

Abstract
Orexigenic hormone ghrelin and anorexic hormone obestatin are encoded by the same preproghrelin gene. While it is known that ghrelin inhibits glucose-stimulated insulin secretion (GSIS), the effect of obestatin on GSIS is unclear. Ghrelin’s effect is mediated by its receptor Growth Hormone Secretagogue Receptor (GHS-R), but the physiologically relevant receptor of obestatin remains debatable. Here we have investigated the effect of obestatin on GSIS in vitro, in vivo and ex vivo, and tested whether obestatin regulates insulin secretion through GHS-R. We found that under hyperglycemic condition, obestatin augments GSIS in rat insulinoma cells (INS-1) and in pancreatic islets from ghrelin −/− mice. Surprisingly, obestatin-induced GSIS was absent in β-cells in which GHS-R was suppressed. Obestatin-induced insulin secretion was abolished in the circulation of Ghsr −/− mice, and in pancreatic islets isolated from Ghsr −/− mice. We also found that obestatin-induced GSIS was attenuated in islets isolated from β-cell-specific Ghsr knockout MIP-Cre/ERT;Ghsrf/f mice. Our data collectively demonstrate that obestatin is a potent insulin secretagogue under hyperglycemic condition, and obestatin’s effect on insulin secretion is mediated by GHS-R in pancreatic β-cells. Our findings reveal an intriguing insight that obestatin and ghrelin have opposing effects on insulin secretion, and both are mediated through ghrelin receptor GHS-R.
Introduction
Type 2 diabetes mellitus (T2DM) is a complex multifactorial disease that is characterized by insulin resistance, loss of pancreatic β-cell mass, and diminished β-cell function. Impairment of β-cell glucose-stimulated insulin secretion (GSIS) is a hallmark of type 2 diabetes. Understanding the regulation of insulin secretion in β-cells is essential for developing therapies for T2DM. Obestatin is a 23-amino-acid anorexic hormone that has been found to exert favorable effects on glucose homeostasis by increasing β cell mass, and by reducing systemic insulin resistance and adipose tissue inflammation1–5. These beneficial effects of obestatin make it an attractive therapeutic candidate for prevention/treatment of T2DM.
Anorexic hormone obestatin is encoded by the preproghrelin gene that also encodes the orexigenic hormone ghrelin6. While high levels of obestatin are expressed in the oxyntic mucosa of the stomach, lower levels of obestatin are also expressed in pancreatic islets1, 7, 8 and some other tissues2, 9–11. In the pancreas, obestatin is expressed in ε-cells of pancreatic islets, while ghrelin is expressed in α, β and ε-cells of the pancreatic islets1, 12–16. It is known that GHS-R is expressed in α, β and δ-cells of the pancreatic islets4, 17–21. Obestatin has been shown to antagonize ghrelin’s effects on appetite, food intake, gastric emptying6, 22, as well as growth hormone secretion23, 24. Obestatin is reduced in obese humans8. It has been reported that obestatin is positively correlated with insulin sensitivity25, and obestatin protects against diet-induced insulin resistance2. More interestingly, it has been shown that obestatin increases β-cell proliferation and survival, and reduces inflammation-induced β-cell apoptosis1. Furthermore, obestatin has been shown to protect against streptozotocin (STZ)-induced β-cell failure in rats26.
We and others have reported that ghrelin inhibits insulin secretion4, 27–31, but the effect of obestatin on insulin secretion is not clear. While some studies showed that obestatin increases insulin secretion in human islets and diet-induced obese mouse islets1, others showed that obestatin inhibits insulin secretion7, 32 or has no effect on insulin secretion33–35. To date, GHS-R is the only identified physiologically relevant receptor for ghrelin; ghrelin is the only ligand that is confirmed to bind to and function through GHS-R. Since the discovery of obestatin, the receptor of obestatin has been controversial. Initially, obestatin was suggested to be the endogenous ligand of orphan G-protein-coupled receptor 39 (GPR39)6, 36. However, subsequent studies failed to confirm specific binding of obestatin to GPR3937–40. Recently, some studies suggested that obestatin regulates survival and proliferation of pancreatic β-cells via glucagon-like peptide-1 receptor (GLP-1R)1, 4, but others failed to detect the direct binding of obestatin to GLP-1R35. Interestingly, evidence does suggest that obestatin binds to a G-protein-coupled receptor (GPC-R)41–44, and GHS-R is a heterotrimeric GPC-R40. Further, it has been shown that obestatin interacts with ghrelin binding sites, and pharmacological blocking of GHS-R abolishes obestatin’s protective effects on cell survival1.
Our study aims to investigate the effect of obestatin on GSIS and determine whether obestatin’s effect on GSIS is mediated through GHS-R. First, to study the effects of obestatin on insulin secretion, we utilized rat insulinoma INS-1 cells, as well as mouse pancreatic islets from ghrelin −/− mice lacking both ghrelin and obestatin. Next, to investigate whether GHS-R mediates obestatin-induced GSIS, we used various model systems: a) in vitro system - INS-1 cells with either GHS-R pharmacologically blocked by GHS-R antagonists YIL781 & JMV2959, or Ghsr transiently suppressed by siRNA. b) in vivo system - Mice with Ghsr deleted globally (Ghsr −/−). c) ex vivo – Isolated islets from Ghsr −/− mice and β-cell-specific Ghsr knockout mice (MIP-Cre/ERT;Ghsrf/f). We demonstrated that obestatin augments insulin secretion in pancreatic β-cells under hyperglycemic condition, and obestatin-induced stimulatory effect on insulin is mediated through GHS-R. To our knowledge, this is the first set of comprehensive evidences showing that obestatin’s effect on GSIS is mediated through ghrelin receptor GHS-R.
Research Design and Methods
Animals
All experiments were approved by the Animal Care Research Committee of the Baylor College of Medicine. Animals were housed under controlled temperature and lighting (75 ± 1 °F; 12 h light-dark cycle) with free access to regular mouse chow and water. Our ghrelin −/−, Ghsr −/− mice on C57BL/6 J background were generated and characterized as we have previously described18, 45. Briefly, ghrelin −/− mice were backcrossed to C57BL/6 J background for 10 generations and Ghsr −/− mice were backcrossed for 15 generations. All mice used in the experiments were age-matched adult congenic males. WT and homozygous knockout mice (ghrelin −/−, Ghsr −/−) were housed and bred in a pathogen-free facility at Baylor College of Medicine, and all methods were performed in accordance with the relevant guidelines and regulations.
Ghsr f/f mice were originally obtained from Taconic Farms and were backcrossed as recently reported46, and then bred with MIP-Cre/ERT47 mice to generate MIP-Cre/ERT;Ghsrf/f. The Ghsr f/f and MIP-Cre/ERT;Ghsrf/f mice were gavaged five times with tamoxifen (4 mg/200 ul) in peanut oil on alternate days.
Reagents
Rat/Mouse obestatin peptide was from Phoenix Pharmaceuticals (Burlingame, CA). The GHS-R antagonists used were YIL781 (Tocris Bioscience, Bristol, UK) and JMV2959 (Calbiochem, San Diego, CA).
Cell Culture and siRNA transfection
INS-1 cells (832/13 cells) were a generous gift from Chris Newgard (Duke University)48. The cells were cultured in RPMI-1640 (Corning Incorporated, NY) containing 11.1 mM D-glucose supplemented with 10% inactivated fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate and 50 µM β-mercaptoethanol48.
For siRNA knockdown of Ghsr expression, INS-1 cells were plated in a 24-well plate at 0.5*10−6 cells/well and cultured until confluent; they were then transfected with scramble/siGhsr and Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as per the manufacturer’s instruction. Briefly, 2.5 μl of 40 μM scramble/siGhsr was added to 60 μl Opti-MEM media (Invitrogen, Carlsbad, CA) in an RNase-free tube. In a separate RNase-free tube, 2.5 μl Lipofectamine2000 (Invitrogen, Carlsbad, CA) was added to 60 μl Opti-MEM media, mixed and incubated for 5 min at room temperature. After 5 min, the scramble/siGhsr mix was combined with the Lipofectamine2000 mix to a total volume of 125 μl. The solution was mixed gently and incubated at room temperature for 20 mins. During the incubation, the RPMI-1640 medium in the 24-well plate was replaced with 125 ul low serum RPMI-1640 media containing 5% FBS and 1% INS-1 supplement without any antibiotic. After 20 min, the scramble/siRNA/Lipofectamine2000 mix was added to the wells. The final siRNA concentration is 0.4 uM in a final volume of 250 ul/well. The plate was then incubated for 4 hours at 37 °C in a humidified incubator with 5% CO2. After 4 hours, 1 ml of RPMI-1640 was added to each well and plate was incubated at 37 °C in the incubator. After overnight culture, the media was replaced with 1 ml of fresh RPMI-1640 media and incubated overnight. 48 hours after the start of transfection, the cells were harvested and used for insulin secretion or RNA/protein analysis. The siRNA sequences were designed with ABI software and synthesized by Sigma-Aldrich Scramble (Sense strand): 5′-CAACAACGAAGCGACAUAAUC-3′; Scramble (Antisense strand): 5′-UUAUGUCGCUUCGUUGUUGUC-3′ or siGhsr (Sense strand) 5′-CCACAAACAGACAGUGAAGUU-3′; siGhsr (Antisense strand): 5′-CUUCACUGUCUGUUUGUGGUU-3′.
Insulin secretion experiments
INS-1 cells (passage numbers 24–40) were plated in a 24-well plate at a density of ~0.5 × 10−6 cells/well and were grown to near confluence. For islet culture experiments, islets were collected using the collagenase method (Collagenase P, Roche, Basel, Switzerland)49 and were cultured overnight. Briefly, 3 ml of 1 mg/ml collagenase P was injected into the opening of the pancreatic duct using a 27 G needle. Pancreas was then removed and placed in 50 ml tube containing 2 ml of the collagenase solution. The tube was later placed in 37 °C water bath for 12–13 minutes with 100–120 shakings/min until the tissues were homogenously digested. 20 ml cold HBSS was subsequently added to stop the digestion and the tubes were centrifuged at 290 g for 30 seconds at 4 °C. The islets were then centrifuged using a Ficoll gradient Histopaque-1077 (Sigma-Aldrich, Kawasaki, Japan) per manufacturer’s instruction. Lastly the islets were handpicked and incubated overnight in RPMI-1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 10 mM Hepes, 2 mM L-glutamine, 1 mM Sodium-pyruvate, 0.05 mM 2-mercaptoethanol and 5.5 mM glucose. The next day, the medium was replaced with Hanks’ Balanced Salt solution (HBSS) pH 7.2, consisting of 114 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.16 mM MgSO4, 20 mM HEPES, 2.5 mM CaCl2, 25.5 mM NaHCO3 0.2% bovine serum albumin and 3.3 mM glucose for 2 hr48. For insulin secretion experiments, cells were incubated in 0.5 ml of the secretion media mentioned above, but containing the indicated glucose concentrations with or without obestatin. For GHS-R antagonist experiments, cells were incubated with 5 µM of antagonist in the HBSS buffer, containing 3.3 mM glucose for 1 hour prior to obestatin treatment; the buffer was then replaced with 500 µl of fresh HBSS containing high glucose (22.2 mM) with the indicated concentrations of obestatin and/or antagonist.
Real-time RT-PCR
Total RNA from islets was isolated using Arcturus PicoPure RNA isolation kit (ABI) following the manufacturer’s instructions. cDNA was synthesized from 250–500 ng RNA using the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Real-time RT-PCR was performed on Bio-Rad real time PCR cycler (Bio-Rad Lab., Hercules, CA) using SYBR Green PCR Master Mix according to the protocol provided by the manufacturer. Relative gene expression levels were normalized by 18S rRNA or β-Actin. The primers specific for mouse GHS-R-1a were as follows: forward primer 5′-GGACCAGAACCACAAACAGACA-3′, reverse primer 5′-CAGCAGAGGATGAAAGCAACA-3′50, and can distinguish the functional receptor GHS-R 1a from truncated receptor GHS-R 1b.
Glucose tolerance testing and Re-feeding experiments
To assess the effect of obestatin on insulin secretion in vivo, mice were fasted overnight (18 hrs) and then injected intraperitoneally (i.p.) with either 0.5 µmol/kg (1.26 mg/kg) obestatin35 or saline, and then oral glucose tolerance test (OGTT) was performed immediately by gavaging 2 g/kg glucose. Blood samples for glucose and insulin measurements were obtained at 0, 15, and 60 minutes after the administration of obestatin and glucose. For re-feeding experiments, the mice were fasted overnight (18 hrs) and were i.p. injected with either 0.5 µmol/kg obestatin or saline, and then provided with chow. Blood glucose and insulin samples were obtained at 0, 15, 30, 60 min after refeeding. Glucose was measured using OneTouch Ultra2 glucometer (LifeScan Inc., Milpitas, CA). Insulin in EDTA-treated plasma was measured using Mouse Insulin ELISA kit (Mercodia, Uppsala, Sweden) per the manufacturer’s instructions.
Statistical analysis
Graph-Pad Prism version 6.0 software was used. Two-way ANOVA with repeated measures or one-way ANOVA was used for statistical analysis. Data are represented as mean ± SEM, and P < 0.05 was considered statistically significant.
Results
Obestatin enhances glucose-stimulated insulin secretion (GSIS)
To determine the effect of obestatin on insulin secretion, we utilized the rat insulinoma INS-1 cells and the pancreatic islets isolated from our preproghrelin knockout mice (ghrelin −/−) where both ghrelin and obestatin coding sequences were deleted45. When INS-1 cells were treated with different concentrations of obestatin, the levels of insulin secretion were similar to controls at 3.3 mM low glucose condition. However, obestatin treatment significantly increased insulin secretion compared to controls at 22.2 mM glucose hyperglycemic condition (Fig. 1A). To exclude the endogenous effects of obestatin and ghrelin, we studied obestatin-induced GSIS in pancreatic islets from ghrelin −/− mice. Islets were treated with obestatin at 22.2 mM glucose condition for 1 hour. Consistent with our observation in INS-1 cells, obestatin-treated islets showed significantly higher insulin secretion (Fig. 1B). Our results collectively show that exogenous obestatin treatment augments glucose-stimulated insulin secretion in β-cells and isolated pancreatic islets.
Figure 1.
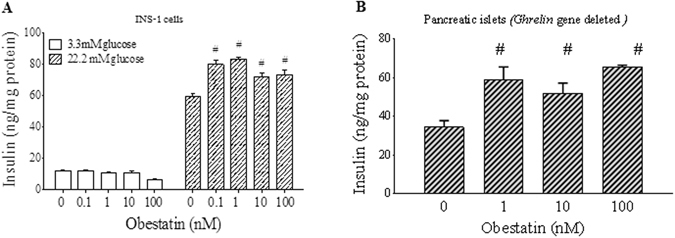
Obestatin increases glucose-stimulated insulin secretion (GSIS). β-cells or pancreatic islets were treated with different doses of obestatin in presence of 3.3 or 22.2 mM glucose for 1 hour. Insulin measured was normalized to total protein content of cells or islets. (A) INS-1 cells. (B) Pancreatic islets isolated from ghrelin −/− mice, and tested under glucose concentration of 22.2 mM. #P < 0.05 obestatin treatment vs saline treatment, per glucose concentration, n = 3–5.
The effect of obestatin on GSIS is mediated by GHS-R in β-cells
The identity of the receptor for obestatin has been elusive. In order to investigate whether the effect of obestatin on insulin secretion is mediated by GHS-R in β−cells, we pharmacologically blocked GHS-R by using GHS-R antagonists YIL781 and JMV2959 in INS-1 cells. The GHS-R antagonists alone showed no effect on insulin secretion. Treatment of 100 nM obestatin significantly increased insulin secretion from treated cells as compared to controls. However, when cells were treated with a combination of obestatin and GHS-R antagonists, the stimulatory effect of obestatin on insulin secretion was eliminated (Fig. 2A).
Figure 2.
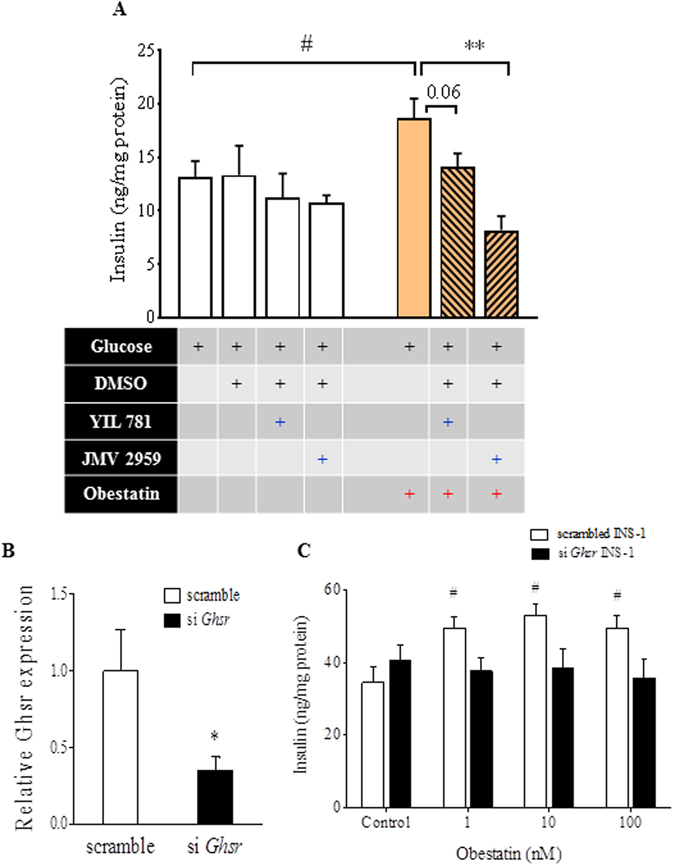
The effect of obestatin on GSIS is mediated by GHS-R in β-cells. (A) INS-1 cells were treated with 5 µM GHS-R antagonists YIL781 or JMV2959 for 1 hour, and insulin secretion was measured at 22.2 mM glucose with or without 100 nM obestatin. #P < 0.05, treatment vs control; *P < 0.05 obestatin vs obestatin with GHS-R antagonist, n = 8–14. (B) GHS-R was transiently knocked down in INS-1 cells and gene expression was measured after 48 hours. (C) Scrambled or siGhsr transfected INS-1 cells were treated with different doses of obestatin at 22.2 mM glucose for 60 minutes. Insulin was normalized to total protein content of cells. #P < 0.05 obestatin treatment vs control for each genotype, *P < 0.05 scrambled vs siGhsr, n = 5.
Next, we suppressed GHS-R expression in INS-1 cells by transiently knocking down Ghsr expression with siGHS-R. Ghsr expression in siGhsr-transfected cells was reduced by 60–70% as compared to scrambled siRNA treated cells after 48 hours (Fig. 2B). The scrambled and siGhsr-transfected cells were treated with 1 nM, 10 nM and 100 nM obestatin in the presence of 22.2 mM glucose for 1 hour. Obestatin-induced GSIS was observed with the scrambled siRNA-treated cells under different concentrations of obestatin, but not in the siGhsr-transfected cells (Fig. 2C). These results indicate that absence of GHS-R attenuates obestatin-induced GSIS in β-cells.
Obestatin-induced insulin secretion is abolished in GHS-R null mice
Our results in INS-1 cells show that absence of GHS-R attenuates obestatin-induced GSIS. To determine the implications of obestatin on insulin secretion in vivo, we further investigated obestatin-induced GSIS in Ghsr −/− mice. We treated overnight-fasted wild-type (WT) and Ghsr −/− mice with either obestatin (0.5 µmol/kg i.p) or saline, and then carried out OGTT. At 15 minutes after glucose administration: 1) obestatin-treated WT mice displayed glucose levels comparable to that of saline-treated WT mice, respectively (Fig. 3A). 2) Insulin in obestatin-treated WT mice increased from 0.37 + 0.2 ng/ml to 4.95 + 0.32 ng/ml, while insulin in saline-injected group only increased from 0.71 + 0.34 ng/ml to 2.36 + 0.38 ng/ml (Fig. 3B). Thus, obestatin-treated WT mice showed significantly higher GSIS compared to those of the saline-injected group. In contrast, glucose and insulin levels were not different between obestatin-injected Ghsr −/− mice and the saline-injected Ghsr −/− mice after 15 minutes of glucose and obestatin administration (Fig. 3C,D).
Figure 3.
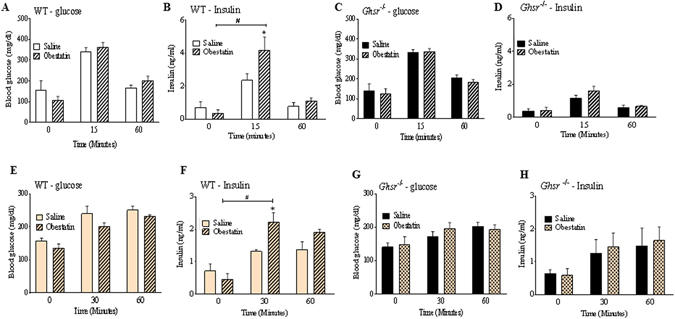
Obestatin-induced insulin secretion is abolished in GHS-R null mice. Overnight-fasted WT and Ghsr −/− mice were i.p. injected with saline or 0.5 µmol/kg obestatin, and 2 g/kg glucose was gavaged. Blood glucose in WT (A) & Ghsr −/− (C) and plasma insulin in WT (B) & Ghsr −/− (D) was measured at 0, 15 and 60 min after injection. #P < 0.05 saline/obestatin treatment at 15 min or 60 min vs saline/obestatin treatment at 0 min, *P < 0.05 saline vs obestatin for each time point, n = 3–5. Overnight-fasted WT and Ghsr−/− mice were fed Ad. Lib. chow diet after saline/obestatin injections. Blood glucose in WT (E) & Ghsr −/− (G) and plasma insulin in WT (F) and Ghsr −/− (H) was measured at 0, 30 and 60 min after injection. #P < 0.05 saline/obestatin treatment at 30 min or 60 min vs saline/obestatin treatment at 0 min, *P < 0.05 saline vs obestatin for same time point, n = 6–8.
To assess the effect of obestatin on insulin secretion under physiological condition, we also investigated the effect of obestatin on insulin secretion in response to regular chow feeding. WT and Ghsr −/− mice were fasted overnight, then i.p. injected with 0.5 µmol/kg obestatin and fed with chow. After 30 minutes of feeding, glucose levels were comparable in both WT and Ghsr −/− mice (Fig. 3E,G). Similar to OGTT, insulin levels in obestatin-treated WT mice were significantly higher when compared to the saline-treated WT mice, with insulin levels at 2.24 + 0.28 ng/ml with obestatin and 1.33 + 0.05 ng/ml with saline at 30 minute time point (Fig. 3F). However, in Ghsr −/− mice, insulin levels were similar between the obestatin- and saline-treated groups at both 30 and 60 minute time points (Fig. 3H).
These data indicate that obestatin increases insulin secretion in WT mice, but this effect is absent in Ghsr −/− mice under both oral glucose challenge and chow feeding. Also note-worthy is that the response of obestatin-induced insulin is rapid, and this effect is diminished within an hour. These studies suggest that obestatin is an import regulator of insulin secretion in vivo and GHS-R is required for obestatin’s action on insulin secretion.
Obestatin’s effect on insulin secretion is mediated directly by GHS-R in pancreatic β-cells
We next validated the necessity of GHS-R for obestatin-induced GSIS in pancreatic islets. We isolated pancreatic islets from WT and Ghsr −/− mice and treated them with obestatin at 22.2 mM glucose. Similar to our findings in the β-cells, obestatin treatment at both obestatin doses increased the insulin secretion of WT islets, but did not affect insulin secretion of Ghsr −/− islets (Fig. 4A). The data further demonstrated that GHS-R mediates obestatin-induced GSIS.
Figure 4.
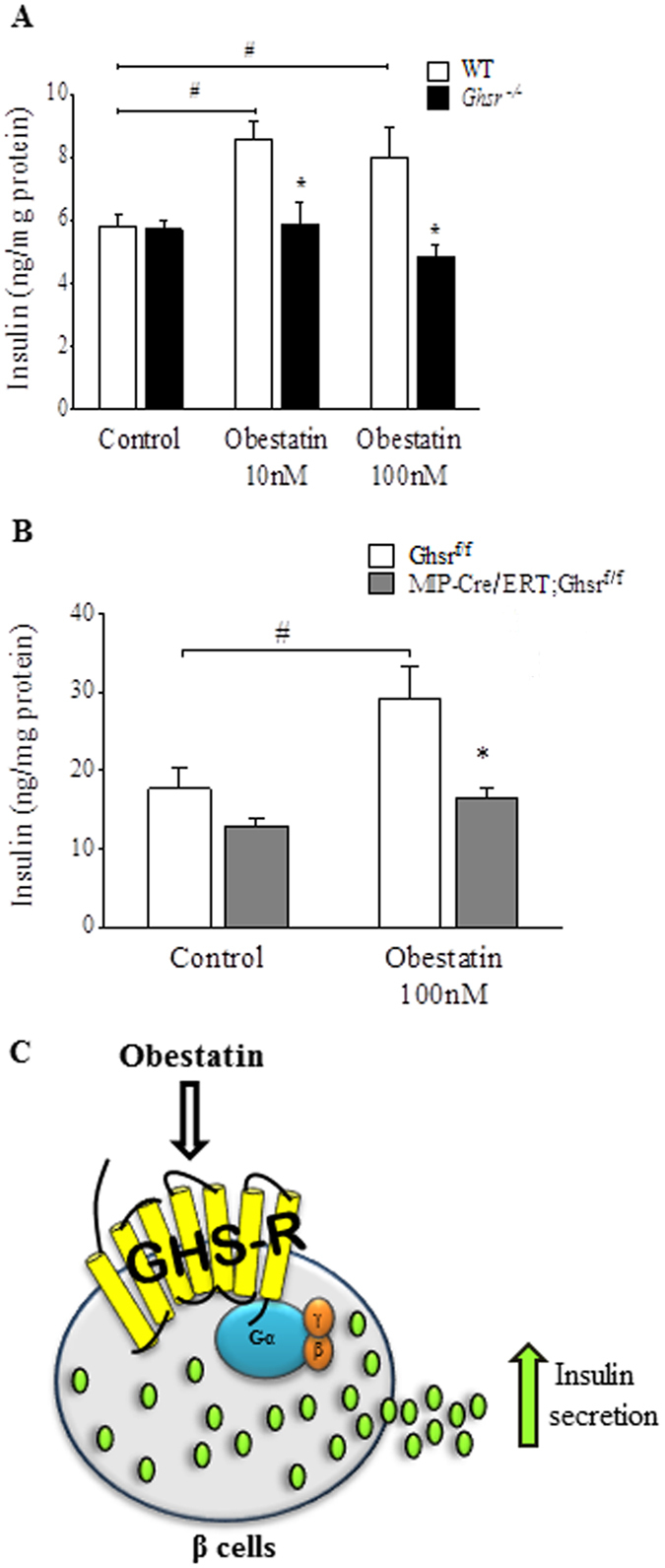
Obestatin-induced insulin secretion is mediated directly by GHS-R expression in pancreatic β-cells. (A) Pancreatic islets from Ghsr −/− mice were treated with different doses of obestatin at 22.2 mM glucose concentration for 1 hour. Insulin was normalized to total protein content of islets. #P < 0.05 obestatin treatment vs saline control for each genotype, *P < 0.05 WT vs Ghsr −/− islets, n = 7–8. (B) Pancreatic islets from Ghsrf/f and MIP-Cre/ERT;Ghsrf/f mice were treated with 100 nM Obestatin at 22.2 mM glucose condition for 1 hour. Insulin was normalized to total protein content of islets. #P < 0.05 obestatin treatment vs control for each genotype, *P < 0.05 Ghsrf/f vs MIP-Cre/ERT;Ghsrf/f islets for each treatment group, n = 6–8. (C) Schematic of obestatin augments glucose-stimulated insulin secretion via GHS-R in β-cells.
Since GHS-R is expressed in α, β and δ-cells of pancreatic islets, we further investigated whether GHS-R in pancreatic β-cells alone determines obestatin’s effect on GSIS. To this end, we generated β-cell-specific Ghsr knockout mice by breeding MIP-Cre/ERT47 with our Ghsrf/f mice51. The MIP-Cre/ERT line is widely accepted as the β-cell-specific Cre line47. To validate the mouse model, we measured Ghsr expression in brain and peripheral tissues. Indeed, the reduced Ghsr expression was detected only in islets (around 30–34%), but not in other tissues tested (Data not shown). The reduction of Ghsr expression in pancreatic islets is likely due to Ghsr deletion in the β-cells, and the remaining GHS-R expression is likely due to the intact GHS-R expression in α and δ-cells of the islets. We isolated islets from tamoxifen-induced Ghsrf/f and MIP-Cre/ERT;Ghsrf/f mice, and treated them with obestatin for 1 hour at high glucose condition. Treatment of Ghsrf/f control islets with 100 nM obestatin significantly increased insulin secretion compared to controls, whereas obestatin treatment of islets from MIP-Cre/ERT;Ghsrf/f mice exhibited no increase of insulin (Fig. 4B). These results indicate that obestatin has direct effect on pancreatic β-cells, and that GHS-R expression in β-cells is required for obestatin-induced GSIS (Fig. 4C).
Discussion
In the current study, we used a comprehensive set of in vitro, ex vivo, and in vivo systems to investigate the effect of obestatin on GSIS, and whether GHS-R mediates the effect of obestatin on insulin secretion. We have found that: 1) Obestatin augments GSIS in INS-1 β-cells and pancreatic islets; 2) Obestatin-induced GSIS is abolished by GHS-R blockade/suppression in β-cells; 3) Obestatin increases insulin secretion in WT mice, but this effect is abolished in GHS-R knockout mice; 4) Obestatin-induced GSIS is absent in pancreatic islets isolated from global and β-cell-specific GHS-R knockout mice. Taken together, our data unambiguously demonstrate that obestatin augments insulin secretion under hyperglycemic state, and the stimulatory effect of obestatin on insulin is mediated by ghrelin receptor GHS-R in pancreatic β-cells.
The effect of obestatin on insulin secretion is controversial. Qader et al. showed that obestatin inhibits insulin secretion in rat and mouse islets at 8.3 mM and 12 mM glucose concentrations, respectively32. Ren et al. showed that obestatin inhibits insulin secretion of pancreatic islets at high glucose concentration7. Egido et al. reported a dual, dose-dependent effect of obestatin on GSIS in perfused rat pancreas at 9 mM glucose: obestatin stimulated insulin secretion at low concentrations of obestatin (3.14 nM and 1 nM), but inhibited insulin secretion at a higher concentration of obestatin (10 nM)3. Several other studies have suggested that obestatin has no effect on insulin secretion7, 33–35.
Islets from the ghrelin −/− mice used in our study lack the coding sequences of both ghrelin and obestatin45, thus obestatin’s effect observed in ghrelin −/− islets is not affected by endogenous ghrelin or obestatin. The stimulatory effect of obestatin on GSIS is intact in ghrelin −/− mice, indicating that obestatin has direct effect on insulin secretion. In our study, injection of WT mice with 0.5 µmol/kg obestatin, and treatments of β-cells with 0.1–100 nM obestatin and pancreatic islets with 1–100 nM obestatin, all increased insulin secretion. Similar to our observations, Granata et al. reported that 100 nM obestatin increases insulin secretion in human islets1. Interestingly they found that obestatin has stimulatory effect on insulin at both high and low glucose concentrations in human islets1, but we only detected the effect under high glucose concentration in mouse islets. The discrepancy between high and low glucose concentrations in studies by us and Granata et al. could be due to difference in species of models studied. Granata et al. showed that the stimulatory effect of obestatin on GSIS persists under high fat diet condition2, and obestatin increases insulin in STZ-induced diabetic rats26. Recently Granata et al. also showed that obestatin enhances the generation of functional β cells in islet-like cell clusters derived from mouse pancreatic precursor cells, and obestatin increases insulin gene expression and C-peptide secretion from these clusters under glucose-stimulated condition52. These findings are well in line with our observations. The dosages of obestatin used in our study are higher than the usual physiological levels of the peptide in the blood, which range from 600 to 800 pg/ml33. However, the concentration of obestatin at the level of the islet is estimated to be much higher than its concentration in circulation, as obestatin is locally secreted from ε-cells in pancreatic islets12. It has been shown that only 70% of the circulating ghrelin originates from the stomach53, whereas the rest is derived from other tissues such as pancreas. The discrepancy between the reports from others and our data could be attributed to: a) species of origin of islets, b) different glycemic conditions and/or obestatin dose, c) the purity and potency of the peptide used in the studies, and/or d) duration of the obestatin treatment.
Similar to the debatable nature of obestatin’s effect on insulin secretion, the receptor mediating the actions of obestatin is even more elusive. Despite the detection of obestatin binding sites in various tissues such as pancreas and white adipose tissues1, 41, 54, obestatin is still considered an orphan ligand because its cognate receptor is uncertain. Originally, Zhang et al. suggested that obestatin’s actions may be mediated by GPR39 receptor, and demonstrated binding of obestatin to CHO cells expressing GPR39 receptor6, 36. Subsequent studies were not able to confirm the finding of Zhang et al. and failed to detect the activation of GPR39 with obestatin treatment38–40. Later, GLP-1R was suggested to mediate the functions of obestatin, promoting β-cell survival and proliferation1. However, experiments using GLP-1R overexpressing β-cells could not confirm the specific binding35. Interestingly, Granata et al. observed that besides GLP-1R, obestatin could also interact with acylated ghrelin binding sites, and obestatin-induced β-cell survival effect was ablated in the presence of GHS-R antagonist [D-Lys3]- GHRP61. GHS-R is a GPCR40, and several studies have suggested that obestatin likely binds and activates a GPCR41–44. It is possible that obestatin possesses low-affinity binding for GHS-R. We demonstrated that obestatin-induced GSIS in INS-1 β-cells is dependent on GHS-R expression; obestatin-induced GSIS is abolished in Ghsr −/− mice, and islets isolated from Ghsr −/− and β-cell-specific Ghsr knockout mice. These results definitively demonstrate that the stimulatory effect of obestatin on insulin secretion is mediated by GHS-R in pancreatic β-cells.
GHS-R is the only recognized biologically relevant receptor of ghrelin55 and is expressed in the brain and peripheral tissues including intestine, endocrine pancreas, and adipose tissues56. We have shown that GHS-R mediates ghrelin’s effects on food intake and growth hormone secretion18. Dezaki et al. has reported that ghrelin-induced inhibition of insulin secretion in β-cells is mediated through GHS-R57. Obestatin and ghrelin, both products of the preproghrelin gene, have opposing effects on insulin secretion as demonstrated by us and others. GSIS can be potentiated or suppressed by hormones and neural substances. Extensive research has established ghrelin’s role in the regulation of insulin release and glycemic control. Ghrelin inhibits glucose-induced insulin release in vivo, and in perfused pancreas and isolated islets57–59. Our data indicate that obestatin has an insulin-stimulatory effect under hyperglycemic condition, which is opposite from the insulin-inhibitory effect of ghrelin. Several reports show that the ratio of obestatin/ghrelin is different in anorexia nervosa and obesity60, which suggests that the obestatin/ghrelin ratio may serve as a biomarker for different nutritional states. We have reported that fasting reduces plasma obestatin, but increases ghrelin in mice61. The stimulatory effect of obestatin and the inhibitory effect of ghrelin on insulin secretion are in line with their anorexic and orexigenic properties.
It is intriguing that both ghrelin and obestatin signal through GHS-R, yet have opposite effects on insulin secretion. GHS-R activation is most commonly associated with coupling of GHS-R to G protein subunit Gαq/11 55, 62. Interestingly, it has been reported that GHS-R couples with Gαi/o in the pancreatic islets to mediate ghrelin’s inhibitory effect on GSIS63. Additionally, GHS-R has been shown to form heterodimers with other GPCRs, such as dopamine receptors DRD1, DRD264, 65, MC3R66 and 5-HT2C 67 to transmit different functions. Park et al. showed that ghrelin inhibits insulin secretion in β cells via GHS-R1a heterodimerization with Somatostatin receptor-5 (SST5), where GHS-R1a couples to Gαi/o instead of Gαq/11 68. It has been reported that GHS-R1a dimerizes with D1 to switch GHS-R1a coupling from Gαq/11 to Gαs in NPY cells of arcuate nucleus69, 70. We thus speculate that obestatin-induced GSIS in β-cells is mediated by the coupling of GHS-R1a to different G protein subunits: a) Gαq/11, to potentiate cytosolic Ca2+ levels subsequently increasing insulin secretion; b) Gαs, through heterodimerization of GHS-R with other GPCR, such as GLP-1R71, 72. While we have demonstrated that GHS-R is required for obestatin’s insulinotropic effect, we did not perform ligand binding assay or test response of obestatin in a heterologous GHS-R expression system. Thus we cannot definitively conclude that obestatin directly binds to GHS-R. Further studies are needed to test the direct binding of obestatin to GHS-R and/or obestatin-induced heterodimerization of GHS-R with other receptors. In depth understanding of the downstream signaling pathways mediating the opposing effects of ghrelin and obestatin in β-cells would be of interest as well.
It has been shown that obestatin plays important roles in the survival and function of pancreatic β-cells. Obestatin treatment improves the survival of β-cells and human islets by reducing apoptosis1. Obestatin enhances insulin sensing of pancreatic islets and streptozotocin-induced β-cells2, 32. Insulin receptor substrate 2 (IRS-2) was reported to reduce the incidence of diabetes in IRS-2-overexpressing non-obese diabetic mice73. It has been suggested that obestatin improves insulin sensitivity by increasing expression and phosphorylation of IRS-21. Our comprehensive in vitro, ex vivo and in vivo dataset collectively demonstrate that obestatin is also a potent insulin secretagogue, and the stimulatory effect of obestatin on insulin secretion is mediated by ghrelin receptor GHS-R in β-cells. Our finding of obestatin’s stimulatory effect on insulin combined with the other beneficial effects of obestatin on β-cells, makes obestatin an extremely attractive multifunctional therapeutic candidate for Type 2 diabetes.
Acknowledgements
We thank Mr. Michael R. Honig at Houston’s Community Public Radio Station KPFT for his editorial assistance. This work is supported in part by a training fellowship from the Burroughs Wellcome Fund - The Houston Laboratory and Population Science Training Program in Gene-Environment Interaction of the University of Texas Health Science Center at Houston (BWF Grant No. 1008200) to Geetali Pradhan. This study was also supported by USDA/CRIS grant 3092-5-001-059 (YS), American Heart Association grants 12IRG9230004 (YS) and 14GRNT18990019 (YS), and American Diabetes Association #1-15-BS-177 (YS). This work was also supported by the USDA National Institute of Food and Agriculture, Hatch project 1010840 (YS). The publication fee is partially supported by Texas A&M University Open Access to Knowlege Fund (OAKFund). Y.S. is the guarantor of this work, has full access to all the data in the study, and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Author Contributions
G.P., J.H.L., P.K. conducted research and analyzed data; G.P., C.-S.W., Y.S. wrote the paper; V.K.Y., S.L.S., S.G. consulted for the study and proofread the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Granata R, et al. Obestatin promotes survival of pancreatic beta-cells and human islets and induces expression of genes involved in the regulation of beta-cell mass and function. Diabetes. 2008;57:967–979. doi: 10.2337/db07-1104. [DOI] [PubMed] [Google Scholar]
- 2.Granata R, et al. Obestatin regulates adipocyte function and protects against diet-induced insulin resistance and inflammation. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26:3393–3411. doi: 10.1096/fj.11-201343. [DOI] [PubMed] [Google Scholar]
- 3.Egido EM, Hernandez R, Marco J, Silvestre RA. Effect of obestatin on insulin, glucagon and somatostatin secretion in the perfused rat pancreas. Regulatory peptides. 2009;152:61–66. doi: 10.1016/j.regpep.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Granata R, Baragli A, Settanni F, Scarlatti F, Ghigo E. Unraveling the role of the ghrelin gene peptides in the endocrine pancreas. Journal of molecular endocrinology. 2010;45:107–118. doi: 10.1677/JME-10-0019. [DOI] [PubMed] [Google Scholar]
- 5.Gesmundo I, Gallo D, Favaro E, Ghigo E, Granata R. Obestatin: a new metabolic player in the pancreas and white adipose tissue. IUBMB life. 2013;65:976–982. doi: 10.1002/iub.1226. [DOI] [PubMed] [Google Scholar]
- 6.Zhang JV, et al. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science. 2005;310:996–999. doi: 10.1126/science.1117255. [DOI] [PubMed] [Google Scholar]
- 7.Ren AJ, et al. Inhibitory effect of obestatin on glucose-induced insulin secretion in rats. Biochem Biophys Res Commun. 2008;369:969–972. doi: 10.1016/j.bbrc.2008.02.146. [DOI] [PubMed] [Google Scholar]
- 8.Zhao CM, Furnes MW, Stenstrom B, Kulseng B, Chen D. Characterization of obestatin- and ghrelin-producing cells in the gastrointestinal tract and pancreas of rats: an immunohistochemical and electron-microscopic study. Cell and tissue research. 2008;331:575–587. doi: 10.1007/s00441-007-0514-3. [DOI] [PubMed] [Google Scholar]
- 9.Volante M, et al. Obestatin in human neuroendocrine tissues and tumours: expression and effect on tumour growth. The Journal of pathology. 2009;218:458–466. doi: 10.1002/path.2551. [DOI] [PubMed] [Google Scholar]
- 10.Gronberg M, Tsolakis AV, Magnusson L, Janson ET, Saras J. Distribution of obestatin and ghrelin in human tissues: immunoreactive cells in the gastrointestinal tract, pancreas, and mammary glands. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2008;56:793–801. doi: 10.1369/jhc.2008.951145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karaoglu A, et al. Expression of obestatin and ghrelin in papillary thyroid carcinoma. Mol Cell Biochem. 2009;323:113–118. doi: 10.1007/s11010-008-9969-0. [DOI] [PubMed] [Google Scholar]
- 12.Prado CL, Pugh-Bernard AE, Elghazi L, Sosa-Pineda B, Sussel L. Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2924–2929. doi: 10.1073/pnas.0308604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Volante M, et al. Expression of ghrelin and of the GH secretagogue receptor by pancreatic islet cells and related endocrine tumors. The Journal of clinical endocrinology and metabolism. 2002;87:1300–1308. doi: 10.1210/jcem.87.3.8279. [DOI] [PubMed] [Google Scholar]
- 14.Wierup N, Yang S, McEvilly RJ, Mulder H, Sundler F. Ghrelin is expressed in a novel endocrine cell type in developing rat islets and inhibits insulin secretion from INS-1 (832/13) cells. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2004;52:301–310. doi: 10.1177/002215540405200301. [DOI] [PubMed] [Google Scholar]
- 15.Date Y, et al. Ghrelin is present in pancreatic alpha-cells of humans and rats and stimulates insulin secretion. Diabetes. 2002;51:124–129. doi: 10.2337/diabetes.51.1.124. [DOI] [PubMed] [Google Scholar]
- 16.Wierup N, Svensson H, Mulder H, Sundler F. The ghrelin cell: a novel developmentally regulated islet cell in the human pancreas. Regulatory peptides. 2002;107:63–69. doi: 10.1016/S0167-0115(02)00067-8. [DOI] [PubMed] [Google Scholar]
- 17.Anderwald-Stadler M, et al. Plasma obestatin is lower at fasting and not suppressed by insulin in insulin-resistant humans. Am J Physiol Endocrinol Metab. 2007;293:E1393–1398. doi: 10.1152/ajpendo.00330.2007. [DOI] [PubMed] [Google Scholar]
- 18.Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4679–4684. doi: 10.1073/pnas.0305930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gnanapavan S, et al. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. The Journal of clinical endocrinology and metabolism. 2002;87:2988–2988. doi: 10.1210/jcem.87.6.8739. [DOI] [PubMed] [Google Scholar]
- 20.DiGruccio MR, et al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Molecular metabolism. 2016;5:449–458. doi: 10.1016/j.molmet.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adriaenssens AE, et al. Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia. 2016;59:2156–2165. doi: 10.1007/s00125-016-4033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen CY, Asakawa A, Fujimiya M, Lee SD, Inui A. Ghrelin gene products and the regulation of food intake and gut motility. Pharmacol Rev. 2009;61:430–481. doi: 10.1124/pr.109.001958. [DOI] [PubMed] [Google Scholar]
- 23.Hassouna R, et al. A natural variant of obestatin, Q90L, inhibits ghrelin’s action on food intake and GH secretion and targets NPY and GHRH neurons in mice. PLoS One. 2012;7:e51135. doi: 10.1371/journal.pone.0051135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zizzari P, Longchamps R, Epelbaum J, Bluet-Pajot MT. Obestatin partially affects ghrelin stimulation of food intake and growth hormone secretion in rodents. Endocrinology. 2007;148:1648–1653. doi: 10.1210/en.2006-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren AJ, et al. Obestatin, obesity and diabetes. Peptides. 2009;30:439–444. doi: 10.1016/j.peptides.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Granata R, et al. Unacylated ghrelin and obestatin increase islet cell mass and prevent diabetes in streptozotocin-treated newborn rats. Journal of molecular endocrinology. 2010;45:9–17. doi: 10.1677/JME-09-0141. [DOI] [PubMed] [Google Scholar]
- 27.Reimer MK, Pacini G, Ahren B. Dose-dependent inhibition by ghrelin of insulin secretion in the mouse. Endocrinology. 2003;144:916–921. doi: 10.1210/en.2002-220819. [DOI] [PubMed] [Google Scholar]
- 28.Broglio F, et al. Ghrelin, a natural GH secretagogue produced by the stomach, induces hyperglycemia and reduces insulin secretion in humans. The Journal of clinical endocrinology and metabolism. 2001;86:5083–5086. doi: 10.1210/jcem.86.10.8098. [DOI] [PubMed] [Google Scholar]
- 29.Egido EM, Rodriguez-Gallardo J, Silvestre RA, Marco J. Inhibitory effect of ghrelin on insulin and pancreatic somatostatin secretion. European journal of endocrinology/European Federation of Endocrine Societies. 2002;146:241–244. doi: 10.1530/eje.0.1460241. [DOI] [PubMed] [Google Scholar]
- 30.Dezaki K, Yada T. Islet beta-cell ghrelin signaling for inhibition of insulin secretion. Methods in enzymology. 2012;514:317–331. doi: 10.1016/B978-0-12-381272-8.00020-9. [DOI] [PubMed] [Google Scholar]
- 31.Sun Y, Asnicar M, Saha PK, Chan L, Smith RG. Ablation of ghrelin improves the diabetic but not obese phenotype of ob/ob mice. Cell metabolism. 2006;3:379–386. doi: 10.1016/j.cmet.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 32.Qader SS, Hakanson R, Rehfeld JF, Lundquist I, Salehi A. Proghrelin-derived peptides influence the secretion of insulin, glucagon, pancreatic polypeptide and somatostatin: a study on isolated islets from mouse and rat pancreas. Regulatory peptides. 2008;146:230–237. doi: 10.1016/j.regpep.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 33.Green BD, Irwin N, Flatt PR. Direct and indirect effects of obestatin peptides on food intake and the regulation of glucose homeostasis and insulin secretion in mice. Peptides. 2007;28:981–987. doi: 10.1016/j.peptides.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 34.Kiewiet RM, Gauna C, van Aken MO, van de Zande B, van der Lely AJ. Bolus administration of obestatin does not change glucose and insulin levels neither in the systemic nor in the portal circulation of the rat. Peptides. 2008;29:2144–2149. doi: 10.1016/j.peptides.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 35.Unniappan S, Speck M, Kieffer TJ. Metabolic effects of chronic obestatin infusion in rats. Peptides. 2008;29:1354–1361. doi: 10.1016/j.peptides.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 36.Moechars D, et al. Altered gastrointestinal and metabolic function in the GPR39-obestatin receptor-knockout mouse. Gastroenterology. 2006;131:1131–1141. doi: 10.1053/j.gastro.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 37.Holst B, et al. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology. 2007;148:13–20. doi: 10.1210/en.2006-0933. [DOI] [PubMed] [Google Scholar]
- 38.Chartrel, N. et al. Comment on “Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake”. Science315, 766; author reply 766, 10.1126/science.1135047 (2007). [DOI] [PubMed]
- 39.Lauwers E, Landuyt B, Arckens L, Schoofs L, Luyten W. Obestatin does not activate orphan G protein-coupled receptor GPR39. Biochem Biophys Res Commun. 2006;351:21–25. doi: 10.1016/j.bbrc.2006.09.141. [DOI] [PubMed] [Google Scholar]
- 40.Tremblay F, et al. Normal food intake and body weight in mice lacking the G protein-coupled receptor GPR39. Endocrinology. 2007;148:501–506. doi: 10.1210/en.2006-1275. [DOI] [PubMed] [Google Scholar]
- 41.Alloatti G, et al. Obestatin affords cardioprotection to the ischemic-reperfused isolated rat heart and inhibits apoptosis in cultures of similarly stressed cardiomyocytes. American journal of physiology. Heart and circulatory physiology. 2010;299:H470–481. doi: 10.1152/ajpheart.00800.2009. [DOI] [PubMed] [Google Scholar]
- 42.Alvarez CJ, et al. Obestatin stimulates Akt signalling in gastric cancer cells through beta-arrestin-mediated epidermal growth factor receptor transactivation. Endocr Relat Cancer. 2009;16:599–611. doi: 10.1677/ERC-08-0192. [DOI] [PubMed] [Google Scholar]
- 43.Dun SL, et al. Distribution and biological activity of obestatin in the rat. The Journal of endocrinology. 2006;191:481–489. doi: 10.1677/joe.1.06944. [DOI] [PubMed] [Google Scholar]
- 44.Pazos Y, Alvarez CJ, Camina JP, Casanueva FF. Stimulation of extracellular signal-regulated kinases and proliferation in the human gastric cancer cells KATO-III by obestatin. Growth factors. 2007;25:373–381. doi: 10.1080/08977190801889313. [DOI] [PubMed] [Google Scholar]
- 45.Sun Y, Ahmed S, Smith RG. Deletion of ghrelin impairs neither growth nor appetite. Mol Cell Biol. 2003;23:7973–7981. doi: 10.1128/MCB.23.22.7973-7981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JH, et al. Neuronal Deletion of Ghrelin Receptor Almost Completely Prevents Diet-Induced Obesity. Diabetes. 2016;65:2169–2178. doi: 10.2337/db15-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wicksteed B, et al. Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010;59:3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hohmeier HE, et al. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424–430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- 49.Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolation from mouse pancreas. Nature protocols. 2009;4:1649–1652. doi: 10.1038/nprot.2009.150. [DOI] [PubMed] [Google Scholar]
- 50.Sun Y, Garcia JM, Smith RG. Ghrelin and growth hormone secretagogue receptor expression in mice during aging. Endocrinology. 2007;148:1323–1329. doi: 10.1210/en.2006-0782. [DOI] [PubMed] [Google Scholar]
- 51.Lee, J. H. et al. Neuronal deletion of ghrelin receptor almost completely prevents diet-induced obesity. Diabetes, doi:10.2337/db15-1587. [DOI] [PMC free article] [PubMed]
- 52.Baragli A, et al. Obestatin enhances in vitro generation of pancreatic islets through regulation of developmental pathways. PLoS One. 2013;8:e64374. doi: 10.1371/journal.pone.0064374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ariyasu H, et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. The Journal of clinical endocrinology and metabolism. 2001;86:4753–4758. doi: 10.1210/jcem.86.10.7885. [DOI] [PubMed] [Google Scholar]
- 54.Zhang W, Chai B, Li JY, Wang H, Mulholland MW. Effect of des-acyl ghrelin on adiposity and glucose metabolism. Endocrinology. 2008;149:4710–4716. doi: 10.1210/en.2008-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kojima M, et al. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 56.Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dezaki K, et al. Blockade of pancreatic islet-derived ghrelin enhances insulin secretion to prevent high-fat diet-induced glucose intolerance. Diabetes. 2006;55:3486–3493. doi: 10.2337/db06-0878. [DOI] [PubMed] [Google Scholar]
- 58.Yada T, et al. Ghrelin signalling in beta-cells regulates insulin secretion and blood glucose. Diabetes, obesity & metabolism. 2014;16(Suppl 1):111–117. doi: 10.1111/dom.12344. [DOI] [PubMed] [Google Scholar]
- 59.Tong J, et al. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes. 2010;59:2145–2151. doi: 10.2337/db10-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hassouna R, Zizzari P, Tolle V. The ghrelin/obestatin balance in the physiological and pathological control of growth hormone secretion, body composition and food intake. Journal of neuroendocrinology. 2010;22:793–804. doi: 10.1111/j.1365-2826.2010.02019.x. [DOI] [PubMed] [Google Scholar]
- 61.Lin L, et al. The suppression of ghrelin signaling mitigates age-associated thermogenic impairment. Aging. 2014;6:1019–1032. doi: 10.18632/aging.100706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. High constitutive signaling of the ghrelin receptor–identification of a potent inverse agonist. Mol Endocrinol. 2003;17:2201–2210. doi: 10.1210/me.2003-0069. [DOI] [PubMed] [Google Scholar]
- 63.Dezaki K, Kakei M, Yada T. Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: novel signal transduction of ghrelin. Diabetes. 2007;56:2319–2327. doi: 10.2337/db07-0345. [DOI] [PubMed] [Google Scholar]
- 64.Kern A, et al. Hippocampal Dopamine/DRD1 Signaling Dependent on the Ghrelin Receptor. Cell. 2015;163:1176–1190. doi: 10.1016/j.cell.2015.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kern A, Albarran-Zeckler R, Walsh HE, Smith RG. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron. 2012;73:317–332. doi: 10.1016/j.neuron.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rediger, A. et al. Functional relevance of MC3R and GHSR heterodimerization in hypothalamic weight regulation. Endocrine Abstracts20HTB3 (2009).
- 67.Schellekens H, et al. Ghrelin’s Orexigenic Effect Is Modulated via a Serotonin 2C Receptor Interaction. ACS chemical neuroscience. 2015;6:1186–1197. doi: 10.1021/cn500318q. [DOI] [PubMed] [Google Scholar]
- 68.Park S, Jiang H, Zhang H, Smith RG. Modification of ghrelin receptor signaling by somatostatin receptor-5 regulates insulin release. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19003–19008. doi: 10.1073/pnas.1209590109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schellekens H, van Oeffelen WE, Dinan TG, Cryan JF. Promiscuous dimerization of the growth hormone secretagogue receptor (GHS-R1a) attenuates ghrelin-mediated signaling. The Journal of biological chemistry. 2013;288:181–191. doi: 10.1074/jbc.M112.382473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kohno D, Gao HZ, Muroya S, Kikuyama S, Yada T. Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes. 2003;52:948–956. doi: 10.2337/diabetes.52.4.948. [DOI] [PubMed] [Google Scholar]
- 71.Buteau J. GLP-1 receptor signaling: effects on pancreatic beta-cell proliferation and survival. Diabetes & metabolism. 2008;34(Suppl 2):S73–77. doi: 10.1016/S1262-3636(08)73398-6. [DOI] [PubMed] [Google Scholar]
- 72.Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacology & therapeutics. 2007;113:546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Norquay LD, et al. Insulin receptor substrate-2 in beta-cells decreases diabetes in nonobese diabetic mice. Endocrinology. 2009;150:4531–4540. doi: 10.1210/en.2009-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]