

Abstract
The citric acid cycle intermediate 2-oxoglutarate (2-OG, a.k.a. alpha-ketoglutarate) links the carbon and nitrogen metabolic pathways and can provide information on the metabolic status of cells. In recent years, it has become exceedingly clear that 2-OG also acts as a master regulator of diverse biologic processes in all domains of life. Consequently, there is a great demand for time-resolved data on 2-OG fluctuations that can’t be adequately addressed using established methods like mass spectrometry-based metabolomics analysis. Therefore, we set out to develop a novel intramolecular 2-OG FRET sensor based on the signal transduction protein PII from Synechococcus elongatus PCC 7942. We created two variants of the sensor, with a dynamic range for 2-OG from 0.1 µM to 0.1 mM or from 10 µM to 10 mM. As proof of concept, we applied the sensors to determine in situ glutamine:2-oxoglutarate aminotransferase (GOGAT) activity in Synechococcus elongatus PCC 7942 cells and measured 2-OG concentrations in cell extracts from Escherichia coli in vitro. Finally, we could show the sensors’ functionality in living human cell lines, demonstrating their potential in the context of mechanistic studies and drug screening.
Introduction
The emerging field of metabolomics has the potential to accurately describe the physiological state of a cell at the time of measurement1, 2. Technological improvements during the last decade have led to more rapid and precise metabolite identification3; however, state of the art HPLC-MS based analyses require extraction of the metabolites from the cells, making the study of detailed temporal metabolite fluctuations a challenging task. Additionally, certain metabolites are not stable under commonly used extraction conditions or their recovery rates are low4. To overcome these drawbacks, a variety of genetically encoded, protein-based sensors were developed. These in vivo/ex vivo sensors allow real-time measurements of high temporal resolution without disrupting the cell. To date, there are hundreds of sensors available, not only for metabolites, but also for cellular aspects like ion concentration, mechanical stress, enzyme and kinase activity, redox potential, etc. (http://biosensor.dpb.carnegiescience.edu/)5. The majority of these sensors utilize the growing number of optimized fluorescence proteins (FPs) to create a Förster resonance energy transfer (FRET) readout6, 7. FRET occurs when donor and acceptor fluorophores with overlapping emission and excitation spectra come in close proximity. Following excitation of the donor, energy is transmitted to the acceptor in a non-radiative manner by means of intermolecular long-range dipole–dipole coupling and emitted by the acceptor. Ligand-binding-induced conformational changes in the sensors containing both, donor and acceptor fluorophres, results in altered FRET efficiency, which can be monitored under a fluorescence microscope or in a fluorimeter8.
One central metabolite of high interest is 2-oxoglutarate (2-OG), which links the carbon and nitrogen metabolic pathways in all domains of life. 2-OG is used as the carbon skeleton for nitrogen assimilatory reactions and has been proposed as a master regulatory metabolite9. It has been shown that the 2-OG pool reacts to changes in extracellular nitrogen availability within minutes and its half-life has been estimated as 0.5 s10–13. Apart from the regulatory PII proteins (see below) 2-OG is sensed by a number of transcription factors9. Furthermore, 2-OG acts as a starvation signal in eukaryotes like S. cerevisiae or the metazoa C. elegans 14, 15. Competitive inhibition of 2-OG-dependent DNA and histone demethylases by the oncometabolite and 2-OG analogue 2-hydroxyglutarate have been shown to be the cause of cancer-specific epigenome and gene expression alterations in glioma and acute myelogenous leukemia16–18. Furthermore, 2-OG levels were proposed to regulate the epigenome of differentiating human pluripotent stem cells19 and embryonic stem cells20, 21. In mice it has recently been shown, that 2-OG acts as a systemic signal providing protection from cardiac ischemia22. In bacteria, the sugar-uptake phosphotransferase system (PTS) in E. coli is regulated by the 2-OG/phosphoenolpyruvate ratio23, 24. The PTS not only promotes sugar transport but is also responsible for activation or inhibition of the adenylate cyclase which produces cyclic AMP, a very important signaling molecule that affects the expression of a vast range of genes25, 26. These examples demonstrate the importance of 2-OG as a regulatory metabolite and underline the need for a functional sensor in living cells, which allows investigations of 2-OG fluctuations with high spatial and temporal resolution.
The small trimeric regulatory protein PII, which is widely distributed in prokaryotes and chloroplasts, is known as a sensor of cellular 2-OG levels27. Binding of 2-OG leads to conformational changes in the protein structure in a concentration dependent manner27–30. These conformational changes modulate the interaction of PII with its regulatory targets31. In previous studies, these interactions have been utilized to create inter-molecular FRET sensors employing cyanobacterial PII proteins and their targets N-acetyl-L-glutamate kinase (NAGK) and PipX. These sensors have successfully been used to expand the knowledge about the 2-OG dependent mode of interaction between PII and its targets32–34. However, FRET sensors using protein-protein interactions have disadvantages, especially for applications in living cells, where different expression rates and protein half-lives have to be taken into account, as well as the increased chance of unwanted side reactions, e.g. by NAGK enzymatic activity. Berg et al. have constructed a PII-based intramolecular FRET sensor to read out fluctuations in ATP/ADP levels, after having inactivated the 2-OG binding site in PII 35. This motivated us to create an enhanced intra-molecular 2-OG FRET sensor making use of the high specificity of PII proteins towards 2-OG.
As a first application we used the 2-OG sensor for an in situ glutamine:2-oxoglutarate aminotransferase (GOGAT) assay. GOGAT catalyzes the reductive transfer of the amide group from glutamine to the carbon backbone of 2-OG, which yields two molecules of glutamate. This is a key reaction in nitrogen assimilation in bacteria and plants36, but studies on GOGAT activity regulation are scarce37, due to the lack of a simple assay. Using the PII-based 2-OG specific FRET sensors, we present here the determination of the in situ fdGOGAT activity in the unicellular cyanobacteria Synechococcus elongatus PCC 7942 (hereafter designated as S. elongatus). Furthermore, we used the sensor to determine 2-OG concentrations in cell extracts from E. coli. Finally, we conducted the first application of a real-time 2-OG sensor in mammalian cells to measure 2-OG fluctuations in human glioblastoma brain-cancer cells and other cell lines. These experiments provide the proof of principle that PII-based 2-OG specific FRET sensors could become useful tools for studying cancer patho-mechanisms and as functional readouts in metabolic-drug screening.
Results and Discussion
Development of the FRET Sensor
On our attempt to construct a 2-OG sensor that employs the conformational change of PII upon 2-OG binding, we made use of the knowledge gained during the development of the PII-NAGK FRET sensor32 and crystal structures of S. elongatus PII 28. Different sensor variants were constructed, most of which use the monomeric (m) cyan FP mCerulean as the FRET donor and the yellow FP Venus as acceptor. The simplest approach was to fuse these FPs to the N and C-terminus of PII. Crystal structures of PII with Mg2+-ATP + 2-OG bound display a conformational change in the C-terminus28: in the ligand free state (PDB: 1QY7) or while interacting with NAGK (PDB: 2V5H), the PII C-terminus adopts a stretched conformation, pointing away from the trimer. By contrast, upon binding of Mg2+-ATP + 2-OG, the C-terminus retracts and folds over the metabolite binding site of the inter-subunit cleft (PDB: 2XUL). With the first variants, we aimed to asses if this conformational change in the C-terminus could be used to create a change in FRET.
To achieve this goal, we had to overcome a problem associated with modifying the N-terminus of bacterial PII proteins. The N-terminus of S. elongatus PII is hidden within the protein and a direct fusion to the buried end leads to misfolding and degradation (data not shown). To solve this problem, we used the non-conserved extended N-terminus of Chlamydomonas rheinhardtii PII that protrudes from the protein38 and fused these six N-terminal amino acids (S-A-F-P-G-V) to the N-terminus of S. elongatus PII. This indeed led to a stably expressed protein. On top of that, we added flexible (FL; L[SGGGG]nSAAA) or stiff helical (HL; A[EAAAK]nA) linkers of 20 or 25 amino acids (Fig. 1, PII-NC1 to 4)39. The Venus FP was fused to the C-terminus of PII via the 12 amino acid long streptavidin affinity tag (Strep-tag), as has been done previously32. The Strep-tag should enable the transfer of the proposed C-terminal movement to the FP and was also used for protein purification. While PII-NC1 and NC3 could be purified in good quantity and quality, PII-NC2 and NC4 were not expressed in E. coli. Probably the helical linker led to misfolding and degradation of these proteins.
Figure 1.
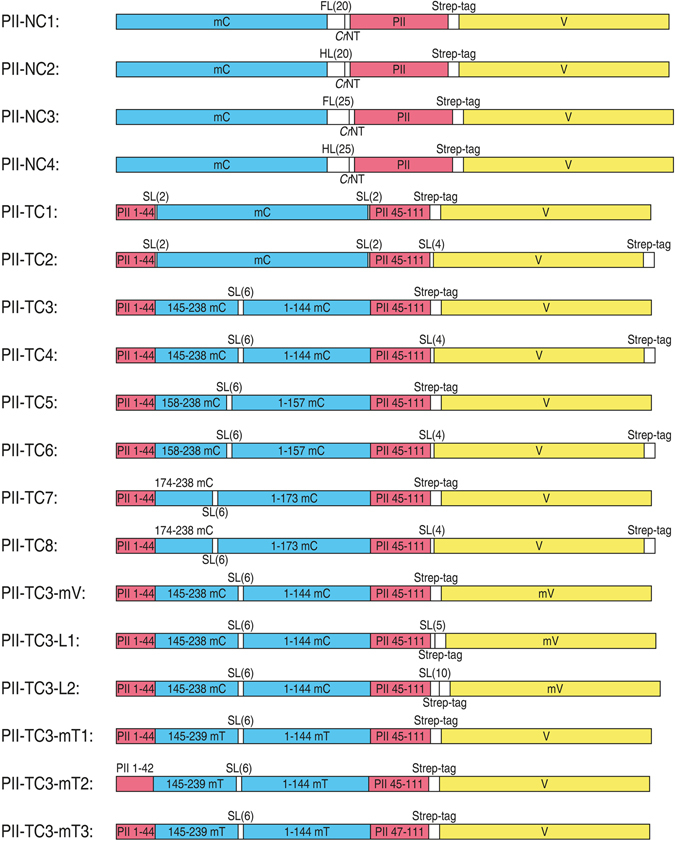
Schematic representation of the FRET-sensor constructs. Red: PII; Blue: mCerulean; Yellow: mVenus; White: linker regions. Flexible (FL) or stiff helical (HL) linkers or short linkers (SL) of different length, Chlamydomonas reinhardtii PII N-terminus (CrNT) or streptavidin affinity tag (Strep-tag). All Domains are presented to scale.
In other variants, we aimed to use the conformational change of the T-loop upon Mg2+-ATP + 2-OG binding to obtain a change in FRET. As shown in the crystal structures mentioned above, the stretched T-loops of ligand-free PII are retracted and folded upon binding of Mg2+-ATP + 2-OG. This folding also prevents the interaction of PII with other receptor proteins like NAGK or PipX30. We devised eight variants to test this approach. In the first two variants (Fig. 1, PII-TC1 and TC2) we inserted mCerulean with its natural C- and N-terminus into the tip of PII’s T-loop between amino acids 44 and 45. Venus was again placed at the C-terminus using either the Strep-tag, or a four amino acid linker (LAAA). FRET is not only influenced by the distance between donor and acceptor, but also by their angle towards each other40. With that in mind, we also used circularly permuted (cp) versions of mCerulean, by connecting its native C- and N-terminus with a short flexible linker (GSGGTG) and using other positions for the fusion into the T-loop, similar to the approach described by Berg et al. for the construction of a PII-based ATP/ADP sensor35. Topell et al. showed that circular permutations of GFP to positions Y145-N144, K158-Q157 as well as G174-D173 yielded correctly folded FPs41. We used these locations for fusion with the T-loop and again varied the linker between the PII C-terminus and Venus (Fig. 1, PII-TC3 to 8).
All tested variants showed strong initial FRET signals, indicating that the FPs were correctly folded and in close proximity. Subsequently 0.1 mM ADP, 0.1 mM ATP or 0.1 mM ATP together with 1 mM 2-OG were added to the solution and the FRET change was measured (Table 1). Most variants showed only small FRET changes, either slightly increasing or decreasing the FRET upon effector molecule addition. Only PII-TC3 showed an 18% drop in FRET. The decreased FRET indicates that the FPs move away from each other, which is consistent with the crystal structures, where the T-loop folds downward upon 2-OG binding, away from the C-terminus.
Table 1.
FRET change after addition of metabolites.
| Sensor | % FRET change after addition of | ||
|---|---|---|---|
| ADP | ATP | ATP + 2-OG | |
| PII-NC1 | n. d. | 0.3 | 4.4 |
| PII-NC3 | n. d. | 3.1 | 3.1 |
| PII-TC1 | −2.0 | −1.1 | −1.7 |
| PII-TC2 | −1.1 | 1.0 | 5.7 |
| PII-TC3 | 0.8 | −0.8 | −18.3 |
| PII-TC4 | −0.4 | 0.8 | −2.5 |
| PII-TC5 | −6.1 | −0.1 | −3.1 |
| PII-TC6 | 1.0 | 0.2 | 3.4 |
| PII-TC7 | −0.6 | −0.8 | −2.3 |
| PII-TC8 | 6.2 | 0.8 | 0.9 |
| PII-TC3-mV | n. d. | n. d. | −15.1 |
| PII-TC3-L1 | n. d. | n. d. | −15.4 |
| PII-TC3-L2 | n. d. | n. d. | −15.6 |
| PII-TC3-mT1 | n. d. | n. d. | −8.3 |
| PII-TC3-mT2 | n. d. | n. d. | 25.8 |
| PII-TC3-mT3 | n. d. | n. d. | −13.3 |
FRET of the purified senor proteins was measured before and after addition of 0.1 mM ADP, 0.1 mM ATP or 0.1 mM ATP + 1 mM 2-OG and the relative FRET change was calculated. (n. d., not determined).
Since PII-TC3 showed the strongest FRET change so far, we tried further modifications of the PII-TC3 variant. GFPs have the tendency to dimerize. To allow the C-terminally attached Venus more flexibility in movement, we added the monomer mutation A206K42 and also elongated the linker between PII and mVenus by five or ten amino acids (Fig. 1, PII-TC3-mV, -L1 and -L2). Even though PII-TC3-mV showed a higher initial FRET value, the relative FRET drop upon addition of ATP + 2-OG was lower compared to PII-TC3. Also the modified linkers did not increase the sensors signal (Table 1). Next, we tried to enhance the TC3 sensor by replacing mCerulean by the advanced mTurquoise2, which shows exceptional quantum yield, brightness, photostability and a fast maturation rate and is thus proposed as an optimal donor for FRET systems43. Additionally, we created two versions with partially deleted T-loops (PII-TC3-mT1 to 3). PII-TC3-mT1 and -mT3 showed lower signal changes than PII-TC3. Interestingly, PII-TC3-mT2, with T-loop amino acids 43 and 44 deleted, showed increased FRET upon addition of ATP + 2-OG. This indicates that the small deletion in the T-loop alters the orientation of the attached FP, demonstrating once again that subtle changes can have a dramatic and unpredictable impact on the signal output of a FRET sensor. A detailed analysis showed that PII-TC3-mT2 was only reacting to high 2-OG concentrations and was not as sensitively responding towards 2-OG as PII-TC3 (data not shown).
After all, PII-TC3 appeared to be the most promising sensor variant for sensitive 2-OG detection. To biochemically characterize PII-TC3, the affinity-tag isolated protein was further purified by size-exclusion chromatography to remove degradation products containing only one of the two FPs. We also optimized the buffer conditions and the workflow to improve TC3’s signal to noise ratio and to be able to use a 96-well plate setup and a small reaction volume. As previously described for PII-NAGK interactions, the proteins can be unstable when buffer conditions are quickly changed32. When the sensor, stored in a 50% glycerol buffer at −20 °C, was quickly mixed with the measurement buffer, it showed a lower signal to noise ratio and sensitivity, possibly due to dissociation of the trimer. To circumvent this problem, we tested a variety of reaction-buffer setups and came up with a mixture containing 15% glycerol, which not only stabilized the sensor and enabled easy preparation of a master mix for 96-well plate application, but also lead to a increase of the signal-to-noise ratio to 25%. Figure 2A shows the 2-OG induced FRET change under these optimized conditions. PII-TC3 displayed a Kd for 2-OG of approximately 3 µM. This is close to the binding constant of the high affinity 2-OG binding site in wild-type PII protein, showing that, despite the insertion of mCerulean into the T-loop, PII-TC3 acts similar to PII-wildtype. However, for possible in vivo applications, where one- to two orders of magnitude higher 2-OG concentrations are expected, this response is too sensitive. Therefore, we constructed a variant of PII-TC3 with a mutation in the vicinity of the 2-OG binding site. Previously, it was shown that mutation of Arg9 reduces the affinity of 2-oxoglutarate binding28. Here, a mutation of arginine 9 to proline (R9P) decreased the affinity of PII for 2-OG 35 times, resulting in a Kd of 91 µM (Fig. 2A). With a dynamic range of the FRET response from 10 µM to 10 mM, this sensor seems to be ideally suited to span the physiological relevant range of 2-OG concentrations. Because ATP and ADP compete for the same binding site on the PII protein, but only ATP enables the binding of 2-OG, we also measured the competitive effect of ADP on the signal output. As shown in Fig. 2B, physiological ATP/ADP ratios (ATP > ADP) do not influence 2-OG sensing.
Figure 2.

Properties of FRET-sensor PII-TC3. (A) Relative NFRET response of PII-TC3 and PII-TC3-R9P to increasing 2-OG concentrations. (B) Influence of different ADP/ATP ratios on the NFRET response of PII-TC3 upon addition of 0.1 mM 2-OG. (C and D) Relative NFRET response of PII-TC3 (C) and PII-TC3-R9P (D), to different metabolites of the TCA cycle. Error bars: ±s.e.m., n = 4.
In the next step, we tested the sensitivity and specificity of TC3 and TC3-R9P. All metabolites of the tricarboxylic acid (TCA) cycle and the 2-OG analog dimethyl-2-OG (dm2-OG) were tested. As shown in Fig. 2C, TC3 is about 460 times more sensitive for 2-OG than for dm2-OG, which gave the second strongest signal. The third strongest signal was induced by oxaloacetate, with a 900 times reduced sensitivity. All other tested metabolites were even less interfering than oxaloacetate, demonstrating the high specificity of the sensor towards 2-OG. Interestingly, increasing NaCl concentrations also lead to a decrease in FRET. Most of the tested compounds were sodium salts or had to be titrated with NaOH to adjust the pH, and therefore might give a stronger response than pure compounds. Similarly, TC3-R9P was also highly specific for 2-OG. It showed an 85 times decreased sensitivity for dm2-OG and responded to other metabolites only at non-physiologically high concentrations (Fig. 2D).
Establishing an in situ fdGOGAT assay using the 2-OG FRET-sensor
One possible application of a 2-OG FRET-sensor is the in situ determination of GOGAT activity. In contrast to previously described approaches, in which the formation of glutamate or consumption of glutamine were measured using HPLC-based amino acid quantification methods44, the FRET sensor based 2-OG quantification is much faster and cost efficient.
The in situ GOGAT assay was established for S. elongatus, which only possesses a ferredoxin-depending GOGAT. The method developed in the present study is based on experiments from Marqués et al. and Kameya et al.44–46. In short, the cells were permeabilized by toluene, the GOGAT substrates 2-OG, glutamine and reductant were added and the mixture incubated at 32 °C. The efficiency of cell permeabilization was tested via fluorescence microscopy using a live/dead staining kit for bacteria, revealing a very efficient permeabilization (Fig. S1). In our experimental setup, the natural reductant ferredoxin was substituted by methyl viologen as electron donor, which was reduced by addition of sodium dithionite. To avoid air-oxidization of the dithionite, we covered the reaction mix with paraffin oil. Samples were taken in 5 to 10 minutes intervals and the reaction was stopped by aeration through intense vortexing and cooling on ice, which is quick and easy to apply. Afterwards, the cells were separated by centrifugation and the supernatant was used for 2-OG quantification via FRET measurements. The components of the GOGAT-reaction mixture itself had no influence on the FRET measurement, since addition of the reaction mix to a 2-OG standard curve did not affect the read-out (Fig. S2).
When all components, including permeabilized cells, glutamine, 2-OG and the electron donor were present in the reaction mix, a decrease of the 2-OG concentration over time could be detected (Fig. 3). Preliminary experiments indicated a linear decline in the first 20 min of the reaction (data not shown) when using 4 * 108 cells (≙6.7 mL of culture at OD750 = 0.3). When glutamine was omitted from the reaction mix, the concentration of 2-OG remained constant (Fig. 3), which indicates the absence of background 2-OG consuming side reactions.
Figure 3.
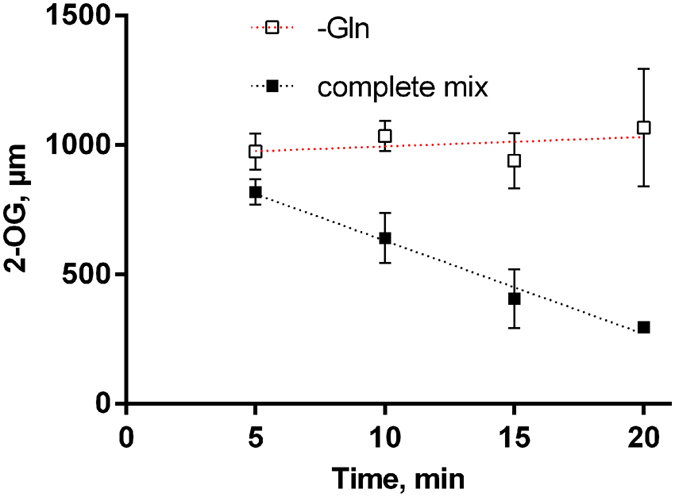
fdGOGAT assay with complete and incomplete reaction mix. S. elongatus cells were grown to an OD750 of 0.3 in nitrate-supplemented medium. The 2-OG concentrations in the reaction mixture was determined by FRET over time. Results are shown for a complete reaction mix (black squares and black dashed line) and the blank mix lacking glutamine (white squares and red dashed line). Error bars: ±s.e.m., n = 3.
Characterization of fdGOGAT in S. elongatus using the in situ fdGOGAT Assay
The glutamine synthetase (GS) - GOGAT cycle represents the connection between the carbon and nitrogen metabolism in bacteria and plants. Whereas GS activity is easy to measure, established GOGAT activity assays are highly elaborate. To determine the fdGOGAT activity with the newly developed FRET-based assay in situ, we used, as a test case, S. elongatus cells grown in presence of nitrate or ammonia and cells that had been shifted to medium without nitrogen sources (Fig. 4). Samples were taken at three different phases of growth.
Figure 4.
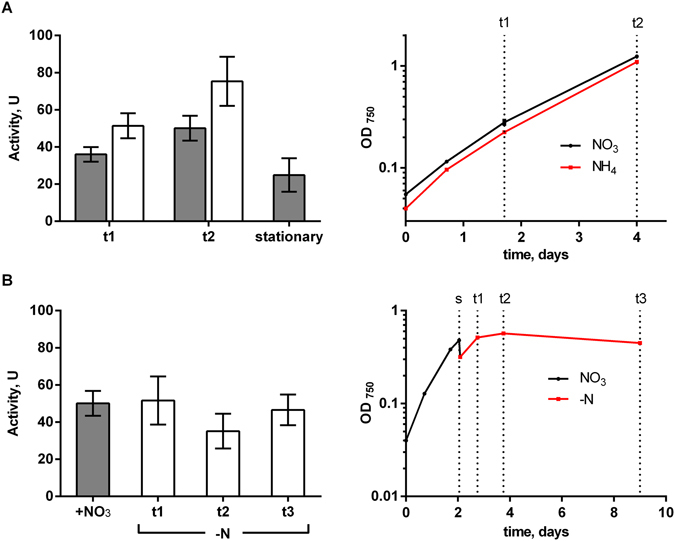
In situ fdGOGAT activity with different nitrogen sources. (A) In situ fdGOGAT activity of S. elongatus cultivated with nitrate (grey bars) or ammonium (white bars). Corresponding growth curves are shown on the right. Stationary cells were cultivated for 9 days to an OD750 of 5.5 to 6.0. (B) fdGOGAT activity of S. elongatus cultivated with nitrate and after shift (s) into nitrogen free medium (t1-3) with corresponding growth curve on the right. Error bars: ±s.e.m., n = 4.
With nitrate as nitrogen source, the in situ fdGOGAT activity increased by approximately 40% from the early exponential (t1) to the late exponential growth phase (t2, Fig. 4A). After the cells entered the stationary phase, fdGOGAT activity decreased to approx. 70% of the initial value, probably in response to the reduced growth rate and declining metabolic activity in the stationary phase. Under ammonium supplemented growth conditions, the fdGOGAT activity was generally 40–50% higher as compared to nitrate conditions (Fig. 4A). Like in the nitrate-supplemented medium, a 47% increase in activity occurred from early exponential to late exponential growth phase.
The higher in situ fdGOGAT activity under ammonium-supplemented growth shows that fdGOGAT is regulated differently to GS. In cyanobacteria and many other prokaryotes, GS is subjected to tight regulation36. In Synechocystis sp. PCC 6803, GS activity is tuned down by addition of ammonia, both at the transcriptional level via the NtcA regulatory system47 as well as post-translationally by interaction with two inactivation factors IF7 and IF1736. Inhibition is reversed by ammonia depletion within 10–20 min. The ammonium-triggered short-term GS inactivation is essential for maintenance of glutamate homeostasis48. When ammonium is in abundance, although GS activity is decreased, more glutamate can be converted to glutamine. Under these conditions, a higher fdGOGAT activity could keep the glutamine/glutamate balance in equilibrium.
Under nitrogen starvation conditions (Fig. 4B), the fdGOGAT activity remains at a constant level. This level is kept constant even after 8 days of starvation, when the cells became chlorotic. This basal activity could ensure sufficiently high nitrogen assimilation capacity via the GS-GOGAT cycle, enabling the chlorotic cells to immediately assimilate any ammonia that becomes available in the environment.
Determination of 2-OG Concentrations in Cell Extracts
Another possibility to use our sensor is to estimate the concentrations of 2-OG in cell-extracts. Metabolite concentrations in living cells are prone to fluctuations and can change very quickly during cell treatments, such as cell harvesting. To extract physiologically relevant quantities of metabolites form cells, the quenching of cellular activity before metabolite extraction is required2. There are several established metabolic inactivation methods. The use of −40 °C cooled 70% methanol rapidly freezes the cells, but also leads to metabolite loss49. Fast centrifugation and subsequent freezing works for heterotrophically growing cells, but is problematic for autotrophically growing cyanobacteria, which quickly adapt their metabolism to darkness. Fast filtration with illumination can solve this problem50. After quenching of enzymatic activities, the metabolites are immediately extracted from the cells. There are several established methods, but not all of them are suitable for 2-OG4: classical methods use mixtures of chloroform, methanol and buffer with 53% 2-OG recovery51, boiling ethanol with 58% recovery52 or pure −40 °C methanol with 82% recovery4. These protocols all include lyophilization of the extracts.
Our first approach was to rapidly filter the cells and immediately freeze the filters in liquid nitrogen. For this purpose, we used 2.5 cm diameter polyether sulfone (PES) membrane filters with a 0.45 µm pore size. The filters were immediately frozen in liquid nitrogen and afterwards briefly grounded. 2-OG was extracted with pure −40 °C methanol or with a mixture of methanol, chloroform and buffer. However, the organic solvents extracted unidentified compounds which interfered with the fluorescence measurements, showing high background fluorescence of more than 50% of the signal. Therefore, we used toluene to permeabilize the cells, after filtration and freezing as above, and measured 2-OG directly in the supernatant using PII-TC3. The cells were vortexed with 1400 rpm for 8 min at 25 °C in the presence of 1% toluene and 4% EtOH (v/v), similar to the protocol described above46. However, the standard deviations were still high. Therefore, we tried an even milder approach without freezing, by sampling the cultures into ice-cooled medium and thus immediately cooling the cells to 0 °C. The cells were then harvested by centrifugation and thereafter quickly permeabilized by toluene/EtOH, yielding reproducible results. The calculated intracellular 2-OG concentrations are shown in Fig. 5. In parallel to the FRET measurements, we determined the 2-OG concentrations by gas chromatography - mass spectrometry (GC-MS), revealing in general 20–30% lower values (for technical detail see Supplementary Information). This is likely due to losses during lyophilization and sample preparation for the GC-MS analyses. The measured intracellular 2-OG concentrations for E. coli cells are around 100 µM, using M9 medium with 19 mM NH4Cl (Fig. 5). These concentrations are in the same range as reported earlier for E. coli growing with high nitrogen supply10, 11. Despite this progress, quantification of cellular 2-OG levels from cell extracts remains problematic due to uncertainties in the extraction method13. The ultimate goal is therefore in vivo detection of cellular 2-OG levels in single cells. For this purpose, the TC3-R9L variant seemed promising and in the following, we applied this sensor to human cell lines, for which microscopic detection of metabolite FRET sensors is documented53.
Figure 5.
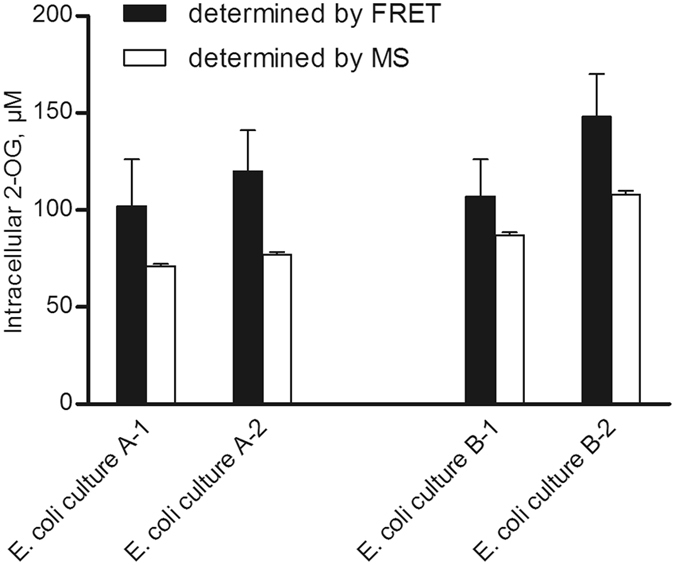
Intracellular 2-OG concentrations of E. coli 20 mL of exponentially growing E. coli culture were rapidly cooled to 0 °C and harvested by centrifugation. Two E. coli cultures (A,B) were used and two samples were taken per culture (1, 2). 2-OG was extracted and intracellular concentrations determined by FRET-sensor (grey; error bars: ±s.e.m. with n = 4) or GC-MS measurements (white; error bar denotes the standard error of the 2-OG calibration curve).
Ex vivo Measurements of 2-OG Fluctuations in Human Cell Lines
As a proof of concept, we tested the TC3-R9P sensor (with a dynamic range between 10 µM – 10 mM) in the human glioblastoma cell line U87-MG. Deregulation of 2-OG metabolism in glioblastoma and other gliomas previously has been shown to be responsible for the aberrant epigenetic gene regulation that characterizes many of these tumors16. While healthy brain cells show 2-OG concentrations in the range of 1–3 mM, brain cancer cells contain drastically reduced concentrations ranging from 100–300 µM16, 54. Accordingly, we transiently transfected these cells with plasmids expressing TC3-R9P under the control of the cytomegalovirus (CMV) promoter. We measured the FRET change after adding either dm2-OG, citrate or glutamine in different concentrations to the cells. The cell-permeable compound dm2-OG is converted into 2-OG inside the cells55. As shown in Fig. 6, the FRET dropped within minutes after addition of the compounds, in a concentration-dependent manner. For example, with 20 mM dm2-OG, a 10% drop in FRET was observed (Fig. 6A; Supplementary Video 1 and 2 ). Theoretically, this drop could be explained by the direct effect of dm2-OG on the sensor; however, since TC3-R9P has an almost two orders of magnitude lower affinity for dm2-OG than 2-OG (Fig. 2D ), and dm2-OG furthermore is rapidly demethylated by esterases in the cell, this FRET change likely represents the accumulation of 2-OG in the cells.
Figure 6.
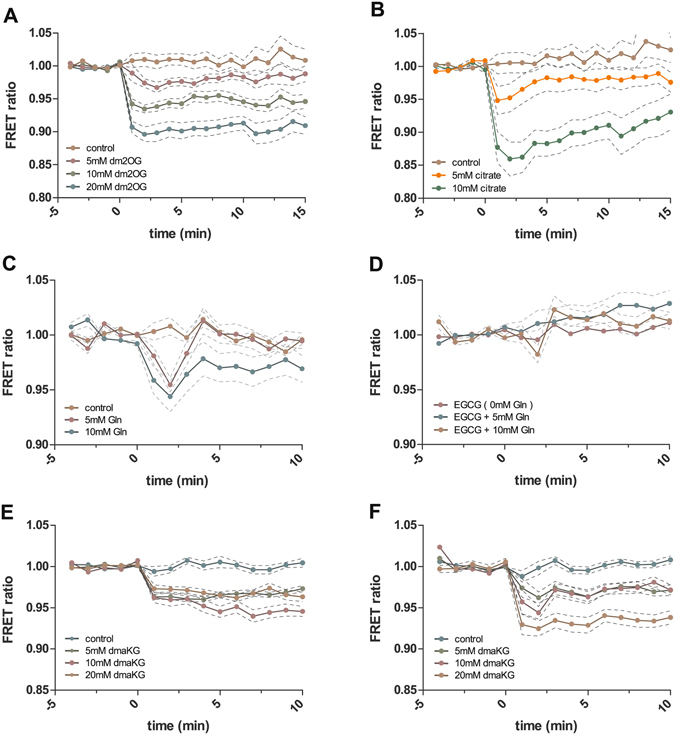
Effect of TCA-cycle metabolites on the FRET signal of TC3-R9P in cultured human cells. (A–D) Various concentrations of the compounds were added to the cell culture media of U87-MG glioblastoma cancer cells at time point zero. 10 to 50 cells were evaluated in each experiment. Average values ± s.e.m. are shown. (A) 2-OG analogue dm2-OG; (B) Citrate; (C) Glutamine; (D) Glutamine following pre-incubation with 100 µM of the glutaminolysis inhibitor epigallocatechin gallate (EGCG). (E) Embryonic kidney cells (HEK293T) treated with dm2-OG. (F) Retinal pigment epithelium cells (RPE1) treated with dm2-OG.
Even more pronounced than dm2-OG, addition of citrate rapidly induced a 15% FRET drop, which is close to the maximal observable FRET change (Fig. 6B ). As the sensor is 3600 times less sensitive for citrate than for 2-OG, the strong drop in FRET can only be generated by the fast conversion of citrate to 2-OG by the TCA cycle. To estimate the absolute changes in 2-OG, we harvested cells 5 minutes after addition of citrate and determined 2-OG amounts by ultra-performance liquid chromatography. Relative to control cells which contained 56.12 ± 4.08 pmol/105 cells (mean ± s.e.m.), addition of 5 mM and 10 mM citrate increased the 2-OG contents to 59.84 ± 1.95 (7%) and 67.77 ± 1.28 pmol/105 cells (21%), respectively. In contrast to dm2-OG addition, the response towards citrate is more dynamic: addition of 10 mM citrate results in a rapid but transient FRET response. This indicates that citrate is actively taken up and rapidly metabolized to 2-OG leading to a peak in intracellular 2-OG levels. Subsequently, the 2-OG concentrations inside the cells return back to equilibrium, as revealed by the sensor. By contrast, in the case of dm2-OG, the FRET signal remains at a decreased steady state level over the measuring time. This indicates that the diffusion of dm2-OG into the cells and subsequent demethylation to 2-OG leads to increased 2-OG levels, which remain constant, since passive dm2-OG diffusion maintains the novel steady state level of 2-OG.
The growth of glioblastoma and many other cancer cell lines is dependent on the uptake of glutamine, which is converted to 2-OG by glutaminolysis. This process involves the enzyme glutamate dehydrogenase 1 (GLUD1), a potential target for anti-cancer therapy, whose activity can be suppressed by the small molecule inhibitor epigallocatechin gallate (EGCG)56. To test whether the TC3-R9P sensor could detect the intracellular increase of 2-OG level due to increased glutamine uptake and conversion to 2-OG by glutaminolysis, we added 5 mM or 10 mM glutamine to U87 MG cells that were cultured in low-glutamine conditions (0.5 mM instead of 2 mM). Immediately following glutamine supplementation we observed a concentration-dependent drop of the FRET ratios suggesting elevated uptake of glutamine by the cells and its conversion to 2-OG (Fig. 6C ). After an initial transient over-accumulation, 2-OG levels returned to a new steady state within a few minutes. When glutaminolysis was blocked by pre-incubation with 100 µM EGCG for three hours, no change in FRET was observed (Fig. 6D) indicating that the additional intracellular 2-OG that accumulated in the inhibitor-free condition (Fig. 6C) indeed was derived from glutamine. These data show that our PII-protein-derived FRET sensors potentially could be used to detect phenotype-relevant changes of 2-OG concentrations in cancer cells, making them useful tools to monitor readout during metabolic drug screening. To test whether the 2-OG sensor also could be used in other types of cells, we measured FRET changes in cell lines that have been derived from embryonic (human embryonic kidney; HEK293T) and differentiated (retinal pigment epithelium; RPE1) tissues. In both cell lines FRET ratios decreased following the addition of dm2-OG (Fig. 6E and F ) suggesting that the TC3-R9P sensor can be used to analyze 2-OG changes in various types of cells.
In summary, with these various applications we could demonstrate the functionality of our FRET based 2-OG sensor for different applications. The TC3 sensor performed very well for in situ GOGAT activity determination. For determining cellular 2-OG concentrations, the final goal was to create a sensor that sensitively reports fluctuating 2-OG concentration in living cells. For this purpose, the R9P variant of the TC3 sensor seems to be a very good starting point. This sensor variant allowed real-time live-cell imaging of 2-OG in cancer, embryonic and differentiated cells, and potentially could also be used as a readout assay in high-throughput drug screening.
For even more sensitive in vivo experiments, the maximal signal change of 25% upon 2-OG addition should be further improved by protein engineering of the FRET sensor, to achieve a more robust signal. As seen in the sensor development, small alterations can have a drastic, unpredictable influence on the FRET output. Accordingly, we suggest a directed evolutionary approach to further improve the FRET sensor. It has been shown before, that a random mutagenesis approach with a well-designed selection mechanism can strongly enhance the capabilities of a FRET sensor57, 58. In view of the emerging importance of 2-OG as effector molecule for central cellular processes, monitoring in vivo fluctuations of 2-OG will lead to a deeper understanding on the role of the metabolism on cell functions.
Material and Method
Cultivation of bacterial and human cell lines
Cyanobacterial strains were cultivated photoautotrophically at a constant illumination of 50 µmol photons m−2 s−1 in BG-11 medium59 supplemented with 5 mM NaHCO3 at 28 °C in flasks shaking at 120 rpm. As nitrogen source, either 17.65 mM NaNO3 or 5 mM NH4Cl, with 5 mM TES/NaOH; pH 8.2 (Roth), was added.
Escherichia coli K-12 substrain MG1655 cells were cultivated in M9 minimal medium containing: 48 mM Na2HPO4, 22 mM KH2PO4, 8.5 mM NaCl, 19 mM NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, 0.4% (w/v) glucose, 0.0001% (w/v) thiamin. Cultures of 100–200 mL volume were grown in 1 L wide-neck flasks at 180 rpm, using silicone sponge closures to ensure aerobic growth. Optical density was measured at 600 nm.
Human glioma cell line U87-MG was cultured at 37 °C, 90% air/10% CO2 in Dulbecco’s Modified Eagle’s medium (DMEM Sigma D5921) without phenol-red, 1000 mg/L glucose, supplemented with 10% FCS (Biochrom #S0115), 2 mM glutamine and 1% penicillin/streptomycin mix. HEK293T cells were cultured in RPMI 1640 Medium, 4500 mg/L glucose, supplemented with 10% FCS, 2 mM glutamine and 1% penicillin/streptomycin mix. hTERT-RPE1 cells were cultured in DMEM/F12 Medium, 4500 mg/L glucose, supplemented with 10% FCS and additional 1% glutamine. All cell lines were transfected with 0.8 µg/mL plasmid DNA using TransIt-LT1 reagent (Mirus). Dimethyl-2-oxoglutarate (dm-2OG, Sigma), citric acid (AppliChem) and glutamine (Gibco 25030) were diluted in DMEM buffered with HEPES solution. For GDH inhibition, cells were pre-incubated 3 h with 100 µM (−)-Epigallocatechin gallate (Sigma E4143).
Cloning and Protein purification
Plasmids were constructed by PCR amplification of PII and FP genes with primers containing overlapping regions. The PCR products were fused with an XbaI and HindIII double digested pASK-IBA3 vector (IBA GmbH, Göttingen, Germany) via isothermal, single-reaction DNA assembly following the protocol by Gibson et al.60. The sensor genes were amplified by standard PCR and cloned into the mammalian expression vector pcDNA3.1 using the BamHI and HindIII restriction sites. The complete sequences of the sensor genes are provided in the Supplementary Information.
PII protein variants were overexpressed in E. coli RB9060 and purified as described earlier using Strep-tag affinity chromatography28. To remove contaminating proteins and degradation products the sensor proteins were further purified by size exclusion chromatography, using an Äkta purifier and a HiLoad 26/600 Superdex 200 prep grade column (GE Healthcare GmbH, Solingen, Germany). The running buffer contained 30 mM Tris (pH 7.5) and 175 mM NaCl. The proteins were stored in a buffer containing 50 mM Tris-HCl pH 7.8, 100 mM KCl, 5 mM MgCl2, 0.5 mM EDTA, 1 mM DTT and 50% glycerol (v/v) at −20 °C.
In situ fdGOGAT Assay
In situ Fd-GOGAT activity was determined according to Marqués et al.44 and Kameya et al.45, with some modifications. For assaying the in situ Fd-GOGAT activity in S. elongatus, 4*108 cells were collected and washed twice with PBS buffer containing 2.7 mM KCl, 1.5 mM KH2PO4, 137 mM NaCl, 8.1 mM Na2HPO4, pH 7.5 at 4 °C. The cell pellet was resuspended in 200 µL reaction buffer containing 20 mM Na3PO4 pH 7.2, 10 mM glutamine, 1 mM 2-OG and 5 mM methyl viologen. Cells were permeabilized by addition of 4.4 µL toluene and constant vortexing at 1400 rpm for 5 min at 32 °C. To insulate the reaction from atmospheric oxygen, the reaction mixture was covered by 500 µL paraffin oil. The reaction was started by addition of 5 mM sodium dithionite, gently mixing and incubating at 32 °C. Reaction aliquots (30 µL) were removed in regular intervals and the reaction was stopped by intensive shaking until the blue color disappeared, indicating oxidation of the electron donor methyl viologen. Aliquots were then centrifuged at 15,000 × g for 10 min at 4 °C and the 2-OG concentration in the supernatant was determined as described below. One unit of activity was defined as µmol 2-OG consumption per min.
Cell Extraction for FRET Measurements
E. coli cultures (20 ml aliquots) were harvested at an optical density (OD600) of 0.5 by pipetting directly out of the shaking culture and mixing with 30 mL of the corresponding culture medium and 30 g ice for rapid quenching of the metabolism. Cells were pelleted by centrifugation at 15,000 × g for 8 min at 0 °C. The cell pellet was resuspended on ice with 94 µL 20% (v/v) ethanol. The cell suspension was transferred into a 2 mL sample tube and 406 µL MilliQ water was added to a final volume of 500 µL with a final ethanol concentration of 3.75% (v/v), as well as 6.25 µL toluene for membrane permeabilization, as described by Gomez Casati et al.46. The mixture was incubated for 8 min at 25 °C under shaking at 1,400 rpm and subsequently quickly cooled on ice and centrifuged at 14,000 × g for 5 min at 0 °C. The clear supernatant was transferred into a fresh tube and centrifuged as before. The supernatant was either directly used for FRET measurements or stored at −20 °C.
Ultra Performance Liquid Chromatography (UPLC) analysis
For determination of intracellular 2-OG content in human glioblastoma cell line, 1 × 105 cells were extracted with 200 μl cold 1 M perchloric acid. Insoluble material was removed by centrifugation for 10 min at 25.000 × g. For derivatization with DMB (1,2-diamino-4,5-methylendioxybenzene), 30 μl extract were mixed with 30 μl DMB derivatization reagent (5 mM DMB, 20 mM sodium hydrosulfite, 1 M 2-mercaptoethanol, 1.2 M HCl) and incubated at 100 °C for 45 min. After 10 min centrifugation, the reaction was diluted with 240 μl 10% acetonitrile. The derivatized ketoacids were separated by reversed phase chromatography on an Acquity HSS T3 column (100 mm × 2.1 mm, 1.7 μm, Waters) connected to an Acquity H-class UPLC system. Prior separation, the column was heated to 40 °C and equilibrated with 5 column volumes of solvent A (0.1% formic acid in 10% acetonitrile) at a flow rate of 0.55 ml/min. Baseline separation of DMB derivates was achieved by increasing the concentration of acetonitrile (B) in buffer A as follows: 2 min 2% B, 4.5 min 15% B, 10.5 min 38% B, 10.6 min 90% B, hold for 2 min, and return to 2% B in 3.5 min. The separated derivates were detected by fluorescence (Acquity FLR detector, Waters; excitation: 367 nm; emission: 446 nm) and quantified using ultrapure standards (Sigma). Data acquisition and processing was performed with the Empower3 software suite (Waters). 2-OG data were globally normalized relative to the mean of all analyzed derivatized metabolites.
In vitro FRET Measurements
FRET measurements were conducted in a buffer containing 50 mM imidazole (pH 7.5), 50 mM KCl, 20 mM MgCl2, 15% glycerol (v/v), 0.5 mM DTT and 5 mM ATP. The sensor protein was carefully added to a final concentration of 100 nM (monomer). For blank measurements the master mix was prepared by adding water instead of the sensor protein. Without mixing, the solution was incubated for 15 min on ice to allow the protein to adapt to the buffer. After that, the solution was mixed by inverting the tube several times and 70 µL (extract measurement) or 90 µL (GOGAT measurement) aliquots were dispensed onto a black non-binding 96-well plate (Greiner Bio-One, Frickenhausen, Germany). 30 µL of the cell extracts or 10 µL of the GOGAT assay supernatant and a 2-OG standard solution were added to the plate. The plate was sealed using an adhesive foil to prevent evaporation. Subsequently the plate was placed in a Tecan Spark 10 M microplate reader (Tecan, Männedorf, Switzerland) at 37 °C. The plate was shaken for 80 s and incubated for 20 min. After the incubation, the plate was again shaken for 20 s, the foil was removed and the FRET signal was measured. Each well was measured in three channels (each 20 nm bandwidth): Venus, excitation: 485 nm, emission: 530 nm; Cerulean: excitation: 435 nm, emission: 480 nm; FRET: excitation: 435 nm, emission: 530 nm. Blank values were subtracted from the measurements in each channel. Following the NFRET formula developed by Xia and Liu61 corrected FRET values were calculated. The correction factors a and b were determined in separate measurements containing only Venus or Cerulean. Each plate contained a standard curve of known 2-OG concentrations and the samples. Using GaphPad Prism 6 (GraphPad Software, San Diego, USA) the unknown concentrations were interpolated from the standard curve. For the calculation of intracellular 2-OG concentrations the intracellular volume of glucose grown E. coli MG1655 was assumed to be 3.3 µL per mL of OD600 = 1 culture, as determined by Volkmer and Heinemann62.
Life-Cell Imaging
Fluorescence measurements were performed in a Leica TCS-SP5 laser scanning confocal microscope equipped with resonant scanners and hybrid photon detectors (HyD) and using a 40x objective (HCX PL APO, NA 1.25, oil-immersion). Cells were excited with a UV diode (405 nm) and emission was detected at 450–490 nm (CFP) and 520–590 nm (FRET). Pictures were acquired every 60 sec. After a baseline of 5 min, media containing metabolites or not were added and changes in fluorescence were recorded for 15 minutes. During imaging, cells were maintained at 37 °C and 5% CO2. Image processing was performed with ImageJ (http://fiji.sc/). After background subtraction and registration, single cells were segmented by threshold and mean intensity was measured for each channel at every time point. The ratio was calculated by dividing the mean intensity of the acceptor (FRET) by the intensity of the donor (CFP). Ratios calculated from every cell were normalized to baseline measurements.
Electronic supplementary material
Acknowledgements
This work was funded by a grant from the Deutsche Forschungsgemeinschaft (Fo195/9-2) and in part by a grant to B.R., provided by the Heidelberg CellNetworks cluster within the framework of the German Excellence Initiative. We’d also like to give special thanks to the National Council for Science and Technology (CONACYT-Mexico) for the financial support provided to Liliana François (CVU 468797). We would like to thank Dr. Joachim Kilian for kind help in GC-MS analysis and to acknowledge the support of the German Cancer Research Center (DKFZ) Light Microscopy Facility.
Author Contributions
J.L. and T.C. developed the FRET sensor. J.L. and P.S. characterized the sensor variants and performed in vitro assays in bacteria. B.W. and J.L. performed in situ fdGOGAT assays. L.F. transfected human cell lines and performed live-cell imaging assays. G.P and R.H performed the UPLC analysis, B.R. and K.F supervised the experiments. J.L, L.F., P.S. and K.F. prepared the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Jan Lüddecke and Liliana Francois contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01440-w
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aretz, I. & Meierhofer, D. Advantages and Pitfalls of Mass Spectrometry Based Metabolome Profiling in Systems Biology. Int J Mol Sci17, doi:10.3390/ijms17050632 (2016). [DOI] [PMC free article] [PubMed]
- 2.Schwarz D, Orf I, Kopka J, Hagemann M. Recent applications of metabolomics toward cyanobacteria. Metabolites. 2013;3:72–100. doi: 10.3390/metabo3010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bingol K, et al. Metabolomics beyond spectroscopic databases: a combined MS/NMR strategy for the rapid identification of new metabolites in complex mixtures. Anal Chem. 2015;87:3864–3870. doi: 10.1021/ac504633z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villas-Boas SG, Hojer-Pedersen J, Akesson M, Smedsgaard J, Nielsen J. Global metabolite analysis of yeast: evaluation of sample preparation methods. Yeast. 2005;22:1155–1169. doi: 10.1002/yea.1308. [DOI] [PubMed] [Google Scholar]
- 5.Jones AM, et al. In vivo biochemistry: applications for small molecule biosensors in plant biology. Curr Opin Plant Biol. 2013;16:389–395. doi: 10.1016/j.pbi.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman RH, Fosbrink MD, Zhang J. Genetically encodable fluorescent biosensors for tracking signaling dynamics in living cells. Chemical reviews. 2011;111:3614–3666. doi: 10.1021/cr100002u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hochreiter B, Garcia AP, Schmid JA. Fluorescent proteins as genetically encoded FRET biosensors in life sciences. Sensors (Basel) 2015;15:26281–26314. doi: 10.3390/s151026281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaper T, Lager I, Looger LL, Chermak D, Frommer WB. Fluorescence resonance energy transfer sensors for quantitative monitoring of pentose and disaccharide accumulation in bacteria. Biotechnol Biofuels. 2008;1:11. doi: 10.1186/1754-6834-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huergo LF, Dixon R. The Emergence of 2-Oxoglutarate as a Master Regulator Metabolite. Microbiol Mol Biol Rev. 2015;79:419–435. doi: 10.1128/MMBR.00038-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan J, et al. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol Syst Biol. 2009;5:302. doi: 10.1038/msb.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radchenko MV, Thornton J, Merrick M. Control of AmtB-GlnK complex formation by intracellular levels of ATP, ADP, and 2-oxoglutarate. J Biol Chem. 2010;285:31037–31045. doi: 10.1074/jbc.M110.153908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodsworth JA, Cady NC, Leigh JA. 2-Oxoglutarate and the PII homologues NifI1 and NifI2 regulate nitrogenase activity in cell extracts of Methanococcus maripaludis. Mol Microbiol. 2005;56:1527–1538. doi: 10.1111/j.1365-2958.2005.04621.x. [DOI] [PubMed] [Google Scholar]
- 13.Yan D, Lenz P, Hwa T. Overcoming Fluctuation and Leakage Problems in the Quantification of Intracellular 2-Oxoglutarate Levels in Escherichia coli. Applied and Environmental Microbiology. 2011;77:6763–6771. doi: 10.1128/AEM.05257-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brauer MJ, et al. Conservation of the metabolomic response to starvation across two divergent microbes. Proc Natl Acad Sci USA. 2006;103:19302–19307. doi: 10.1073/pnas.0609508103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chin RM, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;510:397–401. doi: 10.1038/nature13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gross S, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cascella B, Mirica LM. Kinetic analysis of iron-dependent histone demethylases: alpha-ketoglutarate substrate inhibition and potential relevance to the regulation of histone demethylation in cancer cells. Biochemistry. 2012;51:8699–8701. doi: 10.1021/bi3012466. [DOI] [PubMed] [Google Scholar]
- 19.TeSlaa T, et al. α-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metabolism. 2016;24:485–493. doi: 10.1016/j.cmet.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang, I.-Y. et al. Psat1-Dependent Fluctuations in α-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metabolism24, 494–501, 10.1016/j.cmet.2016.06.014. [DOI] [PubMed]
- 22.Olenchock BA, et al. EGLN1 Inhibition and Rerouting of alpha-Ketoglutarate Suffice for Remote Ischemic Protection. Cell. 2016;165:497. doi: 10.1016/j.cell.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 23.Doucette CD, Schwab DJ, Wingreen NS, Rabinowitz J. D. alpha-Ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nat Chem Biol. 2011;7:894–901. doi: 10.1038/nchembio.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venditti V, Tugarinov V, Schwieters CD, Grishaev A, Clore GM. Large interdomain rearrangement triggered by suppression of micro- to millisecond dynamics in bacterial Enzyme I. Nat Commun. 2015;6:5960. doi: 10.1038/ncomms6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorke B, Stulke J. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol. 2008;6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 26.Park YH, Lee BR, Seok YJ, Peterkofsky A. In vitro reconstitution of catabolite repression in Escherichia coli. J Biol Chem. 2006;281:6448–6454. doi: 10.1074/jbc.M512672200. [DOI] [PubMed] [Google Scholar]
- 27.Huergo LF, Chandra G, Merrick M. PII signal transduction proteins: nitrogen regulation and beyond. FEMS Microbiology Reviews. 2013;37:251. doi: 10.1111/j.1574-6976.2012.00351.x. [DOI] [PubMed] [Google Scholar]
- 28.Fokina O, Chellamuthu V-R, Forchhammer K, Zeth K. Mechanism of 2-oxoglutarate signaling by the Synechococcus elongatus P(II) signal transduction protein. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19760–19765. doi: 10.1073/pnas.1007653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Truan D, et al. A New PII Protein Structure Identifies the 2-Oxoglutarate Binding Site. Journal of Molecular Biology. 2010;400:531–539. doi: 10.1016/j.jmb.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 30.Forchhammer K, Lüddecke J. Sensory properties of the PII signalling protein family. FEBS Journal. 2016;283:425–437. doi: 10.1111/febs.13584. [DOI] [PubMed] [Google Scholar]
- 31.Forchhammer KP., II Signal transducers: novel functional and structural insights. Trends in microbiology. 2008;16:65–72. doi: 10.1016/j.tim.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Lüddecke J, Forchhammer K. From PII Signaling to Metabolite Sensing: A Novel 2-Oxoglutarate Sensor That Details PII - NAGK Complex Formation. PLoS ONE. 2013;8:e83181. doi: 10.1371/journal.pone.0083181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen HL, Bernard CS, Hubert P, My L, Zhang CC. Fluorescence resonance energy transfer based on interaction of PII and PipX proteins provides a robust and specific biosensor for 2-oxoglutarate, a central metabolite and a signalling molecule. FEBS J. 2014;281:1241–1255. doi: 10.1111/febs.12702. [DOI] [PubMed] [Google Scholar]
- 34.Lüddecke J, Forchhammer K. Energy Sensing versus 2-Oxoglutarate Dependent ATPase Switch in the Control of Synechococcus PII Interaction with Its Targets NAGK and PipX. PLoS ONE. 2015;10:e0137114. doi: 10.1371/journal.pone.0137114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berg J, Hung YP, Yellen G. A genetically encoded fluorescent reporter of ATP:ADP ratio. Nat Methods. 2009;6:161–166. doi: 10.1038/nmeth.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muro-Pastor MI, Reyes JC, Florencio FJ. Ammonium assimilation in cyanobacteria. Photosynth Res. 2005;83:135–150. doi: 10.1007/s11120-004-2082-7. [DOI] [PubMed] [Google Scholar]
- 37.Luque, I. & Forchhammer, K. In The Cyanobacteria: Molecular Biology, Genomics and Evolution (eds A. Herrero & E. Flores) Ch. 13, (Caister Academic Press, 2008).
- 38.Ermilova E, et al. PII signal transduction protein in Chlamydomonas reinhardtii: localization and expression pattern. Protist. 2013;164:49–59. doi: 10.1016/j.protis.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Engineering. 2001;14:529–532. doi: 10.1093/protein/14.8.529. [DOI] [PubMed] [Google Scholar]
- 40.Muller SM, Galliardt H, Schneider J, Barisas BG, Seidel T. Quantification of Forster resonance energy transfer by monitoring sensitized emission in living plant cells. Front Plant Sci. 2013;4:413. doi: 10.3389/fpls.2013.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Topell S, Hennecke J, Glockshuber R. Circularly permuted variants of the green fluorescent protein. FEBS Lett. 1999;457:283–289. doi: 10.1016/S0014-5793(99)01044-3. [DOI] [PubMed] [Google Scholar]
- 42.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 43.Goedhart J, et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93% Nature Communications. 2012;3:751. doi: 10.1038/ncomms1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marqués S, Florencio FJ, Candau P. Ammonia assimilating enzymes from cyanobacteria: in situ and in vitro assay using high-performance liquid chromatography. Anal Biochem. 1989;180:152–157. doi: 10.1016/0003-2697(89)90104-8. [DOI] [PubMed] [Google Scholar]
- 45.Kameya M, et al. A novel ferredoxin-dependent glutamate synthase from the hydrogen-oxidizing chemoautotrophic bacterium Hydrogenobacter thermophilus TK-6. J Bacteriol. 2007;189:2805–2812. doi: 10.1128/JB.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomez Casati DF, Aon MA, Cortassa S, Iglesias AA. Measurement of the glycogen synthetic pathway in permeabilized cells of cyanobacteria. FEMS Microbiol Lett. 2001;194:7–11. doi: 10.1016/S0378-1097(00)00484-5. [DOI] [PubMed] [Google Scholar]
- 47.Luque I, Vazquez-Bermudez MF, Paz-Yepes J, Flores E, Herrero A. In vivo activity of the nitrogen control transcription factor NtcA is subjected to metabolic regulation in Synechococcus sp. strain PCC 7942. FEMS Microbiol Lett. 2004;236:47–52. doi: 10.1111/j.1574-6968.2004.tb09625.x. [DOI] [PubMed] [Google Scholar]
- 48.Kustu S, Hirschman J, Burton D, Jelesko J, Meeks JC. Covalent modification of bacterial glutamine synthetase: physiological significance. Mol Gen Genet. 1984;197:309–317. doi: 10.1007/BF00330979. [DOI] [PubMed] [Google Scholar]
- 49.Young JD, Shastri AA, Stephanopoulos G, Morgan JA. Mapping photoautotrophic metabolism with isotopically nonstationary (13)C flux analysis. Metab Eng. 2011;13:656–665. doi: 10.1016/j.ymben.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krall L, Huege J, Catchpole G, Steinhauser D, Willmitzer L. Assessment of sampling strategies for gas chromatography-mass spectrometry (GC-MS) based metabolomics of cyanobacteria. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:2952–2960. doi: 10.1016/j.jchromb.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 51.de Koning W, van Dam K. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal Biochem. 1992;204:118–123. doi: 10.1016/0003-2697(92)90149-2. [DOI] [PubMed] [Google Scholar]
- 52.Gonzalez B, Francois J, Renaud M. A rapid and reliable method for metabolite extraction in yeast using boiling buffered ethanol. Yeast. 1997;13:1347–1355. doi: 10.1002/(SICI)1097-0061(199711)13:14<1347::AID-YEA176>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 53.San Martín A, et al. A Genetically Encoded FRET Lactate Sensor and Its Use To Detect the Warburg Effect in Single Cancer Cells. PLoS ONE. 2013;8:e57712. doi: 10.1371/journal.pone.0057712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thirstrup K, et al. Endogenous 2-oxoglutarate levels impact potencies of competitive HIF prolyl hydroxylase inhibitors. Pharmacol Res. 2011;64:268–273. doi: 10.1016/j.phrs.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 55.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lecumberri E, Dupertuis YM, Miralbell R, Pichard C. Green tea polyphenol epigallocatechin-3-gallate (EGCG) as adjuvant in cancer therapy. Clinical Nutrition. 2013;32:894–903. doi: 10.1016/j.clnu.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 57.Litzlbauer J, et al. Large Scale Bacterial Colony Screening of Diversified FRET Biosensors. PLoS ONE. 2015;10:e0119860. doi: 10.1371/journal.pone.0119860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thestrup T, et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat Methods. 2014;11:175–182. doi: 10.1038/nmeth.2773. [DOI] [PubMed] [Google Scholar]
- 59.Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RY. Generic Assignments, Strain Histories and Properties of Pure Cultures of Cyanobacteria. J Gen Microbiol. 1979;111:1–61. [Google Scholar]
- 60.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 61.Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophysical journal. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Volkmer B, Heinemann M. Condition-dependent cell volume and concentration of Escherichia coli to facilitate data conversion for systems biology modeling. PLoS ONE. 2011;6:e23126. doi: 10.1371/journal.pone.0023126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.