

Abstract
GPR139 is an orphan G protein-coupled receptor expressed in the brain, in particular in the habenula, hypothalamus and striatum. It has therefore been suggested that GPR139 is a possible target for metabolic disorders and Parkinson’s disease. Several surrogate agonist series have been published for GPR139. Two series published by Shi et al. and Dvorak et al. included agonists 1a and 7c respectively, with potencies in the ten-nanomolar range. Furthermore, Isberg et al. and Liu et al. have previously shown that tryptophan (Trp) and phenylalanine (Phe) can activate GPR139 in the hundred-micromolar range. In this study, we produced a mutagenesis-guided model of the GPR139 binding site to form a foundation for future structure-based ligand optimization. Receptor mutants studied in a Ca2+ assay demonstrated that residues F1093×33, H1875×43, W2416×48 and N2717×38, but not E1083×32, are highly important for the activation of GPR139 as predicted by the receptor model. The initial ligand-receptor complex was optimized through free energy perturbation simulations, generating a refined GPR139 model in agreement with experimental data. In summary, the GPR139 reference surrogate agonists 1a and 7c, and the endogenous amino acids l-Trp and l-Phe share a common binding site, as demonstrated by mutagenesis, ligand docking and free energy calculations.
Introduction
G protein-coupled receptors (GPCRs) constitute the largest family of cell surface proteins. The human genome contains approximately 800 GPCR genes1. GPCRs are involved in a broad spectrum of (patho)physiological processes2, 3 related to e.g. vision, neurotransmission, immune responses, and metabolism. GPCRs also constitute one of the most important drug target families as about a third of all approved drugs on the market today target a GPCR4, 5. Intriguingly, 121 GPCRs are non-sensory orphan receptors6 (having unknown endogenous ligands) that could represent yet untapped targets for novel treatments7.
GPR139 is a class A orphan GPCR8 and its mRNA is predominantly expressed in the striatum, habenula and hypothalamus9–12. Attempts to determine the GPR139 protein expression with radioligands have failed9, 13. Consistent cross-species expression of GPR139 mRNA in the striatum9–11, 14 suggests that GPR139 may play a role in locomotor activity. This is also supported by Liu et al. who showed that activation of GPR139 with a surrogate agonist 7c (also known as JNJ-63533054) leads to decreased spontaneous locomotion activity in rats9. One major locomotion pathophysiology is Parkinson’s diseases. MPP+ is a toxin used in animals, which produce the neurological defects observed in Parkinson’s patients by degenerating dopaminergic neurons. A recent study by Andersen et al. showed that GPR139 agonists protect primary dopaminergic neurons against MPP+ in vitro 15. Based on these findings, GPR139 has been hypothesized as a potential target for the treatment of diseases with impaired movement control, e.g. Parkinson’s disease.
Furthermore, the GPR139 mRNA expression in hypothalamus and habenula suggests a role in the regulation of food consumption and/or energy expenditure12. l-Trp and l -Phe 9, 16 activate GPR139, which has therefor been propsed to be a nutrient-sensing receptor9, 16. In support of this hypothesis, the closest homolog GPR142 is also activated by l-Trp and l -Phe 17, 18 and activation of this receptor has been shown to lower blood glucose levels and increase insulin secretion in mice17, 19, 20 making it a new putative target for treatment of diabetes. Since the GPR139 receptor shares the same ligands and is expressed in the hypothalamus it is possible that GPR139 is also involved in the pathophysiology of diabetes. Furthermore, we have very recently shown that the endogenous POMC derived peptides ATCH, α-MSH, and β-MSH, known to be involved in energy homeostasis, also activate GPR139 in vitro 21. Taken together GPR139 has been hypothesized as a potential target for the treatment of the metabolic syndrome e.g. diabetes and eating disorders.
Besides the natural aromatic amino acids l-Trp and l -Phe 9, 16 and the endogenous POMC derived peptides21, GPR139 has been reported to bind surrogate small molecules (e.g. 1a and 7c)13, 22–26. In the present study, an initial homology model of GPR139 in complex with 1a was used to guide a site-directed mutagenesis (SDM) study of the GPR139 ligand binding site, which was performed on ligands 1a, 7c, l-Trp and l -Phe, bearing a common structure-activity relationship (SAR) profile as shown earlier26 and summarized in Fig. 1. The mutations and binding modes were further examined in silico using a ligand-steered homology modeling approach27, in this case based on cycles of molecular dynamics (MD) and free energy perturbation (FEP) on selected alanine mutations28–30 used as a scoring function. From these results, a common interaction profile for the selected GPR139 agonists was confirmed.
Figure 1.
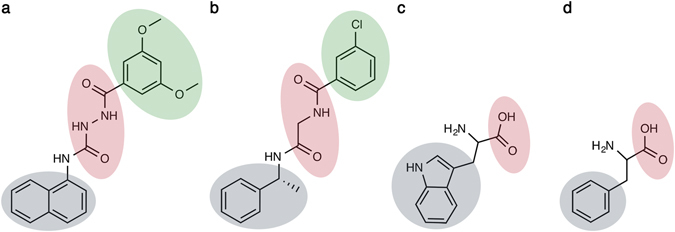
Structure of the GPR139 agonists studied herein. (a) Surrogate agonist 1a from Shi et al.23. (b) Surrogate agonist 7c from Dvorak et al.14, 24. (c,d) Trp and Phe from Isberg et al.16. Coloring denote chemical commonalities (supported by mutations herein); grey: major hydrophobic part, red: polar linkers (1a and 7c) or carboxyls (Phe and Trp), green: hydrophobic element unique for the larger 1a and 7c.
Results
Mutations selected from a preliminary 1a binding mode model
Docking of compound 1a in our preliminary GPR139 structure model indicated four residues; E1053×29, E1083×32, N2717×38 and R2446×51 with putative hydrogen bonds to the ligand linker (Supplementary Fig. 1a). The 1a naphthyl ring displayed a tight fit inside a deep hydrophobic pocket lined by F1093×33, H1875×43, and W2416×48. Binding poses with a flipped ligand orientations were also seen. However, the above-described pose got the highest score (−9.51 compared to −5.29) and is the only one that agrees with the mutation data. Therefore, we decided to only move forward with that and similar poses. Based on this binding pose, we selected 18 binding site mutations (of 12 residues) for pharmacological testing (Supplementary Fig. 1b). The mutations focused solely on residues in the transmembrane region, as none of the extracellular loops were in proximity of the ligand binding site.
Surface expression of GPR139 mutants
Cell surface expression was measured with ELISA using a myc-tag positioned at the extracellular N-terminal of GPR139. The myc-tagged wild-type (WT) receptor displayed equivalent response to 1a in the Fluo-4 Ca2+-assay (EC50 = 670 nM) as compared to the untagged WT receptor (EC50 = 772 nM) (Supplementary Fig. 2). Both R2446×51 mutants (alanine and methionine) lost surface expression, while the expression of H187A5×43 was significantly reduced (36% of WT, Fig. 2). Modestly reduced surface expression (>50%) was observed for the mutants L87A2×64, L87F2×64, Y163A4×61, and N271A7×38 whereas the remaining mutants showed WT-like or even increased surface expression compared to WT (Fig. 2).
Figure 2.
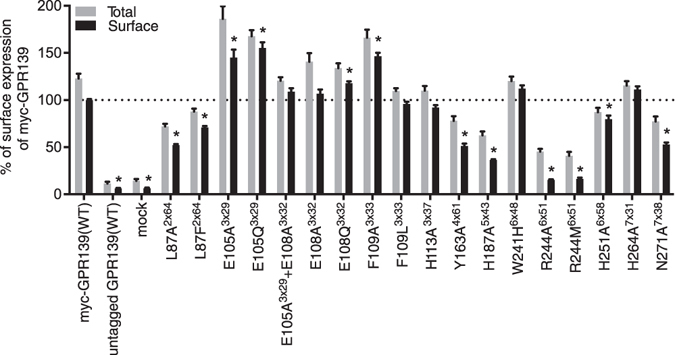
Total and surface expression of GPR139 mutants. Cell surface expression profiles of each of the human GPR139 mutants compared to myc-GPR139(WT). Grey bars = total expression (triton-X treated), black bars = surface expression. Data is mean ± S.E.M. of 4–8 independent experiments performed in triplicates. Statistical analysis was performed on the surface expression using one-way ANOVA followed by Dunnett’s post-hoc test in comparison with the surface expression of myc-GPR139(WT) (*P < 0.01).
The 1a binding site
In vitro mutation effects on ligand potencies
The ability of 1a to activate the mutant receptors was measured in a Fluo-4 Ca2+-assay (Supplementary Fig. 3). Complete loss-of-function was observed for mutants F109A3×33, F109L3×33, and N271A7×38, while 1a displayed markedly reduced potency at W241H6×48 and H187A5×43 (Fig. 3). Noteworthy, for the H187A5×43 and N271A7×38 mutants their lower expression levels (36% and 53%, respectively) could also have contributed to the reduction in potency. However, 1a was equipotent on L87A2×64 indicating that mutants with reduced expression can sometimes still induce a normal functional response. All other mutations showed no significant (>10-fold) change in potency, indicating that these residues are not critical for 1a activity (Table 1 and Supplementary Table 1).
Figure 3.
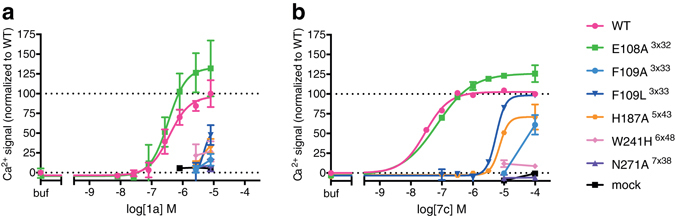
Effects of GPR139 mutations on pharmacological profiles of 1a and 7c. The data demonstrates that the residues F1093×33, H1875×43, W2416×48 and N2717×38 are important for GPR139 activation by 1a and 7c, whereas residue E1083×32 is not. Concentration-response curves of (a) 1a and (b) 7c, on the mutants with an effect (plus WT, mock and E108A3×32). The graphs are one representative (mean ± S.D.) out of three independent experiments performed in (a) triplicates and (b) duplicates. All responses are normalized to myc-GPR139(WT) (0% = buffer, 100% = 8 μM 1a or 100 μM 7c).
Table 1.
GPR139 mutant potencies for 1a, 7c, l-Trp, and l -Phe.
| Mutant | % SE | 1a | 7c | l-Trp | l-Phe | ||||
|---|---|---|---|---|---|---|---|---|---|
| pEC50 ± SEM | Emax ± SEM | pEC50 ± SEM | Emax ± SEM | pEC50 ± SEM | Emax ± SEM | pEC50 ± SEM | Emax ± SEM | ||
| WT | 100 | 6.63 ± 0.08 | 100 | 7.32 ± 0.16 | 100 | 3.70 ± 0.14 | 100 | 3.43 ± 0.22 | 100 |
| E108A3×32 | 109 | 6.68 ± 0.11 | 151 ± 9 | 6.82 ± 0.25 | 112 ± 10 | 3.78§ | 125 | 3.60§ | 127 |
| F109A3×33 | 146 | <5.1 | NE* | <4.0 | 60 ± 3** | <2 | 60 ± 10# | <1.5 | 60 ± 6## |
| F109L3×33 | 96 | <5.1 | 42 ± 16* | 5.40 ± 0.07 | 102 ± 3 | <2 | 60 ± 5# | <1.5 | 72 ± 9## |
| H187A5×43 | 36 | <5.1 | 35 ± 5* | 5.18 ± 0.06 | 60 ± 9 | ND | ND | ND | ND |
| W241H6×48 | 112 | <5.1 | 27 ± 10* | <4.0 | 12 ± 3** | <2 | NE# | <1.5 | NE## |
| N271A7×38 | 53 | <5.1 | NE* | <4.0 | NE** | <2 | 31 ± 5# | <1.5 | 38 ± 9## |
The table displays the mutants with effect on 1a (and E108A3×32), and a percent surface expression (% SE, normalized to WT = 100%) over 35%. The potencies are presented as mean pEC50 ± SEM and mean Emax ± SEM. The potency of 1a was calculated from three independent experiments conducted in triplicates, and normalized to buffer (0%) and 8 μM 1a (100%). The potencies of 7c, Trp and Phe are from three independent experiments conducted in duplicates, and normalized to buffer (0%) and 100 μM 7c or 10 mM Trp or 30 mM Phe (100%), respectively. Remarks: *at 8 μM, **at 100 μM, #at 10 mM, ##at 30 mM, §only one experiment, ND: not determined (not measured) and NE: no effect (loss of activity).
In silico mutation effects on calculated binding affinities
The initial GPR139-1a structural model described above was optimized to take into account the mutagenesis data (see methods). This was done by means of iterative MD simulations, coupled to free energy perturbation (FEP) calculations as a scoring function (Supplementary Figs 4 and 5). The three mutations to alanine that showed a significant effect and were sufficiently expressed were chosen for scoring, i.e. residues F109A3×33, H187A5×43 and N271A7×38. Mutation W241H6×48 also showed drastic effects on ligand potency, but the dynamic role of this residue (activation switch in class A GPCRs31) precluded us from using the data for its mutation to histidine in our model optimization.
The calculated difference in binding free energies for the mutant receptors in the first and second iterations did not correlate well with the experimental mutagenesis data, and the analysis of the trajectories showed unstable protein (iteration 2) and ligand (iteration 1 and 2) conformations. The third MD/FEP iteration showed significant effects for mutants F109A3×33 and N271A7×38, while the H187A5×43 displayed a large standard error of the mean (s.e.m.) (Table 2). For this reason, we ran a final iteration consisting of re-docking and MD/FEP of 1a in the conformation of GPR139 obtained after a similar iteration with ligand 7c (see next section). The obtained final model reproduced the in vitro potency data of 1a by showing positive contributions to the binding free energies (which corresponds to lower ligand affinity) and showed that the FEP scoring approach was able to distinguish between low (iteration 1) and high (iteration 4) quality models.
Table 2.
GPR139 in silico mutant effects of 1a and 7c binding.
| Mutation | Change of in vitro potency | In silico relative binding free energies (ΔΔG kcal/mol) | |||||
|---|---|---|---|---|---|---|---|
| 1a | 7c | 1a | 1a | 7c | |||
| Iteration 1 | Iteration 2 | Iteration 3 | Final model | Final model | |||
| F109A3×33 | Loss of function | >2000 fold decreased | −3.14 ± 0.84 | 0.66 ± 2.02 | 1.25 ± 1.03 | 6.52 ± 0.87 | 6.27 ± 0.66 |
| H187A5×43 | >34 fold decreased | 138 fold decreased | −3.03 ± 1.04 | −1.26 ± 1.60 | 0.56 ± 1.11 | 2.64 ± 0.93 | 1.72 ± 1.29 |
| N271A7×38 | Loss of function | Loss of function | 0.25 ± 0.47 | 4.40 ± 0.54 | 3.73 ± 0.29 | 3.21 ± 0.45 | 2.71 ± 0.34 |
Iterative procedure of model optimization using molecular dynamics and the relative binding free energies obtained by free energy perturbation (FEP) calculations in comparison with in vitro potency as a scoring function. The FEP relative binding free energies that are in agreement with in vitro data are shown in bold.
Binding mode in the receptor model
The 1a naphthyl ring was positioned in a deep hydrophobic pocket lined by F109A3×33, H187A5×43, and W241H6×48 (Fig. 4a); all of which displayed significant effects upon mutation. The available SAR for 1a confirms tight binding of the naphthyl ring, as substitution in the 4, 5 or 7 positions abolishes ligand binding affinities23. The linker in 1a displayed hydrogen bonds to N2717×38 and R2446×51. Notably, the model did not show a hydrogen bond to E1083×32, but instead an indirect interaction via R2446×51. This is in agreement with the mutation data that showed no effect for E108A3×32 and a modest 6-fold potency reduction for E108Q3×32, in which the carboxamide nitrogen may have unfavorable contact with R2446×51.
Figure 4.
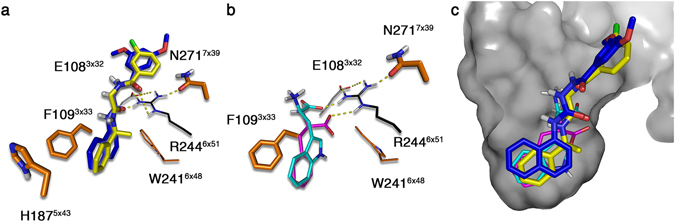
1a, 7c, l-Trp, and l -Phe binding pose models. (a) Binding mode of 1a (blue) and 7c (yellow) and (b) endogenous amino acids l-Trp (cyan) and l -Phe (magenta). Mutations that showed a significant effect when mutated are colored orange. Residues with thick sticks have been mutated in silico and in vitro (F1093×33, H1875×43 and N2717×39) and those with thin sticks in vitro only (W2416×48). The latter was excluded due to the dynamic role of this residue as an activation switch in class A GPCRs25. Residues colored in grey showed no significant changes in potency (E1083×32) and those in black were not expressed respectively (R2446×51). (c) Overlay of all four studies ligands within the GPR139 binding pocket shown as a surface. All tested agonists bind a deep hydrophobic pocket and are shown to undergo hydrogen bonding with R2446×51.
The 7c binding site
In vitro mutation effects on ligand potencies
All mutants that had an effect on 1a also affected 7c potency, although F109L3×33 and H187A5×43 displayed a milder (yet ~100-fold) effect (Table 1).
In silico mutation effects on calculated binding affinities
Compound 7c was docked in the optimized structure of GPR139 obtained from iteration 3 of the GPR139-1a complex, resulting in similar poses for the two ligands. However, during the equilibration phase of the subsequent run of MD/FEP the ligand 7c readjusted its initial pose to bind deeper in the binding pocket. This resulted in a stable conformation that gives calculated energies in excellent agreement with the in vitro results for this ligand (Table 2). Thus, we re-docked 1a in the resulting structure of GPR139 and ran a final (fourth, see above) MD/FEP iteration for this ligand, obtaining excellent qualitative agreement with the experimental data as outlined above (Table 2). The two ligands thus show very similar effects for each of the three mutations considered both in vitro and in silico. Notably, the experimental data on ligand potency for 7c shows a somewhat milder effect for H187A5×43 than F109A3×33 and N271A7×38, which is in line with the lower calculated effect of the H187A5×43 mutation.
Binding mode in receptor model
The final poses for 1a and 7c overlap nearly perfectly (common scaffold RMSD 0.529 Å, Fig. 4a) and they display nearly identical receptor interactions. This reflects their high similarity in both the in vitro and in silico mutants. This confirms a common binding mode for these two ligands.
The l-Trp and l-Phe binding site
In vitro mutation effects on ligand potencies
The mutations displayed very similar effects on the amino acids l-Trp and l -Phe as for 1a and 7c (Table 1 and Supplementary Fig. 6).
Binding mode in receptor model
Both amino acids showed a similar binding mode (Fig. 4b), placing the hydrophobic functional groups of the amino acids deeply in the binding pocket in overlay with hydrophobic moieties of 1a and 7c (Fig. 4c). Furthermore, the charged backbone functionalities overlap with the polar linkers of 1a and 7c.
Discussion
We identified a common binding site for the GPR139 surrogate agonists; 1a, 7c and the endogenous amino acids l-Trp and l -Phe (Fig. 4). The proposed binding pocket consists of a deeply buried hydrophobic region between F1093×33 H1875×43 and W2416×48, and a polar region defined by N2717×38, R2446×51 and E1083×32. The four ligands position themselves in a way that all have an aromatic moiety in the deep hydrophobic region, while their polar (1a and 7c) or charged (amino acids) regions bind to N2717×38 and R2446×51. This is supported by the loss of ligand activity in analogs lacking one of these carbonyls23, 24. E1083×32 is indicated to have only indirect interaction via R2446×51.
The binding mode matches our recent 3D pharmacophore of different series of GPR139 agonists16, 26. One of the two hydrophobic pharmacophore elements harbors the same ligand moieties (1a naphthyl, 7c phenyl and l -Phe/l-Trp sidechains) deep in the hydrophobic part of the binding site. R2446×51 and pharmacophore hydrogen bond acceptor elements both match the polar linkers (1a and 7c) or charged amino acids backbones. The second terminal phenyl in 1a and 7c points to the top of TM2, in contact with Phe832×60 and Leu862×63. A hydrogen bond acceptor functionality in this ring assigned by the pharmacophore model could correspond with the interaction of the methoxy in 1a or the chloride atom in 7c with N2717×38.
FEP calculation dependence of the use of starting structure was traditionally considered a limiting factor for the widespread application of this technique. However, current computational power and the increased robustness of the protocols have made it possible to use the FEP results as a filter to discriminate the most reliable binding mode from a pool of solutions28. In this work, we applied FEP to refine an initial binding mode in an iterative fashion. This iterative protocol allowed for the identification of a binding mode that correlates with and provides satisfactory explanation to the experimental data. As shown in iteration 1, this effect could not be captured with a more simple and intuitive use of docking and modeling.
Conclusions
Our combined in vitro and in silico mutagenesis demonstrates that residues F1093×33, H187A5×43, and N2717×38 are highly important for all studied ligands. We provide (Supplementary Data) highly refined GPR139-ligand complex structure models that fully agree with these data and may serve to guide future mutagenesis or ligand design, including identification of much needed antagonists with increased potency and selectivity. Hence, this first report of the GPR139 binding site paves the way for future studies towards the characterization of GPR139 pharmacology and function.
Methods
Receptor mutagenesis
To study the influence of specific amino acids in the predicted binding pocket on receptor function, the desired mutations were introduced into an N-terminally c-myc-tagged wild type human GPR139 (NM_001002911.3) in the pEGFP-N1 vector (BD Biosciences)32, 33. As previously described33, these plasmids carry the signal peptide of mGluR5 to promote cell surface expression followed by a c-myc epitope to enable detection of total and surface expression by ELISA, followed by an engineered MluI and NotI site for easy insertion of the receptor of interest. Furthermore, the start-codon in the GPR139 sequence was deleted to ensure c-myc-tagging. Mutagenesis was designed in Serial Cloner 2.6 (© Franck Perez [SerialBasics]) and carried out by GenScript (USA). All mutated residues are illustrated in Supplementary Fig. 1b. An untagged GPR139(WT) in pEGFP-N1 was also generated to ensure that the c-myc-tag had no influence on GPR139(WT) function.
Transfection and cell culture
All GPR139 mutants were transfected into HEK293 cells. The HEK293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, 41965) supplemented with 10% dialyzed fetal bovine serum (Gibco, 26400, United States origin), 100 units/mL penicillin and 100 μg/mL streptomycin (Gibco, 15140). HEK293 cells were reverse transfected using Lipofectamine2000 (Invitrogen). More specifically two mixtures were made: 1) 10 ng DNA/well + 25 μL Opti-MEM/well and 2) 250 μg Lipofectamine2000/well + 25 μL Opti-MEM/well. The two mixtures were mixed and 50 μL/well was added directly in black 96-well plates with flat clear bottoms for the Fluo-4 Ca2+-assay (Corning, Falcon, 353219) or in white Opaque 96-well Microplate (PerkinElmer Life, 6005680) for ELISA. 100 μL containing 20,000 cells were added to each well and incubated 48 hours before assays. All plates were coated with poly-d-lysine.
ELISA
Cells were fixed using 50 μL/well 4% paraformaldehyde for 5 minutes (min). The wells were washed twice with DPBS (Gibco, 14190) containing 1 mM Ca2+ (DPBS-Ca), followed by addition of either 50 μL/well 0.1% triton-X (for detection of total expression) or 50 μL/well DPBS-Ca (for detection of surface expression) for 5 min. The wells were again washed twice with DPBS-Ca, followed by the addition of 100 μL/well blocking solution (ddH2O with 3% skim milk, 1 mM Ca2+, 50 mM Trizma hydrochloride solution pH 7.4) and incubated at room temperature (RT) for 30 minutes. After blocking, 75 μL/well 1:1000 c-myc mouse monoclonal antibody (Invitrogen, R950-25) diluted in blocking solution was added to each well and incubated at RT for 45 minutes. Subsequently, each well was washed with 100 μL/well blocking solution and twice with 100 μl/well DPBS-Ca. Then 75 μL/well 1:1500 sheep anti-mouse IgG HRP conjugate (VWR, NA931-100UL) diluted in blocking solution was added, and incubated at RT for 45 minutes. The wells were then washed four times with 100 μL/well blocking solution and four times with 100 μL/well DPBS-Ca. At the end 65 μL/well DPBS-Ca was added, and the detection solution was prepared (SuperSignal ELISA Femto stable peroxidase solution and SuperSignal ELISA Femto luminol enhancer solution (Thermo Fisher Scientific, Waltham, MA), 1:1); 15 μL/well detection solution was added to the plate, and chemiluminescence was measured immediately on an EnSpire reader (PerkinElmer Life and Analytical Sciences).
Ligands
Compound 1a (1a) was kindly provided by H. Lundbeck A/S, Denmark. Compound 7c (Enamine, Z31449867) was tested as a racemate, as the (S)-form described by Janssen et al. was not commercially available. Both compounds were dissolved to 20 mM in DMSO (Sigma, D2650) and subsequently diluted in a HEPES buffer (HBSS (Gibco, 14025) supplemented with 20 mM HEPES + 1 mM MgCl2 + 1 mM CaCl2, pH = 7.4) to a final concentration of 0.5% DMSO in the Fluo-4 Ca2+-assay. The DMSO level was kept constant for all concentrations of both compounds. DMSO was confirmed not to have any activity by itself at this concentration16. l-Trp (T0254) and l -Phe (P2126) were obtained from Sigma-Aldrich and dissolved in buffer.
Fluo-4 Ca2+-assay
The Ca2+ measurements were preformed using the Fluo-4 NW Calcium Assay Kit (Invitrogen, Molecular Probes, F36206) as previously described26. Briefly, the Fluo-4 dye loading solution was prepared according to the manufacturer’s instructions by dissolving it in HEPES-buffer supplemented with 2.5 mM probenecid. 50 μL dye loading solution was added to each well. Cells were incubated with the Fluo-4 dye for 60 min at 37 °C, then washed with 100 μL HEPES-buffer. 100 μL HEPES-buffer supplemented with 2.5 mM probenecid was then added to each well and incubated in 10 minutes at 37 °C before measurement. 33 μL of 1a, 7c, l-Trp or l -Phe (4x concentrated) were added automatically after baseline measurements. Intracellular calcium changes were recorded as indicated on either a NOVOstar (BMG Labtech) at 37 °C with an excitation filter of 485 nm and an emission filters 520 nm or a FlexStation 3 Benchtop Multi-Mode Microplate Reader (Molecular Devices) at 37 °C with an excitation wavelength of 485 nm and emission of 525 nm.
Data analysis and statistics
All pharmacological data have been analyzed by using Prism 6.0 (GraphPad Software Inc., San Diego). Fitting of concentration response curves was performed by non-linear regression log(agonist) vs. response (four parameters). Pooled data are shown with standard error of the mean (S.E.M.) and data showing one representative are shown with standard deviation (S.D.). Changes in potency (pEC50) and span of myc-GPR139(WT) in comparison to untagged GPR139(WT) was statistically analyzed with a paired t-test and significance was accepted at p < 0.05. The ELISA data were normalized to surface expression of myc-GPR139(WT) (100%). The changes in cell surface expression of GPR139 mutants in comparison with WT control and the changes in potency of GPR139 mutants in comparison with WT control were statistically analyzed with one-way analysis of variance (ANOVA) and Dunnett’s post-hoc test.
Homology modeling
The crystal structure of the active human serotonin 2B receptor (5HT2B - PDB: 4IB4)34 was selected as an initial template using the online GPCRdb template selection tool35, 36. The structure has a resolution of 2.7 Å, and the protein sequence similarity between GPR139 and the template structure is 42% in the seven trans-membrane helical (7TM) region. The protein sequences of the template and target were aligned with MEGA637, while utilizing GPCRdb sequence alignment and the main template as a reference for assigning the seven helical tips. The overall receptor structure was built with Modeller9 v.1338, and loops further refined with the LoopModel routine therein implemented. In general, a disulphide bridge between ECL2 Cys45×50 and Cys3×25 is a conserved feature among most Class A GPCRs. However, this feature was not incorporated in our GPR139 model, as both ECL2 and TM3 lack a Cys residue in those positions. To enhance the GPR139 model for subsequent ligand docking, the rotamers of residues that are not conserved between the target and template were manually defined based on the most homologous template from an in-house GPCR position-specific rotamer library that contains rotamers extracted from all published GPCR crystal structures39. Therefore the rotamers of the following residues were refined: Y331×39, L361×42, L862×63, E1053×29, F1093×33, I1845×40, W2416×48, I2486×55, H2516×58, H2647×31, D2687×35, N2717×38, and L2757×42. All rotamers were collected from our in-house GPCR position-specific rotamer library39. On the other hand, the rotamer of R2446×51 – a critical amino acid in the binding site with no available rotamers in our library – was refined based on the rotamer library available in Maestro (Schrödinger Release 2015-3)40. In total, 12 of 18 residues (~67%) were refined in accordance with a similar crystal structure template. The quality of the model was assessed by Ramachandran plots within the PROCHECK webserver41.
Another homology model based on the inactive human opioid kappa receptor (OPRK - PDB: 4DJH)42 was also built. The template structure has a resolution of 2.9 Å, and an overall 7TM similarity of 42% to GPR139. The model was built and its quality assessed using the same software and routines as described above.
Ligand Docking and Molecular Visualization
The two homology models were prepared for docking studies with the Schrödinger Protein Preparation Wizard, including a hydrogen optimization at pH = 7 of the ionisable polar groups using Maestro PROPKA43.
Docking was done with Glide44–46 with default settings. The partial charges of the ligand were assigned by Epik47, 48 using the OPLS_3 force field. Further options were set to allow the rotation of hydroxyl hydrogen atoms in the binding site, enhance the planarity of conjugated π-systems, and to include the Epik state penalties to the scoring calculations. Flexible ligand sampling was applied combined with biased sampling of amide groups (penalization of nonplanar conformations). The docking grid centroid was placed around binding pocket residues E1053×29, F1093×33, W2416×48, and R2446×51, and the cubic grid box sides were set at 10 Å. Subsequently, the resulting receptor–ligand complexes were minimized using the energy minimization tool in MacroModel Schrödinger49. The TNCG (Truncated Newton Conjugate Gradient) minimization method was used with maximum iteration steps set to 5000, and with a convergence gradient of 0.001. Heavy atoms were strained at a ±0.3 Å radius while applying a force constant of 120 kcal/mol/Å2. Poses correlating with the mutation data were used for further MD analysis. All 3D images were produced in PyMOL50.
Membrane insertion and equilibration
Ligand receptor complexes obtained in the previous stage were inserted in the membrane and equilibrated under periodic boundary conditions (PBC) using the PyMemDyn protocol described elsewhere51. Shortly, the starting structure is automatically embedded in a pre-equilibrated membrane consisting of POPC (1-palmitoyl-2-oleoyl phosphatidylcholine) lipids, with the TM bundle aligned to its vertical axis. This hexagonal-prism shaped box is then soaked with bulk water and energy minimized with GROMACS 4.651, 52, using the OPLS-AA force field53 for protein and ligands, combined with the Berger parameters for the lipids54. The same setup is used for a 2.5 ns MD equilibration, where initial restraints on protein and ligand atoms are gradually released as described in detail in ref. 51.
MD and FEP calculations
The MD software Q55 was used for free energy perturbation (FEP) calculations under spherical boundary conditions, using a 25 Å sphere centered on the center of geometry of the ligand. Protein atoms in the boundary of the sphere 22–25 Å outer shell) had a positional restraint of 20 kcal/mol/Å2, while solvent atoms were subject to polarization and radial restrains using the surface constrained all-atom solvent (SCAAS)56, 57 model to mimic the properties of bulk water at the sphere surface. Atoms lying outside the simulation sphere are tightly constrained (200 kcal/mol/Å2 force constant) and excluded from the calculation of non-bonded interactions. Long range electrostatics interactions beyond a 10 Å cut off were treated with the local reaction field method58, except for the atoms undergoing the FEP transformation where no cutoff was applied. Solvent bond and angles were constrained using the SHAKE algorithm59. The consideration of the binding site region under spherical boundary conditions offers several advantages over the alternate periodic boundary conditions for free energy calculations. Besides the obvious reduction in system size, it avoids possible larger scale conformational fluctuations distal to the binding site. Such fluctuations commonly introduce noise and decrease convergence for the free energy calculations, while a spherical system still considers the relevant fluctuations in the binding site, as previously demonstrated60. In the particular case of membrane proteins, we have recently reported a similar effect when increasing the sphere size to consider distal parts of the receptor and membrane environment26.
All titratable residues outside the sphere were neutralized and protonation states of the histidines were manually assigned. Histidine residues H1133×37, H1875×43, H2516×58, and H2647×31 were assigned a hydrogen atom on the δ nitrogen and residues H1323×56, H13734×54, H1803×36, and H1813×37 on the ε nitrogen. Ligand and lipid parameters were obtained from the previous MD stage, whereas residue parameters were translated from the latest version of the OPLSAA force field61.
The sphere was equilibrated for 0.61 nanoseconds, where temperature was increased from 0.1 to 298 K whilst slowly removing a 25 kcal/mol/Å2 restraint on all atoms, and the protein-ligand interactions maintained through a distance restraint (15.0 kcal/mol/Å2 force constant for distance between 1.8 and 2.2 Å) for hydrogen bonds between carbonyl atoms in the ligand and hydrogen atoms in R2446×51 and N2717×38. These restraints were gradually removed during an additional 2 nanosecond equilibration MD, before the data collection period. This phase consisted of 14 replicates with different initial velocities and a 0.25 nanosecond unbiased equilibration period for each amino acid mutation, before applying the FEP protocol for amino acid mutations as previously published28–30. Briefly, a given mutation of any residue to alanine is divided in several smaller subperturbations to allow for a smoother transition between the end-states. Three steps are introduced for groups of atom (charge groups) starting with the group with the highest topological distance (number of atoms) from the protein backbone: (i) removal of partial charges per charge group, (ii) the introduction of a soft core van der Waals potential and (iii) the full annihilation of the atom(s). The last step includes the introduction of the Cβ hydrogen atom of the alanine residue. A mutation consists of eight, seven and five subperturbations of 51 λ windows of 10 ps each for phenylalanine, histidine and asparagine mutations, respectively. In the final models (iterations three and four) we tripled the simulation time to increase the convergence and hysteresis.
Electronic supplementary material
Acknowledgements
D.E.G. acknowledges financial support from the Lundbeck Foundation (R163-2013-16327) and the European Research Council (DE-ORPHAN 639125). H.B.-O. acknowledges financial support from the Lundbeck Foundation. W.J. acknowledges financial support from cost action CM1207; and H.G.T., J.Å. and W.J. from the Swedish research council (VR). The simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC). M.A.S. and A.C.N. acknowledge funding from the Faculty of Health and Medical Sciences.
Author Contributions
A.C.N. and K.B.A. preformed the pharmacological experiments and analysis and W.J., M.A.S., L.F. and V.I. performed the computational analysis. A.C.N., W.J., M.A.S. and L.F. drafted the manuscript, which was commented by all authors. D.E.G. and H.B.-O. designed the study. D.E.G., H.G., J.Å., supervised the computational work and H.B.O. supervised the pharmacological work.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Anne Cathrine Nøhr, Willem Jespers, Mohamed A. Shehata, Hans Bräuner-Osborne and David E. Gloriam contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01049-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hans Bräuner-Osborne, Email: hbo@sund.ku.dk.
David E. Gloriam, Email: david.gloriam@sund.ku.dk
References
- 1.Fredriksson R, Lagerström MC, Lundin L-G, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 2.Granier S, Kobilka B. A new era of GPCR structural and chemical biology. Nat. Chem. Biol. 2012;8:670–673. doi: 10.1038/nchembio.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 4.Rask-Andersen M, Almén MS, Schiöth HB. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 5.Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 1–16 (2016). [DOI] [PMC free article] [PubMed]
- 6.Alexander SP, et al. The concise guide to pharmacology 2013/14: G protein-coupled receptors. Br. J. Pharmacol. 2015;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garland SL. Are GPCRs still a source of new targets? J. Biomol. Screen. 2013;18:947–966. doi: 10.1177/1087057113498418. [DOI] [PubMed] [Google Scholar]
- 8.Gloriam DEI, Schiöth HB, Fredriksson R. Nine new human Rhodopsin family G-protein coupled receptors: identification, sequence characterisation and evolutionary relationship. Biochim. Biophys. Acta. 2005;1722:235–46. doi: 10.1016/j.bbagen.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Liu C, et al. GPR139, an orphan receptor highly enriched in the habenula and septum, is activated by the essential amino acids L-tryptophan and L-phenylalanine. Mol. Pharmacol. 2015;88:911–925. doi: 10.1124/mol.115.100412. [DOI] [PubMed] [Google Scholar]
- 10.Matsuo A, et al. Molecular cloning and characterization of a novel Gq-coupled orphan receptor GPRg1 exclusively expressed in the central nervous system. Biochem. Biophys. Res. Commun. 2005;331:363–9. doi: 10.1016/j.bbrc.2005.03.174. [DOI] [PubMed] [Google Scholar]
- 11.Süsens U, Hermans-Borgmeyer I, Urny J, Schaller HC. Characterisation and differential expression of two very closely related G-protein-coupled receptors, GPR139 and GPR142, in mouse tissue and during mouse development. Neuropharmacology. 2006;50:512–20. doi: 10.1016/j.neuropharm.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Wagner F, Bernard R, Derst C, French L, Veh RW. Microarray analysis of transcripts with elevated expressions in the rat medial or lateral habenula suggest fast GABAergic excitation in the medial habenula and habenular involvement in the regulation of feeding and energy balance. Brain Struct. Funct. 2016;221:4663–4689. doi: 10.1007/s00429-016-1195-z. [DOI] [PubMed] [Google Scholar]
- 13.Kuhne S, et al. Radiosynthesis and characterisation of a potent and selective GPR139 agonist radioligand. RSC Adv. 2016;6:947–952. doi: 10.1039/C5RA21326F. [DOI] [Google Scholar]
- 14.Dvorak, C. A., Liu, C. & Kuei, C. Physiological ligands for GPR139; International Patent WO2014/152917 A2, Janssen Pharmaceutica (2014).
- 15.Andersen KB, Leander Johansen J, Hentzer M, Smith GP, Dietz GPH. Protection of primary dopaminergic midbrain neurons by GPR139 agonists supports different mechanisms of MPP+ and rotenone toxicity. Front. Cell. Neurosci. 2016;10:1–10. doi: 10.3389/fncel.2016.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isberg V, et al. Computer-aided discovery of aromatic L-α-amino acids as agonists of the orphan G protein-coupled receptor GPR139. J. Chem. Inf. Model. 2014;54:1553–1557. doi: 10.1021/ci500197a. [DOI] [PubMed] [Google Scholar]
- 17.Lin HV, et al. GPR142 controls tryptophan-induced insulin and incretin hormone secretion to improve glucose metabolism. PLoS One. 2016;11:1–17. doi: 10.1371/journal.pone.0157298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Carrillo JJ, Lin HV. GPR142 agonists stimulate glucose-dependent insulin secretion via gq-dependent signaling. PLoS One. 2016;11:1–14. doi: 10.1371/journal.pone.0154452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu M, et al. Aminopyrazole-phenylalanine based GPR142 agonists: Discovery of tool compound and in vivo efficacy studies. ACS Med. Chem. Lett. 2013;4:829–834. doi: 10.1021/ml4000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo L, et al. Discovery and optimization of a novel triazole series of GPR142 agonists for the treatment of type 2 diabetes. ACS Med. Chem. Lett. 2016;22:5942–7. doi: 10.1021/acsmedchemlett.6b00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nøhr AC, et al. The orphan G protein-coupled receptor GPR139 is activated by the peptides: Adrenocorticotropic hormone (ACTH), α-, and β-melanocyte stimulating hormone (α-MSH, and β-MSH), and the conserved core motif HFRW. Neurochem. Int. 2016;102:105–113. doi: 10.1016/j.neuint.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu La, et al. Identification of surrogate agonists and antagonists for orphan G-protein-coupled receptor GPR139. J. Biomol. Screen. 2009;14:789–97. doi: 10.1177/1087057109335744. [DOI] [PubMed] [Google Scholar]
- 23.Shi F, et al. Discovery and SAR of a series of agonists at orphan G protein-coupled receptor 139. ACS Med. Chem. Lett. 2011;2:303–306. doi: 10.1021/ml100293q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dvorak CA, et al. Identification and SAR of glycine benzamides as potent agonists for the GPR139 receptor. ACS Med. Chem. Lett. 2015;6:1015–1018. doi: 10.1021/acsmedchemlett.5b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hitchcock, S., Lam, B., Monenschein, H. & Reichard, H. 4-oxo-3,4-dihyroI-1,2,3-benzotriazine modulators of GPR139; US Patent US2016/0145218 A1. Takeda Pharmaceutical Company Limited. (2016).
- 26.Shehata MA, et al. Novel agonist bioisosteres and common structure-activity relationships for the orphan G protein-coupled receptor GPR139. Sci. Rep. 2016;6:36681. doi: 10.1038/srep36681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavasotto CN, et al. Discovery of Novel Chemotypes to a G-Protein-Coupled Receptor through Ligand-Steered Homology Modeling and Structure-Based Virtual Screening. J. Med. Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 28.Boukharta L, Gutiérrez-de-Terán H, Aqvist J. Computational prediction of alanine scanning and ligand binding energetics in G-protein coupled receptors. PLoS Comput. Biol. 2014;10:e1003585. doi: 10.1371/journal.pcbi.1003585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keränen H, Gutiérrez-de-Terán H, Åqvist J. Structural and energetic effects of A2A adenosine receptor mutations on agonist and antagonist binding. PLoS One. 2014;9:e108492. doi: 10.1371/journal.pone.0108492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keränen H, Åqvist J, Gutiérrez-de-Terán H. Free energy calculations of A 2A adenosine receptor mutation effects on agonist binding. Chem. Commun. 2015;51:3522–3525. doi: 10.1039/C4CC09517K. [DOI] [PubMed] [Google Scholar]
- 31.Ahuja S, Smith SO. Multiple Switches in G Protein-Coupled Receptor Activation. Trends Pharmacol. Sci. 2009;30:494–502. doi: 10.1016/j.tips.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Wellendorph P, et al. Deorphanization of GPRC6A: A promiscuous L-α-amino acid receptor with preference for basic amino acids. Mol. Pharmacol. 2005;67:589–597. doi: 10.1124/mol.104.007559. [DOI] [PubMed] [Google Scholar]
- 33.Pagano A, et al. C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABA(B) receptors. J. Neurosci. 2001;21:1189–202. doi: 10.1523/JNEUROSCI.21-04-01189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wacker D, et al. Structural features for functional selectivity at serotonin receptors. Science (80-.) 2013;340:615–9. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munk C, et al. GPCRdb: the G protein-coupled receptor database – an introduction. Br. J. Pharmacol. 2016;16:2195–2207. doi: 10.1111/bph.13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Isberg V, et al. GPCRdb: an information system for G protein-coupled receptors. Nucleic Acids Res. 2016;44:356–364. doi: 10.1093/nar/gkv1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sali A, Blundell T. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 39.Fidom K, et al. A new crystal structure fragment-based pharmacophore method for G protein-coupled receptors. Methods. 2015;71:104–112. doi: 10.1016/j.ymeth.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 40.Bochevarov AD, et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013;113:2110–2142. doi: 10.1002/qua.24481. [DOI] [Google Scholar]
- 41.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 42.Wu H, et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 44.Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 45.Halgren TA, et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 46.Friesner RA, et al. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 47.Greenwood JR, Calkins D, Sullivan AP, Shelley JC. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided. Mol. Des. 2010;24:591–604. doi: 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- 48.Shelley JC, et al. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 49.MacroModel, V 10.9 Schrödinger, LLC (2017).
- 50.The PyMOL Molecular Graphics System, Version 1.4 Schrödinger, LLC (2017).
- 51.Gutiérrez-de-Terán H, Bello X, Rodríguez D. Characterization of the dynamic events of GPCRs by automated computational simulations. Biochem. Soc. Trans. 2013;41:205–12. doi: 10.1042/BST20120287. [DOI] [PubMed] [Google Scholar]
- 52.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 53.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides†. J. Phys. Chem. B. 2001;105:6474–6487. doi: 10.1021/jp003919d. [DOI] [Google Scholar]
- 54.Berger O, Edholm O, Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997;72:2002–13. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marelius J, Kolmodin K, Feierberg I, Åqvist J. Q: a molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998;16:213–225. doi: 10.1016/S1093-3263(98)80006-5. [DOI] [PubMed] [Google Scholar]
- 56.King G, Warshel A. A surface constrained all‐atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 1989;91:3647–3661. doi: 10.1063/1.456845. [DOI] [Google Scholar]
- 57.Marelius, J., Kolmodin, K., Feierberg, I. & Aqvist, J. Q: a molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 16, 213–25, 261 (1998). [DOI] [PubMed]
- 58.Lee FS, Warshel A. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J. Chem. Phys. 1992;97:3100. doi: 10.1063/1.462997. [DOI] [Google Scholar]
- 59.Ryckaert J-P, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 60.Bjelic S, Brandsdal BO, Åqvist J. Cold Adaptation of Enzyme Reaction Rates. Biochemistry. 2008;47:10049–100057. doi: 10.1021/bi801177k. [DOI] [PubMed] [Google Scholar]
- 61.Robertson MJ, Tirado-Rives J, Jorgensen WL. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015;11:3499–3509. doi: 10.1021/acs.jctc.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.