–73. doi: 10.1016/j.expneurol.2013.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loy DN, Crawford CH, Darnall JB, Burke DA, Onifer SM, Whittemore SR. Temporal progression of angiogenesis and basal lamina deposition after contusive spinal cord injury in the adult rat. J Comp Neurol 2002;445(4):308–24. doi: 10.1002/cne.10168 [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Pinilla F, Ying Z, Roy RR, Hodgson J, Edgerton VR. Afferent input modulates neurotrophins and synaptic plasticity in the spinal cord. J Neurophysiol 2004;92(6):3423–32. doi: 10.1152/jn.00432.2004 [DOI] [PubMed] [Google Scholar]
- 21.Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 2008;319(5870):1683–7. doi: 10.1126/science.1152864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez-Pinilla F, Ying Z, Zhuang Y. Brain and spinal cord interaction: protective effects of exercise prior to spinal cord injury. PLoS One 2012;7(2);e32298. doi: 10.1371/journal.pone.0032298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan ZK, Lv G, Wang YF, Li G, Yu DS, Wang YS, et al. . The protective effect of salvianolic acid B on blood-spinal cord barrier after compression spinal cord injury in rats. J Mol Neurosci 2013;51(3):986–93. doi: 10.1007/s12031-013-0083-8 [DOI] [PubMed] [Google Scholar]
- 24.Norenberg MD, Smith J, Marcillo A. The pathology of human spinal cord injury: defining the problems. J Neurotrauma 2004;21(4):429–40. doi: 10.1089/089771504323004575 [DOI] [PubMed] [Google Scholar]
- 25.Tator CH, Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg 1997;86(3):483–92. doi: 10.3171/jns.1997.86.3.0483 [DOI] [PubMed] [Google Scholar]
- 26.Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, Ivanov A, et al. . De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat Med 2009;15(2):185–91. doi: 10.1038/nm.1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han S, Arnold SA, Sithu SD, Mahoney ET, Geralds JT, Tran P, et al. . Rescuing vasculature with intravenous angiopoietin-1 and alpha v beta 3 integrin peptide is protective after spinal cord injury. Brain 2010;133(Pt 4):1026–42. doi: 10.1093/brain/awq034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamauchi T, Lin Y, Sharp FR, Noble-Haeusslein LJ. Hemin induces heme oxygenase-1 in spinal cord vasculature and attenuates barrier disruption and neutrophil infiltration in the injured murine spinal cord. J Neurotrauma 2004;21(8):1017–30. doi: 10.1089/0897715041651042 [DOI] [PubMed] [Google Scholar]
- 29.Haddadi G, Shirazi A, Sepehrizadeh Z, Mahdavi SR, Haddadi M. Radioprotective effect of melatonin on the cervical spinal cord in irradiated rats. Cell J 2013;14(4):246–53. [PMC free article] [PubMed] [Google Scholar]
- 30.Carmeliet P. Angiogenesis in life, disease and medicine. Nature 2005;438(7070):932–6. doi: 10.1038/nature04478 [DOI] [PubMed] [Google Scholar]
- 31.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992;359(6398):843–5. doi: 10.1038/359843a0 [DOI] [PubMed] [Google Scholar]
- 32.Lucitti JL, Mackey JK, Morrison JC, Haigh JJ, Adams RH, Faber JE. Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and a disintegrin and metalloprotease family members 10 and 17. Circ Res 2012;111(12):1539–50. doi: 10.1161/CIRCRESAHA.112.279109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ying Z, Roy RR, Edgerton VR, Gomez-Pinilla F. Exercise restores levels of neurotrophins and synaptic plasticity following spinal cord injury. Exp Neurol 2005;193(2):411–9. doi: 10.1016/j.expneurol.2005.01.015 [DOI] [PubMed] [Google Scholar]
- 34.Vaynman S, Gomez-Pinilla F. License to run: exercise impacts functional plasticity in the intact and injured central nervous system by using neurotrophins. Neurorehabil Neural Repair 2005;19(4):283–95. doi: 10.1177/1545968305280753 [DOI] [PubMed] [Google Scholar]
- 35.Chen M, Xiang Z, Cai J. The anti-apoptotic and neuro-protective effects of human umbilical cord blood mesenchymal stem cells (hUCB-MSCs) on acute optic nerve injury is transient. Brain Res 2013;1532:63–75. doi: 10.1016/j.brainres.2013.07.037 [DOI] [PubMed] [Google Scholar]
- 36.Fouad K, Vavrek R, Cho S. A TrkB antibody agonist promotes plasticity following cervical spinal cord injury in adult rats. J Neurotrauma 2010. http://dx.doi.org/10.1089/neu.2009.1116 (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 37.Gulino R, Dimartino M, Casabona A, Lombardo SA, Perciavalle V. Synaptic plasticity modulates the spontaneous recovery of locomotion after spinal cord hemisection. Neurosci Res 2007;57(1):148–56. doi: 10.1016/j.neures.2006.10.001 [DOI] [PubMed] [Google Scholar]
- 38.Fumagalli F, Madaschi L, Caffino L, Marfia G, Di Giulio AM, Racagni G, et al. . Acute spinal cord injury reduces brain derived neurotrohic factor expression in rat hippocampus. Neuroscience 2009;159(3):936–9. doi: 10.1016/j.neuroscience.2009.01.030 [DOI] [PubMed] [Google Scholar]