

Abstract
Congenital cataract is the most frequent inherited ocular disorder and the most leading cause of lifelong visual loss. The screening of pathogenic mutations can be very challenging in some cases, for congenital cataracts are clinically and genetically heterogeneous diseases. The aim of this study is to investigate the mutation spectrum and frequency of 54 cartaract-associated genes in 27 Chinese families with congenital cataracts. Variants in 54 cataract-associated genes were screened by targeted next-generation sequencing (NGS) and then validated by Sanger sequencing. We identified pathogenic variants in 62.96% (17/27) of families, and over 52.94% (9/17) of these variants were novel. Among them, three are splicing site mutations, four are nonsense mutations, seven are missense mutations, two are frame shift mutations and one is intronic mutation. This included identification of: complex ocular phenotypes due to two novel PAX6 mutations; progressive cortical cataract and lamellar cataract with lens subluxation due to two novel CRYGS mutations. Mutations were also found in rarely reported genes including CRYBA4, CRYBA2, BFSP1, VIM, HSF4, and EZR. Our study expands the mutation spectrum and frequency of genes responsible for congenital cataracts. Targeted next-generation sequencing in inherited congenital cataract patients provided significant diagnostic information.
Introduction
Congenital cataract is the most frequent eye disease and the most leading cause of blindness in childhood, affecting tens of millions of people1, 2. The prevalence of congenital cataracts is approximately 1 to 6 per 10,000 live births, while 27–39% of which are believed to be inherited3. There are autosomal-dominant, autosomal-recessive, and X-linked genetic forms of congenital cataracts, which may be isolated or associated with other ophthalmic abnormalities and syndromic associations4.
So far, more than 40 genes have been reported to be associated with congenital cataracts (Cat-Map; http://cat-map.wustl.edu/)5. These genes code for a variety of lens proteins with structural and chaperone functions, including α-, β-, and γ-crystallins, lens-specific transmembrane gap junction protein genes (GJA3 and GJA8), membrane protein genes (MIP and LIM2), and lens-associated transcription factors (e.g. HSF4, PITX3, MAF, PAX6, and FOXE3). Structural proteins such as the lens-specific beaded filament protein genes (BFSP1 and BFSP2) represent an additional group of proteins that may have mutations leading to cataract formation6. For most of these genes, cataract is the only disease phenotype observed7.
In order to identify the genetic cause of our newly recruited 27 families with congenital cataracts, we applied targeted exome sequencing using SureSelect Target Enrichment Kit. 17 mutations were identified in the 27 families, and 13 mutations were considered to be novel. Mutations were identified in 12 genes and we found a high mutation detection rate of approximately 62.96% in these families.
Results
Next Generation Sequencing
The present study recruited 27 families with congenital cataract. Targeted exome sequencing results of the 27 probands detected 6,024 variants in the 54 known genes (Table S1). Bioinformatics analysis of these mutations revealed that 30 of them are potential pathogenic (Table S2).
Validation by Sanger sequencing
All of 30 mutations are confirmed by Sanger sequencing in probands and available family members. Among them, seventeen mutations were confirmed to be cosegregated with congenital cataracts (Table 1). SIFT predicts substitutions with scores less than 0.05 as deleterious, Polyphen-2 predicts substitutions with scores greater than 0.75 as “probably damaging”. The pedigrees of seventeen families are presented in Fig. 1.
Table 1.
The pathogenic mutations identified in Chinese families with congenital cataract.
| Family ID | Gene | Nucleotide | Amino acid | Mutation type | Status | Bioinformation prediction | Variant in controls | Note | |
|---|---|---|---|---|---|---|---|---|---|
| SIFT | Polyphen-2 | ||||||||
| 1 | CRYBA4 | c.26C > T | p.A9V | missense | Hetero | 0.66 | 0 | 0/100 | Novel |
| 4 | CRYGS | c.53G > A | p.G18D | missense | Hetero | 0 | 0.989 | 0/100 | Novel |
| 5 | CRYBA1 | c.271_273delGGA | p.G91del | flame shift | Hetero | / | / | 0/100 | Novel |
| 6 | HSF4 | c.-497-8C > G | intronic | Hetero | / | / | 0/100 | Novel | |
| 7 | CRYGS | c.224_225GC > TT | p.G75V | missense | Hetero | 0 | 0.999 | 0/100 | Novel |
| 9 | CRYBA1 | c.607C > T | p.Q203X | nonsense | Hetero | 1 | 0.735289 | 0/100 | Novel |
| 10 | EZR | c.1597-7insTAAT | splicing site | Hetero | / | / | 0/100 | Novel | |
| 14 | VIM | c.623A > G | p.Q208R | missense | Hetero | 0.07 | 0.712 | 0/100 | Novel |
| 15 | MIP | c.607-1G > A | splicing site | Hetero | / | / | 0/100 | [8] | |
| 16 | CRYBB2 | c.463C > T | p.Q155X | nonsense | Hetero | 0.01 | 0.641104 | 0/100 | [9] |
| 17 | CRYBB2 | c.452G > A | p.W151X | nonsense | Hetero | 0 | 0.641681 | 0/100 | Novel |
| 18 | CRYBA2 | c.343A > G | p.N115D | missense | Hetero | 0.22 | 0.004 | 0/100 | Novel |
| 19 | BFSP1 | c.625 + 3A > G | splicing site | Hetero | / | / | 0/100 | Novel | |
| 22 | CRYGD | c.70C > A | p.P24T | missense | Hetero | 0.05 | 0.102 | 0/100 | [10] |
| 24 | PAX6 | c.795delA | p.E265fs | flame shift | Hetero | / | / | 0/100 | Novel |
| 26 | CRYGD | c.43C > A | p.R15S | missense | Hetero | 0 | 0.974 | 0/100 | [11] |
| 27 | PAX6 | c.342G > A | p.W114X | nonsense | Hetero | 0 | 0.735284 | 0/100 | Novel |
Figure 1.
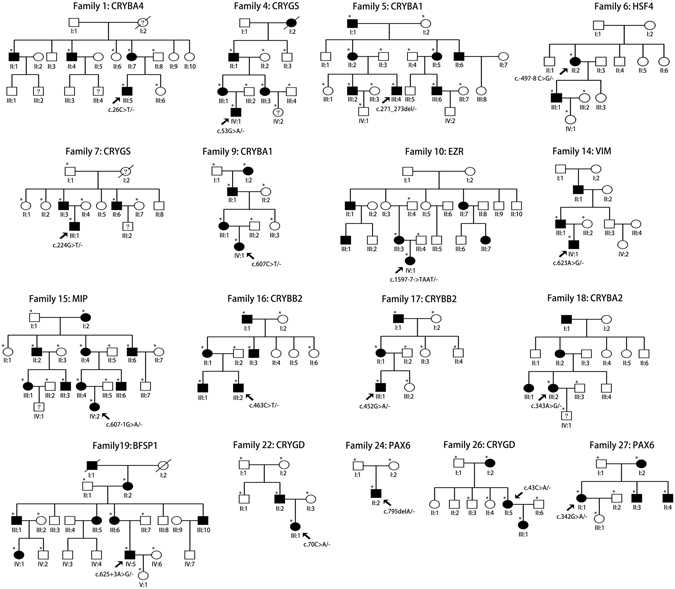
Pedigrees of the families with mutations. Squares indicate men and circles women; black and white symbols represent affected and unaffected individuals, respectively. The proband is marked with an arrow, and asterisks indicate those members enrolled in this study.
Ten of seventeen mutations were identified in crystallin genes, while other seven mutations identified in six genes. Two mutations are in PAX6 (MIM: 607108); three mutations are in cytoskeletal protein (BFSP1 (MIM: 611391), VIM (MIM: 116300), and EZR); and one mutation each in MIP (MIM: 154050) and HSF4 (MIM: 602438). Among seventeen mutations, three are splicing site mutations, four are nonsense mutations, seven are missense mutations, two are frame shift mutations and one is intronic mutation. None of these seventeen mutations was detected in 100 controls. Nine mutations were considered as novel disease-causing mutations (DNA sequencing results provided in Fig. 2); while four have been previously linked to congenital cataracts8–11 (Figure S1). However, four pathogenic mutations (in family 6, 10, 14 and 18) could not be strongly associated with congenital cataracts due to the limited DNA samples of the family members and bioinformation prediction results (Figure S2).
Figure 2.
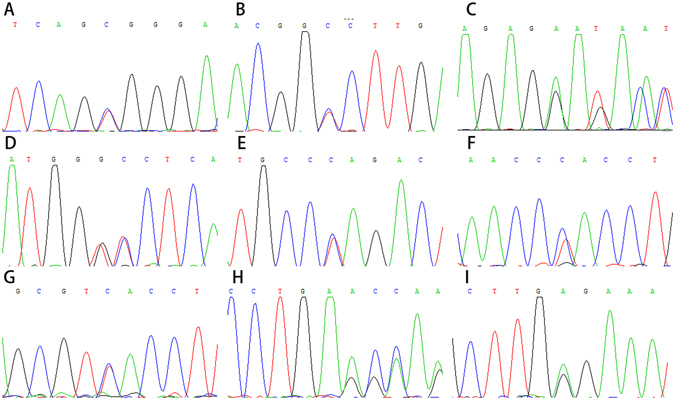
Sequencing results of nine novel disease-causing mutations. (A) Forward sequencing showed c.26C > T mutation of CRYBA4 gene in patients from family 1. (B) Reverse sequencing showed c.53G > A mutation of CRYGS gene in patients from family 4. (C) Forward sequencing showed p.G91del mutation of CRYBA1 gene in patients from family 5. (D) Forward sequencing showed c.224_225GC > TT mutation of CRYGS gene in patients from family 7. (E) Forward sequencing showed c.607C > T mutation CRYBA1 gene in patients from family 9. (F) Reverse sequencing showed c.452G > A of CRYBB2 gene in patientsfamily 17. (G) Reverse sequencing showed c.625 + 3A > G mutation of BFSP1gene in patients from family 19. (H) Forward sequencing showed c.795delA mutation of PAX6 gene in patient from family 24. (I) Forward sequencing showed c.342G > A mutation of PAX6gene in patients family 27.
Clinical findings
All patients in this study had different types of congenital cataracts without other systemic diseases. Other ophthalmic findings of seventeen probands were listed in Table 2. Two families (family 24 and 27) with PAX6 mutation showed aniridia. Twelve phenotypes of probands with congenital cataract were recorded (Fig. 3), while other five probands underwent cataract surgery prior to this study. The phenotype of these families could only determinate by their medical record.
Table 2.
Clinical features of affected probands with variants identified in this study.
| Family ID | Variation | Sex | Age at examination (yrs) | Cataract types | Other clinical finding |
|---|---|---|---|---|---|
| Family 1 | CRYBA4, c.26C > T | M | 38 | Anterior polar cataract | |
| Family 4 | CRYGS, c.53G > A | M | 7 | Cortical and sutural cataract | Progressive |
| Family 5 | CRYBA1, c.271_273delGAG | M | 35 | Zonular Cataracts | |
| Family 6 | HSF4, c.-497-8 C > G | F | 59 | Lamellar, punctate | |
| Family 7 | CRYGS, c.224_225GC > TT | M | 8 | Lamellar cataract | Lens subluxation |
| Family 9 | CRYBA1, c.607C > T | F | 3 | Nuclear cataract | Nystagmus |
| Family 10 | EZR, c.1597-7- > TAAT | F | 3 | Total cataract | Nystagmus |
| Family 14 | VIM, c.623A > G | M | 6 | Posterior polar cataract | |
| Family 15 | MIP, c.607-1G > A | F | 1 | Nuclear cataract | Nystagmus |
| Family 16 | CRYBB2, c.463C > T | M | 6 | Cerulean cataract | |
| Family 17 | CRYBB2, c.452G > A | M | 2 | Cerulean cataract | |
| Family 18 | CRYBA2, c.343A > G | F | 26 | Total cataract | Progressive |
| Family19 | BFSP1, c.625 + 3A > G | M | 23 | Lamellar, punctate | Progressive |
| Family 22 | CRYGD, c.70C > A | F | 2 | Coralliform cataract | Nystagmus |
| Family 24 | PAX6, c.795delA | M | 7 | Coralliform cataract | Nystagmus, aniridia |
| Family 26 | CRYGD, c.43C > A | F | 28 | Coralliform cataract | Nystagmus |
| Family 27 | PAX6, c.342G > A | F | 28 | Anterior and posterior polar cataract | Nystagmus, aniridia |
Figure 3.
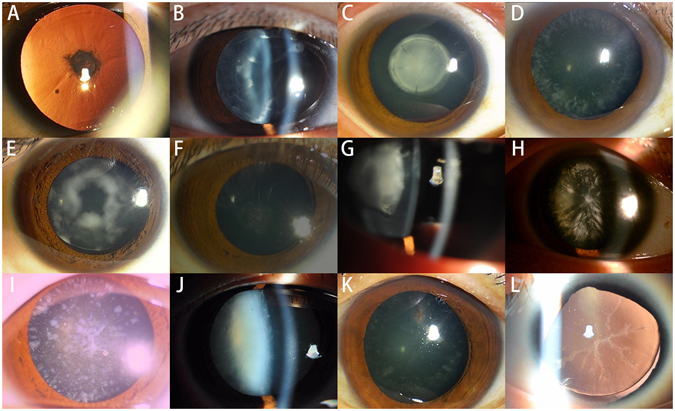
Phenotypes of the probands. (A) Photograph of proband in family 1 presented an anterior polar cataract. (B) Slit-lamp photograph of proband in family 4 showed a progressive cortical and sutural cataract. (C) Photograph of proband in family 5 showed a perinuclear zonular cataract. (D) Photograph of proband of family 6 showed a lamellar cataract with fine punctate opacities involving the cortical area of lens. (E) Photograph of proband in family 7 showed a subluxation of lens with a lamellar cataract. (F) Photograph of proband in family 14 presented a posterior polar cataract. (G) Slit-lamp photograph of proband in family 15 presented a nuclear cataract. (H) Photograph of proband in family 16 presented a cerulean cataract. (I) Photograph of proband in family 17 presented a cerulean cataract. (J) Slit-lamp photograph of proband in family 18 presented a total cataract. (K) Photograph of proband in family 19 showed a lamellar punctate cataract. (L) Photograph of proband in family 24 showed a coralliform cataract with aniridia.
Discussion
More than 40 genes have been associated with congenital cataracts. Screening of these genes in groups of congenital cataract patients showed that the mutation frequencies have great differences12–15. Hansen et al. recruited 28 Danish families with hereditary congenital cataracts, and screened 17 cataract-related genes. He found that mutations in genes encoding crystallins and connexins account for 53.5% of inherited cataracts14. Dave et al. believed that EPHA2 mutations are major contributors to inherited cataracts in South-Eastern Australia12. Sun et al. indicated that mutations in NHS are the common causes of nonsyndromic congenital cataracts and account for 11.8% of the congenital cataracts15. In this study, we performed targeted exome sequencing on probands from 27 families with congenital cataracts. Sequence results indicated that 30 mutations are potentially pathogenic. Sanger sequencing confirmed that seventeen mutations are disease-causing. Our study revealed that mutations in crystallin genes are still the leading causes of nonsyndromic congenital cataracts with a frequency of 37.03%.
Mutations in the Lens-Specific Crystallin Genes
Ten crystallins gene mutations were found in 27 families corresponding to 37.03% of the analyzed families, which is in the same magnitude as the percentage of crystallin mutations in Denmark group (36%)14. However, only 2 crystallin mutations (5%) were identified among 32 families with autosomal dominant congenital cataracts (ADCC) in southeastern Australia16. This difference of results may be influenced by different ethnic background and selection bias of family samples.
Three crystallins gene mutations have been associated with congenital cataracts. CRYGD p.P24T is a hotspot for mutation which has been reported for several times10, 16–19. Previous studies have showed different phenotypes (e.g. coralliform, cerulean, lamellar) of CRYGD p.P24T. Our proband showed a coralliform cataract, which is one of the most common phenotype of this mutation20–23. CRYBB2 p.Q155X is another hotspot for mutation in congenital cataracts13, 24, 25. Phenotypes of this mutation have been described as cerulean cataracts, which is also in correspondence with the proband of family16. CRYGD p.R15S has been reported once by Zhang and colleagues with a phenotype of coraliform cataracts11. The proband of family also present a coralliform cataract. Our results confirmed these recurrent mutations, and further expanded the mutation spectrum of congenital cataracts.
Two novel nonsense mutations CRYBB2 p.W151X and CRYBA1 p.Q203X may terminate the reading frame before the authentic stop codon. Nonsense-mediated decay (NMD) is the process by which mRNAs containing pre-mature termination codons (PTCs) are degraded before production of supposed truncated proteins26, 27. Two CRYGS mutations p.G18D and p.G75V has been detected in two families. The CRYGS p.G18V mutation has been associated with dominant progressive cortical cataract28, and reported to increase the gammaS-crystallin sensitivity to thermal and chemical stress29.
Kingsley et al. suggested that the potential mechanism for CRYGS p.G18V mutation to cause cataract formation is the depletion of the finite αB-crystallin population of the lens30. The results of their study indicated normal association and structural properties of the G18V mutant γS-crystallin under mild conditions, but increased sensitivity stress, which were thus consistent with the progressive nature of the cataracts in the family. The CRYGS p.G18D mutation, located in the same locus of p.G18V, may also alter the sensitivity to thermal and chemical stress, and deplete αB-crystallin of the lens as well. SWISS-MODEL revealed both p.G18V and p.G18D are significantly different from wild type (Fig. 4). The phenotype of CRYGS p.G18D mutation is also progressive cortical and sutural cataract, and this is in accordance with the phenotype p.G18D caused.
Figure 4.
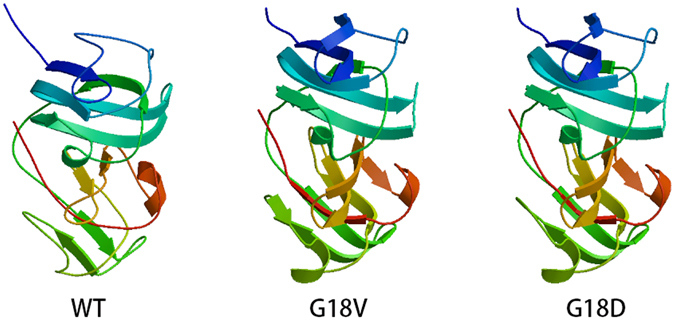
Stuctural modeling of WT, p.G18V and p.G18D crystallin gamma S using SWISS-MODEL.
The novel deletion mutation (c.271_273delGAG) in exon 4 of CRYBA1 was identified in a family with autosomal dominant congenital cataracts. Several deletion mutations have been identified in CRYBA1 gene31, 32 and CRYBA1c.272_274delGAG has been widely reported33–36. Xu indicated that DeltaG91 mutation of CRYBA1altered protein-protein interaction between human lens betaA1-crystallins, and lead to protein insolubilization and contribute to cataracts37. In our study, a novel in-frame deletion of three bp was dcted in exon 4 of CRYBA1 (c.271_273delGAG). Though this is a novel mutation on DNA level, it also leads to a DeltaG91 deletion like c.272_274delGAG mutation dose. Thus, this mutation was predicted to cause the same protein insolubilization of betaA1-crystallins as c.271_273delGAG dose.
The mutation found in CRYBA4 (c.26C > T, p.A9V) is the first cataract-associated CRYBA4 mutation with a dominant pattern. This mutation has been previously detected by Sun et al.15. They suggested that CRYBA4 p.A9V may be the pathogenic mutation of a Chinese family with congenital cataracts. But they cannot be sure due to bioinformation prediction results and limited family members. Our results confirmed that this mutation is cosegregated with congenital cataracts within the family, verified their hypothesis.
Mutations in the cytoskeletal protein
The structural framework of lens cells is determined by the interaction of the cytoskeleton and the crystallins within the cytoplasm. Beaded filament is a type of intermediate filament which is unique to the lens fiber cells6. They are made up of BFSP1 (also called CP115 or filensin) and BFSP2 (also called CP49 or phakinin), highly divergent intermediate filament proteins that combine in the presence of crystallin to form the appropriate beaded structure4. Several different mutations of BFSP2 have been linked to ADCCs38–40, while BFSP1gene mutations have been linked to both autosomal dominant pattern (p.D348N)41 and autosomal recessive pattern (p.T246del74fsX6)42. To date, only these two BFSP1 disease-causing mutations have been reported. Thus, BFSP1 c.625 + 3A > G mutation we detected was the first report of BFSP1 splicing site mutation.
We also detected two cytoskeletal protein mutations EZR c.1597-7insTAAT and VIM p.Q208R. Lin et al. has linked several EZR mutations to age-related cataracts43. The mutation of VIM (p.E151K) is associated with inherited congenital cataracts. The mutant formed an aberrant vimentin cytoskeleton and increased the proteasome activity in transfected cells44. Thus, further investigation of EZR c.1597-7insTAAT and VIM p.Q208R are needed to clarify the pathogenicity of these two mutations.
Mutations in PAX6 gene
Congenital aniridia with cataract is linked to a mutation of the PAX6 genes. Human PAX6 is composed of two DNA-binding domains: the paired domain (PD) of 128 amino acids and the homeodomain (HD) of 61 amino acids separated by a linker region of 79 amino acids, and is followed by a proline, serine, threonine-rich (PST) domain of 79 amino acids which have transcriptional trans-activation function45. It is a highly conserved transcription factor which regulates the tissue-specific expression of various molecules, hormones, and structural proteins. It is required for the development of the nervous system, eyes, nose, pancreas, and pituitary gland46–48.
As a crucial transcriptional factor, PAX6 mutations may affect a broad range of structures during development. Therefore, the phenotypes of different PAX6 mutations can be very diverse. PAX6 mutations is characterized by partial or complete absence of the iris accompanied with other ocular abnormalities such as cataract, glaucoma49, corneal degeneration50, microphthalmia51, foveal hypoplasia52, optic-nerve malformations53. Some individuals with PAX6 mutation developed other systemic diseases such hepatoblastoma, polydactylia54. PAX6 regulates numerous downstream genes, and its expression level is also regulated by several factors during eye development. Thus, the aniridia phenotype may vary even within the family, and the obvious genotype–phenotype correlation was very hard to identified54. However, Lin et al. reviewed the mutations archived in the PAX6 AllelicVariant Database, and found that over three-quarters of aniridia cases are caused by mutations that introduce a PTC into the open reading frame of PAX650. It was widely belived that truncations of Pax6 can usually cause aniridia phenotype, due to haploinsufficiency55. Patients with PAX6 contiguous deletion, may have relatively severe phenotype, including bilateral complete absence of iris and foveal hypoplasia49. The two novel PAX6 mutations detected in our study were p.E265fs and p.W114X. Patient with p.E265fs mutation showed a partial absence of the iris, congenital coralliform cataracts and nystagmus (Fig. 3). This frameshift mutation is very close to p. E265fs. All patients in family 27 with p.W114X mutation showed a complete absence of iris, congenital anterior and posterior polar cataracts, as well as nystagmus. PAX6 nonsense mutations been widely reported (p.Arg240X, p.W100X, p.R103X, etc.), and linked to aniridia with congenital cataract56–58. The phenotypes caused by two PAX6 mutations in this study were in accordance with these previous results. Liu et al. revealed the PAX6 mRNA level was about 50% lower in patients caused by p.A266fs mutation than in unaffected family members, indicating that this mutation caused nonsense-mediated mRNA decay (NMD)59. Since NMD is a common pathogenic mechanism of nonsense and frameshift mutations, we hypothesized that nonsense-mediated decay (NMD) may be the pathogenic mechanism of two PAX6 mutations we identified as well.
In conclusion, our results showed that mutations in the 54 known genes were responsible for about 62.96% of this set of Chinese families with congenital cataracts. And mutations in the crystallin gene were identified in 37.03% of the families. Therefore, we believed that targeted exome sequencing is an efficient method in disease-causing mutation identification.
Materials and Methods
Patient Recruitment
The research protocols of this study adhered to the guidelines of the Declaration of Helsinki and were approved by the Medical Ethics Committees of the Second Affiliated Hospital, College of Medicine, Zhejiang University (Hangzhou, China). Appropriate informed consent from each participant was obtained.
Among 27 families, 24 were diagnosed with congenital cataracts, while 3 were diagnosed with aniridia and congenital cataract. 25 families with family history showed autosomal dominant inheritance, and 2 were sporadic patients. Available individuals indicated in Fig. 1 were given complete physical, ophthalmic examinations. One hundred unrelated healthy subjects from the same ethnic background were recruited as controls. Peripheral blood was collected by venipuncture in EDTA-coated Vacutainer tubes (BD, New Jersey, USA) and stored at −20 °C.
DNA Extraction and Next Generation Sequencing
Genomic DNA of 27 probands was isolated from the 2 ml peripheral blood samples using QIAamp DNA Blood kits (Qiagen, Hilden, Germany). Then the purity and quantity of DNA samples were measured by the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, Massachusetts). Genomic DNA was shearing by CovarisTM system. Then sample preparation by following the manufacturer’s standard procedure using Truseq DNA Sample preparation Kit (Illumina, Inc, San Diego, CA).
The coding exons, flanking regions and promotor regions of 54 genes related to inherited cataracts were selected and captured using a SureSelect Target Enrichment Kit (Agilent technologies, Inc, USA). The kit included 5,721 probes and could enrich about 551 exons and cover about 94.7% targeted regions. The enrichment libraries were sequenced on Illumina HiSeq2000 Sequencer (Illumina, Inc, San Diego, CA); the average sequencing depth was 500-fold.
Bioinformatics Analysis
The low quality reads and adaptor sequences were filtered out with the FASTX program. Picard program was used to remove the PCR duplicates. After high-quality reads were retrieved, the clean data were aligned using BWA program according to human genome parameters (hg19). Subsequently, we determined SNPs using the SOAPsnp program, realigned the reads with BWA, and detected the deletions or insertion (InDels) with the GATK software. After SNPs are identified, we use ANNOVAR to do annotation and classification. Finally, all nonsynonymous variants were evaluated by three algorithms, SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Tester (http://www.mutationtaster.org/).
Expanded Validation
DNA samples of probands were taken for further Sanger sequencing, to confirm the potential pathogenic variants detected by exome sequencing. Polymerase chain reaction (PCR) was performed in a 20 μl reaction system using the primer pairs previously published60 or designed by Primer Premier 6.0 (Table S3). PCR products were isolated using electrophoresis on 3% agarose gels and sequenced using the BigDye Terminator Cycle sequencing kit V 3.1 (ABI–Applied Biosystems; Sangon Co, China) on an ABI PRISM 3730 Sequence Analyzer (ABI). Sequencing results were analyzed using Chromas 2.3.0 and compared with sequences from NCBI human genome database. Confirmed variants were further sequenced in the all available family members and 100 control individuals.
Electronic supplementary material
Acknowledgements
We are grateful to the members of the families for their participation in the study. This work was supported by National Natural Science Foundation of China (No. 81428005), National Natural Science Foundation of China (No. 81371001), Natural Science Foundation of Zhejiang Province (No. LQ13H120002), Zhejiang Key Innovation Team Project of China (No. 2009R50039), Zhejiang Key Laboratory Fund of China (No. 2011E10006), Project of National Clinical Key Discipline of the Chinese Ministry of Health.
Author Contributions
K.Y. and X.H.G. conceived, designed and supervised the research. Y.Z. and J.Y.L. performed the experiments. W.S.Y., S.Z., Y.H.Y., M.H.W., G.Z.S. recruited patients and collected Samples. Y.Z., J.Y.L., S.Z. performed data analyses. Y.Z. and K.Y. wrote the manuscript. All authors have read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Yi Zhai and Jinyu Li contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01182-9
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Apple, D. J., Ram, J., Foster, A. & Peng, Q. Elimination of cataract blindness: a global perspective entering the new millenium. Survey of ophthalmology45 Suppl 1, S1–196 (2000). [PubMed]
- 2.Pascolini D, Mariotti SP. Global estimates of visual impairment: 2010. The British journal of ophthalmology. 2012;96:614–618. doi: 10.1136/bjophthalmol-2011-300539. [DOI] [PubMed] [Google Scholar]
- 3.Haargaard B, Wohlfahrt J, Fledelius HC, Rosenberg T, Melbye M. A nationwide Danish study of 1027 cases of congenital/infantile cataracts: etiological and clinical classifications. Ophthalmology. 2004;111:2292–2298. doi: 10.1016/j.ophtha.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 4.Hejtmancik JF. Congenital cataracts and their molecular genetics. Seminars in cell & developmental biology. 2008;19:134–149. doi: 10.1016/j.semcdb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Molecular vision. 2010;16:2007–2015. [PMC free article] [PubMed] [Google Scholar]
- 6.Huang B, He W. Molecular characteristics of inherited congenital cataracts. European journal of medical genetics. 2010;53:347–357. doi: 10.1016/j.ejmg.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Shiels A, Hejtmancik JF. Genetic origins of cataract. Archives of ophthalmology. 2007;125:165–173. doi: 10.1001/archopht.125.2.165. [DOI] [PubMed] [Google Scholar]
- 8.Jiang J, et al. Identification of a novel splice-site mutation in MIP in a Chinese congenital cataract family. Molecular vision. 2009;15:38–44. [PMC free article] [PubMed] [Google Scholar]
- 9.Gill D, et al. Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Investigative ophthalmology & visual science. 2000;41:159–165. [PubMed] [Google Scholar]
- 10.Santhiya ST, et al. Novel mutations in the gamma-crystallin genes cause autosomal dominant congenital cataracts. Journal of medical genetics. 2002;39:352–358. doi: 10.1136/jmg.39.5.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang LY, et al. A novel gammaD-crystallin mutation causes mild changes in protein properties but leads to congenital coralliform cataract. Molecular vision. 2009;15:1521–1529. [PMC free article] [PubMed] [Google Scholar]
- 12.Dave A, et al. Mutations in the EPHA2 gene are a major contributor to inherited cataracts in South-Eastern Australia. PloS One. 2013;8:e72518. doi: 10.1371/journal.pone.0072518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devi RR, et al. Crystallin gene mutations in Indian families with inherited pediatric cataract. Molecular vision. 2008;14:1157–1170. [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen L, et al. Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Investigative ophthalmology & visual science. 2009;50:3291–3303. doi: 10.1167/iovs.08-3149. [DOI] [PubMed] [Google Scholar]
- 15.Sun W, Xiao X, Li S, Guo X, Zhang Q. Exome sequencing of 18 Chinese families with congenital cataracts: a new sight of the NHS gene. PloS one. 2014;9:e100455. doi: 10.1371/journal.pone.0100455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burdon KP, et al. Investigation of crystallin genes in familial cataract, and report of two disease associated mutations. The British journal of ophthalmology. 2004;88:79–83. doi: 10.1136/bjo.88.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nandrot E, et al. Gamma-D crystallin gene (CRYGD) mutation causes autosomal dominant congenital cerulean cataracts. Journal of medical genetics. 2003;40:262–267. doi: 10.1136/jmg.40.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mackay DS, Andley UP, Shiels A. A missense mutation in the gammaD crystallin gene (CRYGD) associated with autosomal dominant “coral-like” cataract linked to chromosome 2q. Molecular vision. 2004;10:155–162. [PubMed] [Google Scholar]
- 19.Shentu X, et al. Special fasciculiform cataract caused by a mutation in the gammaD-crystallin gene. Molecular vision. 2004;10:233–239. [PubMed] [Google Scholar]
- 20.Jia X, et al. Combinational analysis of linkage and exome sequencing identifies the causative mutation in a Chinese family with congenital cataract. BMC medical genetics. 2013;14:107. doi: 10.1186/1471-2350-14-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanita V, Singh D. A missense mutation in CRYGD linked with autosomal dominant congenital cataract of aculeiform type. Molecular and cellular biochemistry. 2012;368:167–172. doi: 10.1007/s11010-012-1355-2. [DOI] [PubMed] [Google Scholar]
- 22.Yang G, Xiong C, Li S, Wang Y, Zhao J. A recurrent mutation in CRYGD is associated with autosomal dominant congenital coralliform cataract in two unrelated Chinese families. Molecular vision. 2011;17:1085–1089. [PMC free article] [PubMed] [Google Scholar]
- 23.Khan AO, Aldahmesh MA, Ghadhfan FE, Al-Mesfer S, Alkuraya FS. Founder heterozygous P23T CRYGD mutation associated with cerulean (and coralliform) cataract in 2 Saudi families. Molecular vision. 2009;15:1407–1411. [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, et al. Autosomal-dominant cerulean cataract in a chinese family associated with gene conversion mutation in beta-B2-crystallin. Ophthalmic research. 2009;41:148–153. doi: 10.1159/000209668. [DOI] [PubMed] [Google Scholar]
- 25.Litt M, et al. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Human molecular genetics. 1997;6:665–668. doi: 10.1093/hmg/6.5.665. [DOI] [PubMed] [Google Scholar]
- 26.Wen J, Brogna S. Nonsense-mediated mRNA decay. Biochemical Society transactions. 2008;36:514–516. doi: 10.1042/BST0360514. [DOI] [PubMed] [Google Scholar]
- 27.Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Muhlemann O. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. Rna. 2011;17:2108–2118. doi: 10.1261/rna.030247.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun H, et al. Gamma-S crystallin gene (CRYGS) mutation causes dominant progressive cortical cataract in humans. Journal of medical genetics. 2005;42:706–710. doi: 10.1136/jmg.2004.028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Z, Piszczek G, Wingfield PT, Sergeev YV, Hejtmancik JF. The G18V CRYGS mutation associated with human cataracts increases gammaS-crystallin sensitivity to thermal and chemical stress. Biochemistry. 2009;48:7334–7341. doi: 10.1021/bi900467a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kingsley CN, et al. Preferential and specific binding of human alphaB-crystallin to a cataract-related variant of gammaS-crystallin. Structure. 2013;21:2221–2227. doi: 10.1016/j.str.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, et al. Congenital cataracts due to a novel 2bp deletion in CRYBA1/A3. Molecular medicine reports. 2014;10:1614–1618. doi: 10.3892/mmr.2014.2324. [DOI] [PubMed] [Google Scholar]
- 32.Qi Y, et al. A deletion mutation in the betaA1/A3 crystallin gene (CRYBA1/A3) is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Human genetics. 2004;114:192–197. doi: 10.1007/s00439-003-1049-7. [DOI] [PubMed] [Google Scholar]
- 33.Reddy MA, et al. Characterization of the G91del CRYBA1/3-crystallin protein: a cause of human inherited cataract. Human molecular genetics. 2004;13:945–953. doi: 10.1093/hmg/ddh110. [DOI] [PubMed] [Google Scholar]
- 34.Lu S, et al. Two Chinese families with pulverulent congenital cataracts and deltaG91 CRYBA1 mutations. Molecular vision. 2007;13:1154–1160. [PubMed] [Google Scholar]
- 35.Yang G, Zhai X, Zhao J. A recurrent mutation in CRYBA1 is associated with an autosomal dominant congenital nuclear cataract disease in a Chinese family. Molecular vision. 2011;17:1559–1563. [PMC free article] [PubMed] [Google Scholar]
- 36.Gillespie, R. L. et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology121, 2124–2137, e2121–2122, doi:10.1016/j.ophtha.2014.06.006 (2014). [DOI] [PubMed]
- 37.Xu J, et al. Decreasing the homodimer interaction: a common mechanism shared by the deltaG91 mutation and deamidation in betaA3-crystallin. Molecular vision. 2010;16:438–444. [PMC free article] [PubMed] [Google Scholar]
- 38.Ma X, et al. A new mutation in BFSP2 (G1091A) causes autosomal dominant congenital lamellar cataracts. Molecular vision. 2008;14:1906–1911. [PMC free article] [PubMed] [Google Scholar]
- 39.Conley YP, et al. A juvenile-onset, progressive cataract locus on chromosome 3q21-q22 is associated with a missense mutation in the beaded filament structural protein-2. American journal of human genetics. 2000;66:1426–1431. doi: 10.1086/302871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui X, et al. The E233del mutation in BFSP2 causes a progressive autosomal dominant congenital cataract in a Chinese family. Molecular vision. 2007;13:2023–2029. [PubMed] [Google Scholar]
- 41.Wang H, Zhang T, Wu D, Zhang J. A novel beaded filament structural protein 1 (BFSP1) gene mutation associated with autosomal dominant congenital cataract in a Chinese family. Molecular vision. 2013;19:2590–2595. [PMC free article] [PubMed] [Google Scholar]
- 42.Ramachandran RD, Perumalsamy V, Hejtmancik JF. Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Human genetics. 2007;121:475–482. doi: 10.1007/s00439-006-0319-6. [DOI] [PubMed] [Google Scholar]
- 43.Lin Q, et al. Genetic variations and polymorphisms in the ezrin gene are associated with age-related cataract. Molecular vision. 2013;19:1572–1579. [PMC free article] [PubMed] [Google Scholar]
- 44.Muller M, et al. Dominant cataract formation in association with a vimentin assembly disrupting mutation. Human molecular genetics. 2009;18:1052–1057. doi: 10.1093/hmg/ddn440. [DOI] [PubMed] [Google Scholar]
- 45.Mishra R, Gorlov IP, Chao LY, Singh S, Saunders GF. PAX6, paired domain influences sequence recognition by the homeodomain. The Journal of biological chemistry. 2002;277:49488–49494. doi: 10.1074/jbc.M206478200. [DOI] [PubMed] [Google Scholar]
- 46.Fuhrmann S. Eye morphogenesis and patterning of the optic vesicle. Current topics in developmental biology. 2010;93:61–84. doi: 10.1016/B978-0-12-385044-7.00003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kioussi C, et al. Pax6 is essential for establishing ventral-dorsal cell boundaries in pituitary gland development. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14378–14382. doi: 10.1073/pnas.96.25.14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dohrmann C, Gruss P, Lemaire L. Pax genes and the differentiation of hormone-producing endocrine cells in the pancreas. Mechanisms of development. 2000;92:47–54. doi: 10.1016/S0925-4773(99)00324-X. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, et al. Large novel deletions detected in Chinese families with aniridia: correlation between genotype and phenotype. Molecular vision. 2011;17:548–557. [PMC free article] [PubMed] [Google Scholar]
- 50.Lin Y, et al. PAX6 analysis of two sporadic patients from southern China with classic aniridia. Molecular vision. 2012;18:2190–2194. [PMC free article] [PubMed] [Google Scholar]
- 51.Lin Y, et al. PAX6 analysis of one family and one sporadic patient from southern China with classic aniridia. Molecular vision. 2011;17:3116–3120. [PMC free article] [PubMed] [Google Scholar]
- 52.Mirzayans F, Pearce WG, MacDonald IM, Walter MA. Mutation of the PAX6 gene in patients with autosomal dominant keratitis. American journal of human genetics. 1995;57:539–548. [PMC free article] [PubMed] [Google Scholar]
- 53.Azuma N, et al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. American journal of human genetics. 2003;72:1565–1570. doi: 10.1086/375555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yokoi T, et al. Genotype-phenotype correlation of PAX6 gene mutations in aniridia. Human genome variation. 2016;3:15052. doi: 10.1038/hgv.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dubey SK, Mahalaxmi N, Vijayalakshmi P, Sundaresan P. Mutational analysis and genotype-phenotype correlations in southern Indian patients with sporadic and familial aniridia. Molecular vision. 2015;21:88–97. [PMC free article] [PubMed] [Google Scholar]
- 56.Khan AO, Aldahmesh MA. PAX6 analysis of two unrelated families from the Arabian Peninsula with classic hereditary aniridia. Ophthalmic genetics. 2008;29:145–148. doi: 10.1080/13816810802078195. [DOI] [PubMed] [Google Scholar]
- 57.He Y, Pan Z, Luo F. A novel PAX6 mutation in Chinese patients with severe congenital aniridia. Current eye research. 2012;37:879–883. doi: 10.3109/02713683.2012.688165. [DOI] [PubMed] [Google Scholar]
- 58.Jin C, et al. A recurrent PAX6 mutation is associated with aniridia and congenital progressive cataract in a Chinese family. Molecular vision. 2012;18:465–470. [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Q, et al. A novel PAX6 deletion in a Chinese family with congenital aniridia. Gene. 2015;563:41–44. doi: 10.1016/j.gene.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Yu Y, et al. Congenital polymorphic cataract associated with a G to A splice site mutation in the human beta-crystallin gene CRYbetaA3/A1. Molecular vision. 2012;18:2213–2220. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.