

Abstract
Oral drug absorption is a process influenced by the physicochemical and biopharmaceutical properties of the drug and its inter-relationship with the gastrointestinal tract. Drug solubility, dissolution and permeability across intestinal barrier are the key parameters controlling absorption. This review provides an overview of the factors that affect drug absorption and the classification of a drug on the basis of solubility and permeability. The biopharmaceutical classification system (BCS) was introduced in early 90׳s and is a regulatory tool used to predict bioavailability problems associated with a new entity, thereby helping in the development of a drug product. Strategies to combat solubility and permeability issues are also discussed.
Abbreviations: ABC, ATP-binding cassette; AP, absorption potential; API, active pharmaceutical ingredient; ATP, adenosine triphosphate; AZT, azidothymidine; BA/BE, bioavailability/bioequivalence; BCRP, breast cancer resistance protein; BCS, biopharmaceutical classification system; BDDS, biopharmaceutical drug disposition system; BSP, bromosulfophthalein; CD, cyclodextrin; CDER, Centre for Drug Evaluation and Research; CNT, concentrative nucleoside transporter; CNT, Na+-dependent concentrative transporter; CYP, cytochrome P450; D:S, dose:solubility; E217G, estradiol 17β-glucuronide; EMEA, European Medicines Agency; ENT, equilibrative nucleoside transporter; FaSSIF, fasted state simulated intestinal fluid; FATP, fatty acid transporter protein; FDA, U.S. Food and Drug Administration; FeSSIF, fed state simulated intestinal fluid; FIP, International Pharmaceutical Federation; GIS, gastrointestinal simulator; GIT, gastrointestinal tract; GITA, gastrointestinal transit and absorption; GLUT, sodium-independent facilitated diffusion transporter; GRAS, generally recognized as safe; HIV, human immunodeficiency disease; HPC-SL, LBDDS, lipid based drug delivery system; HUGO, Human Genome Organization; ICH, International Council of Harmonization; IDR, intrinsic dissolution rate; IR, immediate release; ISBT, sodium dependent bile salt transporter; MCT, monocarboxylate transporter; MPP, 1-methyl-4-phenylpyridinium; MRP, multidrug resistance associated protein; NLC, nanostructured lipid carrier; NME, new molecular entity; NTCP, sodium-dependent taurocholate co-transporting polypeptide; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; OCT, organic cationic transporter; OCTN, organic cationic/carnitine transporter; OMM, ordered mesoporous material; PAH, p-aminohippurate; PAMPA, parallel artificial membrane permeability assay; Papp, apparent permeability; Peff, effective permeability; PEG, polyethylene glycol; PEI, polyethyleneimine; PEPT, peptide transporter; PGA, polyglycolic acid; P-gp, P-glycoprotein; PLA, poly(lactic acid); PLGA, poly-d,l-lactide-co-glycoside; PMAT, plasma membrane monoamine transport; pMMA, polymethyl methacrylate; PSA, polar surface area; Psi, porous silicon; PVDF, polyvinylidene difluoride; RFC, reduced folate transporter; SDS, sodium dodecyl sulphate; SGLT, sodium dependent secondary active transporter; SIF, simulated intestinal fluid; SLC, solute carrier; SLCO, solute carrier organic anion; SLN, solid lipid nanoparticles; SMVT, sodium dependent multivitamin transporter; SPIP, single pass intestinal perfusion; SUPAC, scale-up and post approval changes; SVCT, sodium-dependent vitamin C transporter; TEOS, tetraethylortho silicate; UWL, unstirred water layer; VDAD, volume to dissolve applied dose; vit. E TPGS, vitamin E tocopherol polyethylene glycol succinate; WHO, World Health Organization
Key words: BCS, Solubility, Permeability, Formulation strategies, Factors affecting absorption
Graphical abstract
Bioavailability of a drug depends on physicochemical properties of the drug and physiological factors and dosage-related factors. Drug absorption occurs from the gut wall by passive diffusion, carrier-mediated uptake and paracellular transport. Several formulation strategies can be used to combat poor bioavailability.
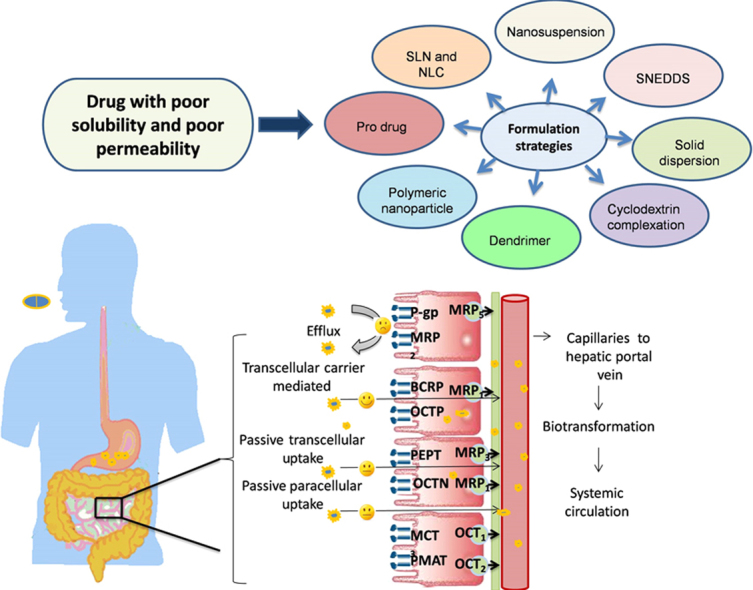
1. Introduction
Peroral administration is the predominantly acceptable route of drug administration owing to its benefits such as self administration with minimal discomfort to patients, which improves patient compliance, makes it cost effective and provides flexibility in design of dosage form1. There are various factors which control of absorption through the oral route and thus affect the bioavailability of a drug. FDA defines bioavailability as “the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action”. A prior knowledge of biopharmaceutical optimization and in vivo availability of drug were first only on focused disintegration time while ignoring fundamental factors like dissolution. Researchers tried to mimic the biological conditions like gut pH, food content and peristalsis for precisely predicting in vivo performance. In the years 1960–1970, several studies were carried out which demonstrated the effect of dissolution, formulation parameters [excipients, slight change in concentration of active pharmaceutical ingredient (API), dosage form, etc.] and food on bioavailability. Bioavailability concerns and quality control consideration further initiated the need for an official dissolution test. The first dissolution test apparatus, basket stirred flask type (USP apparatus I) was introduced in 1970 and subsequently paddle type (USP Apparatus II) in 19782. In vitro tests have been successful in predicting in vivo performance of dosage forms. Despite the complexity of the factors, progress has been made to improve the performance of dosage forms in vivo. Some of the prominent research carried out in this field is listed chronologically in Table 1, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12.
Table 1.
Evolution of Biopharmaceutical Classification System.
| Year | Prominent research | Ref. |
|---|---|---|
| 1897 | Noyes-Whitneys first experiment on dissolution | 3 |
| 1904 | Nernst-Brunner diffusion layer concept | 4 |
| 1931 | Hixon-Crowell model | 5 |
| 1950 | Official disintegration test in USP | 6 |
| 1951 | Danckwert׳s theory | 7 |
| 1961 | Higuchi׳s interfacial barrier model | 8 |
| 1970 | Dissolution apparatus I (Basket type) | 9 |
| 1978 | Dissolution Apparatus II (Paddle type) | 9 |
| 1981 | FIP guidelines for dissolution of solid dosage form | 10 |
| 1985 | General chapter on “drug release” in USP | 9 |
| 1991 | USP dissolution apparatus III “reciprocating cylinder type” | 9 |
| 1995 | USP dissolution apparatus IV “flow through cell” | 9 |
| 1995 | Amidon Gordan introduced BCS | 11 |
| 2000 | FDA introduced BCS guidelines | 12 |
This article reviews the drug absorption process which depends upon drug properties, such as solubility and permeability, physiological factors like pH, regional permeability differences, food effects and formulation factors. Combinatorial chemistry and high throughput screening has led to the development of lead drug compounds having higher molecular weight, poor wetting properties and high lipophilicity, thus placing 40% of lead candidates into Biopharmaceutical Classification System (BCS) class II and class IV. Better understanding of the various factors during the lead optimization phase can help to reduce the cost of development. The other approach to address poor solubility and permeability issues is to modify the drug by using different formulation approaches. This review provides insight on fundamentals of BCS and a literature database of formulation strategies used to manage solubility and permeability problems. These approaches can help in shifting the lead candidate to a better class of BCS.
2. Movement of drug through the gastrointestinal tract
Fig. 1 shows the journey of a drug through the gastrointestinal (GI) tract. The GI tract is a complex system; the first organ which a drug encounters is the stomach, which contains many digestive enzymes, has an acidic environment (pH 1.5–3.5) and very few drugs are absorbed through stomach. The small intestine (duodenum, jejunum, and ileum) is the major site for drug absorption. Dosage form disintegration and dissolution, degradation, binding in the intestinal lumen, intestinal permeation and intestinal and hepatic metabolism controls the pharmacological activity and transition of a drug in the GI tract13, 14. Also, these processes in the GI tract are interlinked and controlled by various factors like physicochemical, physiological and the type of dosage form (tablet, capsule, solution, suspension, emulsion, and gel)15, 16. Physicochemical factors include pKa, solubility, stability, diffusivity, lipophilicity, polar and nonpolar surface area, the presence of hydrogen bonding functionalities, particle size, and crystal form, whereas physiological factors includes GI pH, GI blood flow, gastric emptying, small intestinal transit time, colonic transit time, and absorption mechanisms.
Figure 1.

Journey of drug in gastrointestinal tract.
Achieving sufficient absorption and a reproducible pharmacokinetic profile in humans has become a hurdle in developing oral drug delivery systems due to introduction of high throughput screening and combinatorial chemistry, which has increased the number of lipophilic and higher mass molecules17. A drug exerts its pharmacological effect only when it binds to specific receptor, for which it has to be in solubilized form and then should be able to traverse the intestinal barrier. This implies that only a drug which is in soluble form will be available for the absorption process. Thus solubility and dissolution are the major rate-limiting steps and are key parameters in preformulation studies at the early development phase18, 19. Dissolution is the process by which the drug is released, dissolved and becomes readily available for absorption. The rate of dissolution is greatly affected by the solubility of the API, as explained by Noyes and Whitney׳s postulate (1897)20 which states that “the rate at which solid substance dissolves in its own solution is proportional to difference in the concentration of that solution and concentration of saturated solution. Nernst and Brunner (1904)4, 21 later modified Noyes and Whitney׳s equation by incorporating the value of surface area accessible to dissolution.
| (1) |
where C is amount of drug dissolved (usually in mg or mmol) in time t (s), D is the diffusion coefficient of the drug (cm2/s), S is surface area (cm2), h is thickness of the liquid film, Cs and Cb are the concentrations of the drug at the surface of the particle and the bulk medium, and V is the volume of dissolution medium. The other important factor is “drug permeability” which is referred as ability of drug molecule to permeate through a mucosal barrier into the systemic circulation. The major site for absorption being epithelial cell in gut wall within the small intestine provides a high surface area because of microvilli present. The microvilli have glycoproteins protruding in the luminal fluid and consist of goblet cells which releases mucus, a gel like structure containing water (95%) and mucin which blankets the surface. This glycoprotein and mucous forms an unstirred water layer (UWL) with a thickness of 100 μm22. UWL in GIT is a significant barrier to drug absorption for lipophilic drugs23. Drug transport occurs through the apical side of the enterocyte membrane and then across a mucosal barrier (polarized enterocytes) crossing the basolateral membrane and entering the systemic circulation via the capillary network surrounding the enterocytes and hepatic portal vein. These enterocytes are closely linked, producing the tight junctions. Tight junctions are, in reality, small aqueous-filled pores with dimensions in the range of 3–10 Å which depends upon the membrane type. Drug molecule can cross a biological membrane by transcellular or paracellular mechanisms (passive diffusion). Transcellular absorption occur through enterocytes, which may be by passive diffusion, carrier-mediated transport and endocytosis. Passive diffusion of a drug is driven by concentration gradient wherein drug moves from higher to lower concentration, thus following Fick׳s law of diffusion. Carrier-mediated transport involves the interaction of the carrier protein with drug, which may be either a facilitated or an active process. Facilitated carrier-mediated transport occurs through passage of a drug down its electrochemical gradient without utilization of energy. In contrast, active carrier-mediated transport involves an energy-coupling mechanism that creates an ion/solute gradient across the membrane. The third transcellular absorption mechanism is endocytosis which involves internalization of a drug inside the cell by a forming membrane-bound vesicle known as an endosome. Endocytosis can be divided into two broad categories, i.e., phagocytosis (uptake of large particles) and pinocytosis (uptake of solute and single molecules). The reticuloendothelial system is only capable of phagocytosis, such as by macrophages, neutrophils, monocytes and dendritic cells, while pinocytosis is further classified into clathrin-mediated endocytosis and clathrin-independent endocytosis. Another classification of endocytosis is based on interaction with cellular membrane which involves receptor-mediated endocytosis (drug reacts with a specific surface receptor) and absorption-mediated endocytosis (non-specific interaction with a surface receptor)24. Many compounds can be absorbed by the paracellular route but the process is invariably slower than the transcellular route (surface area of pores versus surface area of the membrane) and is dependent on molecular size due to the finite dimensions of the aqueous pores. Whether a drug diffuses via a passive transcellular mechanism or a paracellular mechanism is determined by both physiological and physicochemical factors25, 26. During transition of a drug through the enterocyte membrane, it may undergo gut membrane metabolism by cytochrome P450 (CYP) enzymes and may encounter both efflux and absorptive transporters27. Specific transporters are expressed in different regions including luminal and basolateral membranes of enterocytes, hepatocytes, renal tubular epithelial cells and other important barrier tissues including the blood–brain barrier, blood–testis barrier and placental barrier28. According to the guidelines of the Human Genome Organization (HUGO) Gene Nomenclature Committee, 229 solute carrier (SLC) family genes and 52 ATP-binding cassette (ABC) genes have been identified29. The major uptake transporters responsible for xenobiotic transport belongs to the two solute carrier (SLC and SLCO) subfamilies. These subfamilies are involved in transport of wide variety of substrates like amino acids, peptides, sugars, vitamins, bile acids, neurotransmitter and xenobiotics. Solute carriers, known to play a relevant role in drug transport in the intestine, include the oligopeptide transporter (PEPT), organic cation/carnitine transporter (OCTN), organic cation transporter (OCT), plasma membrane monoamine transporter (PMAT), organic anion transporting protein (OATP) and the monocarboxylate transporter (MCT)30. The most investigated and highly abundant transporters at the luminal membrane of enterocytes (intestine) are the efflux transporter P-glycoprotein (P-gp), the multidrug resistance associated protein 2 (MRP 2) and the breast cancer resistance protein (BCRP) of the ABC family. Many drugs such as statins, antibiotics, HIV protease inhibitors, immunosuppressant and anticancer drugs are substrates for efflux transporters (P-gp, MRP 2, BCRP) which place them back into the gut lumen from where they can be excreted31, 32. Table 2 shows various membrane transporters, their subfamilies and substrate drugs29, 31, 33. Drug leaving the enterocyte then enters the hepatic portal vein by which it is transported to the liver. Liver either metabolizes it or excrete it unchanged into the bile which is termed hepatic first pass metabolism. Thus, oral bioavailability of a drug is determined by the amount absorbed from GIT (Fa), the fraction escaping first pass extraction by the gut (Fg), and the fraction escaping first pass extraction by the liver (Fh) as given by following Eq. (2):
| (2) |
Table 2.
| Transporter | Subtype (No. of isoform) | Gene Symbol | Localization | Substrate drug |
|---|---|---|---|---|
| Peptide transporter | PEP1 | SLC15A | Apical side of intestine | β-Lactum antibiotic, angiotensin converting enzyme inhibitor, cephalosporins, rennin inhibitor, oseltamivir, thrombin inhibitor, betastin, l-α-methyldopa-phenylalanine, d-phenylglycine-l-α-methyldopa, l-valacyclovir |
| PEPT2 | ||||
| PTR3 | ||||
| hPT-1 | ||||
| PHT1 | ||||
| Nucleoside transporter | CNT1 | SLC28A | Apical side of intestine | Azidothymidine (AZT), zalcitabine, cladribine, cytarabine, gemcitabine, 5ʹ-deoxy-5-flurouridine |
| CNT2 | – | Cladribine, didanosine | ||
| CNT3 | – | 5-Flurouridine, floxuridine, zebularine, gemcitabine, AZT, cladribine | ||
| ENT1 | SLC29A | Cladribine, cytarabine, fludarabine, gemcitabine, zalcitabine, didanosine | ||
| ENT2 | – | AZT, didanosine, gemcitabine | ||
| ENT3 | – | |||
| Organic cation transporters | OCT1/2 | SLC22A | Basolateral side of intestine | Tetraethylammonium (TEA), thiamine, tyramine, tryptamine, N-methylnicotineamide (NMN), choline, spermine, spermidine quinine, d-tubocurarine, procainamide, dopamine, noradrenaline, serotonine, histamine, corticosterone, 1-methyl-4-phenylpyridinium (MPP), desipramine, metformin, acyclovir, ranitidine, memantine |
| OCT 3 | – | – | Dopamine, MPP, TEA, guanidine | |
| OCTN1 | SLC22A | – | TEA, pyrilamine, quinidine, ergothioneine, verapamil | |
| OCTN2 | – | – | Carnitine derivative, betaine, cephaloridine, choline, emetine, pyrilamine, quinidine, TEA, valproate, verapamil, imatinib, ipratropium | |
| OCTN3 | – | – | Carnitine | |
| Organic anion transporter | OAT1 | SLC22A | Apical side of intestine | p-Aminohippurate (PAH), methotrexate, β-lactum antibiotics, non-steroidal anti-inflammatory drugs, antiviral nucleoside analogues |
| OAT2 | – | Methotrexate, PAH, salicylate | ||
| OAT3 | – | Estrone sulfate, ochratoxin A, cimetidine | ||
| OATP1/2 | SLC21A | Bromosulfophthalein (BSP), pravastatin, temocaprilat, estradiol 17β-glucuronide (E217G), enalapril, gadoxetate, dehydroepiandrosterone sulfate | ||
| hOATPs | – | BSP, taurocholate, glycocholate, estrone sulfate, dehydroepiandrosterone sulfate, ouabain, N-methyl quinidine, prostaglandine E2, triiodothyronine, thyroxine, deltorphin II, d-penicillamine, enkephalin, fexofenadine, rifampin, rocuronium, quinidine, methotrexate, pravastatin, digoxin | ||
| Glucose transporter | SGLTs (3) | SLC5A | Apical side of intestine | Inositol, proline, pantothenatem iodide, urea, glucose derivative |
| GLUTs (13) | SLC2A | Basolateral side of intestine | ||
| Vitamin transporter | SVCTs (2) | SLC23A | – | Ascorbic acid derivative |
| RFC1 | SLC19A | Reduced folate derivatives, methotrexate | ||
| SMVT | SLC5A6 | Pantothenate, biotin, lipoate | ||
| Bile acid transporter | NTCP | SLC10A | Basolateral membrane of hepatocyte | Steroids and steroid conjugates, cyclic peptides, bumetanide, BSP |
| ISBT | SLC10A | Ileum brush border membrane | Peptide drugs | |
| Fatty acid transporter | FATPs (6) | SLC27A | Apical side of enterocytes | Long chain fatty acids, like myristate and palmitate |
| Phosphate transporter | SLC17As (4) | SLC17A | Brush border membrane | Foscarnet, fosfomycin |
| SLC34As (2) | SLC34A | |||
| Monocarboxylic acid transporter | MCTs (6) | SLC16A | Apical side of enterocytes | Atorvastatin, valproic acid, pyruvic acid, benzoic acid |
| ABC transporter | MDR1/P-gp | ABCB1 | Apical side of enterocytes | Steroid hormone, bile salts, glycocholate, doxorubicin, ciprofloxacin, etoposide tauroursodeoxycholate, daunorubicin, reserpine, vincristine, vinblastine, valinomycin, cyclosporine, tacrolimus, tandutinib, aldosterone, hydrocortisone, dibucaine, talinolol, digoxin, ivermectin, paclitaxel, grepafloxacin, indinavir, nelfinavir, saquinavir, colchicines, darunavir, flavonoids, glyburide, methotrexate, mitoxantrone, prazosin, temocapril, celiprolol |
| BCRP | ABCG2 | Apical side of enterocytes | Topotecan, irinotecan, doxorubicin, daunorubicin, doxorubicin, imatinib, geftinib, tandutinib, statins, prazosin, glyburide, dipyridamole, quercetin, temocapril, sulfate conjugates, porphyrin, nitrofurantoin, fluroquinolones, zidovudine, lamivudine, efavirenz, ciprofloxacin, rifampicin, sulfasalazine, quercetin, resveratrol conjugates. | |
| MRP2 | ABCC1 | Apical side of enterocytes | Leukotrienes glutathione, 2,4-dinitrophenyl-S-glutathione, bromosulfophthalein, conjugates of bile salts and heavymetals, resveratrol conjugates, naringenin glucoronides, vinblastine, reduced folates, pravastatin, ceftriaxone, ampicillin, grepafloxacin, sulfasalazine, fexofenadine, lopinavir, fosinopril, ochratoxin A, epicatechin, phenols, colchicines, darunavir, flavonoids, methotrexate |
– Not available.
3. Reciprocity between factors affecting drug absorption
The journey of drug through the GI tract as discussed suggests that the successful development of a dosage form depends upon complex functions such as dissolution, intestinal permeability, cellular permeability, binding to plasma proteins, drug distribution, metabolism and disposition. Pharmacokinetic failure of lead candidates occurs because of a combination of one or more factors such as physicochemical properties of drug, physiological barriers or dosage form design which affect solubility and absorption. Physicochemical properties of a drug such as lipophilicity and solubility are the key properties which are affected by molecular weight, melting point, H-bonding, pKa, molecular shape and amphiphilicity34. The pioneer and worldwide-accepted qualitative predictive tool is Lipinski׳s ‘rule of five’ introduced by Christopher Lipinski35 in the 1990s which is the computational filter for rapid evaluation of drug properties. It states that a molecule would be most likely to have poor absorption if it satisfies any two of the following criteria36, 37, 38: a molecular weight (MW) > 500 Da, an octanol–water partition coefficient (clogP)>5 or MlogP> 4.15, the number of H-bond donors is > 5 and the number of H-bond acceptors is > 10. Molecular weight of the compound should be less than 500, preferably uncharged (unionized form) and fairly lipophilic so as to cross the intestinal epithelium passively. A highly lipophilic molecule will stick to membranes and will not cross the intestinal barrier. Though the ‘rule of five’ is an effective tool in early development, it has a particular limitation in that it only holds for molecules which are not substrates of active transporters. In addition to the molecular properties discussed by Lipinski, other properties discussed below are also very important. A simple measure of hydrogen binding capacity (hydrogen bond donors or acceptor) is the polar surface area, defined as the area occupied by nitrogen, oxygen and hydrogen atoms attached to these heteroatoms. Palm et al.39 used dynamic polar surface area (PSAd) to predict poorly absorbed drugs at an early stage of research and development. They found a good correlation (r2=0.94) between PSA and fraction absorbed for 20 drugs and revealed that drug with PSAd >140 will show poor absorption (<10%) while PSAd<60 yields good absorption (> 90%). Clark et al.40 studied the relationship of PSA with intestinal absorption considering only single conformer, suggested that a poorly absorbed molecule will have PSA>140 Å. Daniel et al.41 assessed molecular properties such as polar surface area, hydrogen bond count and number of rotational bond as a simple measures of molecular flexibility for over 1100 drug candidates studied at GlaxoSmithKline and they reported that about 65% of the compounds with seven or fewer rotational bonds have oral bioavailability (F, %) of 20% or more in rats, independent of molecular weight while 75% compounds with more than 10 rotational bonds show oral bioavailability less than 20%. The fewer the number of aromatic rings in the lead molecule, the more chances of it showing good a pharmacokinetic profile. More than three aromatic rings in a molecule relates to a poor pharmacokinetic profile and more chances of failure in drug development. Aromatic heterocycles will have a lesser effect on increasing lipophilicity than carbon-containing aromatics but will increase the PSA which might reduce oral absorption42. Understanding the physicochemical properties of a drug are very important for the successful development of a drug candidate. Several efforts have been carried out to predict the favorable performance of a molecule for drug absorption depending on physicochemical properties. Dressman et al.43 developed an absorption potential (AP) model based on readily available physicochemical property data which could be determined at an earlier phase of drug development and thus provide an excellent tool for initial prediction of absorption, as shown in Eq. (3):
| (3) |
where P is octanol–water partition coefficient, Fnon is the fraction in nonionised form at pH 6.5, S0 is intrinsic solubility, VL is volume of the luminal contents and X0 is the dose administered. Similarly, Yalkowsky et al.44 expressed absorption parameter π based on physicochemical properties like aqueous solubility, Cs, melting point and the octanol–water partition coefficient. Anatomical and physiological conditions like gastrointestinal pH, surface area for absorption, fed and fasted condition, gastric transit, disease state, age, sex also affect the pharmacokinetic behavior45, 46. It is widely accepted fact that the nonionized form of a drug is better absorbed than the ionized form. The unionized form of a drug depends upon the dissociation constant of the drug in the physiological pH range. GI pH changes depending upon the fed or fasted state. In the fasted-state stomach the pH ranges from 1 to 3, duodenum 5–6, and around pH 6–7 in the jejunum and ileum while, in the fed-state stomach the pH ranges in between 4 and 5, 4.5–5.5 in duodenum and 6.5–7.5 in jejunum and ileum. Maximum absorption of drug occurs in jejunum and ileum within 3–5 h, in pH range of 4.5–8 which suggests that weak acids are better absorbed in jejunum while weak bases are better absorbed in the ileum47. Table 3 lists some important variables of fed- and fasted-state conditions. When drug is given orally, the absorption window exists within the GI tract in which drug is efficiently absorbed, and thus gastrointestinal motility and transit time affects absorption. The small intestine is generally regarded as a main site for absorption and hence contact time of drug with small intestinal mucosa will be a key determinant especially for drugs having low permeability, those transported by a carrier-mediated pathway or subjected to intestinal degradation, and for drugs having poor dissolution. Gastric emptying rate also plays an important role in absorption after oral drug administration which is affected by various factors such as intake of fluid volume, pH, and size and density of drug particles. The GI transit and absorption (GITA) model analyzes absorption kinetics of drugs with variable absorption characteristics and shows the importance of GI transit rate in determining the bioavailability of orally administered drugs48. Fujioka et al.49 have predicted the mean plasma concentration time profile of griseofulvin using the GITA model and concluded that longer residence time could lead to higher dissolution and absorption, thus variance in intestinal transit may be responsible for inter-individual differences in vivo. We also reported that gastric emptying rate was not significantly correlated with the absorption and dissolution behavior of griseofulvin. The other important factor responsible for poor bioavailability is the formulation-related factor. Formulation factors include types of excipients (lubricant, glidant, bulking agent, solubilizing agent), formulation process (dry granulation and wet granulation, etc.), and particle size.
Table 3.
Fed and fasted state variables47.
| Position | Fasted state | Fed state |
|---|---|---|
| Stomach | ||
| Fluid volume | 50–100 | Up to 1000 |
| pH | 1–2 | 2–5 |
| Ionic strength | 0.1 | Varying |
| Motility pattern/ intensity | Cyclic/low–high | Continuous/high |
| Surface tension (mN/m) | 40 | Often lower than fasted |
| Osmolarity (mOsm) | 200 | Up to 600 |
| Upper small intestine | ||
| Flow rate (mL/min) | 0.6–1.2 | 2.0–4.2 |
| pH | 5.5–6.5 | 5.5–6.5 |
| Bile acids (mmol/L) | 4-6 | 10–40 |
| Ionic strength | 0.16 | 0.16 |
4. BCS
Solubility and permeability interactions and their impact on intestinal drug absorption are most prominently described by the BCS framed by Amidon et al.11 on the basis of dimensionless numbers. BCS provides drug designers with an opportunity to manipulate the structure or physicochemical properties of lead candidates to achieve better “deliverability”. BCS classification is extensively used by the pharmaceutical companies throughout drug discovery and development and can also help companies save development time and reduce costs50, 51. This system has been adopted by US Food and Drug Administration (FDA), the European Medicines Agency (EMEA) and World Health Organization (WHO) for setting bioavailability/bioequivalence (BA/BE) standards for immediate-release oral drug product approvals52. BCS principles are also included in ICH guidelines for requirements of in vitro dissolution testing as a quality control in manufacturing12.
BCS has classified API and finished dosage form on the basis of simple laboratory test solubility and permeability while dissolution is the final dosage form characteristic. Fundamental behind the BCS are three dimensionless numbers viz. absorption number (An), dissolution number (Dn) and dose number (Do) which predicts the fraction dose absorbed in humans based on physicochemical and physiological factors. An is the ratio of radial absorption rate to axial convection rate. An An value larger than 1 suggests complete absorption. Dissolution number refers to the time required for drug dissolution which is the ratio of the intestinal residence time to the dissolution time. The higher the dissolution number the higher will be the fraction-dose absorbed. Dose number is a criterion for solubility (Do) which is defined as the ratio of dose concentration to drug solubility. A dose number equal to or lower than 1 indicates high solubility and Do > 1 signifies low solubility53.
Drugs are classified as highly soluble and highly permeable (Class I) if they are well absorbed (although systemic availability may be low due to first-pass metabolism), do not have narrow therapeutic index, and dissolution of >85% in <15 min predicts complete absorption. These candidates may qualify for biowaiver of very expensive BA/BE studies. Class I drugs have a high absorption number (An>1.15) and a high dissolution number (Dn>1). For drugs having low solubility and high permeability (Class II), the rate-limiting step in absorption is poor solubility. Hence, a correlation between the in vivo bioavailability and the in vitro solvation can be found. Nonpolar characteristics of a drug are responsible for poor solubility and thus have a higher absorption number. The dissolution rate of water-insoluble compounds is low (Dn<1) while An and Do are high for many class II drugs. Class III compounds are highly soluble with low permeability where the rate-controlling step is absorption while the drug gets solvated very quickly and there is always risk of being excreted without showing any physiological effect. Class IV drugs are compounds which suffer from poor bioavailability and high variability because of low solubility and low permeability. These compounds have lower An and Dn number. Class IV compounds are rarely developed and marketed. Nevertheless, several Class IV drugs do exist. The BCS classes along with in-vitro–in-vivo correlation expectation for immediate release expectation are shown in Fig. 254.
Figure 2.

BCS classification and IVIVC expectation for immediate release dosage form54.
4.1. Solubility boundaries
BCS defines the API as being “highly soluble” when the highest recommended dose is soluble in 250 mL or less of aqueous media over the pH range of 1.2 to 7.5. A volume of 250 mL of aqueous media is taken to mimic the in vivo condition as same amount is present in upper GI tract when administering drug in the fasted state. Solubility is the amount of a substance that has passed into solution when equilibrium is attained between the solution and excess substance at a given temperature and pressure. The BCS solubility definition includes the highest dose strength in volume of 250 mL where dose/solubility (D:S)<250 refers highly soluble drugs but cannot be extended to pediatric patients as the volume does not hold true. Attempts are being carried out to extend the BCS to pediatric BCS55. Various solubility determination methods have been developed to assess solubility at various stages of drug discovery and development. These solubility protocols have been studied such that they are close to the actual solubilization process. Kinetic solubility, semi-equilibrium and the equilibrium solubility method have been developed over the past several years. When determining solubility by the kinetic approach, the drug is in a predissolved state in DMSO and precipitated in aqueous buffer with typical pH 6.5 (intestine) or pH 7.4 (bioassay or blood). Precipitation is detected optically and kinetic solubility is the concentration preceding precipitation. A rule of thumb is to keep the amount of DMSO typically ≤1% to prevent the potential cosolvency effect. Precipitate thus formed can either be removed or can be used as an indicator for direct measuring by nephelometer through light scattering signals or increased UV absorbance due to particle blocking the light from reaching the detector. When filtering the solution for assay, selection of the proper filter membrane is critical as it shows non-specific drug absorption. Hydrophillic PVDF filters are most suitable and nylon filters are not recommended. Another more recent method is dried DMSO or the semi-equilibrium method which subsequently removes DMSO by evaporation thus leaving solid-like material prior to addition of buffer to determine solubility56. Such solubility protocols provide rapid throughput screening, rapid availability of results, are efficient in terms of workload and use API in milligram quantities, thus saving 80% of time compared with the traditional shake-flask method. Though having mentioned these positive attributes questions should be raised as to whether such protocols misguide the chemist during lead optimization. It has been shown to overestimate the solubility when compared to thermodynamic solubility. Thus the value of such techniques are stage-dependent and cannot replace thermodynamic solubility studies which are usually performed at latter stages of discovery57. Thermodynamic solubility, in contrast, is performed by dispersing an excess of powdered drug into a liquid (buffer or water) and is assayed after equilibrium is established. It usually takes 24–48 h to establish equilibrium. To confirm that equilibrium has been achieved, compound solubility has to be constant with time, and hence solubility measurements at several points are necessary. To overcome the disadvantages with conventional shake-flask method, a novel miniaturized shake-flask solubility method streamlined with HPLC analysis has been validated and optimized via test set of 85 marketed drugs and Novartis internal compounds58. Different analytical tools used for detection are discussed in Table 4.
Table 4.
| Analytical method | Solubility determination technique | Advantage | Disadvantage |
|---|---|---|---|
| Light scattering or turbidity | Kinetic solubility | Universal, fast, economical | Interference from certain colored compounds and impurities, sensitive to sedimentation and particle size, low sensitivity, measures precipitates rather than solution concentration |
| UV plate reader | Equilibrium or saturation solubility | High sample coverage, fast, economical, sufficient sensitivity for solubility measurement, good linearity over wide dynamic range | Require UV chromophore, interference from impurities and matrix material |
| LC-UV | Equilibrium or semi-equilibrium solubility | High sample coverage, less interference from impurities and matrix material, sufficient sensitivity for solubility measurement, good linearity over dynamic range | Requires UV chromophore, might need different HPLC method for special compounds, not as fast and economical as UV method |
| LC-MS | Equilibrium or semi-equilibrium solubility | High sensitivity, high selectivity, low interference | Less universal, moderate sample coverage, low dynamic range of linearity, too sensitive to solubility measurement, high maintenance, costly |
4.2. Dissolution boundaries
A drug product is considered to be “rapidly dissolving” when > 85% of the labeled amount of drug substance dissolves within 30 min using USP apparatus I or II in a volume of < 900 mL buffer solutions. As discussed above, the dissolution study is the basis for BCS classification and used to predict the performance of formulations in the gastrointestinal tract (in vivo). The dissolution study is the predictive tool for bioavailability and in vitro–in vivo correlation (IVIVC). Proposed criteria to be used to avoid absorption problems caused by poor dissolution are given in Table 533, 59. Choosing the dissolution system parameter like media, stirring rate, temperature, instrument is based on considerations such as where in the GI tract is drug released from the dosage form, how long the dosage should release the drug and the composition of the fluid. Ideally, physiological conditions should be taken into consideration when choosing the in vitro test condition. Dissolution media listed in USP are dilute hydrochloric acid, USP simulated gastric fluid without enzymes (a pH 4.5 buffer; and a pH 6.8 buffer), simulated intestinal fluid USP without enzymes and surfactant solutions containing polysorbate 80 and sodium lauryl sulphate. These media simulate the osmolarity and pH effect of drug release while surfactant solution increases the solubility of drugs in aqueous media. These media are easily reproducible and routinely used in QC protocols but may not be useful for BCS class II or IV drugs. For such compounds dissolution is rate the limiting step for absorption thus the prediction of in vivo behavior should be based on a well designed in vitro test mimicking the in vivo conditions using biorelevant media. Simulating the in vivo condition using biorelevant media has been used to increase the in vivo predictability60, 61. The International Pharmaceutical Federation (FIP) guidelines published two complex media including a fasted state simulated intestinal fluid (FaSSIF) and fed state simulated intestinal fluid (FeSSIF) which contain the most important physiological amphiphiles, bile salt and lecithin, having pH, buffer capacity and osmolarity of the gut lumen62. Wei et al.63 studied dissolution behavior of glyburide BCS class II drug in apparatus II with various dissolution media FaSSIF, simulated intestinal fluid (SIF) and blank FaSSIF without lecithin and taurocholate (BL-FaSSIF) with constant and dynamic pH conditions (5.0 to 7.5). The change in pH simulates the physiological change in the small intestine and large intestine. Results revealed that micellization prevents glyburide from precipitation despite unfavorable pH and thus concluded that biorelevant media (FaSSIF) are suitable for studying the dissolution rate of BCS class II drugs. Biorelevant media are useful in predicting IVIVC for class II drugs but the complexity and cost has limited its extensive use for industrial applications. Lehto et al.64 studied the dissolution behavior of N74 (BCS class II drug) in biorelevant simple conventional surfactant media containing various concentrations of anionic surfactant, sodium lauryl sulphate and non ionic surfactant polysorbate 80 (Tween) which easily replaced bile salt/lecithin mixture owing to its similar wetting and solubilization behavior. They concluded that the potential of substituting FaSSIF with more simple and cost effective conventional surfactant media which could largely assist in industrial drug development and quality control purposes was possible. Several efforts have also been carried out to mimic the in vivo condition. A novel dissolution apparatus known as a gastrointestinal simulator (GIS) has the potential to be a standard biopredictive tool for in vitro dissolution testing, and consists of three dissolution chambers representing stomach, duodenum and early jejunum chambers. The fluid transit time between those chambers can be adjusted in the range of 1–40 mL/min by peristaltic pumps. Authors have studied the dissolution profile for BCS class II drugs such as pioglitazone and ketoconazole by using GIS system and concluded that buffer capacity, buffer species and pH of the medium have a significant effect on dissolution rate of pioglitazone and ketoconazole65. Similarly, Motz et al.66 have attempted to develop a novel apparatus containing a flow-through dissolution cell (USP apparatus 4) connected with a Caco-2 permeation cell to assess intestinal permeability of drugs containing a solid dosage form. Intrinsic dissolution rate is generally defined as the dissolution rate of a pure drug substance under the condition of constant surface area, agitation or stirring speed, pH and ionic strength of the dissolution medium. Milani et al.67 have studied intrinsic dissolution rate (IDR) and rat intestinal permeability (using SPIP technique) for drugs with different physicochemical properties to evaluate suitability in BCS. Muenster et al.68 have used apparent dissolution rate and volume to dissolve applied dose (VDAD) as a tool to predict in vivo performance of a drug contributing to a successful drug development candidate.
Table 5.
| Factor | Limit |
|---|---|
| Solubility in pH 1–7 | >10 mg/mL at all pH |
| Solubility in pH 1–8 and dose | Complete dose dissolved in 250 mL at all pH |
| Water solubility | >0.1 mg/mL |
| Dissolution rate in pH 1–7 | >1 mg/min cm2 (0.1–1 mg/min cm2 borderline) at all pH |
4.3. Permeability boundaries
BCS defines drug substance as “highly permeable” when the extent of absorption in humans is greater than 90% of an administered dose, based on mass-balance or compared with an intravenous reference dose. Drug is transported through the intestinal barrier via passive diffusion or other parallel transport mechanisms and thus controls effective permeability (Peff) of the drug. Effective permeability (Peff) is generally described in terms of units of molecular movement distance per unit time (e.g., 10−4 cm/s). The drugs with jejunal Peff>1.5×10−4 are completely absorbed independent of transport mechanism69. The FDA BCS guideline describes various methods to predict drug permeability through GI tract. Permeability data is considered valid if it is obtained from in vivo human trials (mass balance pharmacokinetic studies, absolute bioavailability studies, intestinal perfusion methods). Apart from the human trials, in situ permeability studies (e.g., rat intestine), in vivo permeability studies in animal and in vitro permeability studies such as in epithelial cell monolayers, e.g. Caco-2, provide supportive data for permeation70. Drugs with an apparent permeability (Papp) value less than 1×10−6 cm/s are poorly absorbed (0–20% absorbed), drugs with Papp value between 1 and 10×10−6 cm/s are moderately absorbed (20%–70% absorbed) and drugs with Papp value between 10×10−6 cm/s are well absorbed (70%–100% absorbed)71. Wahlang et al.72 assessed the permeability of curcumin using Caco-2 cell monolayers. Curcumin was found to be poorly permeable across the Caco-2 cell monolayer. Poor solubility and poor permeability places curcumin into class IV which helps in designing a drug delivery system for poorly bioavailable molecules.
BCS classification provides a tool to skip the in vivo bioequivalence studies by a simple in vitro dissolution test. In fact for immediate release (IR) solid dosage form which contains rapid dissolving and easily permeating API (BCS class I), bioequivalence studies may not be required because these drugs or drug products behave as a simple solution which is readily absorbed. If two drug products contain same API having similar GI concentration–time under all luminal conditions, then a similar rate and extent of absorption is ensured for these products. Thus, bioequivalence (BE) can be guaranteed based on in vitro dissolution tests rather than doing empirical in vivo human trials. Initially, waivers of in vivo bioequivalence were accepted only for Scale-up and Post Approval Changes (SUPAC) but later were extended to approval of new generic drug products, thereby avoiding the cost of human trials and reducing the cost to develop the generic product. Such a tool shortens the drug development period, economizes the resources and leads to improved product quality. According to the FDA guidance for the industry ‘Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System’, a biowaiver can currently be requested only for solid, orally administered immediate-release products (>85% release in 30 min), containing drugs with a high solubility over the pH range from 1 to 7.5 (D:S<250 mL) and high permeability (fraction absorbed >90%)73. It has also been recommended that biowaiver can also be extended to BCS class III drugs as these behave in vivo like an oral solution and thus their bioavailability would be dependent on gastric emptying rather than on drug product properties. Thus emphasis should be made on an excipient which modifies GI transit (e.g., sugar alcohols) or drug absorption74. BCS can also be used as a signal tool for need of formulation design (for BCS class II, III and IV drugs). Despite its simplicity, BCS has various complexities in the evaluation and investigation of in vitro and in vivo performance of a drug or drug product. Hence, continuous refinements are being done to develop a more science-based mechanistic tool. Recently, Tsume et al.75 subclassified class II and class IV drugs depending upon acidic (a), basic (b) and neutral (c) characteristics in the physiological pH range (pH<7.5) which can serve as a basis for developing in vivo predictive dissolution and absorption methodology. BCS has also lead to development of other systems such as the biopharmaceutical drug disposition system (BDDS) which is a complimentary system focused on drug disposition replacing the permeability criteria. Wu et al.76 in 2005 noticed the fact that highly permeable drugs are eliminated via metabolism while poorly permeable drugs are eliminated unchanged in bile and urine, leading to the development of this system. Solubility criteria for both the systems are the same, permeability has been replaced by route of elimination since it can be determined based on extent of metabolism easily. BDDS serves as a basis for predicting the importance of transporters in determining several pharmacokinetic parameters. BDDS class I compounds are highly soluble and extensively metabolized. Class I drugs are present in gut in high concentration to saturate transporter and may be substrates for either efflux or influx transporters. Transporter effects are minimal and are not clinically important. Class II drugs, being highly permeable, readily cross the gut membrane and hence influx transporters are clinically unimportant. Effects of efflux transporters are predominant as these drugs are poorly soluble preventing the saturation of efflux transporter. Class III and Class IV drugs are highly soluble/poorly metabolized and poorly soluble/poorly metabolized drugs, respectively. Class III compounds suffer from poor permeability and thus influx transporters play an important role and their effect is predominant. Efflux transporters may also be important when permeation occurs through absorptive transporters. Absorptive as well as efflux transporters are both important for absorption of class IV compounds77.
5. Formulation approaches for manipulating solubility and permeability
Despite the number of tools available, there is often a need to compromise with two fundamental issues during lead optimization phase, i.e., drug solubility and drug permeability. Knowledge of BCS can help the formulation scientist to manipulate solubility and permeability issues. BCS class I drugs do not generally have bioavailability issues as these behave as simple solution in vivo and show a very fast increase in blood plasma level. But sometimes a slower and longer-lasting effect is desired which can be achieved by using modified-release dosage forms and polymer-based formulation approaches. Poor water solubility is the important criteria for both oral as well as intravenous administration routes. Almost half of the new molecular entity (NMEs) synthesized annually by pharmaceutical companies are poorly water-soluble, which has reduced the performance of 10% of successfully marketed drugs78. Poorly water-soluble drugs belong to class II and class IV of the BCS. A candidate being poorly water-soluble falls in a different class of BCS because of different physicochemical reasons. Poor solubility may be due to solvation extreme called as “grease ball” having higher logP and lower melting point or due to crystal packing interactions called “brick dust” which has low solubility in lipids but considerable solubility in surfactant. Various approaches have been used to deal with poor solubility and poor permeability issues like particle size reduction79, 80, 81, nanoparticulate systems82, 83, solid dispersion84, 85, crystal modification86, 87, host-guest complexation88, 89 and lipid-based drug delivery systems90 are considered the most successful approaches to improve the rate of dissolution and permeability.
5.1. Nanoparticulate systems
The particle size reduction approach is a widely used technique to increase the rate of dissolution of poorly water-soluble drug by increasing the surface area of drug91. Two basic particle size reduction approaches are micronization and nanonization. Mechanical pulverization (crushing, grinding and milling) is the common techniques for preparation of micron size particles. Dry milling techniques reduce the particle size to 2–5 μm which not always increases the dissolution rate and sometimes may increase agglomeration, increasing the surface area. Thus, surfactants and polymeric materials are needed to prevent particle agglomeration. Larger particle size can lead to capillary blockage and embolism with intravenous administration. Reducing the particle size to the nanorange (< 1000 nm) improves the safety of oral delivery by increasing the distribution uniformity in the GI fluid and avoiding high and prolonged local concentrations83. Reduction of particle size to the nano range increases the dissolution rate by increasing surface area (A), increasing the concentration gradient (Cs–Cb), reducing the diffusional layer distance (h) and increasing the adherence to intestinal membrane92. Reducing the particle size to less than 1 μm increases the saturation solubility as described by Ostwald–Freundlich׳s equation. Nanocrystal and nanosuspension technology can be applied to both BCS class II and class IV drugs. Nanocrystal formulation can be prepared by two methods, i.e., top-down technique (wet milling, high pressure homogenization) and bottom up technique (controlled precipitation). Basically, the top down method is an attrition method where larger crystals (μm) are turned to smaller ones (nm). Hydrophillic polymer or surfactants systems are required to stabilize the nanoparticles. Stabilizer must be capable of wetting the surface of drug nanocrystal thus providing steric and ionic stabilization. Stable nano systems can be obtained when the weight ratio of the drug to stabilizer is 20:1 to 2:1. For increasing the intestinal adhesion surface modification of nanoparticles may be required by using mucoadhesive polymers, as cationic polymers adhere to negative surface of mucin of gastric mucosa93. Surface modification of paclitaxel nanocrystals using synthesized pluronic grafted chitosan copolymer has shown to improve relative bioavailability by modulating intestinal P-gp efflux system94. Similarly, surfactant, like vitamin E tocopherol polyethylene glycol succinate (vit. E TPGS), has also been used to circumvent P-gp mediated drug efflux mechanism95.
Polymeric nanoparticles are also frequently used to improve the therapeutic value of BCS class II and class IV drugs. The most commonly used polymers include synthetic polymers like polyethylene glycol (PEG), N-(2-hydroxypropyl)methacrylamide (HPMA), poly(vinyl pyrrolidone) (PVP), polyethyleneimine (PEI); pseudosynthetic biodegradable polymers like poly-ɛ-caprolactone, poly amino acids, poly(lactic acid) (PLA), polyglycolic acid (PGA) and their copolymers, poly-d,l-lactide-co-glycoside (PLGA); and natural polymers like chitosan, dextrin and hyaluronic acid. The API molecule can be either conjugated onto the polymeric material or can be protected and encapsulated inside the core. These polymeric nanosystems provide controlled or sustained release, prolong residence time, reduce nonspecific distribution and provide protection against proteolytic enzymes and can improve oral bioavailability. Different polymers can be used for preparation of nanoparticles with distinctive surface properties, which will strongly affect structure, properties and biological applications. For example, nanoparticulate systems synthesized using PLGA, polymethacrylates and PEG are considered to be promising delivery systems due to their mucoadhesive properties which drive various interaction forces between nanoparticle and mucus membrane such as hydrogen bonding, van der Waals forces, polymer chain interpenetration, hydrophobic forces and electrostatic/ionic interactions96. PLGA nanoparticles were surface modified using chitosan, vitamin E TPGS, lecithin and Eudragit RS. An ex vivo mucoadhesive study and Caco-2 uptake study revealed enhanced mucoadhesion and cell internalization for chitosan-modified PLGA nanoparticles. This may be due to electrostatic interactions between the positively charged surface of the nanoparticle and negatively charged mucin97. Andrographolide-loaded pH-sensitive nanoparticles prepared by using cationic poly-methacrylate copolymer have been shown to improve bioavailability98. The poly(methacrylic acid and methacrylate) copolymer allows release of load at a specific pH within the gastrointestinal tract. Table 6, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146, 147 reveals a few examples of nanoparticulate systems which improve bioavailability.
Table 6.
Formulation strategies for bioavailability enhancement of poorly water-soluble/absorbable drug.
| Formulation strategy | Technique | Drug | BCS class | Comment | Ref. |
|---|---|---|---|---|---|
| Microcrystals | Antisolvent precipitation | Megestrol acetate | II | Kollidon VA64 and Poloxamer 407 inhibits crystal growth thereby improved dissolution rate in when compared to unprocessed drug. | 99 |
| Microparticles | Rapid expansion of supercritical solution in liquid antisolvent | Fenofibrate | II | Suspension with high drug load stabilized electrostatically using sodium dodecyl sulphate (SDS). | 100 |
| PLGA microparticle | Spray drying | Nimodipine | II | PLGA polymeric microparticles with high drug loading suspended in Tisseel fibrin sealent as an in situ device for the local treatment of vasospasm after subarachnoid hemorrhage. | 101 |
| Nanocrystal | Supercritical antisolvent method | Apigenin | II | Decreased particle size, smooth surface with spherical shape and no substantial change in crystallinity of drug. | 102 |
| Amorphous nanoparticle | Controlled Precipitation technique | Aprepitant | II | Nanostructured formulation stabilized by soluplus and SDS as secondary stabilizer having particle size of less than 100 nm with instantaneous redispersibility. Solubility and PAMPA assay in agreement with in vivo kinetic studies. | 103 |
| pMMA coated thiolated chitosan nanoparticle | Radical polymerization | Docetaxel | II | Tenfold increase in oral bioavailability of nanoparticle formulation may be attributed to mucoadhesion, P-gp efflux inhibition and permeability enhancement effect of thiolated chitosan. | 104 |
| PEG-b-PLA nanoparticle | Flash nanoprecipitation | Doxorubicin | III | Overexpression of P-gp in MDR cell contribute low cellular accumulation. Self-assembled PEG-b-PLA nanoparticle demonstrated higher retention in MDR cell and passive targeting to tumor cell. | 105 |
| Nanocrystal | Combination technology (antisolvent precipitation and microfluidization) | Bexarotene | II | Nanocrystal formulation optimized using L9 orthogonal array stabilized using lecithin and poloxamer 188 for oral and parenteral delivery. | 106 |
| Nanocrystal | Antisolvent precipitation | Carvedilol | II | SDS stabilized nanosuspension demonstrated increased Cmax and AUC while decrease in Tmaxwhen compared with coarse suspension. | 107 |
| Nanocrystal | Wet media Milling | Febuxostat | II | HPMC and vitamin E TPGS stabilized system with 221.6% increase in relative bioavailability. | 108 |
| Nanocrystal | Precipitation-high pressure homogenization method | Nitrendipine | II | Surface modified chitosan nanocrystal stabilized with polyvinyl alcohol (PVA) has better stability and bioavailability compared with unmodified crystals. | 109 |
| Nanocrystal | Wet-milling technology | Tranilast | II | Hydroxy propyl cellulose-SL and SDS stabilized redispersible system exhibited improvement in the dissolution behavior under acidic conditions and enhancing the therapeutic potential of tranilast to treat liver dysfunction. | 110 |
| Solid nanodispersion | Dry media milling | Ingliforib, celecoxib furosemide | II, IV | Novel formulation approach combining two technologies,i.e.,solid dispersion and nanocrystal, and stabilized with PVP K12 and SDS. | 111 |
| Solid dispersion | Spray drying technique | Tacrolimus | II | The formulation containing drug–Eudragit E exhibited higher drug solubility as it inhibits reprecipitation in neutral pH condition. | 112 |
| Solid dispersion | Lyophillization technique | Atorvastatin | II | Solid dispersion formulation containing skimmed milk as a carrier in varying ratio has shown 33 fold increase in solubility as compared to pure drug and 3-fold increase in lipid lowering potential. | 113 |
| Solid dispersion | Lyophillization technique | TMC-240 (HIV protease inhibitor) | IV | Inulin based SD combined with ritonavir to improve permeation through intestinal wall. | 114 |
| Solid dispersion | Solvent evaporation | Pioglitazone | II | SD prepared by amorphous polymer (PVP K30 and PVP K90) and semicrystalline polymer (PEG 6000 and F68). Further concluding amorphous polymer being more suitable as it is more effective at inhibiting crystallization rates. | 115 |
| Solid dispersion | Wet milling followed by freeze drying | Tranilast | II | Nanocrystal TL-loaded SD formulation containing HPC-SL and SDS was found to have better dissolution and pharmacokinetic behaviors and thus bioavailability with high photochemical stability. | 116 |
| Cyclodextrin complexation | Lyophillization technique | Acetazolamide | IV | Amorphous HP-β-CD/drug complex prepared with and without triethanolamine (salt formation significantly increased the HP-β-CD solubilizing power) shows enhanced dissolution rate thus improving intraocular pressure lowering effect. | 117 |
| Cyclodextrin complexation | Blending, co-grinding, kneading, coevaporation | Clonazepam | II | Co-grinded product with methylated-β-CD was found to be best carrier for improving the solubility and dissolution rate of drug. | 118 |
| Cyclodextrin complexation | Kneading method | Ibuprofen | II | Tablet (direct compression) and pellet (extrusion/spheronization) formulated by drug/β-CD complex have shown high solubility and dissolution rate when compared with reference and marketed formulation. | 119 |
| SNEDDS | Vortexing | Lurasidone | II | SNEDDS prepared using Capmul MCM, Tween 80 and glycerol as oil phase, surfactant and co-surfactant system respectively with enhanced oral bioavailability with no food effect. | 120 |
| SMEDDS | Vortexing | Puerarin | II | SMEDDS containing castor oil (oil), cremophore EL (emulsifier) and 1,2-propanediol (co-emulsifier) was pelletised via extrusion-spheronization technique to form SMEDDS sustained release pellets. | 121 |
| SNEDDS | Vortexing | Cinnarazine | II | SNEDDS containing sesame oil (oil phase), cremophore RH40 (surfactant), oleic acid (surfactant) and brij 97 (co-surfactant). Food effect on cinnarazine could be significantly reduced by dosing either as SNEDDS capsule or tablet. | 122 |
| SNEDDS | Pre-concentrate preparation method | Amiodarone and talinolol | II | SNEDDS resulted in higher and less variable AUC and Tmax. SNEDDS increases the solubilization, reduces intraenterocyte metabolism, reduced P-gp efflux and does not cause intestinal tissue damage. | 123 |
| SNEDDS | Vortexing | Cefpodoxime proxetil | IV | SNEDDS containing campul MCM (oil), Tween 80 as surfactant, TPGS as co surfactant which was further pelletised has shown to improve solubilization which improves the permeability by 10-fold and bioavailability by 4-fold. | 124 |
| SNEDDS | Vortexing | Valsartan | II | Solid-SNEDDS system was prepared containing campul MCM (oil), labrasol (surfactant) and Tween 20 (co-surfactant) SNEDDS adsorbed on the solid carrier (Sylysia 350) and compressed into tablet. The system has shown 3.5-fold increase in dissolution rate of drug due to enhanced solubility. | 125 |
| SNEDDS | Vortexing | Ziprasidone | II | SNEDDS prepared using campul MCM (oil phase), labrasol (surfactant) and PEG 400 (co surfactant) which was further used to prepare sustained release pellets showed prolonged action with enhanced bioavailability. | 126 |
| SMEDDS | Vortexing | Pioglitazone | II | SMEDDS prepared using cottonseed oil, Tween 80 as surfactant and PEG as co-surfactant has been used to improve rate of dissolution of pioglitazone 2- to 3-fold when compared with commercial tablet. | 127 |
| SMEDDS | Vortexing | Furesemide | IV | SMEDDS was developed using oleic acid based heterolipid as oil phase, solutol HS 15 as surfactant and ethanol as co surfactant. It significantly improved solubility of furesemide as compared to parent oil, oliec acid. | 128 |
| SMEDDS | Vortexing | Baicalein | II | SMEDDS formulation containing capryliccapric triglyceride, cremophor RH40 and transcutol P has shown significantly higher release rate and 200.7% increase in relative bioavailability compared with that of the baicalein suspension. | 129 |
| SLN | Hot emulsification/ solidification method | Paclitaxel | IV | SLN prepared by hot homogenization technique as a carrier showed higher cellular uptake demonstrating higher efficacy in cancer cell death. | 130 |
| NLC | Melt emulsification homogenization | Montelukast | II | NLC prepared using precirol ATO-5 and capryol-90 and d,l-pyrolidonecarboxylic acid salt of l-cocyl arginine ethyl ester surfactant showed 143-fold improvement in bioavailability. | 131 |
| NLC | High pressure homogenization | Saquinavir | IV | Three NLC based formulation containing precirol ATO5, miglyol 812 as lipid phase and different concentration of poloxamer 188 and Tween 80 as aqueous phase. NLC enhanced SQV permeability and circumvented P-gp efflux. | 132 |
| Mesoporous silica (SBA-15) | – | Fenofibrate | II | Drug-silica formulation has shown significant increase in dissolution rate when compared with micronized fenofibrate which was attributed to high surface area and decreased in crystallinity of drug after absorption onto silica. | 133 |
| Mesoporous silica MCM-41 | – | Furesemide | IV | Drug inclusion into MCM-41 mesoporous displayed enhancement in dissolution rate with complete release in 90 min and enhanced photochemical stability. | 134 |
| Mesoporous silica MCM-41 | – | Piroxicam | II | Inclusion of poor soluble drug in MCM-41 improved the dissolution rate due to lack of crystallinity and extremely high surface area of siliceous material. | 135 |
| Mesoporous silica | – | Itraconazole | II | Itraconazole loaded into ordered mesoporous silica have shown significantly improved AUC, decreased Tmax. OMS formulation compares well with the marketed product (sporanox) thus considered as better carrier. | 136 |
| Porous silicon based microparticles | – | Antipyrene, ibuprofen, griseofulvin, ranitidine, furosemide | II, IV | Drug loaded in mesoporous silicon microparticle increased the dissolution rate and reduced the pH dependency dissolution. | 137 |
| Micelle | Thin film hydration | Amphotericin | II | Self-assembled lecithin-based mixed polymeric micelle containing pluronic, kolliphor RH40,TPGS and DSPE-PEG2K showed 2.18- and 1.50-fold increased in bioavailability when administered i.v. and orally. | 138 |
| Micelle | Dialysis method | Paclitaxel | IV | Pluronic F127, P188 and heparin-all-trans-retinoid acid conjugate mixed micelle exhibited higher AUC, Cmax and 5- to 6-fold increase in effective permeability. | 139 |
| Co-crystal | – | Quercetin | II | Quercetin–caffeine, quercetin–caffeine–methanol, quercetin–isonicotinamide and quercetin–theobromine dihydrate co-crystals exhibited pharmacokinetic properties that are vastly superior than quercetin alone. | 140 |
| Co-crystal | Anti-solvent crystallization | Indomethacin | II | Saccharine-indomethacin cocrystals were hygroscopic and found to have significantly higher dissolution rate than pure indomethacin. | 141 |
| Co-crystal | Anti-solvent crystallization | Diflunisal | II | Nicotinamide–diflunisal cocrystal improves intrinsic dissolution rate by 20%. | 142 |
| Co-crystal | Anti-solvent crystallization | Ibuprofen | II | Highly soluble molecule in crystallographic pattern of ibuprofen enhances the solubility more than 7.5 times. | 143 |
| Co-crystal | Anti-solvent crystallization | Ezetimibe | II | Benzoic acid and salicylic acid ezetimibe co-crystal showed significant enhancement in the dissolution profile as compared to pure ezetimibe. | 144 |
| Dendrimer | – | Camptothecin | II | G.4 and G.3.5 PAMAM dendrimer increased camptothecin solubilization in simulated gastric fluid and caused 2-fold to 3-fold increase in oral absorption suggested increased bioavailability. | 145 |
| Dendrimer | – | Famotidine, indomethacin, amphotericin | II | G.5 PPI dendrimer–drug complex demonstrated increase in solubility due to hydrophobic and electrostatic interactions for acidic, basic and amphoteric drug. | 146 |
| Dendrimer | – | Ketoprofen | II | PAMAM dendrimer was found to improve solubility of ketoprofen. Solubility of ketoprofen was found to be proportional to dendrimer concentration. | 147 |
–not applicable.
5.2. Lipid-based drug delivery systems and solid lipid-based drug delivery systems
Lipid based drug delivery system (LBDDS) has shown a great promise to deliver poorly soluble and poorly permeable candidates and appear to be a “one key fits all” system as it strikes all rate-limiting steps to absorption. Oral bioavailability of water-insoluble lipophilic drug may be enhanced when it is co-administered with a fat-rich meal which has lead to realization of lipids as a means to enhance solubilization in the gastrointestinal tract. Lipid-based formulations have a positive influence on drug absorption by increasing solubilization capacity, preventing drug precipitation on intestinal dilution, enhancing membrane permeability, inhibiting efflux transporters, reducing CYP enzymes, stimulating secretion of chylomicrons and improving lymphatic transport148, 149. These are a diverse group of formulations sharing some common features. They are categorized into five classes by Colin Pouton150 which includes excipients like triglycerides, mono- and diglycerides, water-insoluble surfactants, water-soluble surfactants and cosolvents as shown in Table 7, 150 along with their key features. Proper screening of excipients and logical design of formulations is important as LBDDS involves many complex biological processes like digestion of lipid excipients, formation of different colloid phases during lipid digestion, inhibition of efflux systems.
Table 7.
Lipid based formulation classification system150.
| Excipient in formulation | Content of formulation (%, w/w) |
||||
|---|---|---|---|---|---|
| Type I | Type II | Type IIIA | Type IIIB | Type IV | |
| Oils: triglycerides or mixed mono and diglycerides | 100 | 40–80 | 40–80 | <20 | – |
| Water-insoluble surfactants (HLB<12) | – | 20–60 | – | – | 0–20 |
| Water-soluble surfactants (HLB>12) | – | – | 20–40 | 20–50 | 30–80 |
| Hydrophillic co-solvents (e.g., PEG or propylene glycol) | – | – | 0–40 | 20–50 | 0–50 |
| Characteristic | Non dispersing; requires digestion | SEDDS without water soluble component | SEDDS/SMEDDS with water soluble component | SMEDDS with water soluble component and low oil content | Oil free formulation based on surfactants and cosolvent |
| Advantages | GRAS status; simple; excellent capsule compatibility | Unlikely to lose solvent capacity on dispersion | Clear or almost clear dispersion; drug absorption without digestion | Clear dispersion; drug absorption without digestion | Good solvent capacity for many drugs; disperses to micellar solution |
| Disadvantages | Formulation has poor solvent capacity unless drug is highly lipophilic | Turbid o/w dispersion (particle size: 0.25–2 μm) | Possible loss of solvent capacity on dispersion; less easily digested | Likely loss of solvent capacity on dispersion | Loss of solvent capacity on dispersion; may not be digestible |
–not applicable.
Lipid colloidal drug delivery systems like liposomes, niosomes, solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) are also widely used to enhance bioavailability and also provide controlled release of active compound151, 152. These nanostructures are widely used due to their versatility, biocompatibility and low cytotoxicity. Liposomes are self-assembled spherical vesicles consisting of one or several concentric phospholipid bilayer with an aqueous core inside. Despite great interest in drug delivery, this system is associated with problems like poorer entrapment efficiency, expense, and physicochemical instability due to phospholipid hydrolysis or oxidation. An alternative to such technical difficulties is niosome; self-assembled structures consisting of nonionic surfactants of alkyl or dialkyl polyglycerol ether class with cholesterol. Although structure, properties and in vivo performance of liposomes and niosomes are similar, they bypass disadvantages like chemical instability and cost, thus making them suitable for industrial manufacturing153. In the early 1990s, SLN were developed as an alternative system to liposome and noisome with clear advantages of great kinetic stability, rigid morphology thus capable of modulating drug release, and good production scalability with wide potential applications. SLN are nanospheres made from solid lipid with a mean diameter between 50–1000 nm, while NLC are next generation lipidic carriers with a mixture of solid–lipid and liquid–lipid. The basic fundamentals of NLC matrix are to create imperfections in highly ordered crystal matrix of solid lipid thus increasing the payload of API and preventing the expulsion of drug during storage154. Some of the remarkable researches in LBDDS are exemplified in Table 6.
5.3. Solid dispersion
Solid dispersion (SD) technology is dispersion of one or more API in an inert matrix at the molecular level, which was defined in the early 1970s155. Active ingredients could exist in solubilized form, amorphous state or the crystalline state in an amorphous or crystalline-inert matrix. Solid dispersion could dissolve drug immediately in contact in the GI fluid which lead to saturated or supersaturated solutions for rapid absorption, and excess drug could precipitate in GI fluid in very finely divided state. Solid dispersions exist in various forms like eutectic mixtures, crystalline solid solution, amorphous solid solution, amorphous solid suspension, and controlled-release solid dispersion. Eutectic mixtures consists of two compounds which are completely miscible in the liquid state but to a very limited extent in the solid state. In a eutectic mixture, the melting point of the mixture is lower than the melting point of the drug and carrier and is preferable because both drug and carrier will crystallize simultaneously in the cooling process, resulting in a dispersed state of drug in carrier, thus enhancing the dissolution rate. Second is a solid solution which contains microfine crystalline or amorphous API in crystalline solid dispersion. Particle size of the API is reduced in such a system and the dissolution rate is determined by the dissolution rate of the carrier. In such systems API molecules are distributed substitutionally or interstitially in which the API molecule can either substitute for the carrier molecules in the crystal lattice or fit into the interstices between the solvent molecules in the crystal lattice84. In the case of an amorphous solid dispersion API molecules are dispersed molecularly but irregularly in the amorphous carrier and are classified into either an amorphous solid solution or an amorphous solid suspension. In an amorphous solid solution (glass solution) the drug and amorphous carrier are completely miscible to form a molecularly homogenous mixture while in the case of amorphous solid suspension small API particles in an amorphous state are dispersed in the amorphous carrier156. Various synthetic or natural origin cellulose polymers have been utilized as carriers for solid dispersion. Surfactants and emulsifiers can also be used as a carrier or additives in solid dispersion which show significant improvement in overcoming the problems with amorphous solid dispersion in which a super saturation state of the drug may cause precipitation of drug and decreases the concentration in vitro and in vivo. Several methods have been used to prepare solid dispersion such as the fusion method, solvent evaporation and melting solvent method85. Numerous studies have demonstrated the marked enhancement in oral bioavailability obtained by solid dispersion technology as shown in Table 6.
5.4. Cyclodextrin complexation
Cyclodextrins (CD) are cyclic oligosaccharides derived from starch containing six (α-CD), seven (β-CD), eight (γ-CD), nine (δ-CD), ten (ε-CD) or more (α-1,4)-linked α-d-glucopyranose units. Due to the chair conformation of the glucopyranose units, the CD takes the shape of a truncated cone or torus rather than a perfect cylinder. The primary hydroxyl groups are located on the narrow side of the cone while secondary hydroxyl group are located on the wider side. The central cavity of the CD molecule is lined with skeletal carbons and ethereal oxygen of glucose which provide it with a lipophilic character. Due to these chemical properties, CDs are able to form inclusion complexes with many drugs thereby increasing the drug solubility. No covalent bonds are formed or broken during drug/CD complex formation. CD derivatives of pharmaceutical interest include the hydroxypropyl derivatives of β- and γ-CD, the randomly methylated β-CD, sulfobutylether β-CD and the so-called branched CD such as glucosyl-β-CD89. CD intervention is applicable mostly to BCS class II and class IV drug compounds to alter their properties and possibly shift them to a better class of BCS. Numerous studies have reported the enhancement of oral bioavailability of poorly water-soluble drugs by the CD inclusion complex (Table 6).
5.5. Micelles
Micelles are nano self-assemblies of amphipathic surfactant/polymers with the hydrophobic part making the core and the hydrophilic part forming the outer shell of assembly. These colloidal structures have hydrodynamic diameter typically in range of 20–80 nm. These structures solubilize poorly water-soluble drugs in a hydrophobic core while the hydrophilic shell provides the protection against micelle–protein interaction which contributes to longer blood circulation and stability. Prolonged circulation allows maintaining the required therapeutic level of drug157. Along with micellar solubilization of poorly soluble drugs, self-assembled systems also have various advantages like cellular internalization, subcellular localization and ligand-mediated targeting158.
5.6. Pharmaceutical co-crystals
Pharmaceutical co-crystals are simply a system in which at least one of the molecular components is an API in conjunction with another type of molecule called a co-former in the same crystal lattice159. The Office of Pharmaceutical Science Center for Drug Evaluation and Research (CDER) at the FDA has also issued guidance for Pharmaceutical co-crystals160. For ionisable compounds׳ (anionic or cationic) salt formation is the cheapest and simplest strategy to improve the solubility with extreme purity, manufacturability (flow property) and stability. With non-ionisable compounds and with compounds having pKa in a range where salt formation is not possible a pharmaceutical co-crystal is good alternative. Traditionally, solid-state polymorphic forms of an API are classified as either crystalline, amorphous, or solvate and hydrate forms. Co-crystals are distinguishable from these traditional pharmaceutical solid-state forms. Unlike polymorphs, which contain only the API within the crystal lattice, co-crystals are composed of an API with a neutral guest compound (referred as a conformer) in the crystal lattice. Similarly, unlike salts, where the components in the crystal lattice are in an ionized state, a co-crystal׳s components are in a neutral state and interact via nonionic interactions. The non-API component in the co-crystal (co-former) would be substances which are non-toxic with no adverse effects and would appear on a Generally Recognized as Safe (GRAS) list. Co-crystal solubility may or may not be greater than that of the API and generally a co-former׳s solubility tends to increase co-crystal solubility161. Pharmaceutical cocrystallization has emerged as a novel way to improve solubility and dissolution rate of poorly soluble APIs as addressed by various researchers. Sanphui et al.162 prepared niclosamide co-crystal with theophylline which showed good dissolution (5-fold) compared to the API. Mcnamara et al.163 prepared glutaric acid co-crystal for a poorly soluble API which showed an improved dissolution rate by 18-fold when compared with drug.
5.7. Prodrug
A prodrug is a masked form of a drug that is designed to be activated once introduced into the body. A prodrug is viewed as a bioreversible strategy of optimizing the delivery properties by maintaining the original active structure. The rationale behind using this approach is to optimize drug-like properties (manipulating with ADMET—absorption, distribution, metabolism, elimination and toxicity) as well as prolonging the commercial life cycle of potential drug candidate. The prodrug approach is widely used to increase solubility and dissolution rate as well as improving the permeability of BCS class II and BCS class IV drug. Prodrug strategy is an alternative to all above techniques to improve solubility and dissolution rate by attaching the ionizable group or polar neutral groups such as phosphates, amino acids or sugar moieties164. However, increasing the hydrophilicity is always interlinked with poor permeation characteristics, and hence balance should be maintained between both. Practically insoluble drug cyclosporine A was converted to a water-soluble prodrug to develop a water-based concentrated eye drop for treatment of ocular disease165. Two water insoluble drugs doxorubicin and dexmethasone were delivered in combination using a macromolecule prodrug strategy having antitumor activity. Dexmethasone was conjugated with a water-soluble polysaccharide which has amphiphilic character such that doxorubicin can be encapsulated in the hydrophobic core of the micelle166. The classical example is a phosphate prodrug of the HIV protease inhibitor amprenavir. Amprenavir was formulated as a 150 mg capsule which required 8 capsules to achieve a 1600-mg dose twice daily, which was a clear disadvantage over other anti-AIDS drugs. The secondary alcohol group of amprenavir was phosphorylated to produce fosamprenavir. Fosamprenavir is 10-fold more soluble than amprenavir and formulated as a 700 mg tablet with reduced dosing to 2 tablets twice a day making it a great commercial success167, 168.
5.8. Mesoporous material
Increasing the effective surface area of a poorly soluble drug in contact with the dissolution medium enhances the dissolution rate which can be achieved by loading drug onto the mesoporous material. Porous materials are classified as per IUPAC as microporous (pore diameter <2 nm), macroporous (pore diameter >50 nm) and mesoporous material (pore diameter 2–50 nm). Typical mesoporous material for drug delivery includes porous silicon (Psi) mesoporous silica material such as SBA-15, MCM-48 and MSU, and some inorganic material like mesoporous Al2O3, TiO2, carbon and hydroxycarbonate apatite169, 170, 171. Mesoporous material have an ordered unidirectional network (hexagonal, cubic or lamellar structure) with extremely large surface area, pore volume, narrow pore size distribution, strong absorption ability, high drug loading capacity and chemical inertness thus improving the dissolution kinetics for poorly soluble drugs99, 172, 173. Particle size and surface area of ordered mesoporous materials (OMM) influences drug loading and dissolution rate enhancement. It has been reported that larger silica particles results in slower drug release because of longer mesopore length174. PSi is produced by chemical stain etching, metal assisted etching and electrochemical etching method175. Mesoporous silicas are usually synthesized by interaction between negatively charged silica tetraethylortho silicate (TEOS) or sodium silicate and a positively charged surfactant (quaternary ammonium salt) micelle that act as a structure-directing agent which leads to formation of a ordered structure. The surfactant is removed by calcinations or extraction, leaving a porous silicate network176. Several model drugs from BCS class II and BCS class IV have been tested using mesoporous material as a carrier shown in Table 6.
6. Conclusions
This review discusses the drug absorption process through oral route. Better understanding of the physicochemical properties of a drug and its relationship with physiology of body can predict API bioavailability during the development phase. Further, we have also tried to explain the fundamentals of BCS which is the decision support system. It can be useful in selection of proper formulation strategies and or proper excipient selection to deal with solubility and permeability issues. For solubility/dissolution-related hurdles techniques like particle size reduction, solid dispersion, complexation, micellization, co-crystal formation can be feasible approaches. Poor permeability issues can be tackled through lipid-based systems, various surface modifications, macromolecule architects like dendritic structures or polymeric conjunctions. Thus, for BCS class II drugs techniques solving solubility/dissolution issue can be beneficial while, for BCS IV compounds combining both the strategies, viz., solubility and permeability solving techniques can be more preferable.
Acknowledgment
We thank Department of Science and Technology, New Delhi for providing financial assistance under Women Scientist Scheme-A (SR/WOS-A/LS-248/2012).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Bergström C.A., Holm R., Jørgensen S.A., Andersson S.B., Artursson P., Beato S. Early pharmaceutical profiling to predict oral drug absorption: current status and unmet needs. Eur J Pharm Sci. 2014;57:173–199. doi: 10.1016/j.ejps.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 2.Dokoumetzidis A., Macheras P. A century of dissolution research: from noyes and whitney to the biopharmaceutics classification system. Int J Pharm. 2006;321:1–11. doi: 10.1016/j.ijpharm.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Noyes A.A., Whitney W.R. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19:930–934. [Google Scholar]
- 4.Brünner E. Reaktionsgeschwindigkeit in heterogen systemen. Z Phys Chem. 1904;47:56–102. [Google Scholar]
- 5.Hixson A.W., Crowell J.H. Dependence of reaction velocity upon surface and agitation. Ind Eng Chem. 1931;23:923–931. [Google Scholar]
- 6.United States Pharmacopoeia (USPXV). Rockville, Maryland: The United States Pharmacopoeial Convection; 1950.
- 7.Danckwerts P.V. Significance of liquid-film coefficients in gas absorption. Ind Eng Chem. 1951;43:1460–1467. [Google Scholar]
- 8.Higuchi T. Rate of release of medicaments from ointment bases containing drugs in suspension. J Pharm Sci. 1961;50:874–875. doi: 10.1002/jps.2600501018. [DOI] [PubMed] [Google Scholar]
- 9.Brown W.E., Marques M.R. USP and dissolution—20 years of progress. Dissolut Technol. 2014;2014:24–27. [Google Scholar]
- 10.Federation International Pharmaceutique FIP guidelines for dissolution testing of solid oral products. Die Pharm Ind. 1981;42:334–343. [Google Scholar]
- 11.Amidon G.L., Lennernäs H., Shah V.P., Crison J.R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 12.Dahan A., Miller J.M., Amidon G.L. Prediction of solubility and permeability class membership: provisional BCS classification of the world’s top oral drugs. AAPS J. 2009;11:740–746. doi: 10.1208/s12248-009-9144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez M.N., Amidon G.L. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol. 2002;42:620–643. doi: 10.1177/00970002042006005. [DOI] [PubMed] [Google Scholar]
- 14.Pavurala N., Achenie L.E. A mechanistic approach for modeling oral drug delivery. Comp Chem Eng. 2013;57:196–206. [Google Scholar]
- 15.Lennernäs H. Human intestinal permeability. J Pharm Sci. 1998;87:403–410. doi: 10.1021/js970332a. [DOI] [PubMed] [Google Scholar]
- 16.Yu L.X., Lipka E., Crison J.R., Amidon G.L. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19:359–376. doi: 10.1016/0169-409x(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 17.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 18.Azarmi S., Roa W., Löbenberg R. Current perspectives in dissolution testing of conventional and novel dosage forms. Int J Pharm. 2007;328:12–21. doi: 10.1016/j.ijpharm.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Alsenz J., Kansy M. High throughput solubility measurement in drug discovery and development. Adv Drug Deliv Rev. 2007;59:546–567. doi: 10.1016/j.addr.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Noyes A.A., Whitney W.R. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1987;19:930–934. [Google Scholar]
- 21.Dressman J.B., Amidon G.L., Reppas C., Shah V.P. Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms. Pharm Res. 1998;15:11–22. doi: 10.1023/a:1011984216775. [DOI] [PubMed] [Google Scholar]
- 22.Bansil R., Turner B.S. Mucin structure, aggregation, physiological functions and biomedical applications. Curr Opin Colloid Interface Sci. 2006;11:164–170. [Google Scholar]
- 23.Behrens I., Stenberg P., Artursson P., Kissel T. Transport of lipophilic drug molecules in a new mucus-secreting cell culture model based on HT29-MTX cells. Pharm Res. 2001;18:1138–1145. doi: 10.1023/a:1010974909998. [DOI] [PubMed] [Google Scholar]
- 24.Sahay G., Alakhova D.Y., Kabanov A.V. Endocytosis of nanomedicines. J Control Release. 2010;145:182–195. doi: 10.1016/j.jconrel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith D.A., van de Waterbeemd H. Pharmacokinetics and metabolism in early drug discovery. Curr Opin Chem Biol. 1999;3:373–378. doi: 10.1016/s1367-5931(99)80056-8. [DOI] [PubMed] [Google Scholar]
- 26.Lipka E., Crison J., Amidon G.L. Transmembrane transport of peptide type compounds: prospects for oral delivery. J Control Release. 1996;39:121–129. doi: 10.1016/0168-3659(95)00145-x. [DOI] [PubMed] [Google Scholar]
- 27.Petri N., Borga O., Nyberg L., Hedeland M., Bondesson U., Lennernas H. Effect of erythromycin on the absorption of fexofenadine in the jejunum, ileum and colon determined using local intubation in healthy volunteers. Int J Clin Pharmacol Ther. 2006;44:71–79. doi: 10.5414/cpp44071. [DOI] [PubMed] [Google Scholar]
- 28.Sai Y. Biochemical and molecular pharmacological aspects of transporters as determinants of drug disposition. Drug Metab Pharmacokinet. 2005;20:91–99. doi: 10.2133/dmpk.20.91. [DOI] [PubMed] [Google Scholar]
- 29.Estudante M., Morais J.G., Soveral G., Benet L.Z. Intestinal drug transporters: an overview. Adv Drug Deliv Rev. 2013;65:1340–1356. doi: 10.1016/j.addr.2012.09.042. [DOI] [PubMed] [Google Scholar]
- 30.Minuesa G., Huber-Ruano I., Pastor-Anglada M., Koepsell H., Clotet B., Martinez-Picado J. Drug uptake transporters in antiretroviral therapy. Pharm Ther. 2011;132:268–279. doi: 10.1016/j.pharmthera.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Kunta J.R., Sinko P.J. Intestinal drug transporters: in vivo function and clinical importance. Curr Drug Metab. 2004;5:109–124. doi: 10.2174/1389200043489144. [DOI] [PubMed] [Google Scholar]
- 32.Varma M.V., Peruma O.P., Panchagnula R. Functional role of P-glycoprotein in limiting peroral drug absorption: optimizing drug delivery. Curr Opin Chem Biol. 2006;10:367–373. doi: 10.1016/j.cbpa.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 33.Abrahamsson B., Lennernäs H. Application of the biopharmaceutic classification system now and in the future. In: van de Waterbeemd H., Lennernäs H., Artursson P., editors. Drug bioavailability: estimation of solubility, permeability, absorption and bioavailability. Wiley-VCH; Weinheim: 2003. pp. 495–531. [Google Scholar]
- 34.Curatolo W. Physical chemical properties of oral drug candidates in the discovery and exploratory development settings. Pharm Sci Technol Today. 1998;1:387–393. [Google Scholar]
- 35.Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 36.van de Waterbeemd H., Smith D.A., Beaumont K., Walker D.K. Property-based design: optimization of drug absorption and pharmacokinetics. J Med Chem. 2001;44:1313–1333. doi: 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]
- 37.Lipinski C., Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 38.Wenlock M.C., Austin R.P., Barton P., Davis A.M., Leeson P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J Med Chem. 2003;46:1250–1256. doi: 10.1021/jm021053p. [DOI] [PubMed] [Google Scholar]
- 39.Palm K., Stenberg P., Luthman K., Artursson P. Polar molecular surface properties predict the intestinal absorption of drugs in humans. Pharm Res. 1997;14:568–571. doi: 10.1023/a:1012188625088. [DOI] [PubMed] [Google Scholar]
- 40.Clark D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J Pharm Sci. 1999;88:807–814. doi: 10.1021/js9804011. [DOI] [PubMed] [Google Scholar]
- 41.Veber D.F., Johnson S.R., Cheng H.Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 42.Ritchie T.J., Macdonald S.J. The impact of aromatic ring count on compound developability—are too many aromatic rings a liability in drug design? Drug Discov Today. 2009;14:1011–1020. doi: 10.1016/j.drudis.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 43.Dressman J.B., Amidon G.L., Fleisher D. Absorption potential: estimating the fraction absorbed for orally administered compounds. J Pharm Sci. 1985;74:588–589. doi: 10.1002/jps.2600740523. [DOI] [PubMed] [Google Scholar]
- 44.Yalkowsky S.H., Johnson J.L., Sanghvi T., Machatha S.G. A ‘rule of unity’ for human intestinal absorption. Pharm Res. 2006;23:2475–2481. doi: 10.1007/s11095-006-9000-y. [DOI] [PubMed] [Google Scholar]
- 45.Fleisher D., Li C., Zhou Y., Pao L.H., Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration: clinical implications. Clin Pharmacokinet. 1999;36:233–254. doi: 10.2165/00003088-199936030-00004. [DOI] [PubMed] [Google Scholar]
- 46.Freire A.C., Basit A.W., Choudhary R., Piong C.W., Merchant H.A. Does sex matter? The influence of gender on gastrointestinal physiology and drug delivery. Int J Pharm. 2011;415:15–28. doi: 10.1016/j.ijpharm.2011.04.069. [DOI] [PubMed] [Google Scholar]
- 47.Avdeef A. Physicochemical profiling (solubility, permeability and charge state) Curr Top Med Chem. 2001;1:277–351. doi: 10.2174/1568026013395100. [DOI] [PubMed] [Google Scholar]
- 48.Kimura T., Higaki K. Gastrointestinal transit and drug absorption. Biol Pharm Bull. 2002;25:149–164. doi: 10.1248/bpb.25.149. [DOI] [PubMed] [Google Scholar]
- 49.Fujioka Y., Metsugi Y., Ogawara K.I., Higaki K., Kimura T. Evaluation of in vivo dissolution behavior and GI transit of griseofulvin, a BCS class II drug. Int J Pharm. 2008;352:36–43. doi: 10.1016/j.ijpharm.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 50.Cook J.A., Bockbrader H. An industrial implementation of the biopharmaceutics classification system. Dissolut Technol. 2002;9:6–9. [Google Scholar]
- 51.Cook J.A., Davit B.M., Polli J.E. Impact of biopharmaceutics classification system-based biowaivers. Mol Pharm. 2010;7:1539–1544. doi: 10.1021/mp1001747. [DOI] [PubMed] [Google Scholar]
- 52.Committee for Proprietary Medical Products (CPMP). Note for guidance on the investigation of bioavailability and bioequivalence. CPMP/EWP/QWP/1408/98. 2001: London. p. 1–18. Available from: 〈http://www.vivodevelopment.com/En/guidline_PDF/EMEA%20Note%20for%20Guidance%20on%20BE.pdf〉
- 53.Oh D.M., Sinko P.J., Amidon G.L. Predicting oral drug absorption in humans: a macroscopic mass balance approach for passive and carrier-mediated compounds. In: D׳Argenio D.Z., editor. Advanced methods of Pharmacokinetic and Pharmacodynamic Systems Analysis. Springer; New York: 1991. pp. 3–11. [Google Scholar]
- 54.Sakore S., Chakraborty B. In vitro–in vivo correlation (IVIVC): a strategic tool in drug development. J Bioequiv Availab. 2011;2011:S3. [Google Scholar]
- 55.Batchelor H.K., Kendall R., Desset-Brethes S., Alex R., Ernest T.B. Application of in vitro biopharmaceutical methods in development of immediate release oral dosage forms intended for paediatric patients. Eur J Pharm Biopharm. 2013;85:833–842. doi: 10.1016/j.ejpb.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 56.Alelyunas Y.W., Liu R., Pelosi-Kilby L., Shen C. Application of a dried-DMSO rapid throughput 24-h equilibrium solubility in advancing discovery candidates. Eur J Pharm Sci. 2009;37:172–182. doi: 10.1016/j.ejps.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 57.Saal C., Petereit A.C. Optimizing solubility: kinetic versus thermodynamic solubility temptations and risks. Eur J Pharm Sci. 2012;47:589–595. doi: 10.1016/j.ejps.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 58.Zhou L., Yang L., Tilton S., Wang J. Development of a high throughput equilibrium solubility assay using miniaturized shake-flask method in early drug discovery. J Pharm Sci. 2007;96:3052–3071. doi: 10.1002/jps.20913. [DOI] [PubMed] [Google Scholar]
- 59.Hörter D., Dressman J.B. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 1997;25:3–14. doi: 10.1016/s0169-409x(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 60.Lue B.M., Nielsen F.S., Magnussen T., Schou H.M., Kristensen K., Jacobsen L.O. Using biorelevant dissolution to obtain IVIVC of solid dosage forms containing a poorly-soluble model compound. Eur J Pharm Biopharm. 2008;69:648–657. doi: 10.1016/j.ejpb.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 61.Klein S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010;12:397–406. doi: 10.1208/s12248-010-9203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aiache J.M., Aoyagi N., Blume H., Dressman J., Friedel H.B., Grady I.T. FIP guidelines for dissolution testing of solid oral products. Dissolut Technol. 1997;4:5–14. [Google Scholar]
- 63.Wei H., Löbenberg R. Biorelevant dissolution media as a predictive tool for glyburide a class II drug. Eur J Pharm Sci. 2006;29:45–52. doi: 10.1016/j.ejps.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 64.Lehto P., Kortejärvi H., Liimatainen A., Ojala K., Kangas H., Hirvonen J. Use of conventional surfactant media as surrogates for FaSSIF in simulating in vivo dissolution of BCS class II drugs. Eur J Pharm Biopharm. 2011;78:531–538. doi: 10.1016/j.ejpb.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 65.Tsume Y., Amidon G.L., Takeuchi S. Dissolution effect of gastric and intestinal pH fora BCS class II drug, pioglitazone: new in vitro dissolution system to predict in vivo dissolution. J Bioequiv Availab. 2013;5:224–227. [Google Scholar]
- 66.Motz S.A., Schaefer U.F., Balbach S., Eichinger T., Lehr C.M. Permeability assessment for solid oral drug formulations based on Caco-2 monolayer in combination with a flow through dissolution cell. Eur J Pharm Biopharm. 2007;66:286–295. doi: 10.1016/j.ejpb.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 67.Zakeri-Milani P., Barzegar-Jalali M., Azimi M., Valizadeh H. Biopharmaceutical classification of drugs using intrinsic dissolution rate (IDR) and rat intestinal permeability. Eur J Pharm Biopharm. 2009;73:102–106. doi: 10.1016/j.ejpb.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 68.Muenster U., Pelzetter C., Backensfeld T., Ohm A., Kuhlmann T., Mueller H. Volume to dissolve applied dose (VDAD) and apparent dissolution rate (ADR): tools to predict in vivo bioavailability from orally applied drug suspensions. Eur J Pharm Biopharm. 2011;78:522–530. doi: 10.1016/j.ejpb.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 69.Lennernäs H. Human in vivo regional intestinal permeability: importance for pharmaceutical drug development. Mol Pharm. 2014;11:12–23. doi: 10.1021/mp4003392. [DOI] [PubMed] [Google Scholar]
- 70.Kukes VG, Ramenskaya GV, Vasilenko GF, Vasilenko KS, Krasnykh LM, Savchenko AY, et al. Federal Service on Surveillance in Healthcare and Social Development. Methodological recommendations for drug manufacturers on in vitro equivalence test for generic drug products according to biowaiver procedure. 2010. Available from: 〈http://www.fip.org/files/fip/BPS/BCS/Biowaiver%20guidance%20(ru)%20-%20ENG%20March%202011.pdf〉
- 71.Lennernäs H. Regional intestinal drug permeation: biopharmaceutics and drug development. Eur J Pharm Sci. 2013;57:333–341. doi: 10.1016/j.ejps.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 72.Wahlang B., Pawar Y.B., Bansal A.K. Identification of permeability-related hurdles in oral delivery of curcumin using the Caco-2 cell model. Eur J Pharm Biopharm. 2011;77:275–282. doi: 10.1016/j.ejpb.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 73.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. Guidance for industry. 2000. Available from: 〈http://www.fda.gov/downloads/Drugs/.../Guidances/ucm070246.pdf〉
- 74.Blume H.H., Schug B.S. The biopharmaceutics classification system (BCS): class III drugs—better candidates for BA/BE waiver? Eur J Pharm Sci. 1999;9:117–121. doi: 10.1016/s0928-0987(99)00076-7. [DOI] [PubMed] [Google Scholar]
- 75.Tsume Y., Mudie D.M., Langguth P., Amidon G.E., Amidon G.L. The biopharmaceutics classification system: subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur J Pharm Sci. 2014;57:152–163. doi: 10.1016/j.ejps.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu C.Y., Benet L.Z. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 77.Custodio J.M., Wu C.Y., Benet L.Z. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60:717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shah A.K., Agnihotri S.A. Recent advances and novel strategies in pre-clinical formulation development: an overview. J Control Release. 2011;156:281–296. doi: 10.1016/j.jconrel.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 79.Rabinow B.E. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3:785–796. doi: 10.1038/nrd1494. [DOI] [PubMed] [Google Scholar]
- 80.Gao L., Liu G., Ma J., Wang X., Zhou L., Li X. Drug nanocrystals: in vivo performances. J Control Release. 2012;160:418–430. doi: 10.1016/j.jconrel.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 81.Kalepu S., Nekkanti V. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm Sin B. 2015;5:442–453. doi: 10.1016/j.apsb.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Basavaraj S., Betageri G.V. Can formulation and drug delivery reduce attrition during drug discovery and development—review of feasibility, benefits and challenges. Acta Pharm Sin B. 2014;4:3–17. doi: 10.1016/j.apsb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Desai P.P., Date A.A., Patravale V.B. Overcoming poor oral bioavailability using nanoparticle formulations—opportunities and limitations. Drug Discov Today. 2012;9:e87–e95. doi: 10.1016/j.ddtec.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 84.Huang Y., Dai W.G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B. 2014;4:18–25. doi: 10.1016/j.apsb.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vo C.L., Park C., Lee B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85:799–813. doi: 10.1016/j.ejpb.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 86.Blagden N., de Matas M., Gavan P.T., York P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv Drug Deliv Rev. 2007;59:617–630. doi: 10.1016/j.addr.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 87.Elder D.P., Holm R., de Diego H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int J Pharm. 2013;453:88–100. doi: 10.1016/j.ijpharm.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 88.Semalty A. Cyclodextrin and phospholipid complexation in solubility and dissolution enhancement: a critical and meta-analysis. Expert Opin Drug Deliv. 2014;11:1255–1272. doi: 10.1517/17425247.2014.916271. [DOI] [PubMed] [Google Scholar]
- 89.Brewster M.E., Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 2007;59:645–666. doi: 10.1016/j.addr.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 90.Mu H., Holm R., Müllertz A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int J Pharm. 2013;453:215–224. doi: 10.1016/j.ijpharm.2013.03.054. [DOI] [PubMed] [Google Scholar]
- 91.Chen H., Khemtong C., Yang X., Chang X., Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today. 2011;16:354–360. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 92.Dinarvand M., Kiani M., Mirzazadeh F., Esmaeili A., Mirzaie Z., Soleimani M. Oral delivery of nanoparticles containing anticancer SN38 and hSET1 antisense for dual therapy of colon cancer. Int J Biol Macromol. 2015;78:112–121. doi: 10.1016/j.ijbiomac.2015.03.066. [DOI] [PubMed] [Google Scholar]
- 93.Kharia A.A., Singhai A.K. Development and optimisation of mucoadhesive nanoparticles of acyclovir using design of experiments approach. J Microencapsul. 2015;32:521–532. doi: 10.3109/02652048.2015.1010457. [DOI] [PubMed] [Google Scholar]
- 94.Sharma S., Verma A., Pandey G., Mittapelly N., Mishra P.R. Investigating the role of pluronic-g-cationic polyelectrolyte as functional stabilizer for nanocrystals: impact on paclitaxel oral bioavailability and tumor growth. Acta Biomater. 2015;26:169–183. doi: 10.1016/j.actbio.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 95.Gadadare R., Mandpe L., Pokharkar V. Ultra rapidly dissolving repaglinide nanosized crystals prepared via bottom-up and top-down approach: influence of food on pharmacokinetics behavior. AAPS PharmSciTech. 2015;16:787–799. doi: 10.1208/s12249-014-0267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lai S.K., Wang Y.Y., Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61:158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Durán-Lobato M., Muñoz-Rubio I., Holgado M.A., Alvarez-Fuentes J., Fernández-Arévalo M., Martín-Banderas L. Enhanced cellular uptake and biodistribution of a synthetic cannabinoid loaded in surface-modified poly(lactic-co-glycolic acid) nanoparticles. J Biomed Nanotechnol. 2014;10:1068–1079. doi: 10.1166/jbn.2014.1806. [DOI] [PubMed] [Google Scholar]
- 98.Chellampillai B., Pawar A.P. Improved bioavailability of orally administered andrographolide from pH-sensitive nanoparticles. Eur J Drug Metab Pharmacokinet. 2011;35:123–129. doi: 10.1007/s13318-010-0016-7. [DOI] [PubMed] [Google Scholar]
- 99.Cho E., Cho W., Cha K.H., Park J., Kim M.S., Kim J.S. Enhanced dissolution of megestrol acetate microcrystals prepared by antisolvent precipitation process using hydrophilic additives. Int J Pharm. 2010;396:91–98. doi: 10.1016/j.ijpharm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 100.Dalvi S.V., Azad M.A., Dave R. Precipitation and stabilization of ultrafine particles of Fenofibrate in aqueous suspensions by RESOLV. Powder Technol. 2013;236:75–84. [Google Scholar]
- 101.Bege N., Renette T., Endres T., Beck-Broichsitter M., Hänggi D., Kissel T. In situ forming nimodipine depot system based on microparticles for the treatment of posthemorrhagic cerebral vasospasm. Eur J Pharm Biopharm. 2013;84:99–105. doi: 10.1016/j.ejpb.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 102.Zhang J., Huang Y., Liu D., Gao Y., Qian S. Preparation of apigenin nanocrystals using supercritical antisolvent process for dissolution and bioavailability enhancement. Eur J Pharm Sci. 2013;48:740–747. doi: 10.1016/j.ejps.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 103.Angi R., Solymosi T., Ötvös Z., Ordasi B., Glavinas H., Filipcsei G. Novel continuous flow technology for the development of a nanostructured Aprepitant formulation with improved pharmacokinetic properties. Eur J Pharm Biopharm. 2014;86:361–368. doi: 10.1016/j.ejpb.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 104.Saremi S., Dinarvand R., Kebriaeezadeh A., Ostad S.N., Atyabi F. Enhanced oral delivery of docetaxel using thiolated chitosan nanoparticles: preparation, in vitro and in vivo studies. Biomed Res Int. 2013;2013:150478. doi: 10.1155/2013/150478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tam Y.T., Chow K.K. AH. Fabrication of doxorubicin nanoparticles by controlled antisolvent precipitation for enhanced intracellular delivery. Colloids Surf B Biointerfaces. 2015;139:249–258. doi: 10.1016/j.colsurfb.2015.12.026. [DOI] [PubMed] [Google Scholar]
- 106.Chen L., Wang Y., Zhang J., Hao L., Guo H., Lou H. Bexarotene nanocrystal-oral and parenteral formulation development, characterization and pharmacokinetic evaluation. Eur J Pharm Biopharm. 2014;87:160–169. doi: 10.1016/j.ejpb.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 107.Liu D., Pan H., He F., Wang X., Li J., Yang X. Effect of particle size on oral absorption of carvedilol nanosuspensions: in vitro and in vivo evaluation. Int J Nanomed. 2015;10:6425–6434. doi: 10.2147/IJN.S87143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ahuja B.K., Jena S.K., Paidi S.K., Bagri S., Suresh S. Formulation, optimization and in vitro-in vivo evaluation of febuxostat nanosuspension. Int J Pharm. 2015;478:540–552. doi: 10.1016/j.ijpharm.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 109.Quan P., Shi K., Piao H., Piao H., Liang N., Xia D. A novel surface modified nitrendipine nanocrystals with enhancement of bioavailability and stability. Int J Pharm. 2012;430:366–371. doi: 10.1016/j.ijpharm.2012.04.025. [DOI] [PubMed] [Google Scholar]
- 110.Onoue S., Yamamoto K., Kawabata Y., Yamada S. In vitro/in vivo characterization of nanocrystalline formulation of tranilast with improved dissolution and hepatoprotective properties. Eur J Pharm Biopharm. 2013;85:952–957. doi: 10.1016/j.ejpb.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 111.Nkansah P., Antipas A., Lu Y., Varma M., Rotter C., Rago B. Development and evaluation of novel solid nanodispersion system for oral delivery of poorly water-soluble drugs. J Control Release. 2013;169:150–161. doi: 10.1016/j.jconrel.2013.03.032. [DOI] [PubMed] [Google Scholar]
- 112.Yoshida T., Kurimoto I., Yoshihara K., Umejima H., Ito N., Watanabe S. Aminoalkyl methacrylate copolymers for improving the solubility of tacrolimus. I: evaluation of solid dispersion formulations. Int J Pharm. 2012;428:18–24. doi: 10.1016/j.ijpharm.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 113.Choudhary A., Rana A.C., Aggarwal G., Kumar V., Zakir F. Development and characterization of an atorvastatin solid dispersion formulation using skimmed milk for improved oral bioavailability. Acta Pharm Sin B. 2012;2:421–428. [Google Scholar]
- 114.Visser M.R., Baert L., Klooster G.V., Schueller L., Geldof M., Vanwelkenhuysen I. Inulin solid dispersion technology to improve the absorption of the BCS Class IV drug TMC240. Eur J Pharm Biopharm. 2010;74:233–238. doi: 10.1016/j.ejpb.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 115.Shi N.Q., Lei Y.S., Song L.M., Yao J., Zhang X.B., Wang X.L. Impact of amorphous and semicrystalline polymers on the dissolution and crystallization inhibition of pioglitazone solid dispersions. Powder Technol. 2013;247:211–221. [Google Scholar]
- 116.Kawabata Y., Yamamoto K., Debari K., Onoue S., Yamada S. Novel crystalline solid dispersion of tranilast with high photostability and improved oral bioavailability. Eur J Pharm Sci. 2010;39:256–262. doi: 10.1016/j.ejps.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 117.Mora M.J., Tártara L.I., Onnainty R., Palma S.D., Longhi M.R., Granero G.E. Characterization, dissolution and in vivo evaluation of solid acetazolamide complexes. Carbohydr Polym. 2013;98:380–390. doi: 10.1016/j.carbpol.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 118.Mennini N., Bragagni M., Maestrelli F., Mura P. Physico-chemical characterization in solution and in the solid state of clonazepam complexes with native and chemically-modified cyclodextrins. J Pharm Biomed Anal. 2014;89:142–149. doi: 10.1016/j.jpba.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 119.Salústio P.J., Feio G., Figueirinhas J.L., Cabral-Marques H.M., Costa P.C., Pinto J.F. Release profile of ibuprofen in β-cyclodextrin complexes from two different solid dosage forms. Powder Technol. 2012;221:245–251. [Google Scholar]
- 120.Miao Y., Sun J., Chen G., Lili R., Ouyang P. Enhanced oral bioavailability of lurasidone by self-nanoemulsifying drug delivery system in fasted state. Drug Dev Ind Pharm. 2016;42:1234–1240. doi: 10.3109/03639045.2015.1118496. [DOI] [PubMed] [Google Scholar]
- 121.Zhang Y., Wang R., Wu J., Shen Q. Characterization and evaluation of self-microemulsifying sustained-release pellet formulation of puerarin for oral delivery. Int J Pharm. 2012;427:337–344. doi: 10.1016/j.ijpharm.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 122.Christiansen M.L., Holm R., Kristensen J., Kreilgaard M., Jacobsen J., Abrahamsson B. Cinnarizine food-effects in beagle dogs can be avoided by administration in a Self Nano emulsifying drug delivery system (SNEDDS) Eur J Pharm Sci. 2014;57:164–172. doi: 10.1016/j.ejps.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 123.Elgart A., Cherniakov I., Aldouby Y., Domb A.J., Hoffman A. Improved oral bioavailability of BCS class 2 compounds by self nano-emulsifying drug delivery systems (SNEDDS): the underlying mechanisms for amiodarone and talinolol. Pharm Res. 2013;30:3029–3044. doi: 10.1007/s11095-013-1063-y. [DOI] [PubMed] [Google Scholar]
- 124.Bajaj A., Rao M.R., Khole I., Munjapara G. Self-nanoemulsifying drug delivery system of cefpodoxime proxetil containing tocopherol polyethylene glycol succinate. Drug Dev Ind Pharm. 2013;39:635–645. doi: 10.3109/03639045.2012.683440. [DOI] [PubMed] [Google Scholar]
- 125.Beg S., Swain S., Singh H.P., Patra C.N., Rao M.E. Development, optimization, and characterization of solid self-nanoemulsifying drug delivery systems of valsartan using porous carriers. AAPS PharmSciTech. 2012;13:1416–1427. doi: 10.1208/s12249-012-9865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miao Y., Chen G., Ren L., Pingkai O. Characterization and evaluation of self-nanoemulsifying sustained-release pellet formulation of ziprasidone with enhanced bioavailability and no food effect. Drug Deliv. 2015;21:1–10. doi: 10.3109/10717544.2014.950768. [DOI] [PubMed] [Google Scholar]
- 127.Hyma P., Abbulu K. Formulation and characterisation of self-microemulsifying drug delivery system of pioglitazone. Biomed Prev Nutr. 2013;3:345–350. [Google Scholar]
- 128.Kalhapure R.S., Akamanchi K.G. Akamanchi. Oleic acid based heterolipid synthesis, characterization and application in self-microemulsifying drug delivery system. Int J Pharm. 2012;425:9–18. doi: 10.1016/j.ijpharm.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 129.Liu W., Tian R., Hu W., Jia Y., Jiang H., Zhang J. Preparation and evaluation of self-microemulsifying drug delivery system of baicalein. Fitoterapia. 2012;83:1532–1539. doi: 10.1016/j.fitote.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 130.Videira M.A., Arranja A.G., Gouveia L.F. Experimental design towards an optimal lipid nanosystem: a new opportunity for paclitaxel-based therapeutics. Eur J Pharm Sci. 2013;49:302–310. doi: 10.1016/j.ejps.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 131.Patil-Gadhe A., Pokharkar V. Montelukast-loaded nanostructured lipid carriers: part I oral bioavailability improvement. Eur J Pharm Biopharm. 2014;88:160–168. doi: 10.1016/j.ejpb.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 132.Beloqui A., Solinís M.A., Gascón A.R., del Pozo-Rodríguez A., Rieux A., Préat V. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J Control Release. 2013;166:115–123. doi: 10.1016/j.jconrel.2012.12.021. [DOI] [PubMed] [Google Scholar]
- 133.Sanganwar G.P., Gupta R.B. Dissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxide. Int J Pharm. 2008;360:213–218. doi: 10.1016/j.ijpharm.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 134.Ambrogi V., Perioli L., Pagano C., Latterini L., Marmottini F., Ricci M. MCM-41 for furosemide dissolution improvement. Microp Mesop Mater. 2012;147:343–349. [Google Scholar]
- 135.Ambrogi V., Perioli L., Marmottini F., Giovagnoli S., Esposito M., Rossi C. Improvement of dissolution rate of piroxicam by inclusion into MCM-41 mesoporous silicate. Eur J Pharm Sci. 2007;32:216–222. doi: 10.1016/j.ejps.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 136.Mellaerts R., Mols R., Jammaer J.A., Aerts C.A., Annaert P., van Humbeeck J. Increasing the oral bioavailability of the poorly water soluble drug itraconazole with ordered mesoporous silica. Eur J Pharm Biopharm. 2008;69:223–230. doi: 10.1016/j.ejpb.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 137.Salonen J., Laitinen L., Kaukonen A.M., Tuura J., Björkqvist M., Heikkilä T. Mesoporous silicon microparticles for oral drug delivery: loading and release of five model drugs. J Control Release. 2005;108:362–374. doi: 10.1016/j.jconrel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 138.Chen Y.C., Su C.Y., Jhan H.J., Ho H.O., Sheu M.T. Physical characterization and in vivo pharmacokinetic study of self-assembling amphotericin B-loaded lecithin-based mixed polymeric micelles. Int J Nanomed. 2015;10:7265–7274. doi: 10.2147/IJN.S95194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dahmani F.Z., Yang H., Zhou J., Yao J., Zhang T., Zhang Q. Enhanced oral bioavailability of paclitaxel in pluronic/LHR mixed polymeric micelles: preparation, in vitro and in vivo evaluation. Eur J Pharm Sci. 2012;47:179–189. doi: 10.1016/j.ejps.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 140.Smith A.J., Kavuru P., Wojtas L., Zaworotko M.J., Shytle R.D. Cocrystals of quercetin with improved solubility and oral bioavailability. Mol Pharm. 2011;8:1867–1876. doi: 10.1021/mp200209j. [DOI] [PubMed] [Google Scholar]
- 141.Basavoju S., Boström D., Velaga S.P. Indomethacin-saccharin cocrystal: design, synthesis and preliminary pharmaceutical characterization. Pharm Res. 2008;25:530–541. doi: 10.1007/s11095-007-9394-1. [DOI] [PubMed] [Google Scholar]
- 142.Évora A.O., Castro R.A., Maria T.M., Silva M.R., Ter Horst J.H., Canotilho J. A thermodynamic based approach on the investigation of a diflunisal pharmaceutical co-crystal with improved intrinsic dissolution rate. Int J Pharm. 2014;466:68–75. doi: 10.1016/j.ijpharm.2014.02.048. [DOI] [PubMed] [Google Scholar]
- 143.Soares F.L., Carneiro R.L. Evaluation of analytical tools and multivariate methods for quantification of co-former crystals in ibuprofen-nicotinamide co-crystals. J Pharm Biomed Anal. 2014;89:166–175. doi: 10.1016/j.jpba.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 144.Mulye S.P., Jamadar S.A., Karekar P.S., Pore Y.V., Dhawale S.C. Improvement in physicochemical properties of ezetimibe using a crystal engineering technique. Powder Technol. 2012;222:131–138. [Google Scholar]
- 145.Sadekar S., Thiagarajan G., Bartlett K., Hubbard D., Ray A., McGill L.D. Poly(amido amine) dendrimers as absorption enhancers for oral delivery of camptothecin. Int J Pharm. 2013;456:175–185. doi: 10.1016/j.ijpharm.2013.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gupta U., Agashe H.B., Jain N.K. Polypropylene imine dendrimer mediated solubility enhancement: effect of pH and functional groups of hydrophobes. J Pharm Pharm Sci. 2007;10:358–367. [PubMed] [Google Scholar]
- 147.Cheng Y., Xu T., Fu R. Polyamidoamine dendrimers used as solubility enhancers of ketoprofen. Eur J Med Chem. 2005;40:1390–1393. doi: 10.1016/j.ejmech.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 148.Yáñez J.A., Wang S.W., Knemeyer I.W., Wirth M.A., Alton K.B. Intestinal lymphatic transport for drug delivery. Adv Drug Deliv Rev. 2011;63:923–942. doi: 10.1016/j.addr.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Carrière F. Impact of gastrointestinal lipolysis on oral lipid based formulations and bioavailability of lipophilic drugs. Biochimie. 2016;125:297–305. doi: 10.1016/j.biochi.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 150.Pouton C.W. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29:278–287. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 151.Kumar G.P., Rajeshwarrao P. Nonionic surfactant vesicular systems for effective drug delivery-an overview. Acta Pharm Sin B. 2011;1:208–219. [Google Scholar]
- 152.Pandita D., Kumar S., Poonia N., Lather V. Solid lipid nanoparticles enhance oral bioavailability of resveratrol, a natural polyphenol. Food Res Int. 2014;62:1165–1174. [Google Scholar]
- 153.Das S., Chaudhury A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech. 2011;12:62–76. doi: 10.1208/s12249-010-9563-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Iqbal M.A., Md S., Sahni J.K., Baboota S., Dang S., Ali J. Nanostructured lipid carriers system: recent advances in drug delivery. J Drug Target. 2012;20:813–830. doi: 10.3109/1061186X.2012.716845. [DOI] [PubMed] [Google Scholar]
- 155.Chiou W.L., Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60:1281–1302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 156.van der Mooter G. The use of amorphous solid dispersions: a formulation strategy to overcome poor solubility and dissolution rate. Drug Discov Today. 2012;9:e79–e85. doi: 10.1016/j.ddtec.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 157.Lu Y., Park K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int J Pharm. 2013;453:198–214. doi: 10.1016/j.ijpharm.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gaucher G., Dufresne M.H., Sant V.P., Kang N., Maysinger D., Leroux J.C. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109:169–188. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 159.Good D.J., Rodríguez-Hornedo N. Solubility advantage of pharmaceutical cocrystals. Cryst Growth Des. 2009;9:2252–2264. [Google Scholar]
- 160.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Regulatory classification of pharmaceutical co-crystals. Guidance for industry. 2013. Available from: 〈http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM516813.pdf〉
- 161.Jayasankar A., Roy L., Rodríguez-Hornedo N. Transformation pathways of cocrystal hydrates when coformer modulates water activity. J Pharm Sci. 2010;99:3977–3985. doi: 10.1002/jps.22245. [DOI] [PubMed] [Google Scholar]
- 162.Sanphui P., Kumar S.S., Nangia A. Pharmaceutical cocrystals of niclosamide. Cryst Growth Des. 2012;12:4588–4599. [Google Scholar]
- 163.McNamara D.P., Childs S.L., Giordano J., Iarriccio A., Cassidy J., Shet M.S. Use of a glutaric acid cocrystal to improve oral bioavailability of a low solubility API. Pharm Res. 2006;23:1888–1897. doi: 10.1007/s11095-006-9032-3. [DOI] [PubMed] [Google Scholar]
- 164.Vig B.S., Huttunen K.M., Laine K., Rautio J. Amino acids as promoieties in prodrug design and development. Adv Drug Deliv Rev. 2013;65:1370–1385. doi: 10.1016/j.addr.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 165.Rodriguez-Aller M., Guillarme D., El Sanharaw M., Behar-Cohen F., Veuthey J.L., Gurny R. In vivo distribution and ex vivo permeation of cyclosporine A prodrug aqueous formulations for ocular application. J Control Release. 2013;170:153–159. doi: 10.1016/j.jconrel.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 166.Li N.N., Lin J., Gao D., Zhang L.M. A macromolecular prodrug strategy for combinatorial drug delivery. J Colloid Interface Sci. 2014;417:301–309. doi: 10.1016/j.jcis.2013.11.061. [DOI] [PubMed] [Google Scholar]
- 167.Falcoz C., Jenkins J.M., Bye C., Hardman T.C., Kenney K.B., Studenberg S. Pharmacokinetics of GW433908, a prodrug of amprenavir, in healthy male volunteers. J Clin Pharmacol. 2002;42:887–898. doi: 10.1177/009127002401102803. [DOI] [PubMed] [Google Scholar]
- 168.Vierling P., Greiner J. Prodrugs of HIV protease inhibitors. Curr Pharm Des. 2003;9:1755–1770. doi: 10.2174/1381612033454441. [DOI] [PubMed] [Google Scholar]
- 169.Bimbo L.M., Sarparanta M., Santos H.A., Airaksinen A.J., Mäkilä E., Laaksonen T. Biocompatibility of thermally hydrocarbonized porous silicon nanoparticles and their biodistribution in rats. ACS Nano. 2010;4:3023–3032. doi: 10.1021/nn901657w. [DOI] [PubMed] [Google Scholar]
- 170.Wang F., Hui H., Barnes T.J., Barnett C., Prestidge C.A. Oxidized mesoporous silicon microparticles for improved oral delivery of poorly soluble drugs. Mol Pharmacol. 2010;7:227–236. doi: 10.1021/mp900221e. [DOI] [PubMed] [Google Scholar]
- 171.Kapoor S., Hegde R., Bhattacharyya A.J. Influence of surface chemistry of mesoporous alumina with wide pore distribution on controlled drug release. J Control Release. 2009;140:34–39. doi: 10.1016/j.jconrel.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 172.Zhang Y., Zhang J., Jiang T., Wang S. Inclusion of the poorly water-soluble drug simvastatin in mesocellular foam nanoparticles: drug loading and release properties. Int J Pharm. 2011;410:118–124. doi: 10.1016/j.ijpharm.2010.07.040. [DOI] [PubMed] [Google Scholar]
- 173.Cao X., Deng W., Fu M., Zhu Y., Liu H., Wang L. Seventy-two-hour release formulation of the poorly soluble drug silybin based on porous silica nanoparticles: in vitro release kinetics and in vitro/in vivo correlations in beagle dogs. Eur J Pharm Sci. 2013;48:64–71. doi: 10.1016/j.ejps.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 174.Chen Z., Li X., He H., Ren Z., Liu Y., Wang J. Mesoporous silica nanoparticles with manipulated microstructures for drug delivery. Colloids Surf B Biointerfaces. 2012;95:274–278. doi: 10.1016/j.colsurfb.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 175.Korotcenkov G., Cho B.K. Silicon porosification: state of the art. Crit Rev Solid State Mater Sci. 2010;35:153–160. [Google Scholar]
- 176.Xu W., Riikonen J., Lehto V.P. Mesoporous systems for poorly soluble drugs. Int J Pharm. 2013;453:181–197. doi: 10.1016/j.ijpharm.2012.09.008. [DOI] [PubMed] [Google Scholar]