

Abstract
O-linked β-N-acetylglucosamine transferase (OGT) is an essential human enzyme that glycosylates numerous nuclear and cytoplasmic proteins on serine and threonine. It also cleaves Host cell factor 1 (HCF-1) by a mechanism in which the first step involves glycosylation on glutamate. Replacing glutamate with aspartate in an HCF-1 proteolytic repeat was shown to prevent peptide backbone cleavage, but whether aspartate glycosylation occurred was not examined. We report here that OGT glycosylates aspartate much faster than it glycosylates glutamate in an otherwise identical model peptide substrate; moreover, once formed, the glycosyl aspartate reacts further to form a succinimide intermediate that hydrolyzes to produce the corresponding isoaspartyl peptide. Aspartate to isoaspartate isomerization in proteins occurs in cells, but was previously thought to be exclusively non-enzymatic. Our findings suggest it may also be enzyme-catalyzed. In addition to OGT, enzymes that may catalyze aspartate to isoaspartate isomerization include PARPs, enzymes known to ribosylate aspartate residues in the process of poly(ADP-ribosyl)ation.
TOC image
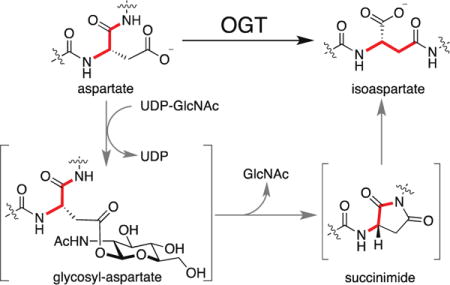
OGT is an essential mammalian enzyme known to catalyze two distinct post-translational modifications (PTMs) using the same active site.1 One is the transfer of N-acetylglucosamine to Ser/Thr residues of nuclear and cytoplasmic proteins,2 and the other is cleavage of Host cell factor 1 (HCF-1),3 a transcriptional co-regulator found in several chromatin-associated complexes.4 Ser/Thr O-GlcNAcylation affects localization, stability, and enzymatic activity of a wide variety of proteins and may serve to link cell state to glucose levels;2 HCF-1 cleavage is required for cell cycle progression.3,5 HCF-1 cleavage occurs via glycosylation on glutamate followed by on-enzyme formation of an internal pyroglutamate that undergoes spontaneous hydrolysis.6 Glycosylation is a common feature of both OGT-catalyzed PTMs. Nevertheless, the discovery that OGT can glycosylate glutamate was surprising because there are major steric and electronic differences between Ser/Thr and Glu side chains. Given OGT’s capacity to glycosylate glutamate, we wondered whether it could also glycosylate aspartate. Here we report that OGT indeed catalyzes aspartate glycosylation, and we show that this modification leads to isomerization of the aspartyl residue to isoaspartate (isoAsp). Isoaspartate formation is a physiologically relevant phenomenon thought to occur spontaneously as proteins age;7 this is the first report of enzyme-catalyzed isoaspartate formation. Our findings raise the possibility that some Asp to isoAsp isomerization events in cells result from enzymatic modification of aspartate side chains.8
We assessed glycosylation on aspartate using a variant of a peptide previously employed to investigate the mechanism of HCF-1 cleavage by OGT.6 The cleavage peptide, E10-32 (Figure 1a), contains a threonine-rich region that binds to the tetratricopeptide repeat domain of OGT and a cleavage region consisting of ‘CET’, with the site of cleavage being the amide bond between Cys9 and Glu10.9 While the residues adjacent to the glutamate are not required for cleavage, the glutamate itself is indispensable.3,6,9–10 We replaced glutamate with aspartate (D10-32) and incubated this peptide with UDP-GlcNAc and OGT for varying amounts of time before analyzing the reaction mixture by high-resolution LC-MS. A glycopeptide product [M+GlcNAc, 892.3978 (z = 4+)] formed within a minute of initiating the reaction and continued to accumulate for approximately fifteen minutes before decreasing. When aspartate was replaced with alanine (A10-32), we did not observe this glycopeptide (Figure 1b, S1). We therefore concluded that glycosylation had occurred on the aspartate side chain.
Figure 1.
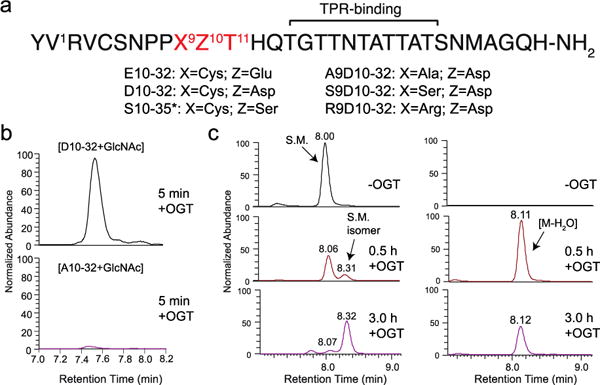
An aspartate-containing substrate is modified by OGT. (a) Sequences of peptides used in this study. The ‘*’ denotes that S10-35 bears 3 N-terminal lysine residues not found in the other peptides. (b) EIC traces for [M+GlcNAc] for both D10-32 (top) and A10-32 (bottom) after 5 min incubation with OGT. (c) EIC traces of 841.6279 [M] (left panel) and 837.1253 [M-H2O] (right panel) for D10-32 show conversion of starting material (S.M.) over time to a species isobaric with starting material (S.M. isomer) and to an anhydropeptide.
The decrease in glycopeptide abundance over time suggested that the glycopeptide had converted to other species. Further analysis of the reaction mixtures revealed the appearance of an anhydropeptide [M-H2O, 837.1253 (z = 4+)] as well as a peptide with the same exact mass (isobaric) as the starting material but with a different retention time by LC (henceforth S.M. isomer)(Figure 1c, S2). Formation of both species was found to depend on the presence of both OGT and UDP-GlcNAc (Figure S3). A time course showed that glycopeptide appeared before anhydropeptide, which in turn appeared before the starting material isomer (Figure S4). From these observations we concluded that the anhydropeptide had formed from the glycosyl aspartate.
We considered three pathways by which the glycosyl aspartate could progress to an anhydropeptide (Figure S5), and the two deemed most likely are shown in Figure 2a. To test whether the anhydropeptide was a cyclic thioester (pathway “a”), we replaced Cys9 with alanine (A9D10-32), serine (S9D10-32) or arginine (R9D10-32). These substitutions were previously shown to be compatible with glutamate glycosylation and cleavage.6,9 For each peptide we observed both an anhydropeptide and a peak isobaric with the corresponding starting material (R9D10-32, Figure 2b; A9D10-32 and S9D10-32, Figure S6). Therefore, the anhydropeptide observed in reactions with D10-32 was not a cyclic thioester. The other pathway (Figure 2a, pathway “b”) leads to a succinimide. Succinimides are known to form spontaneously from some aspartic acid-containing peptides, but are hydrolytically unstable under physiological conditions (t1/2 = ~138 min at pH 7.4 and 37 °C).11 Hydrolysis can occur at either of the succinimide carbonyl groups to regenerate aspartic acid or produce the corresponding isoaspartate-containing peptide (Figure 3a). The regiochemistry of attack favors the formation of isoaspartate.12 As isoaspartate and aspartate are isomeric, hydrolysis of an intermediate succinimide would explain the isobaric peaks observed in reactions of the D10 peptides with OGT.
Figure 2.
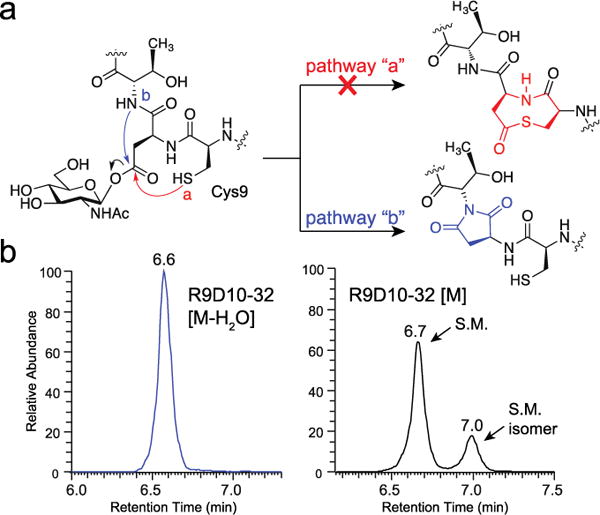
The anhydropeptide formed from glycosyl aspartate breakdown is a succinimide. (a) Two plausible pathways for the breakdown of a glycosyl aspartate intermediate. (b) EIC traces for the anhydropeptide (left), [M-H2O] = 850.3983 (4+), and starting material (right), [M] = 854.9009 (4+), for the R9D10-32 peptide after treatment with OGT for 60 min.
Figure 3.
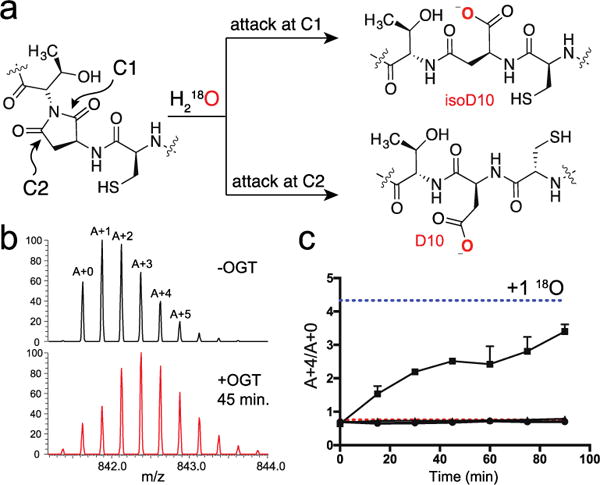
18O is incorporated into D10-32 by OGT. (a) Succinimide hydrolysis can occur at either C1 or C2 to produce isoaspartate and aspartate, respectively. (b) Isotope envelope (z = 4+) for D10-32 prior to (top) and 45 minutes after treatment with OGT and 18O-water (bottom). At 45 minutes, two peaks were present in the EIC; the isotope envelope shown is averaged over both peaks. Isotopomers are labeled A+0 through A+5; vertical axes are normalized. (c) A plot of A+4/A+0 over time for D10-32 treated with wild-type OGT and 18O water (■), with wild-type OGT and 16O water (●), and with OGT K842M and 18O water (▲). The red and blue dotted lines denote the expected ratio for incorporation of zero and one 18O, respectively. All points are mean ± s.e.m. (n=2).
Incorporation of 18O is commonly employed to demonstrate the presence of a succinimide within a polypeptide.13 We used an isotope model to determine the relative peak intensities expected for zero, one or two molar equivalents of 18O being incorporated into the D10-32 peptide (Figure S7); we then conducted a time course experiment using 18O-water. A time-dependent shift in the charge envelope occurred when we treated D10-32 with UDP-GlcNAc and wild-type OGT (Figure 3b, 3c), but not when we added UDP-GlcNAc and a catalytically inactive OGT mutant, K842M (Figure 3c).14 We also did not observe a shift when the reaction was conducted in 16O-water (Figure 3c) or when the corresponding glutamate-containing peptide, E10-32, was treated with OGT (Figure S8). We quantified the shift in the charge envelope for D10-32 by measuring the ratio of each isotopomer to the monoisotopic peak at each time point and found that isotope ratios were most consistent with the incorporation of a single 18O atom into the peptide (Figure 3c, S9).
We mapped the site of 18O incorporation using MS-MS (Figure S10, Tables S1–4). Compared to fragment ions for reactions carried out in 16O water, no shift was observed for b ions up to b9 or y ions up to y11, which was as far as we could detect; however, a systematic shift of +2 Da was observed for each ion from b12 through b20. These data showed that a single 18O atom had been incorporated within the tripeptide sequence spanning residues 9 to 11 (Figure S10). Taken together with our other results, we concluded that 18O had been incorporated via hydrolysis of a succinimide produced from a glycosyl aspartate. We measured a first order rate constant for succinimide hydrolysis, obtained from the rate of 18O incorporation, of 0.0042 min−1 (t1/2 = ~163 min) (Figure S11), which is comparable to rates previously measured for the spontaneous hydrolysis of succinimides in solution under similar conditions.11
To confirm that the isobaric peak observed in our reactions was an isoaspartyl peptide, as would be expected from hydrolysis of a succinimide intermediate, we purchased an authentic peptide standard, isoD10-32. This peptide had the same retention time as the isobaric peptide peak observed in the OGT reactions with D10-32 (Figure S12). Isoaspartate formation is a physiologically relevant phenomenon and cells contain an enzyme, protein isoaspartyl methyl transferase (PIMT), that repairs isomerized proteins by selectively transferring a methyl group from SAM to the carboxylic acid of isoaspartate. Methylation promotes succinimide formation (Figure 4a), and repeated cycles of methylation and succinimide hydrolysis eventually convert isoaspartate back to aspartate even though hydrolysis favors the former.15
Figure 4.
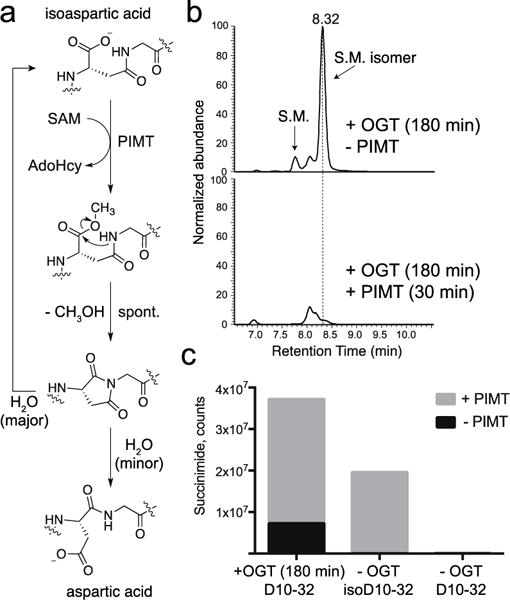
OGT converts aspartic acid to isoaspartate. (a) PIMT catalyzes isoaspartate methylation, producing a succinimide that hydrolyzes to a mixture of aspartate and isoaspartate. (b) EIC traces for 841.6279 (4+), corresponding to D10-32 starting material and isomer. Traces are after treatment with OGT alone (top) or with OGT followed by PIMT (bottom). (c) Normalized succinimide abundance for OGT and PIMT treated D10-32 (left), pure isoD10-32 (middle) or pure D10-32 (right); abundances were measured by integrating the respective EIC traces for succinimide, 837.1253 (4+).
We treated the authentic peptides D10-32 and isoD10-32 with PIMT and SAM, and only isoD10-32 reacted (Figure S13). We next treated a sample of D10-32, which had previously been incubated with OGT and UDP-GlcNAc to produce the putative isoaspartyl peptide, with PIMT/SAM; the peptide was consumed rapidly (Figure 4b) concomitant with a substantial increase in succinimide abundance (Figure 4c). Therefore, we concluded that OGT catalyzes isomerization of D10-32 to isoD10-32.
We have shown that OGT can glycosylate aspartate in addition to Ser/Thr and glutamate, but whether OGT glycosylates any cellular proteins on aspartate is currently unknown. To assess whether aspartate glycosylation is efficient compared with glycosylation on serine or glutamate, we measured the rate of starting material disappearance for three nearly identical peptide substrates with different glycosyl acceptor residues (S10-35, D10-32, and E10-32). The serine peptide reacted approximately 5-fold faster than the aspartyl peptide and at least 100-fold faster than the glutamyl peptide (Figure S14). Based on structural data supported by measurements of reaction rate constants for an OGT mutant, we have previously proposed that glycosylation on glutamate is disfavored by a repulsive interaction with aspartate D554 in OGT.6 The nucleophile in D10-32 would not approach the D554 carboxylate as closely, which may explain its faster rate of glycosylation compared with E10. Given that glycosylation on aspartate is relatively fast for a model peptide substrate, the possibility exists that OGT may glycosylate some proteins on aspartate.
Until now, isoaspartate formation was known only as a non-enzymatic process associated with aging proteins.7,11a,12,16 Our findings show that OGT can catalyze aspartate to isoaspartate isomerization in a model substrate, and one might expect that other enzymes that glycosylate aspartate will also promote this process as long as the local sequence permits succinimide formation. While we do not yet know if OGT modifies aspartate in any cellular proteins, we note that PARPs naturally glycosylate aspartic acid residues;8,17 this activity may, therefore, lead to succinimide intermediates in some cases. If so, it is worth considering whether isoaspartate formation is always an unwanted side reaction or may serve other functions.18
Supplementary Material
Acknowledgments
J.J. is a National Science and Engineering Research Council (NSERC) of Canada PGS-M and PGS-D3 fellowship recipient. This work was supported by NIH grant R01 GM094263. We thank Sunia A. Trauger (Harvard) for assistance with mass spectrometry.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Notes
The authors declare no competing interest.
References
- 1.(a) Janetzko J, Walker S. J Biol Chem. 2014;289:34424. doi: 10.1074/jbc.R114.604405. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Levine ZG, Walker S. Annu Rev Biochem. 2016;85:631. doi: 10.1146/annurev-biochem-060713-035344. [DOI] [PubMed] [Google Scholar]
- 2.(a) Bond MR, Hanover JA. J Cell Biol. 2015;208:869. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hardiville S, Hart GW. Cell Metab. 2014;20:208. doi: 10.1016/j.cmet.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, Herr W. Cell. 2011;144:376. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 4.(a) Parker JB, Yin H, Vinckevicius A, Chakravarti D. Cell Rep. 2014;9:967. doi: 10.1016/j.celrep.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tyagi S, Chabes AL, Wysocka J, Herr W. Mol Cell. 2007;27:107. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 5.(a) Julien E, Herr W. EMBO J. 2003;22:2360. doi: 10.1093/emboj/cdg242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mangone M, Myers MP, Herr W. PloS one. 2010;5:e9020. doi: 10.1371/journal.pone.0009020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janetzko J, Trauger SA, Lazarus MB, Walker S. Nat Chem Biol. 2016;12:899. doi: 10.1038/nchembio.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin Z, Dimitrijevic A, Aswad DW. Neurobiol Aging. 2015;36:1029. doi: 10.1016/j.neurobiolaging.2014.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hassa PO, Haenni SS, Elser M, Hottiger MO. Microbiol Mol Biol Rev. 2006;70:789. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S. Science. 2013;342:1235. doi: 10.1126/science.1243990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kapuria V, Rohrig UF, Bhuiyan T, Borodkin VS, van Aalten DM, Zoete V, Herr W. Genes Dev. 2016;30:960. doi: 10.1101/gad.275925.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Geiger T, Clarke S. J Biol Chem. 1987;262:785. [PubMed] [Google Scholar]; (b) McFadden PN, Clarke S. Proc Natl Acad Sci USA. 1987;84:2595. doi: 10.1073/pnas.84.9.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guttler BH, Cynis H, Seifert F, Ludwig HH, Porzel A, Schilling S. Amino Acids. 2013;44:1205. doi: 10.1007/s00726-012-1454-0. [DOI] [PubMed] [Google Scholar]
- 13.(a) Grossenbacher H, Marki W, Coulot M, Muller D, Richter WJ. Rapid Commun Mass Spectrom. 1993;7:1082. doi: 10.1002/rcm.1290071205. [DOI] [PubMed] [Google Scholar]; (b) Terashima I, Koga A, Nagai H. Anal Biochem. 2007;368:49. doi: 10.1016/j.ab.2007.05.012. [DOI] [PubMed] [Google Scholar]; (c) Liu M, Cheetham J, Cauchon N, Ostovic J, Ni W, Ren D, Zhou ZS. Anal Chem. 2012;84:1056. doi: 10.1021/ac202652z. [DOI] [PubMed] [Google Scholar]
- 14.Schimpl M, Zheng X, Borodkin VS, Blair DE, Ferenbach AT, Schuttelkopf AW, Navratilova I, Aristotelous T, Albarbarawi O, Robinson DA, Macnaughtan MA, van Aalten DM. Nat Chem Biol. 2012;8:969. doi: 10.1038/nchembio.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Griffith SC, Sawaya MR, Boutz DR, Thapar N, Katz JE, Clarke S, Yeates TO. J Mol Biol. 2001;313:1103. doi: 10.1006/jmbi.2001.5095. [DOI] [PubMed] [Google Scholar]; (b) Furuchi T, Sakurako K, Katane M, Sekine M, Homma H. Chem Biodivers. 2010;7:1337. doi: 10.1002/cbdv.200900273. [DOI] [PubMed] [Google Scholar]
- 16.Stephenson RC, Clarke S. J Biol Chem. 1989;264:6164. [PubMed] [Google Scholar]
- 17.(a) Zhang Y, Wang J, Ding M, Yu Y. Nat Methods. 2013;10:981. doi: 10.1038/nmeth.2603. [DOI] [PubMed] [Google Scholar]; (b) Daniels CM, Ong SE, Leung AK. J Proteome Res. 2014;13:3510. doi: 10.1021/pr401032q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P. Nat Commun. 2014;5:4426. doi: 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Martello R, Leutert M, Jungmichel S, Bilan V, Larsen SC, Young C, Hottiger MO, Nielsen ML. Nat Commun. 2016;7:12917. doi: 10.1038/ncomms12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Corti A, Curnis F. J Cell Sci. 2011;124:515. doi: 10.1242/jcs.077172. [DOI] [PubMed] [Google Scholar]; (b) van Loo B, Permentier HP, Kingma J, Baldascini H, Janssen DB. FEBS Lett. 2008;582:1581. doi: 10.1016/j.febslet.2008.04.001. [DOI] [PubMed] [Google Scholar]; (c) Biterge B, Richter F, Mittler G, Schneider R. Sci Rep. 2014;4:6674. doi: 10.1038/srep06674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.