

Abstract
Reactive Oxygen Species (ROS) are key mediators of ischemia-reperfusion injury but also required for the induction of the stress response that limits tissue injury and underlies the protection provided by ischemic-preconditioning protocols. Liver steatosis is an important risk factor for liver transplant failure. Liver steatosis is associated with mitochondrial dysfunction and excessive mitochondrial ROS production. Studies aiming at decreasing the sensibility of the steatotic liver to ischemia-reperfusion injury using pre-conditioning protocols, have shown that the steatotic liver has a reduced capacity to respond to these protocols. Recent studies indicate that these effects are related to a reduced capacity of the steatotic liver to respond to elevated ROS levels following reperfusion by inducing a compensatory response. This failure to respond to ROS is associated with reduced levels of antioxidants, mitochondrial damage, hepatocyte cell death, activation of the immune system and induction of pro-fibrotic mediators.
Keywords: ROS, Liver, Ischemia-reperfusion, Ischemic preconditioning, Steatosis
Graphical abstract
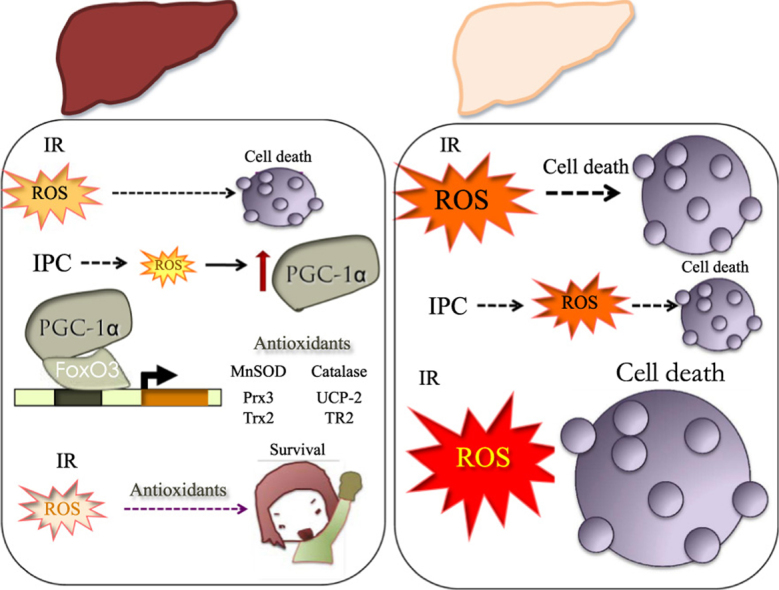
Response to IR in normal vs steatotic liver. Induction of antioxidant systems in the normal liver facilitates survival in response to preconditioning protocols. Reduced PGC-1α activity in the steatotic liver limits antioxidant induction and results in extensive hepatocyte cell death. IR, Ischemia-Reperfusion; IPC, Ischemic Preconditioning; and ROS, Reactive Oxygen Species.
1. Liver transplants
The prevalence of the steatotic liver is rapidly increasing worldwide reaching almost 30% of the total population in some countries [1], [2], with incidences rapidly rising in the pediatric population [3]. Hepatic steatosis is largely considered the hepatic manifestation of the metabolic syndrome [4], although, in fact, recent studies have shown that it actually precedes the development of metabolic syndrome [5] and its prevalence is expected to rise along with the number of overweight and obese people [6]. Traditionally considered a benign condition, a plethora of studies have more than clearly demonstrated that the steatotic liver is more sensitive than the lean liver to a vast array of pathological insults, and the most prevalent and significant risk factor for the development of NAFLD [7], [8], [9].
The particular sensitivity of the steatotic liver has been dramatically attested in the transplant field [10]. Liver and kidney are the most commonly transplanted organs. Liver transplantation is the only treatment option for end-stage liver disease, acute hepatic failure, hepatocellular carcinoma (HCC), hilar cholangiocarcinoma and several other disorders. While the number of patients requiring a liver transplant increases, the number of livers donated following cardiac death (DCD) or brain death (DBD) donated in Europe has not increased at the same pace. Furthermore, the number of livers coming from healthy young people, deceased in fatal car accidents has decreased. Organ shortage has been compensated in Asian countries with increasing numbers of living-donor liver transplantation (LDLT). In western countries the limitation has fostered the need to extend donor's criteria, in particular the degree of steatosis considered acceptable has been increased [11].
Steatosis is an important risk factor in liver surgery due to the special sensitivity of the steatotic liver to ischemia reperfusion injury (IRI), [12], which impairs liver regeneration and is a major cause of liver damage [13]. The exact mechanism responsible for the increased susceptibility is not fully understood, but clearly involves mitochondrial dysfunction. The steatotic liver produces higher levels of mitochondrial ROS [14], both baseline and in response to ischemia reperfusion (IR), reduced levels of antioxidants [15] and has a reduced capacity to recover normal ATP levels following reperfusion, leading to enhanced cell death [16]. A recent study further showed that the steatotic liver has reduced electron transport chain (ETC) complex I (CI) activity both baseline and in response to IR, and more importantly, CI activity did not recover even following long reperfusion times [13]. The CI complex is a major site of generation of superoxide in the mitochondria [17].
Despite improved allograft preservation techniques many livers continue to go unused and many studies have attempted to develop protocols that improve the safety of steatotic liver transplants with limited success. A particular focus of attention has been the ischemic preconditioning protocol (IPC) first described in the heart [18]. The liver is conditioned with a brief ischemic period followed by reperfusion, prior to sustained ischemia. IPC boost the resistance of the liver to a longer ischemic insult. It has been used and tested in many experimental and some clinical contexts but so far it has not shown general clinical benefit and its use is not clinically implemented [19]. The molecular pathways involved in IPC have been extensively characterized. IPC suppresses neutrophil infiltration, decreases the production of oxygen free radicals and increases antioxidant activity in the hepatocyte, prevents the activation of the apoptotic cascade and the occlusion of microvessels [20]. Importantly, it has been suggested that all these effects are mediated through the protection of mitochondrial function [21].
2. Ischemic-preconditioning
Among the many possible strategies to rescue steatotic livers before transplantation in clinical and experimental studies [22], important efforts have focused on the applicability of IPC protocols to the steatotic liver that would allow their use in transplants [23]. However, despite some limited improvement in some liver damage biomarkers (ie ATL), the lack of significant positive effect in clinical outcome have so far prevented its translation to the clinical setting, possibly related to the failure to recover the mitochondrial CI activity noted for the steatotic liver following IR [13] and the associated loss in ROS homeostasis.
Evidence for oxidative stress might seem an almost irrelevant or non-specific finding, since there is a plethora of pathological conditions associated with oxidative stress, in fact all that involve inflammation, that the correlation may seem of little clinical relevance. However, accumulated data indicate otherwise. Increased ROS levels following the reperfusion phase have been shown to be the main triggering factor responsible for the induction of hepatocyte cell death following IR, and are therefore, the key mediators of acute liver failure following liver transplant [24], [25]. Furthermore, sustained low-grade oxidative stress has been proposed to promote pathological progression to liver fibrosis and could be associated to an increased risk of mid-term graft failure [26].
During the ischemic phase, ATP depletion is compensated through the increase in the anaerobic catabolism of glucose, producing an increase in lactate levels and a drop in the intracellular pH. This drop activates the Na+/H+ anti-porter, and the ensuing increase in intracellular Na+ levels cannot be compensated due to the limiting ATP concentrations, leading to an increase in the intracellular levels of Ca2+, and facilitates the activation of pro-apoptotic pathways. Once perfusion is restored the sudden increase in O2 levels produces a mitochondrial burst of ROS, that together with the elevated Ca2+ and the low pH can induce the opening of the MTP and result in cell apoptosis [27].
Following reperfusion, ROS induce inflammation in two phases, an initial or acute phase, and a late or sub-acute phase [28]. The acute phase corresponds to the first 6 h following reperfusion, in this stage mitochondrial ROS seem essential for the activation of the Kuppfer cells, that will later mediate the release of proinflammatory cytokines [29].
The sub-acute phase is the inflammatory stage, characterized by a massive infiltration of neutrophils, release of cytotoxic and proinflammatory mediators, and activation of the mesenchymal stem cells, that can contribute to the development offibrosis. During this stage, the main source of ROS are the NOX enzymes and work as amplifiers of the inflammatory reaction [26].
At the molecular level, elevated ROS levels boost the inflammatory cascade, at least partially through the direct activation of the transcription factor NF-κB [30], although there are other indirect mechanisms through which ROS can also promote a pro-inflammatory status increasing the levels/activity of TGF-β, TNFα and IL-1ß among others [31].
3. ROS, preconditionong mediators
However, ROS do not only have deleterious effects, ROS dependent signaling is a crucial mediator of ischemic preconditioning. Therefore, increased ROS production following IR is a necessary step during liver preconditioning [32]. ROS trigger many cellular responses that could potentially contribute to ischemic preconditioning. In particular, ROS facilitates the activation of AMPK and stabilization of the transcription factor HIF-1α, resulting in a shift in the cellular metabolism, that makes the liver more glycolytic and less dependent on O2. This compensatory effect facilitates survival immediately following reperfusion, since during reperfusion not all the tissue recovers immediately a normal oxygen tension and capillaries tend to collapse [33]. Recovery of normal tissue perfusion depends on the formation of new microvessels, a process that can take about two weeks to be successfully completed and that largely depends on the HIF-1α dependent activation of the angiogenesis mediator VEGF-A [34], [35].
The picture that emerges is that ROS play a double edged role in IR, on one hand promotes apoptotic cell death and induces inflammatory mediators, on the other hand, facilitates cell survival in hypoxic conditions and induces antioxidant defenses.
ROS dependent IRI is to a large extent dependent on the tissue levels of antioxidants. In fact, reduced levels of the antioxidant proteins MnSOD, catalase and GPx have been associated with larger lesions while forced increase of antioxidants has the opposite effect [36], [37]. Conversely, ROS exposure can result in the induction/activation of antioxidant systems through the activation of the transcription factors nuclear factor erythroid 2-related factor 2 (Nrf2) [38], [39] and the transcriptional coactivator PGC-1α have been shown to be activated/induced in response to oxidative stress and drive the increased expression of antioxidant systems in hepatocytes [40], [41], [42].
Nrf2 is considered a master regulator of cellular redox homeostasis [39]. Under normal conditions Keap1 binds to Nrf2 in the cytoplasm, and promotes its degradation by the proteasome. Exposure to ROS leads to Keap1, Nrf2 stabilization and nuclear translocation where it binds to the anti-oxidant response element (ARE) to induce the expression a set of antioxidant genes [43]. Is has been proposed that following IR induction of Nrf2 plays a key role in the control of the inflammatory response, reducing apoptotic cell death [44].
PGC-1α is a master regulator of mitochondrial biogenesis and activity [45]. Oxidative catabolism is associated with increased mitochondrial production of superoxide. To prevent oxidative stress in conditions of high mitochondrial oxidative activity PGC-1α also controls the expression of several antioxidant proteins [41]. PGC-1α is highly expressed in the liver where it controls both oxidative catabolism and gluconeogenesis in response to reduced nutrient availability [46]. PGC-1α levels are highly sensitive to the nutritional status being induced by starvation and inhibited by feeding [47]. Importantly, in the steatotic liver PGC-1α levels are reduced [48] and this downregulation results in a reduction of antioxidant systems in the liver [49]. This fact, together with the observation of a protective role of PGC-1α in IR tolerance in the liver [49] and several other tissues (ie heart, kidney, the central nervous system (CNS)) [50], [51], [52] suggest that PGC-1α regulation of antioxidant systems can be relevant in the enhanced sensitivity of the steatotic liver to IR. This concept is supported by the observation that in terms of liver damage following IR, PGC-1α KO mice behave similarly to WT mice with steatotic livers [49].
4. ROS homeostasis
In order to elucidate the reason why the steatotic liver, that produces more ROS than the normal liver, cannot precondition, some studies have focused on the evaluation of the activity of antioxidant enzymes and found conflicting results, with some enzymes being over-expressed in the steatotic liver and some others with reduced levels in steatosis [15]. Importantly, several studies have shown the positive effects of antioxidants against IR injury [53] suggesting that the steatotic liver has lost its capacity to modulate the production of ROS and hence of using ROS as signaling mediators. The study by Sanchez Ramos et al. [49] showed that while the normal liver responds to IR inducing PGC-1α and antioxidant gene expression, and this induction is further enhanced by preconditioning, the steatotic liver shows a significant reduction in its capacity to induce antioxidant gene expression in response to IR and IPC protocols. Furthermore, the study supports the notion that this induction depends on PGC-1α, since PGC-1α KO mice also fail to induce PGC-1α and antioxidant gene expression in response to IR.
The work by C. Sanchez et al. has demonstrated that the down-regulation of the transcriptional co-activator PGC-1α in the steatotic liver limits its capacity to control antioxidant enzyme levels and to modulate them in response to IR. In the healthy liver, in response to IR, PGC-1α levels are induced and this induction results in the augmentation of the cellular antioxidant defenses. In steatosis, not only PGC-1α levels are reduced but the steatotic liver fails to induce PGC-1α and its target antioxidant genes in response to IR, resulting in enhanced liver damage. Aiming to understand why PGC-1α levels are regulated in response to IR, the same study analyzed why the steatotic liver fail to induce them in response to IR.
5. ROS control following hypoxia
It is not at all clear how ROS work in preconditioning to protect the mitochondria from depolarization following IR and how do they work to attenuate the inflammatory response. Recent identification of NDUFA4L2 (NADH dehydrogenase [ubiquinone] 1 alpha subcomplex, 4-like 2) as an hypoxia sensitive inhibitor of complex I has served to demonstrate the relevance of a pathway that controls mitochondrial ROS production in response to ischemia [54]. Taking into account that NDUFA4L2 is a HIF-1α induced protein, HIF-1α stabilization is ROS sensitive [55] and HIF-1α is induced following IR in the liver [56], it is tempting to speculate that its induction must also be ROS sensitive, what would provide a mechanism that could explain ROS dependent mitochondrial protection. An important open question is what happens in the fatty liver, where ROS are produced but do not seem to exert their protective effects while they do exert their damaging effects. Activation of HIF-1α does not seem to be impaired in the steatotic liver, in fact HIF-1α activity is higher in the fatty liver than in the normal liver [57], [58].
PGC-1α levels are also regulated by oxygen tension [59]. This regulation could seem a paradoxical effect, since oxidative metabolism requires oxygen and limiting oxygen availability may result in mitochondrial dysfunction. The physiological relevance of this regulation seems related to the PGC-1α dependent induction of VEGF-A levels that promote angiogenesis to increase tissue perfusion and allow oxidative metabolism [60]. However, C. Sanchez and col. [49] showed that severe hypoxic conditions actually down-regulate PGC-1α levels at least in cell culture conditions while restoration of normal oxygen tension results in the induction of PGC-1α levels, indicating that induction of PGC-1α following IR in vivo, is likely to occur during the reperfusion phase.
Taking into account that reperfusion is associated with a burst in mitochondrial ROS, and that is has been previously demonstrated that ROS induce PGC-1α expression, is likely that ROS are key mediators in PGC-1α induction following IR, and the lack of an adaptive response in the steatotic liver also relate to the inability in this context of ROS to induce PGC-1α (Fig. 1).
Fig. 1.

Response to IR in normal vs steatotic liver. Induction of antioxidant systems in the normal liver facilitates survival in response to preconditioning protocols. Reduced PGC-1α activity in the steatotic liver limits antioxidant induction and results in extensive hepatocyte cell death. IR, Ischemia-Reperfusion; IPC, Ischemic Preconditioning; and ROS, Reactive Oxygen Species.
The association of liver steatosis with oxidative stress results in a low-grade chronic inflammatory profile, that is generally associated with poor inflammatory resolution profiles, with a limited M2 macrophage activation [61], [62]. In contrast, acute exposure to ROS promotes concomitant activation of M1 and M2 macrophages and expression of resolution cytokines [63], [64]. Although the mechanisms are only partially elucidated, these effects could be also relevant in ROS dependent preconditioning effects and its failure in the steatotic liver. Importantly, M2 activation has been shown to be strongly dependent on the activation of oxidative phosphorylation and mitochondrial function [65], feasibly via PGC-1α.
6. Translation into the clinical setting
The challenge now is to translate this knowledge into clinical applications. There are some clinical studies in human patients focused in the role of oxidative stress in IRI and postoperative complications in patients after an orthotopic liver transplant (OLT). Since we are not able to measure the levels of ROS in humans due to their short lifetime, others strategies are followed in these clinical studies, as measure oxidative markers, such as lipid peroxidation; the levels of glutathione or enzymatic antioxidants, such as superoxide dismutase (SOD), catalase or peroxiredoxins in blood samples or liver biopsies [66].
Recent clinical researches show that also in humans during ischemia the amounts of ROS increase resulting in oxidative stress imbalance as evidenced by an increase in oxidative stress markers and antioxidant response [67], [68], [69] and this oxidative imbalance persist in late post-operative stage (1 year after liver transplant) [70]. Additionally, as noted above, steatotic livers have poor prognosis in liver transplant that is associated to evidence of mitochondrial dysfunction and enhanced production of ROS [71].
The gene expression profile of transplanted liver versus normal liver shows an induction of stress response genes in transplanted livers, as well as an increase of liver metabolism and inflammatory response. When transplanted livers subjects were analyzed for the correlation of transaminase levels, as hepatic injury marker and the expression of oxidative stress genes, it was found that oxidative stress response significantly correlated with liver injury [72] most probably due to the generation of higher amounts of ROS in these livers. Related to this study, it has also been described that the levels of lipid peroxidation increase at 1 h post-operation and are maintained over basal levels up to 5 days following liver transplant. Lipid peroxidation in turn correlates with the upregulation of enzymatic antioxidants such as superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx) and glutathione reductase (GR) [69]. Furthermore, the level of lipid peroxidation also correlates with pre-operative hepatic injury markers such as bilirubin, AST, ALT, model of end-stage liver disease score (MELD score), International Normalized Ratio (INR) and with expression of pro-inflammatory cytokines [73].
From this perspective, several strategies have been devised to decrease hepatic transplant failure due to oxidative stress trying to avoid the generation of ROS during ischemia and reperfusion. These strategies include ischemic preconditioning (IPC), antioxidant therapy and graft preservation in external perfusion machines. Even though the protective effect of IPC in IRI has been amply demonstrated in pre-clinical studies, clinical studies have shown conflicting results in normal and steatotic livers [23], [74]. Some studies support the protective effect of IPC in human liver ischemia [75], [76], [77] even in steatotic livers, while other studies indicate that IPC do not have clinical relevance in liver transplantation [78], [79] suggesting that new approaches are needed to facilitate the use of IPC protocols in the clinical setting.
Early evidence on the role of ROS in IR injury led to the evaluation of antioxidant administration as a possible therapeutical approach. Glutathione is the major antioxidant system and some precursors of glutathione have been evaluated as IRI treatment, such as N-Acetylcysteine (NAC) or L-Alanyl-Glutamine with conflicting results. NAC was described as a possible antioxidant treatment against IRI [80] but failed when is administrated during graft cold preservation [81], [82], moreover, other studies do not support the protective role of NAC in IRI [83]. In a meta analysis of several clinical studies on NAC treatment the authors concluded that NAC administration improved transaminase levels but without patient or graft survival correlation [84]. L-Alanyl-Glutamine has promising results decreasing lipid peroxidation after liver transplantation [85] but more clinical results are required. Another antioxidant tested for IRI is melatonin, an antioxidant produced in the pineal gland that showed a protective effect against IRI, supraphysiological doses of melatonin improve the general outcome after liver resection [86], [87]. Finally, there is a study on Propofol, an anesthetic compound with antioxidant features, in IRI of liver transplant recipients. The authors showed an improvement on lipids peroxidation in the recipients anesthetized with Propofol [88]. Probably, antioxidant therapy could improve their efficacy with a synergistic approach, using antioxidants both in recipients and grafts without forgetting the important role of ROS in cell signaling.
Graft preservation during ischemia is a critical step in liver transplant, since high amounts of ROS are produced at this stage [89]. Storage solutions are a good way to treat ROS production during graft ischemia. A review of different preservation solutions correlates poor prognosis with those livers preserved in solutions with less antioxidants [90]. In other studies, the authors attribute the protection effect of Institut Georges Lopez preservation solution (IGL-1) on their antioxidant effect, decreasing ROS levels and mitochondrial damage [91]. In addition, storage solution supplemented with a recombinant Manganese Superoxide Dismutase (rMnSOD) showed efficiency reducing O−2 levels in human liver biopsies from donors of hepatic transplantation [92].
In the last decade a novel technology, a hypothermic preservation machine, was developed and implemented to avoid the deleterious effects of ischemia in organs transplantation. First studied in kidney grafts, some liver studies with hypothermic machine preservation (HMP) were realized. HMP showed an improvement of graft function after transplantation, with less hepatic injury markers [93] and later were described the increase of several antioxidants markers, as upregulation of SOD or HIFα [94]. Regarding the temperature storage in the preservation machine, an improvement of mitochondrial function of excluded livers for transplantation was described [95].
7. Conclusions
In sum, oxidative stress has a harmful effect in hepatic IRI in humans as seen in animal models. Limiting ROS production with different approaches or with a synergistic approach may improve the graft function and recipient survival. However, antioxidant treatments aiming at reducing liver injury have not reached the clinical stage. This failure could be related to the fact that ROS are signaling mediators, whose absence can be as detrimental as their excess. In fact, antioxidant treatment prevents ischemic preconditioning, which can also result from the “dual” nature of many antioxidants. They are in fact molecules that can also work as pro-oxidants depending on the redox status of the environment, being strongly modulated by the levels of reducing equivalents, oxygen, pH and they can also propagate pro-oxidant effects. The obvious alternative, the induction of PGC-1α levels also has strong caveats, as demonstrated by studies where over-expression of PGC-1α resulted in the loss of metabolic plasticity. For example, the tolerance of a tissue to hypoxia depends on its ability to sustain a glycolytic non-oxidative metabolism, and forced mitochondrial activity would result in mitochondrial induced cell death.
Author disclosure statement
No competing financial interests exist.
Acknowledgements
This work was supported by grants from the Spanish ‘‘Ministerio de Economía y Competitividad’’ (Grant numbers SAF2015-63904-R and SAF2015-71521-REDC).
References
- 1.Bellentani S., Saccoccio G., Masutti F., Croce L.S., Brandi G., Sasso F., Cristanini G., Tiribelli C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med. 2000;132:112–117. doi: 10.7326/0003-4819-132-2-200001180-00004. [DOI] [PubMed] [Google Scholar]
- 2.Browning J.D., Szczepaniak L.S., Dobbins R., Nuremberg P., Horton J.D., Cohen J.C., Grundy S.M., Hobbs H.H. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 3.Navarro-Jarabo J.M., Ubina-Aznar E., Tapia-Ceballos L., Ortiz-Cuevas C., Perez-Aisa M.A., Rivas-Ruiz F., Andrade R.J., Perea-Milla E. Hepatic steatosis and severity-related factors in obese children. J. Gastroenterol. Hepatol. 2013;28:1532–1538. doi: 10.1111/jgh.12276. [DOI] [PubMed] [Google Scholar]
- 4.Angulo P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 5.Lonardo A., Ballestri S., Marchesini G., Angulo P., Loria P. Nonalcoholic fatty liver disease: a precursor of the metabolic syndrome. Dig. Liver Dis. 2015;47:181–190. doi: 10.1016/j.dld.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 6.Desroches S., Lamarche B. The evolving definitions and increasing prevalence of the metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007;32:23–32. doi: 10.1139/h06-095. [DOI] [PubMed] [Google Scholar]
- 7.Koo S.H. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013;19:210–215. doi: 10.3350/cmh.2013.19.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Persico M., Iolascon A. Steatosis as a co-factor in chronic liver diseases. World J. Gastroenterol. 2010;16:1171–1176. doi: 10.3748/wjg.v16.i10.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powell E.E., Jonsson J.R., Clouston A.D. Steatosis: co-factor in other liver diseases. Hepatology. 2005;42:5–13. doi: 10.1002/hep.20750. [DOI] [PubMed] [Google Scholar]
- 10.Nocito A., El-Badry A.M., Clavien P.A. When is steatosis too much for transplantation? J. Hepatol. 2006;45:494–499. doi: 10.1016/j.jhep.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 11.Jadlowiec C.C., Taner T. Liver transplantation: current status and challenges. World J. Gastroenterol. 2016;22:4438–4445. doi: 10.3748/wjg.v22.i18.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vetelainen R., van Vliet A., Gouma D.J., van Gulik T.M. Steatosis as a risk factor in liver surgery. Ann. Surg. 2007;245:20–30. doi: 10.1097/01.sla.0000225113.88433.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu M.J., Premkumar R., Hickey A.J., Jiang Y., Delahunt B., Phillips A.R., Bartlett A.S. Steatotic livers are susceptible to normothermic ischemia-reperfusion injury from mitochondrial complex-I dysfunction. World J. Gastroenterol. 2016;22:4673–4684. doi: 10.3748/wjg.v22.i19.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selzner M., Rudiger H.A., Sindram D., Madden J., Clavien P.A. Mechanisms of ischemic injury are different in the steatotic and normal rat liver. Hepatology. 2000;32:1280–1288. doi: 10.1053/jhep.2000.20528. [DOI] [PubMed] [Google Scholar]
- 15.Liu W., Baker S.S., Baker R.D., Zhu L. Antioxidant mechanisms in nonalcoholic fatty liver disease. Curr. Drug Targets. 2015;16:1301–1314. doi: 10.2174/1389450116666150427155342. [DOI] [PubMed] [Google Scholar]
- 16.Caraceni P., Domenicali M., Vendemiale G., Grattagliano I., Pertosa A., Nardo B., Morselli-Labate A.M., Trevisani F., Palasciano G., Altomare E., Bernardi M. The reduced tolerance of rat fatty liver to ischemia reperfusion is associated with mitochondrial oxidative injury. J. Surg. Res. 2005;124:160–168. doi: 10.1016/j.jss.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Grivennikova V.G., Vinogradov A.D. Generation of superoxide by the mitochondrial complex I. Biochim. Biophys. Acta. 2006;1757:553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 18.Murry C.E., Jennings R.B., Reimer K.A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 19.Gurusamy K.S., Gonzalez H.D., Davidson B.R. Current protective strategies in liver surgery. World J. Gastroenterol. 2010;16:6098–6103. doi: 10.3748/wjg.v16.i48.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Datta G., Fuller B.J., Davidson B.R. Molecular mechanisms of liver ischemia reperfusion injury: insights from transgenic knockout models. World J. Gastroenterol. 2013;19:1683–1698. doi: 10.3748/wjg.v19.i11.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rolo A.P., Teodoro J.S., Peralta C., Rosello-Catafau J., Palmeira C.M. Prevention of I/R injury in fatty livers by ischemic preconditioning is associated with increased mitochondrial tolerance: the key role of ATPsynthase and mitochondrial permeability transition. Transpl. Int. 2009;22:1081–1090. doi: 10.1111/j.1432-2277.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q., Izamis M.L., Xu H., Berendsen T., Yarmush M., Uygun K. Strategies to rescue steatotic livers before transplantation in clinical and experimental studies. World J. Gastroenterol. 2013;19:4638–4650. doi: 10.3748/wjg.v19.i29.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu M.J., Vather R., Hickey A.J., Phillips A.R., Bartlett A.S. Impact of ischaemic preconditioning on experimental steatotic livers following hepatic ischaemia-reperfusion injury: a systematic review. HPB. 2015;17:1–10. doi: 10.1111/hpb.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reiniers M.J., van Golen R.F., van Gulik T.M., Heger M. Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver. Antioxid. Redox Signal. 2014;21:1119–1142. doi: 10.1089/ars.2013.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun K., Liu Z.S., Sun Q. Role of mitochondria in cell apoptosis during hepatic ischemia-reperfusion injury and protective effect of ischemic postconditioning. World J. Gastroenterol. 2004;10:1934–1938. doi: 10.3748/wjg.v10.i13.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richter K., Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res. 2016;365:591–605. doi: 10.1007/s00441-016-2445-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vardanian A.J., Busuttil R.W., Kupiec-Weglinski J.W. Molecular mediators of liver ischemia and reperfusion injury: a brief review. Mol. Med. 2008;14:337–345. doi: 10.2119/2007-00134.Vardanian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elias-Miro M., Jimenez-Castro M.B., Rodes J., Peralta C. Current knowledge on oxidative stress in hepatic ischemia/reperfusion. Free Radic. Res. 2013;47:555–568. doi: 10.3109/10715762.2013.811721. [DOI] [PubMed] [Google Scholar]
- 29.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. J. Gastroenterol. Hepatol. 2011;26(Suppl 1):173–179. doi: 10.1111/j.1440-1746.2010.06592.x. [DOI] [PubMed] [Google Scholar]
- 30.Nichols T.C. NF-kappaB and reperfusion injury. Drug News Perspect. 2004;17:99–104. doi: 10.1358/dnp.2004.17.2.829042. [DOI] [PubMed] [Google Scholar]
- 31.Mukhopadhyay P., Horvath B., Zsengeller Z., Batkai S., Cao Z., Kechrid M., Holovac E., Erdelyi K., Tanchian G., Liaudet L., Stillman I.E., Joseph J., Kalyanaraman B., Pacher P. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants. Free Radic. Biol. Med. 2012;53:1123–1138. doi: 10.1016/j.freeradbiomed.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alchera E., Dal Ponte C., Imarisio C., Albano E., Carini R. Molecular mechanisms of liver preconditioning. World J. Gastroenterol. 2010;16:6058–6067. doi: 10.3748/wjg.v16.i48.6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyashita T., Nakanuma S., Ahmed A.K., Makino I., Hayashi H., Oyama K., Nakagawara H., Tajima H., Takamura H., Ninomiya I., Fushida S., Harmon J.W., Ohta T. Ischemia reperfusion-facilitated sinusoidal endothelial cell injury in liver transplantation and the resulting impact of extravasated platelet aggregation. Eur. Surg. 2016;48:92–98. doi: 10.1007/s10353-015-0363-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bockhorn M., Goralski M., Prokofiev D., Dammann P., Grunewald P., Trippler M., Biglarnia A., Kamler M., Niehues E.M., Frilling A., Broelsch C.E., Schlaak J.F. VEGF is important for early liver regeneration after partial hepatectomy. J. Surg. Res. 2007;138:291–299. doi: 10.1016/j.jss.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 35.Knudsen A.R., Kannerup A.S., Gronbaek H., Andersen K.J., Funch-Jensen P., Frystyk J., Flyvbjerg A., Mortensen F.V. Effects of ischemic pre- and postconditioning on HIF-1alpha, VEGF and TGF-beta expression after warm ischemia and reperfusion in the rat liver. Comp. Hepatol. 2011;10:3. doi: 10.1186/1476-5926-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka J., Malchesky P.S., Omokawa S., Goldcamp J.B., Harasaki H., Vogt D.P., Broughan T.A., Nose Y. Effects of prostaglandin I2, superoxide dismutase, and catalase on ischemia-reperfusion injury in liver transplantation. ASAIO Trans. 1990;36:M600–603. [PubMed] [Google Scholar]
- 37.Amersi F., Buelow R., Kato H., Ke B., Coito A.J., Shen X.D., Zhao D., Zaky J., Melinek J., Lassman C.R., Kolls J.K., Alam J., Ritter T., Volk H.D., Farmer D.G., Ghobrial R.M., Busuttil R.W., Kupiec-Weglinski J.W. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J. Clin. Investig. 1999;104:1631–1639. doi: 10.1172/JCI7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kudoh K., Uchinami H., Yoshioka M., Seki E., Yamamoto Y. Nrf2 activation protects the liver from ischemia/reperfusion injury in mice. Ann. Surg. 2014;260:118–127. doi: 10.1097/SLA.0000000000000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ke B., Shen X.D., Zhang Y., Ji H., Gao F., Yue S., Kamo N., Zhai Y., Yamamoto M., Busuttil R.W., Kupiec-Weglinski J.W. KEAP1-NRF2 complex in ischemia-induced hepatocellular damage of mouse liver transplants. J. Hepatol. 2013;59:1200–1207. doi: 10.1016/j.jhep.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jager S., Handschin C., Zheng K., Lin J., Yang W., Simon D.K., Bachoo R., Spiegelman B.M. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 41.Valle I., Alvarez-Barrientos A., Arza E., Lamas S., Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez-Ramos C., Tierrez A., Fabregat-Andres O., Wild B., Sanchez-Cabo F., Arduini A., Dopazo A., Monsalve M. PGC-1alpha regulates translocated in liposarcoma activity: role in oxidative stress gene expression. Antioxid. Redox Signal. 2011;15:325–337. doi: 10.1089/ars.2010.3643. [DOI] [PubMed] [Google Scholar]
- 43.Krajka-Kuzniak V., Paluszczak J., Baer-Dubowska W. The Nrf2-ARE signaling pathway: an update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2016;69:393–402. doi: 10.1016/j.pharep.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 44.Rao J., Qian X., Li G., Pan X., Zhang C., Zhang F., Zhai Y., Wang X., Lu L. ATF3-mediated NRF2/HO-1 signaling regulates TLR4 innate immune responses in mouse liver ischemia/reperfusion injury. Am. J. Transplant. 2015;15:76–87. doi: 10.1111/ajt.12954. [DOI] [PubMed] [Google Scholar]
- 45.Summermatter S., Handschin C. PGC-1alpha and exercise in the control of body weight. Int. J. Obes. 2012;36:1428–1435. doi: 10.1038/ijo.2012.12. [DOI] [PubMed] [Google Scholar]
- 46.Herzig S., Long F., Jhala U.S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P., Spiegelman B., Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 47.Barroso E., Rodriguez-Calvo R., Serrano-Marco L., Astudillo A.M., Balsinde J., Palomer X., Vazquez-Carrera M. The PPARbeta/delta activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1alpha-Lipin 1-PPARalpha pathway leading to increased fatty acid oxidation. Endocrinology. 2011;152:1848–1859. doi: 10.1210/en.2010-1468. [DOI] [PubMed] [Google Scholar]
- 48.Aharoni-Simon M., Hann-Obercyger M., Pen S., Madar Z., Tirosh O. Fatty liver is associated with impaired activity of PPARgamma-coactivator 1alpha (PGC1alpha) and mitochondrial biogenesis in mice. Lab Investig. 2011;91:1018–1028. doi: 10.1038/labinvest.2011.55. [DOI] [PubMed] [Google Scholar]
- 49.Sanchez Ramos C., Prieto I., Tierrez A., Laso J., Valdecantos M.P., Bartrons R., Rosello-Catafau J., Monsalve M. PGC-1alpha downregulation in the steatotic liver enhances ischemia-reperfusion injury and impairs ischemic preconditioning. Antioxid. Redox Signal. 2017 doi: 10.1089/ars.2016.6836. [DOI] [PubMed] [Google Scholar]
- 50.Funk J.A., Schnellmann R.G. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol. Appl. Pharmacol. 2013;273:345–354. doi: 10.1016/j.taap.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gehrau R.C., Mas V.R., Dumur C.I., Suh J.L., Sharma A.K., Cathro H.P., Maluf D.G. Donor hepatic steatosis induce exacerbated ischemia-reperfusion injury through activation of innate immune response molecular pathways. Transplantation. 2015;99:2523–2533. doi: 10.1097/TP.0000000000000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo Y., Zhu W., Jia J., Zhang C., Xu Y. NMDA receptor dependent PGC-1alpha up-regulation protects the cortical neuron against oxygen-glucose deprivation/reperfusion injury. J. Mol. Neurosci. 2009;39:262–268. doi: 10.1007/s12031-009-9196-5. [DOI] [PubMed] [Google Scholar]
- 53.Evans Z.P., Mandavilli B.S., Ellett J.D., Rodwell D., Fariss M.W., Fiorini R.N., Schnellmann R.G., Schmidt M.G., Chavin K. Vitamin E succinate enhances steatotic liver energy status and prevents oxidative damage following ischemia/reperfusion. Transplant. Proc. 2009;41:4094–4098. doi: 10.1016/j.transproceed.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tello D., Balsa E., Acosta-Iborra B., Fuertes-Yebra E., Elorza A., Ordonez A., Corral-Escariz M., Soro I., Lopez-Bernardo E., Perales-Clemente E., Martinez-Ruiz A., Enriquez J.A., Aragones J., Cadenas S., Landazuri M.O. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14:768–779. doi: 10.1016/j.cmet.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 55.Movafagh S., Crook S., Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J. Cell Biochem. 2015;116:696–703. doi: 10.1002/jcb.25074. [DOI] [PubMed] [Google Scholar]
- 56.Cursio R., Miele C., Filippa N., Van Obberghen E., Gugenheim J. Liver HIF-1 alpha induction precedes apoptosis following normothermic ischemia-reperfusion in rats. Transplant. Proc. 2008;40:2042–2045. doi: 10.1016/j.transproceed.2008.05.037. [DOI] [PubMed] [Google Scholar]
- 57.Zaouali M.A., Ben Mosbah I., Boncompagni E., Ben Abdennebi H., Mitjavila M.T., Bartrons R., Freitas I., Rimola A., Rosello-Catafau J. Hypoxia inducible factor-1alpha accumulation in steatotic liver preservation: role of nitric oxide. World J. Gastroenterol. 2010;16:3499–3509. doi: 10.3748/wjg.v16.i28.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nath B., Levin I., Csak T., Petrasek J., Mueller C., Kodys K., Catalano D., Mandrekar P., Szabo G. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology. 2011;53:1526–1537. doi: 10.1002/hep.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu L., Wang Q., Zhang L., Fang Z., Zhao F., Lv Z., Gu Z., Zhang J., Wang J., Zen K., Xiang Y., Wang D., Zhang C.Y. Hypoxia induces PGC-1alpha expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010;20:676–687. doi: 10.1038/cr.2010.46. [DOI] [PubMed] [Google Scholar]
- 60.Shoag J., Arany Z. Regulation of hypoxia-inducible genes by PGC-1 alpha. Arterioscler. Thromb. Vasc. Biol. 2010;30:662–666. doi: 10.1161/ATVBAHA.108.181636. [DOI] [PubMed] [Google Scholar]
- 61.Rimessi A., Previati M., Nigro F., Wieckowski M.R., Pinton P. Mitochondrial reactive oxygen species and inflammation: molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016;81:281–293. doi: 10.1016/j.biocel.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 62.Tan H.Y., Wang N., Li S., Hong M., Wang X., Feng Y. The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid. Med. Cell Longev. 2016;2016:2795090. doi: 10.1155/2016/2795090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sasaki H., Yamamoto H., Tominaga K., Masuda K., Kawai T., Teshima-Kondo S., Rokutan K. NADPH oxidase-derived reactive oxygen species are essential for differentiation of a mouse macrophage cell line (RAW264.7) into osteoclasts. J. Med. Investig. 2009;56:33–41. doi: 10.2152/jmi.56.33. [DOI] [PubMed] [Google Scholar]
- 64.Choi H.K., Kim T.H., Jhon G.J., Lee S.Y. Reactive oxygen species regulate M-CSF-induced monocyte/macrophage proliferation through SHP1 oxidation. Cell Signal. 2011;23:1633–1639. doi: 10.1016/j.cellsig.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 65.Hall C.J., Sanderson L.E., Crosier K.E., Crosier P.S. Mitochondrial metabolism, reactive oxygen species, and macrophage function-fishing for insights. J. Mol. Med. 2014;92:1119–1128. doi: 10.1007/s00109-014-1186-6. [DOI] [PubMed] [Google Scholar]
- 66.Czubkowski P., Socha P., Pawlowska J. Oxidative stress in liver transplant recipients. Ann. Transplant. 2011;16:99–108. [PubMed] [Google Scholar]
- 67.Muffak-Granero K., Olmedo C., Villegas T., Comino A., Becerra A., Villar J.M., Fundora Y., Garrote D., Bueno P., Ferron J.A. Perioperative values of glutathione peroxidase activity and malondialdehyde levels in enolic cirrhotic recipients of a liver transplant. Transplant. Proc. 2012;44:2071–2073. doi: 10.1016/j.transproceed.2012.07.071. [DOI] [PubMed] [Google Scholar]
- 68.Villegas T., Olmedo C., Muffak-Granero K., Comino A., Garrote D., Bueno P., Ferron J.A. Perioperative levels of glutathione reductase in liver transplant recipients with hepatitis C virus cirrhosis. Transplant. Proc. 2012;44:1542–1544. doi: 10.1016/j.transproceed.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 69.Hassan L., Bueno P., Ferron-Celma I., Ramia J.M., Garrote D., Muffak K., Garcia-Navarro A., Mansilla A., Villar J.M., Ferron J.A. Time course of antioxidant enzyme activities in liver transplant recipients. Transplant. Proc. 2005;37:3932–3935. doi: 10.1016/j.transproceed.2005.10.088. [DOI] [PubMed] [Google Scholar]
- 70.Augusto V.S., Rodrigues A.J., Reis G.S., Silveira A.P., de Castro e Silva O., Jr., Mente E.D., Jordao A.A., Jr., Evora P.R. Evaluation of oxidative stress in the late postoperative stage of liver transplantation. Transplant. Proc. 2014;46:1453–1457. doi: 10.1016/j.transproceed.2013.12.058. [DOI] [PubMed] [Google Scholar]
- 71.Tashiro H., Kuroda S., Mikuriya Y., Ohdan H. Ischemia-reperfusion injury in patients with fatty liver and the clinical impact of steatotic liver on hepatic surgery. Surg. Today. 2014;44:1611–1625. doi: 10.1007/s00595-013-0736-9. [DOI] [PubMed] [Google Scholar]
- 72.Defamie V., Cursio R., Le Brigand K., Moreilhon C., Saint-Paul M.C., Laurens M., Crenesse D., Cardinaud B., Auberger P., Gugenheim J., Barbry P., Mari B. Gene expression profiling of human liver transplants identifies an early transcriptional signature associated with initial poor graft function. Am. J. Transplant. 2008;8:1221–1236. doi: 10.1111/j.1600-6143.2008.02249.x. [DOI] [PubMed] [Google Scholar]
- 73.Tsai Y.F., Liu F.C., Sung W.C., Lin C.C., Chung P.C., Lee W.C., Yu H.P. Ischemic reperfusion injury-induced oxidative stress and pro-inflammatory mediators in liver transplantation recipients. Transplant. Proc. 2014;46:1082–1086. doi: 10.1016/j.transproceed.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 74.Chu M.J., Vather R., Hickey A.J., Phillips A.R., Bartlett A.S. Impact of ischemic preconditioning on outcome in clinical liver surgery: a systematic review. Biomed. Res. Int. 2015;2015:370451. doi: 10.1155/2015/370451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Clavien P.A., Selzner M., Rudiger H.A., Graf R., Kadry Z., Rousson V., Jochum W. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann. Surg. 2003;238(843–850) doi: 10.1097/01.sla.0000098620.27623.7d. (discussion 851-842) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clavien P.A., Yadav S., Sindram D., Bentley R.C. Protective effects of ischemic preconditioning for liver resection performed under inflow occlusion in humans. Ann. Surg. 2000;232:155–162. doi: 10.1097/00000658-200008000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hahn O., Blazovics A., Vali L., Kupcsulik P.K. The effect of ischemic preconditioning on redox status during liver resections--randomized controlled trial. J. Surg. Oncol. 2011;104:647–653. doi: 10.1002/jso.21907. [DOI] [PubMed] [Google Scholar]
- 78.Zapata-Chavira H.A., Cordero-Perez P., Casillas-Ramirez A., Escobedo-Villarreal M.M., Perez-Rodriguez E., Torres-Gonzalez L., Camara-Lemarroy C., Hernandez-Guedea M.A., Caballero-Mendoza E., Munoz-Espinosa L.E. Is ischemic preconditioning a useful therapeutic strategy in liver transplantation? Results from the first pilot study in Mexico. Arch. Med. Res. 2015;46:296–302. doi: 10.1016/j.arcmed.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 79.Andreani P., Hoti E., de la Serna S., degli Esposti D., Sebagh M., Lemoine A., Ichai P., Saliba F., Castaing D., Azoulay D. Ischaemic preconditioning of the graft in adult living related right lobe liver transplantation: impact on ischaemia-reperfusion injury and clinical relevance. HPB. 2010;12:439–446. doi: 10.1111/j.1477-2574.2010.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bucuvalas J.C., Ryckman F.C., Krug S., Alonso M.H., Balistreri W.F., Kotagal U. Effect of treatment with prostaglandin E1 and N-acetylcysteine on pediatric liver transplant recipients: a single-center study. Pediatr. Transplant. 2001;5:274–278. doi: 10.1034/j.1399-3046.2001.005004274.x. [DOI] [PubMed] [Google Scholar]
- 81.Khan A.W., Fuller B.J., Shah S.R., Davidson B.R., Rolles K. A prospective randomized trial of N-acetyl cysteine administration during cold preservation of the donor liver for transplantation. Ann. Hepatol. 2005;4:121–126. [PubMed] [Google Scholar]
- 82.M. Aliakbarian, S. Nikeghbalian, S. Ghaffaripour, A. Bahreini, M. Shafiee, M. Rashidi, Y. Rajabnejad, Effects of N-acetylcysteine addition to University of Wisconsin solution on the rate of ischemia-reperfusion injury in adult orthotopic liver transplant. Exp. Clin. Transplant. (2015). [DOI] [PubMed]
- 83.Robinson S.M., Saif R., Sen G., French J.J., Jaques B.C., Charnley R.M., Manas D.M., White S.A. N-acetylcysteine administration does not improve patient outcome after liver resection. HPB. 2013;15:457–462. doi: 10.1111/hpb.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jegatheeswaran S., Siriwardena A.K. Experimental and clinical evidence for modification of hepatic ischaemia-reperfusion injury by N-acetylcysteine during major liver surgery. HPB. 2011;13:71–78. doi: 10.1111/j.1477-2574.2010.00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barros M.A., Vasconcelos P.R., Souza C.M., Andrade G.M., Moraes M.O., Costa P.E., Coelho G.R., Garcia J.H. L-alanyl-glutamine attenuates oxidative stress in liver transplantation patients. Transplant. Proc. 2015;47:2478–2482. doi: 10.1016/j.transproceed.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 86.Nickkholgh A., Schneider H., Sobirey M., Venetz W.P., Hinz U., Pelzl le H., Gotthardt D.N., Cekauskas A., Manikas M., Mikalauskas S., Mikalauskene L., Bruns H., Zorn M., Weigand M.A., Buchler M.W., Schemmer P. The use of high-dose melatonin in liver resection is safe: first clinical experience. J. Pineal Res. 2011;50:381–388. doi: 10.1111/j.1600-079X.2011.00854.x. [DOI] [PubMed] [Google Scholar]
- 87.Schemmer P., Nickkholgh A., Schneider H., Sobirey M., Weigand M., Koch M., Weitz J., Buchler M.W. PORTAL: pilot study on the safety and tolerance of preoperative melatonin application in patients undergoing major liver resection: a double-blind randomized placebo-controlled trial. BMC Surg. 2008;8:2. doi: 10.1186/1471-2482-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsai Y.F., Lin C.C., Lee W.C., Yu H.P. Propofol attenuates ischemic reperfusion-induced formation of lipid peroxides in liver transplant recipients. Transplant. Proc. 2012;44:376–379. doi: 10.1016/j.transproceed.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 89.Bhogal R.H., Curbishley S.M., Weston C.J., Adams D.H., Afford S.C. Reactive oxygen species mediate human hepatocyte injury during hypoxia/reoxygenation. Liver Transpl. 2010;16:1303–1313. doi: 10.1002/lt.22157. [DOI] [PubMed] [Google Scholar]
- 90.Adam R., Delvart V., Karam V., Ducerf C., Navarro F., Letoublon C., Belghiti J., Pezet D., Castaing D., Le Treut Y.P., Gugenheim J., Bachellier P., Pirenne J., Muiesan P., Eltr contributing centres, t. E. L. I. T. A. Compared efficacy of preservation solutions in liver transplantation: a long-term graft outcome study from the European liver transplant registry. Am. J. Transplant. 2015;15:395–406. doi: 10.1111/ajt.13060. [DOI] [PubMed] [Google Scholar]
- 91.Zaouali M.A., Ben Abdennebi H., Padrissa-Altes S., Alfany-Fernandez I., Rimola A., Rosello-Catafau J. How Institut Georges Lopez preservation solution protects nonsteatotic and steatotic livers against ischemia-reperfusion injury. Transplant. Proc. 2011;43:77–79. doi: 10.1016/j.transproceed.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 92.Hide D., Ortega-Ribera M., Fernandez-Iglesias A., Fondevila C., Salvado M.J., Arola L., Garcia-Pagan J.C., Mancini A., Bosch J., Gracia-Sancho J. A novel form of the human manganese superoxide dismutase protects rat and human livers undergoing ischaemia and reperfusion injury. Clin. Sci. 2014;127:527–537. doi: 10.1042/CS20140125. [DOI] [PubMed] [Google Scholar]
- 93.Guarrera J.V., Henry S.D., Samstein B., Odeh-Ramadan R., Kinkhabwala M., Goldstein M.J., Ratner L.E., Renz J.F., Lee H.T., Brown R.S., Jr., Emond J.C. Hypothermic machine preservation in human liver transplantation: the first clinical series. Am. J. Transplant. 2010;10:372–381. doi: 10.1111/j.1600-6143.2009.02932.x. [DOI] [PubMed] [Google Scholar]
- 94.Henry S.D., Nachber E., Tulipan J., Stone J., Bae C., Reznik L., Kato T., Samstein B., Emond J.C., Guarrera J.V. Hypothermic machine preservation reduces molecular markers of ischemia/reperfusion injury in human liver transplantation. Am. J. Transplant. 2012;12:2477–2486. doi: 10.1111/j.1600-6143.2012.04086.x. [DOI] [PubMed] [Google Scholar]
- 95.Bruinsma B.G., Yeh H., Ozer S., Martins P.N., Farmer A., Wu W., Saeidi N., Op den Dries S., Berendsen T.A., Smith R.N., Markmann J.F., Porte R.J., Yarmush M.L., Uygun K., Izamis M.L. Subnormothermic machine perfusion for ex vivo preservation and recovery of the human liver for transplantation. Am. J. Transplant. 2014;14:1400–1409. doi: 10.1111/ajt.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]