

SUMMARY
PBRM1 is the second most commonly mutated gene after VHL in clear cell renal cell carcinoma (ccRCC). However, the biological consequences of PBRM1 mutations for kidney tumorigenesis are unknown. Here, we find that kidney-specific deletion of Vhl and Pbrm1, but not either gene alone, results in bilateral, multifocal, transplantable clear cell kidney cancers. PBRM1 loss amplified the transcriptional outputs of HIF1 and STAT3 incurred by Vhl deficiency. Analysis of mouse and human ccRCC revealed convergence on mTOR activation, representing the third driver event after genetic inactivation of VHL and PBRM1. Our study reports a physiological preclinical ccRCC mouse model that recapitulates somatic mutations in human ccRCC and provides mechanistic and therapeutic insights into PBRM1 mutated subtypes of human ccRCC.
eTOC Blurb
Nargund et al. present a three-step process in the pathogenesis of mouse and human clear cell kidney cancer. After the loss of VHL, the loss of SWI/SNF tumor suppressor protein PBRM1/BAF180 further activates HIF1/STAT3 signaling in mouse kidney and positions mTORC1 activation as the preferred third driver event.
INTRODUCTION
The estimated new kidney cancer cases diagnosed in the world and the United Sates every year are ~300,000 and ~63,000, respectively (Global Burden of Disease Cancer et al., 2015; Siegel et al., 2016). Clear cell renal cell carcinoma (ccRCC) is the most common subtype (75%) and is lethal when metastasized (Rini et al., 2009). The Von Hippel-Lindau (VHL) tumor suppressor gene is the most frequently mutated gene in ccRCC (Gnarra et al., 1994; Linehan et al., 1995), and its complete loss constitutes an early, truncal oncogenic driver event. VHL is the substrate recognition of an E3 ligase that labels Hypoxia-inducible factor (HIF) 1α and 2α with ubiquitin for degradation (Kaelin, 2007; Majmundar et al., 2010; Masson and Ratcliffe, 2014; Semenza, 2013). Thus, human ccRCC is highly vascular due to uncontrolled activation of HIFα targets that regulate angiogenesis. Thereby, anti-VEGF/VEGFR agents are effective, first-line treatment for metastatic ccRCC (mRCC) (Rini et al., 2009; Voss et al., 2013).
VHL inactivation was the only known oncogenic driver in ccRCC (Gnarra et al., 1994) until recent large-scale cancer genomic projects uncovered prevalent mutations including PBRM1/BAF180 (29–41%), SETD2 (8–12%), BAP1 (6–10%), and KDM5C (4–7%) (Cancer Genome Atlas Research, 2013; Hakimi et al., 2013a; Pena-Llopis et al., 2012; Sato et al., 2013; Varela et al., 2011). Remarkably, these genes encode chromatin and epigenetic regulatory proteins, and most mutations are predicted to result in functional loss, favoring their roles as tumor suppressors (Hakimi et al., 2013b). PBRM1, the second most commonly mutated gene in all stages of ccRCC (Hsieh et al., 2016), encodes BRG1-associated factor (BAF) 180, the defining subunit of the ~2 MDa Polybromo BAF (PBAF) SWI/SNF complex (Varela et al., 2011). The SWI/SNF chromatin remodeling complexes are macromolecular machineries, which utilizes ATP to mobilize nucleosome and thereby modulate chromatin structure (Biegel et al., 2014; Clapier and Cairns, 2009). They regulate critical cellular processes, including cell cycle, cell fate, cell death, metabolism and DNA repair (Hargreaves and Crabtree, 2011). Interestingly, pan-cancer genomics have uncovered epigenetic regulators including SWI/SNF proteins as a major class of cancer genes (Dawson and Kouzarides, 2012). Mutations of individual SWI/SNF subunits have been detected in ~20% of human cancers and they displayed preferential enrichment of mutations among cancer types (Helming et al., 2014; Kadoch et al., 2013; Marquez et al., 2014). For example, PBRM1 is most highly mutated in ccRCC, SMARCB1 (BAF47) in pediatric rhabdoid tumors, and ARID1A (BAF250A) in ovarian clear-cell carcinoma (Biegel et al., 2014), implicating underlying tissue tropism for disarming specific tumor suppressor gene (TSG) during tumorigenesis (Wei and Hsieh, 2015).
The knowledge of PBRM1 in mammalian biology is limited. Mice with straight knockout of Pbrm1 resulted in embryonic lethality at E11.5 due to heart defects (Huang et al., 2008; Wang et al., 2004), whereas mice with T lymphocyte specific knockout of Pbrm1 exhibited normal thymus and peripheral T cell development (Wurster et al., 2012). In vitro studies demonstrated that PBRM1 activated p21 upon irradiation in breast cancer cell lines (Xia et al., 2008) and participated in p53-induced replicative senescence in fibroblasts (Burrows et al., 2010), and PBRM1 knockdown enhanced proliferation and migration of kidney cancer cell lines (Varela et al., 2011). However, the in vivo tumor suppressor function of PBRM1 has not been established and how PBRM1 loss-of-function promotes tumorigenesis remains unclear.
The long latency (>30 years) for VHL germline mutated patients to develop ccRCC (Fisher et al., 2014; Kaelin, 2007) and the inability of Vhl deficiency to induce ccRCC in mice (Kapitsinou and Haase, 2008) suggest that additional genetic/epigenetic events are probably needed for the development of ccRCC (Wei and Hsieh, 2015). PBRM1 loss-of-function is one of the most likely candidates given its high mutation frequency (~40%) in human ccRCC (Hsieh et al., 2016). Here, we created kidney-specific deletion of Pbrm1 and/or Vhl mice to study the tumor suppressor role of PBRM1 and sought to establish a physiological mouse kidney cancer model that recapitulates human ccRCC.
RESULTS
Genetic Deletion of Pbrm1 in Mouse Kidney Results in Hydronephrosis
As Pbrm1 (BAF180) deletion in mice incurred embryonic lethality (Wang et al., 2004), we deleted the conditional Pbrm1F allele (Wurster et al., 2012) in the mouse kidney using a transgenic Cre recombinase line Ksp-Cre (Shao et al., 2002) that has been widely utilized to model kidney cancer in mice (Adam et al., 2011; Baba et al., 2008; Chen et al., 2008; Igarashi, 2004). The expression of Cre from the Ksp-Cre is driven by the kidney specific Cadherin 16 promoter, which begins expression at embryonic day 14.5 in epithelial cells of the developing kidney and genitourinary tract, and continues to be expressed in tubular epithelial cells in adults. Pbrm1F/FKsp-Cre mice were born at expected Mendelian ratio. To monitor if Pbrm1 loss results in any gross kidney abnormality, serial abdominal magnetic resonance imaging (MRI) was performed on a large cohort of mice. Obstructive hydronephrosis, enlarged kidneys containing fluid-filled renal pelvis, and non-neoplastic masses at the ureteropelvic junction or proximal ureter were detected in some Pbrm1F/FKsp-Cre mice (Figures 1A and S1A). Among 53 Pbrm1F/FKsp-Cre and 27 Pbrm1+/+Ksp-Cre (denoted as WT thereafter) mice examined, 18 (34%) Pbrm1F/FKsp-Cre while 0 WT mice developed hydronephrosis (Figure 1B). Observed hydronephrosis exhibited preponderance of female over male and left over right (Figures 1C and 1D), and could be detected by MRI as early as 6 months of age (Figure S1B). At necropsy, the volume of hydronephrotic Pbrm1F/FKsp-Cre kidneys was at 2–5 times of normal appearing kidneys (Figure 1E), whereas the creatinine of aged Pbrm1F/FKsp-Cre and WT mice was comparable (Figure 1F).
Figure 1. Pbrm1F/FKsp-Cre Mice Develop Obstructive Hydronephrosis.

(A) Representative MRI images of unilateral or bilateral severe hydronephrosis. The non-neoplastic mass at the proximal ureter is marked by arrow. (B) Incidence of hydronephrosis in WT and Pbrm1F/FKsp-Cre mice. Cohorts of animals at 12 months of age on average were randomly selected for MRI scanning. ***, P < 0.001 (Fischer’s Exact). (C) Incidence of hydronephrosis in the Pbrm1F/FKsp-Cre mice based on gender. (D) Location distribution of hydronephrosis in Pbrm1F/FKsp-Cre mouse kidneys. (E) Kidney volume in WT and Pbrm1F/FKsp-Cre mice. *, P < 0.05 (Mann Whitney). (F) Serum creatinine levels of mice in (E). ns denotes not statistically significant.
Genetic Deletion of Vhl and Pbrm1 in Mouse Kidney Results in Polycystic Kidney Disease and Increased Mortality
As neither Vhl nor Pbrm1 deletion alone caused kidney tumors, VhlF/FPbrm1F/FKsp-Cre mice were generated to investigate the genetic interaction between Vhl and Pbrm1 deficiency in kidney cancer pathogenesis. The survival of 325 mice (36 WT, 30 VhlF/FKsp-Cre, 129 Pbrm1F/FKsp-Cre, and 130 VhlF/FPbrm1F/FKsp-Cre) was monitored, which revealed a markedly increased mortality in VhlF/FPbrm1F/FKsp-Cre mice and a moderately increased mortality in VhlF/FKsp-Cre mice (Figure 2A). Remarkably, abdominal MRI detected diffuse polycystic kidney disease (PKD) in 30% (17/56) of 6–9 months old and in 67% (14/21) of 10–14 months old VhlF/FPbrm1F/FKsp-Cre mice, whereas only 1 of 14 aged VhlF/FKsp-Cre mice (12–16 months old) developed PKD with a few scattered cysts (Figures 2B, 2C, and S2). To investigate cystic changes, we performed histological analysis on the kidneys of VhlF/FPbrm1F/FKsp-Cre mice at different ages (3–13 months). Both tubular and glomerular cysts were present in young mice (Figure S3). Of note, scattered cystic anomalies of kidneys have been described in hereditary VHL patients and are implicated as preneoplastic lesions (Mandriota et al., 2002; Neumann and Zbar, 1997; Walther et al., 1995). Consistent with a prior report, mild hydronephrosis was also observed in VhlF/FKsp-Cre mice (Figure 2E) (Frew et al., 2008). Elevated serum creatinine was observed in the majority of aged VhlF/FPbrm1F/FKsp-Cre mice (Figure 2E), which could be accountable for their early demise.
Figure 2. VhlF/FPbrm1F/FKsp-Cre Mice Develop Polycystic Kidney Disease and exhibit Premature Mortality.

(A) Kaplan-Meier survival curve of WT, VhlF/FKsp-Cre, Pbrm1F/FKsp-Cre, and VhlF/FPbrm1F/FKsp-Cre mice. (B) Incidence of polycystic kidney disease in WT, VhlF/FKsp-Cre, Pbrm1F/FKsp-Cre, and VhlF/FPbrm1F/FKsp-Cre. Age and number of animals in each group are specified. (C) Representative MRI and gross images of kidneys of the indicated genotypes. (D) Kidney volumes of WT, VhlF/FKsp-Cre, and VhlF/FPbrm1F/FKsp-Cre mice. Numbers of kidneys measured in each group (n) are indicated. **, P =0.0096; ***, P < 0.0001 (Mann Whitney). (E) Serum creatinine levels in WT, VhlF/FKsp-Cre, and VhlF/FPbrm1F/FKsp-Cre mice (the same as 1D). ***, P < 0.001 (Mann Whitney).
VhlF/FPbrm1F/FKsp-Cre Mice Develop Multifocal, Clear Cell Kidney Cancer
Serial MRI examination of kidney in VhlF/FPbrm1F/FKsp-Cre mice recognized patterns of imaging changes from normal through progressive cystic abnormality to increased multifocal nodularity with decreased cystic appearance (Figures S2 and S3). Gross examination of 58 kidneys (29 mice, 8–17 months old) at necropsy showed diffusely cystic changes in 58.33% (21/36) of VhlF/FPbrm1F/FKsp-Cre kidneys, and none in VhlF/FKsp-Cre (n = 8), Pbrm1F/FKsp-Cre (n = 10), and WT (n = 4) kidneys (Table S1). Histologic examination revealed sheets of tumor cells interspersed within a highly vascularized stroma in 33.33% (12/36) VhlF/FPbrm1F/FKsp-Cre kidneys, whereas no tumors were noted in 22 kidneys of the other genotypes (Table S1). Notably, all the tumors were observed in VhlF/FPbrm1F/FKsp-Cre mice after 10 months of age with a 50% tumor incidence (12/24). The tumor cells displayed central features of human ccRCC, including clear cytoplasm and positive membranous staining of Carbonic Anhydrase IX (CA-IX), a target of HIF1 (Mandriota et al., 2002; Semenza, 2013) (Figures 3A). Consistent with the known aberrant HIF1 activation in the absence of VHL, weak, sporadic CA-IX staining was detected in VhlF/FKsp-Cre, but not in Pbrm1F/FKsp-Cre or WT kidneys (Figure 3A). To compare these mouse tumors with human RCCs, we performed gene expression profile analysis of these mouse tumors in comparison to human TCGA clear cell RCC (KIRC) and chromophobe RCC (KICH) kidney cancers, and demonstrated that VhlF/FPbrm1F/FKsp-Cre tumors resemble KIRC but not KICH (Figure 3B). Data suggests that human clear cell RCC arises from proximal tubule (Chen et al., 2016). Accordingly, we investigated the cell type origin of VhlF/FPbrm1F/FKsp-Cre mouse tumors. Staining for lotus tetragonolobus lectin (LTL) that marks proximal convoluted tubule and for Tamm-Horsfall protein (THP) that marks distal convoluted tubule was performed. Consistent with human ccRCC originating from proximal tubule, our VhlF/FPbrm1F/FKsp-Cre mouse tumors were stained positive for LTL but not THP (Figure 3C). CD45 staining did not detect increased lymphocyte infiltrate of these VhlF/FPbrm1F/FKsp-Cre tumors (Figure 3C). Consistent with human ccRCC reports, these tumors were positive for CD31 staining that marks endothelial cells (Figure 3C). We also performed Oil Red O staining of the fresh frozen section of these mouse tumors to evaluate lipid content in our the clear cell mouse tumors (Figure S4). Furthermore, the presence of high glycogen in these tumors was confirmed by PAS-D staining (Figure S4). Of note, our VhlF/FPbrm1F/FKsp-Cre mouse tumors did not directly originate from cystic lesions (Figure S5A). Higher proliferation index (Ki-67 staining) was observed in these Vhl and Pbrm1 doubly deficient clear cell kidney tumors whereas no alteration in cell death was detected by immunohistochemistry for cleaved caspase-3 and TUNEL assays (Figures S5B, S5C). Quantitative PCR with reverse transcription (qRT-PCR) demonstrated greatly reduced Vhl and Pbrm1 expression in VhlF/FPbrm1F/FKsp-Cre kidney tumors (Figure S5D). Of note, we did not detect local tumor invasion into adjacent tissues or distant metastasis to lungs, livers, bones, and lymph nodes in the examined tumor-bearing VhlF/FPbrm1F/FKsp-Cre mice.
Figure 3. VhlF/FPbrm1F/FKsp-Cre Mice Develop Multifocal CA-IX Positive Clear Cell Kidney Cancers.

A) Representative gross images (column1), histopathological images (column 2), and immunohistochemistry of CA-IX (columns 3 and 4) of WT, VhlF/FKsp-Cre, Pbrm1F/FKsp-Cre, and VhlF/FPbrm1F/FKsp-Cre kidneys. Tumor and cyst are indicated by white and black arrows, respectively. T and N denote Tumor and adjacent Normal, respectively. Scale bars are at 50μm, 100μm, or 200μm as indicated. (B) Heatmap of inter-sample correlations (red, positive) between mRNA profiles of TCGA human RCC tumors (columns, TCGA KIRC and KICH data) and VhlF/FPbrm1F/FKsp-Cre mouse kidney tumors (rows). (C) Representative images of immunofluorescence of LTL (column 1), and immunohistochemistry of THP (column 2), CD45 (column 3) and CD31 (column 4) in VhlF/FPbrm1F/FKsp-Cre tumors and adjacent non-tumor tissues.
Vhl and Pbrm1 Doubly Deficient Clear Cell Kidney Tumors Are Transplantable
To assess the tumor initiating capacity of the de novo Vhl and Pbrm1 doubly deficient kidney tumors, we transplanted 12 tumor fragments from two VhlF/FPbrm1F/FKsp-Cre kidneys into the sub-renal capsules of 12 kidneys of 6 immunocompromised NOD/SCID/IL2Rγnull (NSG) mice. All the recipient kidneys, except for the one animal that died prematurely of known cause, showed large visible kidney tumors upon dissection at 10–12 months after transplantation (Figure 4A). In 2 of the transplanted cases, we also observed tumor invasion into organs such as liver (Figure 4B). Furthermore, tumors that developed in the transplanted NSG mouse could be further successfully propagated into a NSG mouse (Figure 4A). Together, these data demonstrated the malignant potential of VhlF/FPbrm1F/FKsp-Cre kidney tumors. Histology of these tumor allografts showed the same histological features, positive CA-IX staining as donor tumors, and genotypes (Figure 4). Importantly, this step-wise, morphological progression observed in VhlF/FPbrm1F/FKsp-Cre kidneys from normal appearance through cystic changes (~6 months) to ccRCC formation (~10 months) offered an opportunity to temporally dissect the mechanisms by which PBRM1 loss cooperates with VHL loss to initiate the development of ccRCC.
Figure 4. VhlF/FPbrm1F/FKsp-Cre Mice Tumors Are Transplantable and Invasive.

(A) Representative gross images (column 1), histopathological images (column 2), and immunohistochemistry of CA-IX (column 3) of donor VhlF/FPbrm1F/FKsp-Cre kidney tumors (row1), primary allograft kidney tumors (row2), and secondary allograft kidney tumors (row3). (B) Representative histopathological image (top panel), and immunohistochemistry of CA-IX (bottom panel) of the transplanted invasive tumors. (C) PCR genotyping of WT kidney, and donor and allograft VhlF/FPbrm1F/FKsp-Cre tumors.
Gene Expression Profiling of WT, Vhl-deficient, Pbrm1-deficient, or Vhl and Pbrm1 Doubly Deficient Mouse Renal Cortices Identifies Distinct Clusters
To determine why double deficiency of Vhl and Pbrm1 but not single deficiency of either gene resulted in ccRCC, we performed gene expression profiling of RNA isolated from renal cortices of 12-week-old WT, VhlF/FKsp-Cre, Pbrm1F/FKsp-Cre, and VhlF/FPbrm1F/FKsp-Cre mice. Kidneys from 12-week-old mice were chosen to avoid potentially confounding transcriptional changes secondary to cystic anomalies that normally manifest after 6 months of age. Of note, none of the 12-week-old mouse kidneys displayed discernible macroscopic or microscopic cystic abnormalities at necropsy regardless of genotypes. Genes differentially expressed in at least one genotype were identified (Table S2a) and subjected to unsupervised hierarchical clustering analysis, which revealed three distinct clusters (Figure 5A). Cluster I showed enrichment of genes that were upregulated in Vhl-deficient renal cortices and further upregulated in Vhl and Pbrm1 doubly deficient renal cortices. In contrast, Cluster III showed enrichment of genes that were downregulated in Vhl and Pbrm1 doubly deficient renal cortices. Each cluster was then analyzed and visualized using ClueGO (Bindea et al., 2009) to interrogate functionally grouped gene ontology and pathway networks (Figure 5B and Table S2b). Pathways over-represented that are highly pertinent to known ccRCC pathogenesis were HIF1 and JAK-STAT pathway genes in Cluster I and oxidative phosphorylation (OXPHOS) genes in Cluster III (Figure 5B), whereas no pathway was significantly enriched in Cluster II.
Figure 5. PBRM1 Loss Amplifies the Transcriptional Outputs of HIF1 and STAT3 Incurred by VHL Loss.

(A) Heat map of genes with significantly different expression in the renal cortices of WT, VhlF/FKsp-Cre, Pbrm1F/FKsp-Cre, and VhlF/FPbrm1F/FKsp-Cre mice at 12 weeks of age. Unsupervised hierarchical agglomerative clustering identified three distinct clusters using Pearson correlation and average linkage as similarity measures for pairs of genes and pairs of inchoate clusters, respectively. (B) Clusters I and III were tested for pathway enrichment and presented using ClueGO. (C) Inferred HIF and STAT motif activities across the indicated genotypes. *, P = 0.035; **, P = 0.0022 (one sided t-test). (D) The mRNA levels of the indicated genes from the indicated genotypes were assessed by qRT-PCR. Data were normalized against GAPDH (mean ± s.d., n=3 independent experiments). *, P < 0.05; **, P < 0.005 (Student’s t-test).
PBRM1 Loss Amplifies the Transcriptional Outputs of HIF1 and STAT3 Incurred by VHL Loss
HIF1α is stabilized upon VHL loss and plays a central role in the pathogenesis of ccRCC. It was reported that HIF1 induces PKM2 to activate STAT3, which in turn induces HIF1α expression (Demaria and Poli, 2012; Luo and Semenza, 2012). Furthermore, it is known that HIF1 and STAT3 cooperate to activate the expression of HIF1 targets including genes involved in angiogenesis (Jung et al., 2005). Consequently, the intricate interplay between HIF1 and STAT3 establishes a feed-forward amplification loop to maximize target gene expression. Our discovery that HIF1 and JAK-STAT pathway genes were enriched in Vhl-deficient renal cortices and further enriched in Vhl and Pbrm1 doubly deficient renal cortices raises a hypothesis in which PBRM1 prevents the amplification of the HIF1 and STAT3 transcriptional outputs that are initiated upon VHL loss.
To test this, we identified HIF1 and STAT3 motifs from differentially expressed genes to determine the strength of regulation of the targets by individual transcription factor binding motifs (TFBMs). RNA expression data were analyzed using Integrated System for Motif Activity Response Analysis (ISMARA) (Balwierz et al., 2014), which produced an output denoting the inferred activity of HIF1 and STAT3 motifs in every sample. An increase in the HIF1 (P = 0.08) and STAT3 (P = 0.017) motif activities was detected when comparing VhlF/FKsp-Cre to WT kidneys, and a marked increase in both HIF1 (P = 0.00076) and STAT3 (P = 0.00018) outputs was identified when comparing VhlF/FPbrm1F/FKsp-Cre to WT kidneys. In contrast, no changes in HIF1 (P = 0.49) or STAT3 (P = 0.98) motif activity were observed when comparing Pbrm1F/FKsp-Cre to WT kidneys. Remarkably, the effects of Pbrm1 deletion on HIF1 and STAT3 targets manifested only under the premise of Vhl loss, resulting in a further increase of the HIF1 (P = 0.035) and STAT3 (P = 0.0022) motif activities when comparing VhlF/FPbrm1F/FKsp-Cre to VhlF/FKsp-Cre kidneys (Figure 5C). To further validate these findings, we performed qRT-PCR analyses on Hif1a, Stat3, and representative HIF1 target genes (Pdk1 and Egln3) as well as STAT3 target genes (Socs3, Il4r, and Il6r) (Figures 5D and S6A-S6D), which demonstrated consistent results among different gene expression assays. Of note, we did not see any transcriptional upregulation of HIF2α. In summary, our data suggest that PBRM1, a SWI/SNF complex protein, could function like a transcriptional resistor to prevent uncontrolled self-perpetuating amplification of the HIF1 and STAT3 transcriptional outputs incurred by Vhl deficiency. To further interrogate this working hypothesis, qRT-PCR was performed on NIH3T3 cells with knockout of Vhl, Pbrm1, or both using CRISPR-Cas9. Indeed, the loss of PBRM1 further enhances HIF1 and the STAT3 signaling that was primed upon the loss of VHL (Figure S6G).
The Expression of OXPHOS Genes Is Markedly Downregulated in Vhl and Pbrm1 Doubly Deficient Mouse Kidneys
In contrast to Cluster I which encompasses up-regulated genes in the VhlF/FPbrm1F/FKsp-Cre kidneys, Cluster III mainly consists of significantly down-regulated genes (Figure 5A) within which most enriched are OXPHOS genes (Figure 5B). The significant down-regulation of nuclear encoded OXPHOS genes was further confirmed by qRT-PCR of genes involved in different electron-transport chain complexes, including Ndufa2 (Complex I), Sdhd (Complex II), Cox5a (Complex IV), and Atp4a (Complex V) (Figures 5D, S6E, and S6F). HOMER analysis was performed to determine if specific transcription factors might have directly mediated such repression. However, we did not detect enrichment of any pertinent TFBMs within the Cluster III genes. It has been shown that HIF1 inhibits mitochondrial biogenesis and respiration through down-regulation of PGC1β transcription in RCC4, a VHL-deficient human kidney cancer cell line (Zhang et al., 2007). However, neither gene expression profiling nor qRT-PCR detected significant down-regulation of Pgc1β in VhlF/FPbrm1F/FKsp-Cre kidneys (Figure S6F). TCGA pan-kidney cancer analysis on common human kidney cancer types, including KIRC (Kidney Renal Clear), KIRP (Kidney Renal Papillary), and KICH (Kidney Chromophobe), demonstrated a significant down-regulation of OXPHOS genes in ccRCC, but not in papillary RCC (pRCC) or chromophobe RCC (chRCC) (Chen et al., 2016).
Vhl and Pbrm1 Doubly Deficient Clear Cell Kidney Tumors Display Hyperactive mTORC1 Signaling
The observation that clear cell kidney tumors occurred in VhlF/FPbrm1F/FKsp-Cre mice after a long latency period suggests the involvement of additional genetic and/or epigenetic events. To investigate whether transcriptional aberrations might be responsible, RNA-Seq was performed on VhlF/FPbrm1F/FKsp-Cre kidney tumors and age-matched WT renal cortices. The complex pathological changes observed in the aged VhlF/FPbrm1F/FKsp-Cre mouse kidneys precluded the isolation of adjacent normal renal cortices for comparison.
Differentially expressed genes were identified (Table S3) and subjected to unsupervised hierarchical clustering analysis, which identified two clusters denoting tumors and normal controls (Figure 6A). These genes were analyzed and visualized with ClueGO to interrogate functionally grouped gene ontology and pathway networks (Figure 6B and Table S4). Gene Set Enrichment Analysis (GSEA) revealed up-regulation of HIF1 and STAT3 pathways, and down-regulation of OXPHOS pathway in VhlF/FPbrm1F/FKsp-Cre tumors (Figure 6C), consistent with the findings observed in the 12-week-old VhlF/FPbrm1F/FKsp-Cre renal cortices (Figure 5B). Significantly, dysregulation of the mTOR signaling pathway was shown in the Vhl and Pbrm1 doubly deficient tumors but not in the 12-week-old VhlF/FPbrm1F/FKsp-Cre renal cortices (Figures 5B, 6B, and 6C, and Table S4). To validate RNA-sequencing findings, qRT-PCR focused on the HIF1 and mTOR pathway genes was performed (Figures 6E and 6F), which demonstrated consistent findings among gene expression analysis platforms. We also performed immunohistochemistry on VhlF/FPbrm1F/FKsp-Cre mouse tumors to assess HIF1 and mTOR signaling. A significant nuclear accumulation of HIF1, a weak nuclear accumulation of HIF2, and an expression of GLUT1, a HIF target, were detected in these tumors (Figure S7A). Furthermore, increased phosphorylation of 4E-BP1 and S6K, two well-established mTORC1 substrates, were detected in VhlF/FPbrm1F/FKsp-Cre tumors but not adjacent normal looking kidney tissues (Figures 6D and S7B). In contrast, the phosphorylation of ERK was not increased in the Vhl and Pbrm1 doubly deficient kidney tumors (Figure 6D).
Figure 6. Vhl and Pbrm1 Doubly Deficient Clear Cell Kidney Tumors Display Hyperactive mTORC1 Signaling.

(A) Heat map of genes with significantly different expression in age-matched WT kidneys (n=4) and VhlF/FPbrm1F/FKsp-Cre tumors (n=5) based on unsupervised hierarchical agglomerative clustering. (B) The genes that were significantly, differentially expressed in VhlF/FPbrm1F/FKsp-Cre T/N (FDR < 0.05) were tested for enrichment and represented using ClueGO. (C) GSEA plots of the ranked list of differentially expressed genes in VhlF/FPbrm1F/FKsp-Cre kidney tumors (T) and WT normal kidneys (N) generated using three gene sets: curated HIF targets, KEGG JAK STAT signaling pathway, KEGG oxidative phosphorylation pathway, and KEGG mTOR Pathway enrichment. (D) Immunohistochemistry of phosphorylated-4E-BP1 (p4E-BP1) at threonine 37/46 (column 1), phosphorylated S6K (pS6K) at serine 240/244 (column 2), and phosphorylated ERK1/2 (pERK) at threonine 202/tyrosine 204 in VhlF/FPbrm1F/FKsp-Cre tumors. T and N denote Tumor and adjacent Normal, respectively. Scale bars are at 100μm or 200μm as indicated. (E–F) The mRNA levels of Ddit4, Ldha, Hk2, Glut1, Tsc1 and Tsc2 in VhlF/FPbrm1F/FKsp-Cre tumors (n=6) and WT kidneys (n=4) were assessed by qRT-PCR. Data were normalized against GAPDH (mean ± s.d.). *, P < 0.05; **, P < 0.005; ***, P < 0.0005 (Student’s t-test).
It is noteworthy that Ddit4 or Redd1, a transcriptional target of HIF1 (Brugarolas et al., 2004), was significantly upregulated in Vhl and Pbrm1 doubly deficient kidney tumors (Figure 6E). REDD1 is known to suppress mTORC1 activity by releasing TSC2 from its inhibitor 14–3–3 (Brugarolas et al., 2004; DeYoung et al., 2008). Several lines of clinical evidence support the importance of mTORC1 activation in the pathobiology of human ccRCC, including the known therapeutic benefit of administering mTORC1 inhibitors in treating metastatic ccRCC (Voss et al., 2014; Wei and Hsieh, 2015) and the observed prevalent mTORC1 pathway activation in human ccRCC (Linehan et al., 2010; Robb et al., 2007). Induction of REDD1 by HIF1 may activate a tumor suppressor checkpoint that restrains the oncogenic potential of HIF1. Hence, activation of mTORC1 activity through additional genetic/epigenetic events may be required for the initiation of ccRCC in Vhl and Pbrm1 doubly deficient renal epithelial cells. In fact, significant down-regulation of Tsc1 and Tsc2 was demonstrated in Vhl and Pbrm1 doubly deficient kidney tumors (Figure 6F), which would activate mTORC1 even when REDD1 was upregulated. In summary, the emergent mTORC1 activation detected in the Vhl and Pbrm1 doubly deficient ccRCC may represent a prerequisite oncogenic driver event in the pathogenesis of ccRCC once kidney epithelial cells lost VHL and PBRM1.
Analyses of Mouse and Human ccRCC Reveal Convergence on the mTOR Pathway Activation
Contrary to the inability of Vhl deficiency to initiate ccRCC in mice, the Hif1α-M3 transgenic model (Hif1α-M3 TRACK) where kidney-specific overexpression of a non-degradable as well as transcriptionally active mutant HIF1α-M3 (P402A, F564A, N803A) resulted in renal cysts and small clear cell tumors in aged (14–22 months) mice (Fu et al., 2015; Fu et al., 2011). Of note, oxygen-dependent hydroxylation of P402 and F564 of HIF1α by prolyl hydroxylase domain enzymes (PHDs) and of N803 by Factor Inhibiting HIF (FIH) enhances the HIF-VHL interaction, leading to its degradation (Keith et al., 2012; Masson and Ratcliffe, 2014). Furthermore, hydroxylation of N803 by FIH also disrupts the interaction between HIF and transcription coactivator p300/CBP, thereby inhibiting HIF1-mediated transcription (Masson and Ratcliffe, 2014). Although VHL loss leads to the stabilization of HIF1α and HIF2α, oxygen-dependent and FIH1-mediated asparaginyl hydroxylation of HIFs prevents the recruitment of p300/CBP, which helps explain why Vhl deficiency alone is insufficient for kidney tumor initiation.
To further determine whether mTORC1 activation might be a mechanistically preferred node after the hyperactivation of HIF1 during the pathogenesis of ccRCC, we resorted to the transcriptomic data of the Hif1α-M3 TRACK mouse model. Indeed, GSEA revealed up-regulation of the mTOR signaling pathway (Figure 7A). As expected, up-regulation of HIF1 and JAK/STAT signaling pathways, and down-regulation of the OXPHOS pathway were also seen in the Hif1α-M3 TRACK mouse model (Figure 7A). We further compared the transcriptomics of the VhlF/FPbrm1F/FKsp-Cre tumors to those of the Hif1α-M3 TRACK mouse model (Figure 7B). Within the 2430 differentially expressed genes shared between these two models, enrichment in the mTOR and HIF1 pathways was evident (Figures 7B and 7C, and Table S5). The shared mTOR pathway aberration between these two different mouse ccRCC models supports the convergence on mTORC1 activation once HIF1 becomes hyperactive (Figures 7A, 7B, and 7C).
Figure 7. Analyses of Mouse and Human ccRCC Reveal Convergence on the mTOR Pathway Activation.

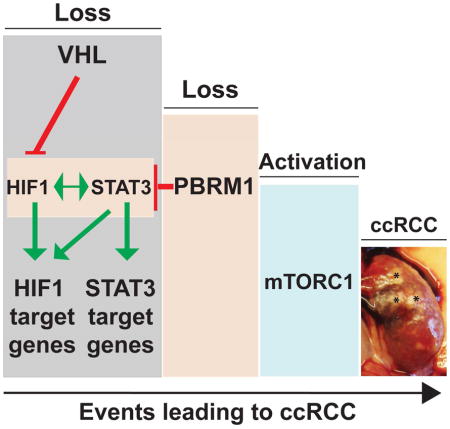
(A) GSEA plots of the ranked list of differentially expressed genes in Hif1α-M3 TRACK mouse tumors (T) and normal (N) generated using four gene sets: curated HIF targets, KEGG JAK STAT signaling pathway, KEGG oxidative phosphorylation pathway, and KEGG mTOR Pathway. (B) Venn diagram of differentially expressed genes in VhlF/FPbrm1F/FKsp-Cre tumors (T) vs. WT normal (N) (T/N), Hif1α-M3 TRACK T/N samples, and human VHLmtPBRM1mt ccRCC tumors vs. normal. (C) The differentially expressed genes at the intersect of VhlF/FPbrm1F/FKsp-Cre T/N and Hif1α-M3 TRACK T/N were tested for enrichment and presented using ClueGO. (D) GSEA plots of the ranked list of differentially expressed genes in human VHLmtPBRM1mt ccRCC tumors versus normal kidneys generated using four gene sets: curated HIF targets, KEGG JAK STAT signaling pathway, KEGG oxidative phosphorylation pathway, and KEGG mTOR Pathway. (E) The shared differentially expressed genes in VhlF/FPbrm1F/FKsp-Cre T/N, Hif1α-M3 TRACK T/N, and TCGA-KIRC VHLmtPBRM1mt T/N were tested for enrichment and presented using ClueGO. (F) Model depicts the chronological sequences of genetic and signaling events during the pathogenesis of Vhl and Pbrm1 doubly deficient ccRCC.
To determine whether this observation could be extended into human ccRCC bearing both VHL and PBRM1 mutations, we first compiled differentially expressed genes in human VHL and PBRM1 mutated ccRCC from the TCGA-KIRC dataset. Consistent with the findings observed in mouse ccRCC, GSEA of these differentially expressed genes revealed up-regulation of HIF1, JAK/STAT3 and mTOR pathways, and the down-regulation of OXPHOS pathway (Figure 7D). Next, we compared the differentially expressed genes identified in human VHL and PBRM1 mutated ccRCC to those shared between the two mouse models, which resulted in the identification of 1772 genes that were shared among these three ccRCC models (Figure 7B). Within this shared gene set, HIF1 and mTOR pathway genes were statistically enriched again (Figure 7E and Table S6). Taken together, our study favors a scenario in which a sequence of at least three distinct genetic/epigenetic events including the loss of VHL, the loss of PBRM1, and the subsequent activation of mTORC1 are required for the development of ccRCC (Figure 7F).
DISCUSSION
VHL is the most commonly mutated gene in human ccRCC and its mutation serves as the initial driver event in the pathogenesis of ccRCC (Linehan et al., 1995). However, genetic deletion of Vhl in mice is insufficient to initiate kidney tumors (Haase et al., 2001; Kapitsinou and Haase, 2008), favoring the involvement of additional genetic/epigenetic events. Such events remained elusive till the discovery of additional 3p21 tumor suppressor genes commonly mutated in human ccRCC, i.e. PBRM1, SETD2, and BAP1 (Hakimi et al., 2013b). Although PBRM1 is the second most commonly mutated gene in human ccRCC, whether and how PBRM1 loss contributes to the pathogenesis of ccRCC are unknown. Through tissue-specific deletion of both Vhl and Pbrm1 (VhlF/FPbrm1F/FKsp-Cre), we created a clear cell kidney cancer mouse model that recapitulates histopathological and molecular features of human ccRCC and elucidated how PBRM1 functions as a tumor suppressor in ccRCC.
The VhlF/FPbrm1F/FKsp-Cre mice developed preneoplastic polycystic kidney lesions at ~6 months and multifocal ccRCC at ~10 months, suggesting that loss of Vhl and Pbrm1 in kidney predisposes to ccRCC. The human pan-cancer genomics identified SWI/SNF complexes as commonly mutated genes (~20%) across cancer types with preferential enrichment of individual mutations in specific cancer types (Helming et al., 2014; Kadoch et al., 2013; Marquez et al., 2014), which presents challenges and opportunities in broadening our knowledge on how chromatin remodeling ATPase complexes function as tumor suppressors. Among the SWI/SNF complexes, SMARCB1 (BAF47) is the best-characterized tumor suppressor that regulates cell cycle and antagonizes PRC2 complex (Helming et al., 2014). Expression profiling of 12-week-old VhlF/FPbrm1F/FKsp-Cre renal cortices revealed the tumor suppressor role of PBRM1 in preventing the self-perpetuating over-amplification of the HIF1 pathway through limiting the intricate feed-forward interplay between HIF1 and STAT3 upon VHL loss (Demaria and Poli, 2012; Jung et al., 2005; Luo and Semenza, 2012). The unexpected tumor suppressor function of PBRM1 is analogous to an electrical resistor in preventing power overdrive, in which PBRM1 restrains the HIF1 and STAT3 transcription outputs from over-amplification upon the loss of VHL (Figure 7F).
The observation that clear cell kidney tumors occurred in VhlF/FPbrm1F/FKsp-Cre mice after a long latency period suggests the involvement of additional signaling aberrations. Immunohistochemical and transcriptomic analyses demonstrated that activation of mTORC1 instead of ERK pathway as the preferred third event. Ample evidences support the importance of mTORC1 activation in the pathobiology of human ccRCC, e.g. mTORC1 pathway activation is prevalent in human ccRCC (Linehan et al., 2010; Robb et al., 2007) and mTOR inhibitors are standard of care in treating metastatic ccRCC (Voss et al., 2014; Wei and Hsieh, 2015). As REDD1, a key transcriptional target of HIF1, negatively regulates mTORC1 through activation of TSC1/TSC2 (Brugarolas et al., 2004; DeYoung et al., 2008), it is foreseeable that mTORC1 activation could be a bottleneck for tumors originating from a hyperactive HIF1 signaling. Consistent with this working hypothesis, both the VhlF/FPbrm1F/FKsp-Cre and the published HIF1α-M3 TRACK mouse models (Fu et al., 2011) developed ccRCC after a long latency period and showed convergence on mTORC1 activation (Figure 7F). Moreover, mTORC1 pathway activation was also observed in human ccRCC carrying mutations of VHL and PBRM1 (Figure 6E). Of note, multi-regional sequencing of a hereditary VHL syndrome patient also detected the mutations of PBRM1 and the convergence of mTORC1 pathway activation (Fisher et al., 2014). These findings are consistent with a recurrent oncogenic theme in which many oncogenes, such as c-MYC, BRAF, and MLL-fusions, activate both oncogenic signaling and intrinsic tumor suppressor checkpoints (Liu et al., 2014; Lowe et al., 2004; Maertens et al., 2013). Therefore, abrogating the built-in intrinsic tumor suppressor activities of individual oncogenes is essential for tumorigenesis. It was recently reported that homozygous deletion of Vhl and Bap1 in mouse kidney resulted in early lethality (<1 month), and some mice (within a cohort of 7) carrying homozygous deletion of Vhl and heterozygous deletion of Bap1 developed tumor micronodules (0.25–1.8mm) with unknown tumor incidence, transplantability, and molecular characteristics (Wang et al., 2014). Notably, complete BAP1 inactivation is observed in human ccRCC (Pena-Llopis et al., 2012). How heterozygous loss of Bap1 cooperates with Vhl loss to initiate kidney tumorigenesis in mice remains intriguing (Wang et al., 2014).
The suppression of OXPHOS genes observed in the VhlF/FPbrm1F/FKsp-Cre ccRCC model is consistent with the global metabolomics reported on ccRCC (Hakimi et al., 2016), which lends further support for the notion that kidney cancer is a metabolic disease that manifests with massive metabolic reprogramming (Hakimi et al., 2016; Linehan et al., 2010). Inhibitors of mTORC1, the key cellular complex integrating nutrient and growth factor signaling to promote anabolic metabolism, are standard of care for metastatic ccRCC (Voss and Hsieh, 2016). However, a wide range of clinical outcomes has been observed. Interestingly, genomic study of RECORD-3, a large clinical trial randomized kidney cancer patients to either VEGFR or mTORC1 inhibitors, demonstrated that ccRCC with mutant PBRM1 associates with longer progression free survival (PFS) on everolimus, an mTORC1 inhibitor, at 12.8 months than those with wild-type PBRM1 at 5.5 months (Hsieh et al., 2015; Hsieh et al., 2016). In parallel, the VhlF/FPbrm1F/FKsp-Cre ccRCC and human VHL and PBRM1 mutated ccRCC shared mTORC1 pathway aberration. Both clinical data and this study support a model in which mTORC1 activation constitutes the preferred third driver event during ccRCC tumorigenesis immediately after genetic inactivation of VHL and PBRM1 (Voss and Hsieh, 2016) (Figure 7F), which serves as an example of preferential pathway convergent evolution of a given cancer type (Voss and Hsieh, 2016; Wei and Hsieh, 2015) that could have predictive values for selecting patients of a given cancer genotype with matched targeted therapies.
EXPERIMENTAL PROCEDURES
More detailed information is available in Supplemental Experimental Procedures.
Mice
Baf180F/F) mice were obtained from Dr. Wang Zhong (Wurster et al., 2012). Animal experiments were performed in accordance to the Institutional Animal Care and Use Committee (IACUC) at MSKCC.
Mouse MRI
Mice MRI scans were carried out on either 200 or 300 MHz Bruker 4.7T or 7T Biospec scanners (Bruker Biospin MRI GmbH, Ettlingen, Germany) equipped with 640 mT/m ID 115 mm and 300 mT/m ID 200 mm gradients respectively (Resonance Research, Inc., Billerica, MA).
RNA Isolation and Microarray Analysis
Total RNA was isolated using TRIzol (Life Technologies) and cleaned up using Qiagen column DNase digestion. RNA samples were prepared from three month old Ksp-Cre, VhlF/FKsp-Cre, Pbrm1F/FKsp-Cre, and VhlF/FPbrm1F/FKsp-Cre mice. Microarray was performed by Integrated Genomics Operation (IGO) at MSKCC.
RNA Sequencing and Analysis
Total RNA was process by the IGO using TruSeq RNA Sample Prep kit according to the manufacturer’s recommendation. Gene ontology (GO) analysis of mouse microarray and RNA Sequencing data were performed with ClueGO.
Motif Activity Analysis
To analyze activities of transcription factor binding motifs (TFBM) using RNA-seq data, we used the Integrated System for Motif Activity Response Analysis (ISMARA).
Supplementary Material
Highlights.
PBRM1 is a bona fide tumor suppressor in the pathogenesis of ccRCC.
PBRM1 prevents self-perpetuating amplification of HIF1/STAT3 signaling in Vhl−/− cell.
Loss of Vhl and Pbrm1 in mouse kidney results in multifocal, transplantable ccRCC.
In ccRCC, mTORC1 activation is the third driver event after loss of VHL and PBRM1.
Significance.
Clear cell renal cell carcinoma (ccRCC) is the most common, lethal subtype (75%) of kidney cancer. ccRCC is associated with the prevalent loss of VHL and ensuing aberrant HIF activation. However, VHL loss alone is insufficient to cause ccRCC. PBRM1, a SWI/SNF complex gene, is mutated in ~40% of ccRCC. SWI/SNF component genes are frequently mutated in ~20% of all human cancers. However, how SWI/SNF proteins act as tumor suppressors is unclear. Here we generate a VhlF/FPbrm1F/FKsp-Cre mouse model that recapitulates the molecular and histopathological features of human ccRCC, present three sequential driver events that are central to the pathogenesis of ccRCC, and provide a physiological preclinical model for studying kidney cancer biology and therapeutics, which could have therapeutic significance.
Acknowledgments
This work was supported by The Jill and Jeffrey Weiss Fund to the Cure of Kidney Cancer and J. Randall & Kathleen L. MacDonald Kidney Cancer Research Fund. We thank Dr. Mihaela Lupu, Mr. Dov Winkleman and Ms. Smrutiben A. Mehta for their technical support, and Dr. Victor Reuter for the initial assessment of mouse renal tumors. The MRI and LCP cores were supported by P30CA008748.
Footnotes
Supplemental information includes supplemental Experimental Procedures, 8 figures and 8 tables and can be found with this article online at
AUTHOR CONTRIBUTIONS
M.B. and Z.D. initiated the study. A.M.N., C.G.P. and Y.D. designed and conducted experiments, and analyzed data. A.M.N. is responsible for molecular, C.G.P for phenotypic, and Y.D. for allograft studies. Y.X., T.O., S.T. and C.E.R. conducted some experiments. P.I.W., H.U.O. and A.A.H. analyzed data. C.L.L. and J.A.K. performed MRI. O.A. evaluated MRI. S.M. and S.K.T. performed histopathological examination. C.S.L., N.W. and W.L. supervised data analyses. J.J.H. conceived and directed the research study. E.H.C. designed research and supervised the project. J.J.H. and E.H.C. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Furihata M, Hong SB, Tessarollo L, Haines DC, Southon E, Patel V, Igarashi P, Alvord WG, Leighty R, et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008;100:140–154. doi: 10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balwierz PJ, Pachkov M, Arnold P, Gruber AJ, Zavolan M, van Nimwegen E. ISMARA: automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res. 2014;24:869–884. doi: 10.1101/gr.169508.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. 2014;166C:350–366. doi: 10.1002/ajmg.c.31410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows AE, Smogorzewska A, Elledge SJ. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc Natl Acad Sci U S A. 2010;107:14280–14285. doi: 10.1073/pnas.1009559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhang Y, Senbabaoglu Y, Ciriello G, Yang L, Reznik E, Shuch B, Micevic G, De Velasco G, Shinbrot E, et al. Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell Rep. 2016;14:2476–2489. doi: 10.1016/j.celrep.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Futami K, Petillo D, Peng J, Wang P, Knol J, Li Y, Khoo SK, Huang D, Qian CN, et al. Deficiency of FLCN in mouse kidney led to development of polycystic kidneys and renal neoplasia. PLoS One. 2008;3:e3581. doi: 10.1371/journal.pone.0003581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Demaria M, Poli V. PKM2, STAT3 and HIF-1alpha: The Warburg’s vicious circle. JAKSTAT. 2012;1:194–196. doi: 10.4161/jkst.20662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14–3–3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R, Horswell S, Rowan A, Salm MP, de Bruin EC, Gulati S, McGranahan N, Stares M, Gerlinger M, Varela I, et al. Development of synchronous VHL syndrome tumors reveals contingencies and constraints to tumor evolution. Genome Biol. 2014;15:433. doi: 10.1186/s13059-014-0433-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew IJ, Thoma CR, Georgiev S, Minola A, Hitz M, Montani M, Moch H, Krek W. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J. 2008;27:1747–1757. doi: 10.1038/emboj.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Minton DR, Zhang T, Nanus DM, Gudas LJ. Genome-Wide Profiling of TRACK Kidneys Shows Similarity to the Human ccRCC Transcriptome. Mol Cancer Res. 2015;13:870–878. doi: 10.1158/1541-7786.MCR-14-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Wang G, Shevchuk MM, Nanus DM, Gudas LJ. Generation of a mouse model of Von Hippel-Lindau kidney disease leading to renal cancers by expression of a constitutively active mutant of HIF1alpha. Cancer Res. 2011;71:6848–6856. doi: 10.1158/0008-5472.CAN-11-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Burden of Disease Cancer, C. Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015;1:505–527. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A. 2001;98:1583–1588. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi AA, Chen YB, Wren J, Gonen M, Abdel-Wahab O, Heguy A, Liu H, Takeda S, Tickoo SK, Reuter VE, et al. Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. European urology. 2013a;63:848–854. doi: 10.1016/j.eururo.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi AA, Pham CG, Hsieh JJ. A clear picture of renal cell carcinoma. Nat Genet. 2013b;45:849–850. doi: 10.1038/ng.2708. [DOI] [PubMed] [Google Scholar]
- Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W, et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell. 2016;29:104–116. doi: 10.1016/j.ccell.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming KC, Wang X, Roberts CW. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell. 2014;26:309–317. doi: 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh JJ, Chen DPW, Chen YB, Redzematovic A, Marker M. Identification of efficacy biomarkers in a large metastatic renal cell carcinoma (mRCC) cohort through next generation sequencing (NGS): Results from RECORD-3. J Clin Oncol. 2015:33. [Google Scholar]
- Hsieh JJ, Chen D, Wang PI, Marker M, Redzematovic A, Chen YB, Selcuklu SD, Weinhold N, Bouvier N, Huberman KH, et al. Genomic Biomarkers of a Randomized Trial Comparing First-line Everolimus and Sunitinib in Patients with Metastatic Renal Cell Carcinoma. European urology. 2016 doi: 10.1016/j.eururo.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Gao X, Diaz-Trelles R, Ruiz-Lozano P, Wang Z. Coronary development is regulated by ATP-dependent SWI/SNF chromatin remodeling component BAF180. Dev Biol. 2008;319:258–266. doi: 10.1016/j.ydbio.2008.04.020. [DOI] [PubMed] [Google Scholar]
- Igarashi P. Kidney-specific gene targeting. J Am Soc Nephrol. 2004;15:2237–2239. doi: 10.1097/01.ASN.0000136298.09488.D7. [DOI] [PubMed] [Google Scholar]
- Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, Lee JW, Choi S, Park JW, Ye SK, Chung MH. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005;19:1296–1298. doi: 10.1096/fj.04-3099fje. [DOI] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–173. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- Kapitsinou PP, Haase VH. The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death Differ. 2008;15:650–659. doi: 10.1038/sj.cdd.4402313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan WM, Lerman MI, Zbar B. Identification of the von Hippel-Lindau (VHL) gene. Its role in renal cancer. JAMA. 1995;273:564–570. [PubMed] [Google Scholar]
- Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature reviews Urology. 2010;7:277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Westergard TD, Cashen A, Piwnica-Worms DR, Kunkle L, Vij R, Pham CG, DiPersio J, Cheng EH, Hsieh JJ. Proteasome inhibitors evoke latent tumor suppression programs in pro-B MLL leukemias through MLL-AF4. Cancer Cell. 2014;25:530–542. doi: 10.1016/j.ccr.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23:560–566. doi: 10.1016/j.tem.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3:338–349. doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Molecular cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, Wykoff CC, Maher ER, Harris AL, Ratcliffe PJ, Maxwell PH. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–468. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- Marquez SB, Thompson KW, Lu L, Reisman D. Beyond Mutations: Additional Mechanisms and Implications of SWI/SNF Complex Inactivation. Front Oncol. 2014;4:372. doi: 10.3389/fonc.2014.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014;2:3. doi: 10.1186/2049-3002-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann HP, Zbar B. Renal cysts, renal cancer and von Hippel-Lindau disease. Kidney international. 1997;51:16–26. doi: 10.1038/ki.1997.3. [DOI] [PubMed] [Google Scholar]
- Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44:751–759. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009;373:1119–1132. doi: 10.1016/S0140-6736(09)60229-4. [DOI] [PubMed] [Google Scholar]
- Robb VA, Karbowniczek M, Klein-Szanto AJ, Henske EP. Activation of the mTOR signaling pathway in renal clear cell carcinoma. J Urol. 2007;177:346–352. doi: 10.1016/j.juro.2006.08.076. [DOI] [PubMed] [Google Scholar]
- Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–867. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. The Journal of clinical investigation. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol. 2002;13:1837–1846. doi: 10.1097/01.asn.0000016444.90348.50. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MH, Hakimi AA, Pham CG, Brannon AR, Chen YB, Cunha LF, Akin O, Liu H, Takeda S, Scott SN, et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:1955–1964. doi: 10.1158/1078-0432.CCR-13-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MH, Hsieh JJ. Therapeutic Guide for mTOuRing through the Braided Kidney Cancer Genomic River. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MH, Hsieh JJ, Motzer RJ. Novel approaches targeting the vascular endothelial growth factor axis in renal cell carcinoma. Cancer J. 2013;19:299–306. doi: 10.1097/PPO.0b013e31829d5cff. [DOI] [PubMed] [Google Scholar]
- Walther MM, Lubensky IA, Venzon D, Zbar B, Linehan WM. Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: clinical implications. J Urol. 1995;154:2010–2014. discussion 2014–2015. [PubMed] [Google Scholar]
- Wang SS, Gu YF, Wolff N, Stefanius K, Christie A, Dey A, Hammer RE, Xie XJ, Rakheja D, Pedrosa I, et al. Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc Natl Acad Sci U S A. 2014;111:16538–16543. doi: 10.1073/pnas.1414789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhai W, Richardson JA, Olson EN, Meneses JJ, Firpo MT, Kang C, Skarnes WC, Tjian R. Polybromo protein BAF180 functions in mammalian cardiac chamber maturation. Genes Dev. 2004;18:3106–3116. doi: 10.1101/gad.1238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei EY, Hsieh JJ. A river model to map convergent cancer evolution and guide therapy in RCC. Nature reviews Urology. 2015;12:706–712. doi: 10.1038/nrurol.2015.260. [DOI] [PubMed] [Google Scholar]
- Wurster AL, Precht P, Becker KG, Wood WH, 3rd, Zhang Y, Wang Z, Pazin MJ. IL-10 transcription is negatively regulated by BAF180, a component of the SWI/SNF chromatin remodeling enzyme. BMC Immunol. 2012;13:9. doi: 10.1186/1471-2172-13-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Nagase S, Montia AG, Kalachikov SM, Keniry M, Su T, Memeo L, Hibshoosh H, Parsons R. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res. 2008;68:1667–1674. doi: 10.1158/0008-5472.CAN-07-5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.